

Epstein-Barr virus (EBV) is a causative agent of various malignancies and may also contribute to autoimmune disease. The latent and episomal form of the virus is known to drive EBV-associated oncogenesis. Persistence of the viral episome in proliferating tumor cells requires the interaction of Epstein-Barr virus nuclear antigen 1 (EBNA1) with the viral origin of plasmid replication (oriP). The dyad symmetry (DS) element in oriP is the essential minimal replicator of oriP. Here we report the X-ray crystal structure of EBNA1 bound to DS. The structure reveals a previous unrecognized interface formed between dimers of EBNA1 necessary for cooperative DNA binding, recruitment of cellular replication machinery, and replication function. These findings provide new insights into the mechanism of EBNA1 function at the replication origin and new opportunities to inhibit EBV latent infection and pathogenesis.
KEYWORDS: DNA replication, dyad symmetry, EBNA-1, EBNA1, EBV, Epstein-Barr virus, OriP, X-ray crystallography
ABSTRACT
Epstein-Barr virus is associated with several human malignancies, including nasopharyngeal carcinoma, gastric cancer, and lymphoma. Latently infected cells carry a circularized EBV episome where the origin of replication (oriP) is comprised of two elements: the family of repeats (FR) and dyad symmetry (DS). The viral protein Epstein-Barr virus (EBV) nuclear antigen 1 (EBNA1) binds to FR and DS to promote EBV episome maintenance and DNA replication during latent infection in proliferating cells. EBNA1 binding to the DS constitutes a minimal origin of DNA replication. Here we report the crystal structure of two EBNA1 DNA-binding domain dimers bound to a DS half-site. This structure shows that the DNA is smoothly bent, allowing for stabilizing interactions between the dimers. The dimer-dimer interface requires an intricate hydrogen bonding network involving residues R491 and D581. When this interface is disrupted, we note loss of stable dimer-dimer complex formation on the DNA, compromised oriP-containing plasmid replication in cells, and impaired recruitment of the MCM3 complex to the oriP. Surface conservation analysis reveals that these residues are part of a larger conserved surface that may be critical for recruitment of replication machinery to the oriP. Our results reveal a new region of EBNA1 critical for its activity and one that may be exploited by targeted small molecules to treat EBV-associated disease.
IMPORTANCE Epstein-Barr virus (EBV) is a causative agent of various malignancies and may also contribute to autoimmune disease. The latent and episomal form of the virus is known to drive EBV-associated oncogenesis. Persistence of the viral episome in proliferating tumor cells requires the interaction of Epstein-Barr virus nuclear antigen 1 (EBNA1) with the viral origin of plasmid replication (oriP). The dyad symmetry (DS) element in oriP is the essential minimal replicator of oriP. Here we report the X-ray crystal structure of EBNA1 bound to DS. The structure reveals a previous unrecognized interface formed between dimers of EBNA1 necessary for cooperative DNA binding, recruitment of cellular replication machinery, and replication function. These findings provide new insights into the mechanism of EBNA1 function at the replication origin and new opportunities to inhibit EBV latent infection and pathogenesis.
INTRODUCTION
Epstein-Barr virus (EBV) establishes a stable, latent infection in proliferating B lymphocytes and cancers of lymphoid and epithelial origin (1). During latent infection, the viral genome is maintained as a covalently closed, circular, multicopy DNA molecule referred to as the viral episome (2, 3). Episome maintenance requires both DNA replication and faithful segregation of replicated genomes (4, 5). The episomal origin of replication, termed oriP, is a 1.7-kb region that contains two functional elements: the family of repeats (FR) and dyad symmetry (DS) (5–7). The FR region is responsible for proper maintenance of the episomes during cell division, while the DS is essential for efficient plasmid replication (8, 9). The only EBV-encoded protein necessary for episome replication and maintenance is Epstein-Barr virus nuclear antigen 1 (EBNA1) (9).
The C terminus of EBNA1 (residues 459 to 607) is responsible for the sequence-specific DNA binding and dimerization of the protein (10–13). The EBNA1 DNA binding and dimerization domain (DBD) recognizes an 18-bp palindromic sequence that is found in several copies at both the FR and DS elements (11, 14–18). Crystal structures show the dimeric DBD to contain an eight-stranded antiparallel β barrel flanked by three α helices on each side (10, 19), which is structurally similar in fold to the DBD of human papillomavirus (HPV) protein E2 (10, 20) and Kaposi sarcoma-associated herpesvirus (KSHV) LANA (21). In addition to the α helices that interact with the major groove of DNA, the N terminus makes sequence-specific contacts with the minor groove, thereby clenching the DNA from both sides (12).
The DS element contains four EBNA1 dimer binding sites in a paired pattern (8) (Fig. 1A). Sites 1 and 2 and sites 3 and 4 are separated by 3 bp (8). In turn, 15 bp separate sites 1 and 2 from sites 3 and 4. Nonamer sites capable of binding telomere repeat binding factors 1 and 2 (TRF1 and TRF2) are found before site 1, after site 4 and between sites 2 and 3 (8, 22). An intact DS is sufficient for plasmid replication in EBNA1-expressing cells, but a DS half-site can still achieve DNA replication, albeit at lower levels (8, 9). EBNA1 has no intrinsic catalytic activity (16) and cannot initiate replication alone (22–24). It is thought to mediate its function in part by recruitment of cellular replication machinery (4).
FIG 1.

Structure of EBNA1 dimer-dimer on DS34. (A) Duplex DNA corresponding to the entire DS element. Sites 1 to 4 are labeled and shaded in blue. The 3-bp separations between sites 4 and 3 and sites 2 and 1 are colored in purple. Orange shading indicates TRF1 and TRF2 binding sites. (B) Duplex DNA used in crystallization of the complex. The same coloring as in panel A is used. Underlined bases in the TRF sites are those that had discernible electron density in the crystal structure. (C) Crystal structure of EBNA1-DNA complex. The DNA coloring is the same as in panel A. One EBNA1 dimer is colored in shades of green (chain A, darker; chain B, lighter); the other dimer is colored in shades of yellow (chain C, darker; chain D, lighter). (D) The two dimer-dimer/DNA complexes of the asymmetric unit. DimerDimer1 is the focus of the paper. DimerDimer2 is rotated 90° into the plane of the page relatively to DimerDimer1. The protein is green and in cartoon representation; DNA is in stick representation. PGP loops are indicated with arrows.
EBNA1 binding to the DS has four important characteristics. First, electrophoretic mobility shift assays (EMSA) show that EBNA1 binding incurs large, smooth bends in the DNA with predictions varying from 70 to 90° (4, 19). Second, the 3-bp spacing between sites 1 and 2 and sites 3 and 4 is crucial for replication and suggests a precise interaction between EBNA1 dimers on the DNA (4, 8, 25, 26). Third, EBNA1 dimers assemble on the DS in a cooperative manner (8, 27). EBNA1 has the highest affinity for sites 1 and 4, but the affinity for sites 2 and 3 is increased upon EBNA1 binding to sites 1 and 4 (4, 27). Cooperativity is dependent upon residues within EBNA1’s DBD (27). Finally, the DNA structure is predicted to change so as to accommodate the closely packed EBNA1 dimers (19).
To understand the molecular details of how EBNA1 binding to the DS initiates replication, we determined the X-ray crystal structure of an EBNA1 dimer-dimer bound to a DS half-site containing EBNA1 binding sites 4 and 3. The structure reveals an important hydrogen bonding network between EBNA1 dimers and provides a molecular rationale for the DNA bending and the strict requirement of the 3-bp separation between EBNA1 binding sites. As predicted from our new structure, site-directed mutagenesis of two key residues in the dimer-dimer interface (R491 and D581) resulted in the loss of cooperative binding to paired DS sites, loss of DNA replication activity, and impaired recruitment of DNA replication machinery in cell-based assays. These findings indicate that EBNA1 dimer-dimer interactions are critical for the formation of a functional origin of DNA replication at the DS element of oriP.
RESULTS
Structure of an EBNA1 dimer-dimer bound to dyad symmetry half-site.
The structure of a dimer-dimer EBNA1/DNA complex was solved by molecular replacement with a resolution of 3.01 Å (Fig. 1 and Table 1). The complex crystallized in space group P2 with two dimer-dimer EBNA1/DNA complexes per asymmetric unit (Fig. 1C). The two complexes are nearly identical, with a root mean square deviation (RMSD) of 0.261 Å2 (Table 1); the predominant difference involves the DNA ends. DNA corresponding to sites 3 and 4 of the DS element (referred to as DS34) was used for crystallization and included the adjacent TRF binding sites (Fig. 1B). The second complex in the asymmetric unit (DimerDimer_2) lacks discernible electron density for several nucleotides at each end of the duplex relative to the first complex (DimerDimer_1 [Fig. 1D]). Thus, our discussion focuses on the first complex, DimerDimer_1, which has the most complete DNA sequence observed in the electron density map.
TABLE 1.
Crystallographic statistics
| Parameter | Value(s) for EBNA1 dimer-dimer |
|---|---|
| Resolution range (Å) | 50 – 3.0 (3.05 – 3.00) |
| Space group | P 1 21 1 |
| Unit cell (a, b, c [Å]; α, β, γ [°]) | 64.1, 283.4, 63.9, 90.0, 89.8, 90.0 |
| Total no. of reflections | 476,302 |
| No. of unique reflections | 44,937 |
| Multiplicity | 10.6 (8.5) |
| Completeness (%) | 99.6 (98.7) |
| I/σ(I) | 8.1 (0.16) |
| Rsym | 0.159 (2.682) |
| Rfactor | 0.2434 |
| Rfree | 0.2815 |
| RMS | |
| Bonds | 0.004 |
| Angles | 0.678 |
| Ramachandran (%) | |
| Favored | 97.84 |
| Outliers | 1.98 |
| Clashscore | 8.69 |
Each EBNA1 dimer has the same overall structure and binds to DNA in the manner as previously reported (10, 12, 19). Briefly, each subunit is comprised of three α helices and four anti-parallel β strands. Upon dimerization, the β strands create an 8-stranded antiparallel β barrel. Each N-terminal loop runs parallel to the DNA helical axis and then falls into the minor groove of the DNA to grip the DNA tightly from either side (Fig. 2A and B). The Cα backbone comparison between each of the four dimers in the asymmetric unit as well as every other published EBNA1 dimer structure (10, 12, 19, 28) (apo and DNA bound) is less than 1 Å2 (see Table S1 in the supplemental material). These data indicate that no structural rearrangement of the dimers is necessary to accommodate the proteins at the DS half-site.
FIG 2.

Protein and DNA analysis. (A and B) The structures of the PGP loops and N-terminal arms are highlighted from 90° rotational views. (C) DNA alone shown in stick representation; coloring is the same as in Fig. 1A. Helical axes from center are represented as red lines. The angle between the axes measures at 120°. (D) The DNA from the complex (yellow) and the structure of Bochkarev et al. (PDB code 1B3T) (gray) shown in surface representation. The DNA for the structure of Bochkarev et al. (PDB code 1B3T) is shown directly under the analogous EBNA1 consensus sequence in the longer DNA. The same degree of unwinding is visually evident in both. (E) (Top) Complex with 4-bp separation modeled on the DNA, with one dimer in taupe and the other in blue. (Bottom) Complex with 2-bp separation modeled on the DNA, one dimer in taupe and the other in red.
The DNA is smoothly bent, which allows the two dimers to interact productively when bound to the DNA. The approximate angle between the DNA arc is 120° (Fig. 2C). The EBNA1 consensus sequence deviations from B-form seen here are similar to those previously reported (PDB code 1B3T) by Bochkarev et al. (12) (Fig. 2D). The unbound DNA duplex ends, which correspond to the TRF binding sites, are most similar in structure to B-form DNA. The functional requirement for 3-bp spacing between EBNA1 binding sites is depicted by modeling 4 or 2 bp (4) (Fig. 2E). It is clear from the modeling that 4-bp spacing prevents dimer-dimer interactions, while 2-bp spacing leads to steric clashing at the dimer-dimer interface.
The majority of the EBNA1 dimer/DNA contacts are as previously reported (12, 19); however, differences are observed for the side chains of two amino acids. In the structure reported by Bochkarev et al. (PDB code 1B3T) (12), N480 sits just out of hydrogen bond reach to both ends of the DNA duplex. In this structure, the DNA extends beyond the EBNA1 consensus sequence so that N480 can contact the phosphate backbone (Fig. 3A). This interaction is observed at the dimer-dimer interface and the outer faces of the complex. Next, R538 in the structure reported by Bochkarev et al. (PDB code 1B3T) contacts the phosphate backbone near the middle of the EBNA1 consensus sequence. The side chain remains proximal to the same area of DNA in the new dimer-dimer structure; however, its contacts vary depending on which side of the DNA helical axis the amino acids sit. R538 chain A (green) and R538 chain D (light yellow) are on the same side of the DNA helical axis, and both donate hydrogen bonds to the phosphate backbone in a manner similar to that seen in the structure reported by Bochkarev et al. (PDB code 1B3T) (Fig. 3B). However, on the opposite face of this axis lie R538 chain B (light green) and R538 chain C (yellow), neither of which contacts the DNA.
FIG 3.
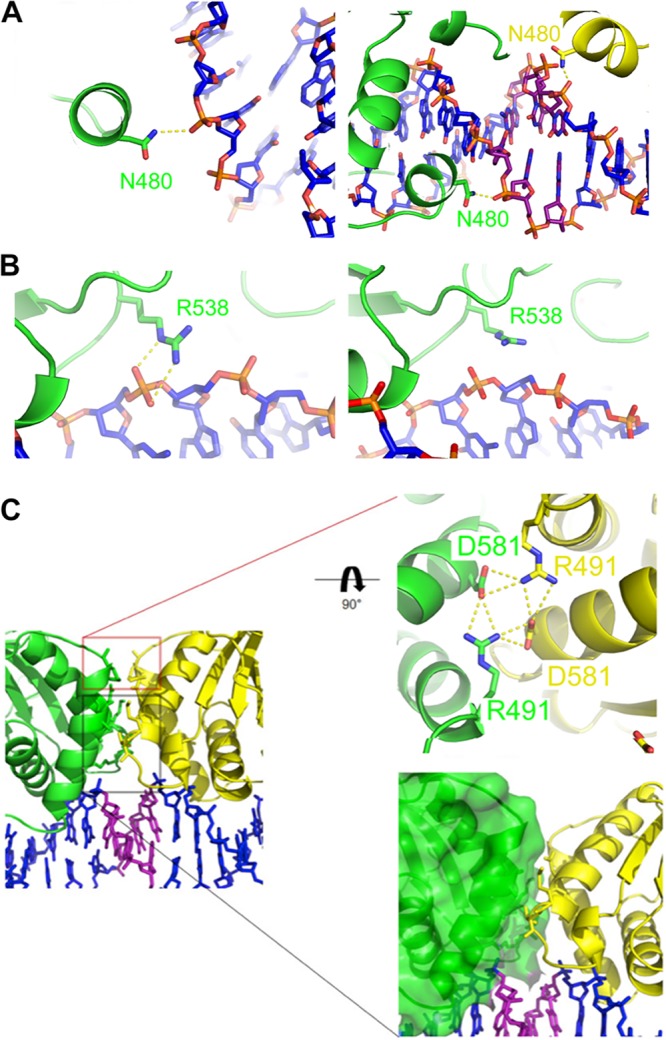
The dimer-dimer interface. (A) (Left) N480 from chain B (distal to the dimer-dimer interface) interaction with DNA. (Right) N480 from chain A (green) and chain C (yellow) interaction with DNA at the dimer-dimer interface. (B) (Left) R538 from chain A (green) interaction with DNA. (Right) R538 from chain B (green) lies above the DNA, similar to the structure of Bochkarev et al. (PDB code 1B3T). Protein and DNA are colored as in Fig. 1. (C) (Left) Close-up of the EBNA1 dimer-dimer interface. (Upper right) Close-up of interface between chain A (green) and chain C (yellow). D581 and R491 form a hydrogen bonding network at the dimer-dimer interface. (Lower right) Distal nonpolar interacting region at the dimer-dimer interface.
The dimer-dimer interface.
The newly identified dimer-dimer interface is symmetrical and buries 395 Å2 of surface area (Fig. 3C, left). The interface has two parts that we characterize based on proximity to the DNA: a proximal predominantly nonpolar interaction (Fig. 3C, bottom box) and a distal hydrogen bonding network (Fig. 3C, top box). Several residues from the first and third α helices of opposing subunits come together just above the 3-bp separation of DS3 and DS4. The lower van der Waals interacting region involves the side chains of L488, L582, M584, and T585 from both polypeptide chains and main chain atoms from A487. Above this set of interactions sits an intricate hydrogen bonding network between R491 and D581. Each D581 accepts a hydrogen bond across the dimer-dimer interface from the opposing R491. Additionally, D581 and R491 of the same chain form a hydrogen bond, thus creating a four-way hydrogen bonding network.
UniProt currently reports the polypeptide sequence for 24 EBNA1 variants. R491, D581, and L488 are strictly conserved among these variants. Because R491 and D581 are intimately involved in the dimer-dimer interface, are strictly conserved, and contribute the only polar interactions at this location, mutational studies focused on these two residues.
Mutations in the dimer-dimer interface impair complex formation on DS DNA.
The wild type (WT) and five single point mutations of the EBNA1 DNA binding domain were expressed in Escherichia coli and purified as hexahistidine-SUMO (small ubiquitin-like modifier) tag fusion proteins (Fig. 4A). Mutants were chosen to disrupt the hydrogen bonding network between the dimers by changing the side chain’s charge (R491E and D581H), replacement with a nonpolar residue (R491A and D581A), or putting a sterically larger amino acid at the interface (D581E).
FIG 4.
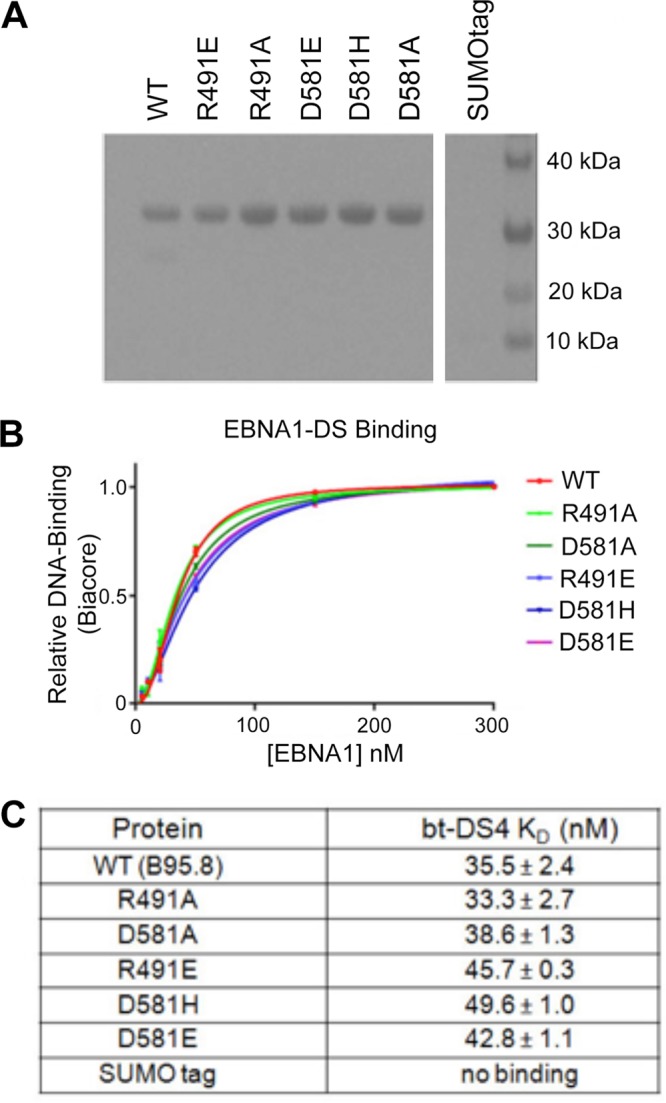
The EBNA1 mutants. (A) Coomassie gel of purified 6×His-SUMO tagged mutant proteins used in Biacore and EMSA studies. (B) Kd plots (fraction of complex versus [EBNA]) for all 6×His-SUMO-EBNA1 proteins binding to biotinylated DS4 DNA on the Biacore. Biacore sensorgrams used to make these plots are in Fig. S1. (C) Observed Kds for EBNA1s binding to DS4 were calculated by Biacore evaluation software. Biacore sensorgrams used to make these plots are in Fig. S1 (left).
None of these residues are reported to contact either the DNA phosphate backbone or a specific nucleotide, so the dimers’ affinity for an EBNA1 consensus sequence was predicted to be unaffected. To test this, we calculated a dissociation constant (Kd) for each mutant binding to the DS binding site DS4 via Biacore technology. DS4 has previously been shown to have the highest affinity of the EBNA1 binding sites in DS (8). 5′-Biotin-labeled DNA corresponding to the 18-bp binding site was coupled to flow cell 2 of a streptavidin (SA) chip; flow cell 1 served as a blank. Concentrations of fusion protein ranging from 0 nM to 300 nM were tested for binding by Biacore (Fig. 4B and Fig. S1). As predicted, the Kd values were all very similar to those for the WT (Fig. 4B and C). The D581H mutant was the weakest-binding protein of the mutants, but its Kd value of 49.6 nM is marginally weaker than that of the WT.
To next test these mutants’ ability to make stable dimer-dimer complexes on DNA, we used two techniques: Biacore and EMSA. Again, 5′-biotinylated DNA corresponding to DS34 was coupled to a Biacore flow cell. Similar concentrations of each SUMO-EBNA1 protein were tested as described above (Fig. 5 and Fig. S1). Deviations from WT behavior were detected (Fig. 5, graphs). WT EBNA1 binds in a cooperative manner with a Hill coefficient of 1.96. Cooperative binding for the D581H and D581A mutants was mildly affected, with Hill coefficients of 1.66 and 1.57, respectively. However, the R491E, R491A, and D581E mutants were significantly impaired, with Hill values ranging from 1.18 to 1.37. This loss of cooperativity between the dimers indicates either that both dimers can get on the DNA but not productively interact with each other or that only one dimer is bound to the DNA at a time.
FIG 5.

Biacore and EMSA results for EBNA1 mutants binding to DS3 + 4. (A to F) The left sides of the panels show Biacore Kd plots (fraction of complex versus [EBNA]) for all 6×His-SUMO-EBNA1 proteins binding to biotinylated DS34 on the Biacore. Hill coefficients (h) were determined with GraphPad Prism and are indicated for each mutant. Biacore sensorgrams used to make these plots are in Fig. S1. The right sides of the panels show EMSA results for IRDye700-labeled DS34 binding to each 6×His-SUMO-EBNA1 mutant. Concentrations of protein are listed for each well. Unbound DNA, DNA plus dimer, and DNA plus dimer-dimer bands are indicated for each gel. Vertical lines indicate an unstable or aggregated complex. (A) WT EBNA1, (B) R491E, (C) R491A, (D) D581E, (E) D581H, (F) D581A.
We then analyzed the binding of the mutant dimers to DS34 by EMSA (Fig. 5, gel images). Similar to Biacore analysis, strong differences between the WT and the mutants were apparent. The WT formed dimer-dimer pairs on the DS34 DNA; very little DS34 with only one dimer bound is seen in the gel. The D581E mutant, which had the lowest Hill coefficient in Biacore, preferentially formed one dimer bound to DS34. At higher concentrations, a dimer-dimer complex formed but migrated as a smeared band, suggesting that the complex was either not stable or less compact or aggregated improperly (Fig. 5B to F, vertical lines). Similar observations were made in gels for the R491E and R491A mutants. The D581H mutant showed the most cooperative binding, with a Hill coefficient with DS34 at 1.66. These data indicate that mutations in the dimer-dimer interface compromise cooperative DNA binding in both EMSA and Biacore assays and that most radical mutations, D581E and R491E, have the most severe effect on cooperativity.
Dimer-dimer interphase mutants have compromised replication and binding activity in cells.
EBNA1 protein binding to the DS is essential for successful EBV episome replication. We asked if these dimer-interface-disrupting mutants could replicate a plasmid containing the oriP element (Fig. 6). A derivative of the pREP10 plasmid containing the oriP and full-length EBNA1 (B95.8) lacking the GA repeats was used to generate each of the five mutants by site-directed mutagenesis. Plasmids were transfected into 293T cells and cells were grown for 3 days. EBNA1 proteins were assayed by Western blotting and shown to be expressed as similar levels (Fig. 6A). Plasmid DNA was recovered from the cells by Hirt lysate preparations. Recovered DNA was either uncut to measure total DNA input or cut by DpnI to leave only plasmids replicated within the mammalian cells. Southern blots were made from gels containing both input and DpnI cut samples and then probed with 32P-labeled plasmid (Fig. 6B) and quantified (Fig. 6C). We found that all the dimer-dimer interface mutants were impaired for replication activity. Importantly, the R481E and D591E mutants were most severely compromised for DNA replication, consistent with their more significant disruption of cooperative DNA binding in EMSA and Biacore (Fig. 5). We also measured the ability of these EBNA1 mutants for episome maintenance using a colony formation assay (Fig. 6D). This assay reflects that ability to maintain stable episomes over many cell generations under hygromycin selection. We found that all of the EBNA1 dimer-dimer interface mutants had reduced episome maintenance relative to that of WT EBNA1, with those of the R491E and D58E mutants the most severely reduced, consistent with loss of function in DNA replication and cooperative DNA binding.
FIG 6.

OriP-dependent DNA replication and colony formation assays. (A) Western blot for transfected 293T cells used for DNA replication assay probed for EBNA1 and the loading control, actin. (B) Representative Southern blot results for all mutants. Input uncut DNA (left panel) and DpnI-resistant replicated DNA (right panel) are indicated. (C) Quantitation of Southern blot results normalized to WT activity. Error bars indicate 1 standard deviation. All mutant activity is statistically significant compared to WT activity (P < 0.05). (D) Colony formation assay for each EBNA1 mutant plasmid in pREP10 using hygromycin resistance and quantified relative to WT EBNA1. Error bars indicate standard deviation and P values of <0.05.
We next asked if two most severely impaired mutants, the R491E and D581E mutants, were able to bind to oriP and recruit key protein complexes for DNA replication at oriP (Fig. 7). We first verified that the EBNA1 WT and R491A and D581E mutants were expressed at similar levels (Fig. 7A) and were significantly impaired for DNA replication (Fig. 7B and C). We then assayed these mutants for their behavior in chromatin immunoprecipitation (ChIP) for binding at the DS and FR regions of OriP and recruiting cellular replication helicase protein MCM3 (Fig. 7D). We found that both the R491E and D581E mutants bound to the FR and DS regions of OriP but not to the control ampicillin gene (AMP). The D581A mutant was reduced ∼2 fold relative to WT EBNA1 at both DS and FR. Strikingly, both mutants were severely defective in their ability to recruit MCM3 to the DS or FR compared with WT EBNA. These findings indicated that mutations in the EBNA1 dimer-dimer interface that disrupt DNA replication function correspond with a similar disruption in the recruitment of the MCM complex to OriP.
FIG 7.

DNA replication and ChIP assay. 293T cells were transfected with plasmids for OriP control lacking the EBNA1 gene (pHEBO) or OriP vectors in pREP10 expressing WT EBNA1 or the R481E or D591E mutant. (A) Western blot for EBNA1 and the loading control, actin. (B) Southern blot analysis of plasmid DNA replication assay for EcoRV-linearized (left) and EcoRV- and DpnI-digested (right) DNA probed with 32P-labeled OriP DNA. (C) Quantification of Southern blot DNA replication assay for WT and mutant EBNA1 shown in panel B. (D) ChIP assay with antibodies to IgG, EBNA1, or MCM3 and assayed by quantitative PCR (qPCR) at AMP, DS, and FR regions of the pREP10 plasmid.
Modeling of mutant EBNA1 dimer-dimer surfaces.
Cooperative EBNA1 binding to DS is unlikely to fully account for replication origin function. To gain further insight into other potential interaction surfaces, we mapped amino acid conservation on the surface of the EBNA1 dimer-dimer complex (Fig. 8A). Areas responsible for DNA binding are predictably conserved in this representation (Fig. 8A, red). The PGP loops, which are known to be highly conserved and critical for EBNA1 replication function (29), form stable and prominent wing-like structures extending perpendicular to the DNA axis. The surface 180° from the DNA binding region is also highly conserved and not known to be essential for EBNA1 function or mediate interactions. Based on the observed patch of conservative amino acids, this surface is also likely to mediate important functional interactions. Interesting, nestled in the center of this area is the surface created by R491 and D581. We then used Chimera to model the interfaces of the mutants (Fig. 8B). In contrast to the WT smooth interface, all the mutants are pitted and uneven. Taken together with the failure of mutant EBNA1s to bind cooperatively, stimulate DNA replication, and recruit MCM3 to the DNA, it is likely that these residues are key to both stable binding of EBNA1 to the DNA and creating a surface or architecture that helps bring cellular replication machinery to the site.
FIG 8.
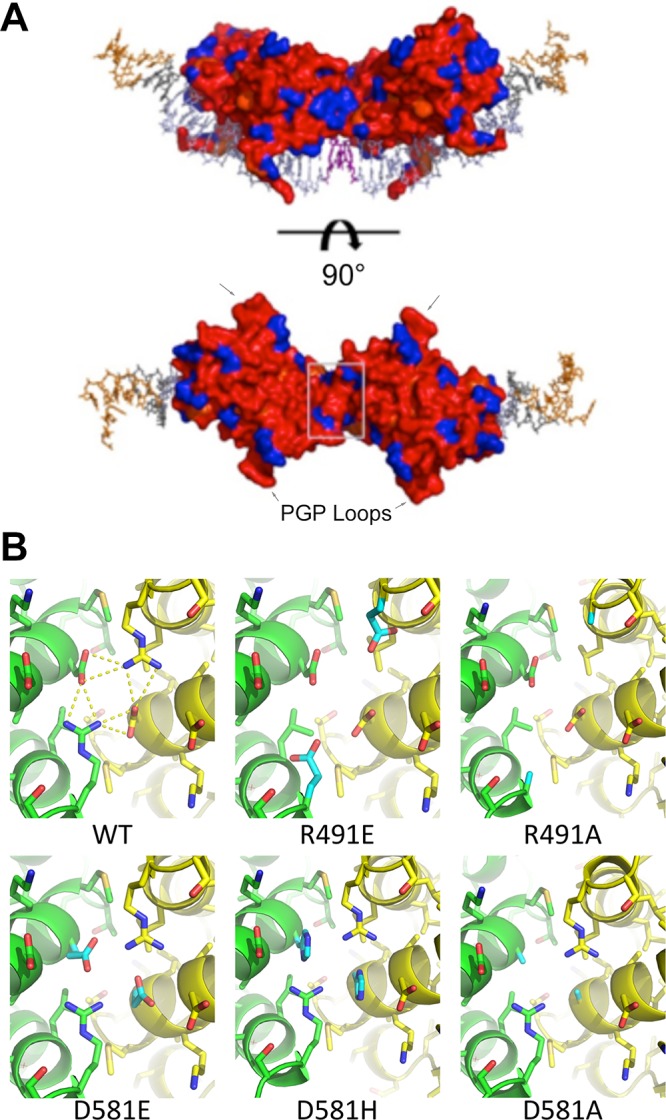
Surface conservation of EBNA1. (A) DNA is shown in stick representation and colored as in Fig. 1A. Both EBNA dimers are shown in surface representation and rainbow colored according to conservation of amino acid. Red and orange are highly conserved; blue is not conserved. The surface created by R491 and D581 is boxed in light gray. (B) Modeled mutation of amino acids 491 and 581 of the WT and each mutant. The WT structure reveals a network of hydrogen bonding at the dimer-dimer interface. Mutated residues are colored cyan.
DISCUSSION
Latent infection of EBV requires the continued expression and proper function of the viral protein EBNA1 (4). The C terminus of EBNA1 is responsible for dimerization and binding to DNA consensus sequences at the EBV episome’s oriP and required for the initiation of DNA replication by recruiting cellular origin recognition complex (ORC) and MCM helicase complex (10–13, 30). The DS element contains four EBNA1 consensus binding sequences in a paired orientation with each half-site (sites 1 and 2 and sites 3 and 4) representing a minimal origin of DNA replication (4, 8, 27). Previous structural studies focused on a single EBNA1 dimer bound to an idealized consensus sequence (12, 19). Here we present the crystal structure of an EBNA1 DBD bound to sites 3 and 4 of the DS element, which represents the first higher-order structure of EBNA1 bound to the viral minimal origin of DNA replication at OriP.
Our data shows that the EBNA1 protein when bound to DS DNA does not undergo any structural changes compared with previously reported EBNA1 protein structures (Table S1). Instead, the DNA is strongly bent (Fig. 1C). This is consistent with biochemical results from Bashaw and Yates (4), who published EMSA data showing that EBNA1 binding induced a large DNA bend with the center at the midway point between the two EBNA1 binding sites. The differences in observed bending angles may reflect differences in solution studies relative to crystallographic methods used in this study, as well as different DNA substrates. Previous studies have also shown that interactions between EBNA1 bound to paired DS sites were necessary for EBNA1-mediated DNA bending and untwisting (31, 32).
Bashaw and Yates also demonstrated a strict need for the 3-bp separation between DS sites; insertions or deletions of 1 to 2 bp between paired DS sites resulted in loss of replication activity (4). Our new dimer-dimer crystal structure provides a molecular rationale of the explicit need for a 3-bp separation between sites on the DS (Fig. 2E). With 4 bp between the consensus sites, one dimer is rotated away from the other and results in a loss of contact between dimers. The DNA could bend more to allow for contact, but the resulting interface would be small and would not likely provide enough stability to such a constrained structure. In contrast, when 1 bp is removed, the second dimer sterically clashes with its partner. A 3-bp separation provides the necessary orientation to create a biologically relevant and stabilizing interface between the dimers.
The Kaposi’s sarcoma-associated herpesvirus (KSHV) episome maintenance protein LANA, like EBNA1, was also found to bind cooperatively to multiple recognition sites in the KSHV terminal repeat origin of plasmid replication (33). Like EBNA1, LANA dimers bound with similar spacing between dimer binding sites and induced a smooth bending of the DNA around the protein oligomers. Spacing between LANA-LANA dimers was shown to be important for maintaining LANA proteins in the same phase along the DNA axis (33). Similar to EBNA1, the LANA oligomeric interaction interface has been found to be important for DNA replication and episome maintenance function, suggesting that this structure may be involved in recruiting cellular factors critical for replication origin formation (34).
We identified two residues at the interface that are essential to a successful dimer-dimer interaction: R491 and D581. Mutating these residues resulted in impaired dimer-dimer formation on the DNA and reduced replication of an oriP-containing plasmid. These data are consistent with results from Bashaw and Yates, who also effectively ruined the EBNA1 dimers’ ability to cross talk by inserting/deleting DNA base pairs (4).
The DS element has four binding sites of various affinities; sites 1 and 4 are high affinity, while sites 2 and 3 are lower affinity (27). Binding of EBNA1 to sites 1 and 4 increases the affinity of the protein for sites 2 and 3 (27). All residues necessary for this cooperative activity of EBNA1 are within the DNA binding domain (27). Mutating residue R491 or D581 resulted in loss of EBNA1 cooperative binding in biochemical studies. Our structure highlights R491 and D581 as the molecular basis for EBNA1 cooperative binding to DS half-sites.
In addition to cooperative binding to DS, the dimer-dimer interface may contribute more directly to origin replication function. It is possible that the interaction interface between R491 and D581 creates a binding surface for cellular proteins required for DNA replication. Mutations of R491A and D581A were defective in the recruitment of MCM3 as measured by ChIP assay (Fig. 7D). Both the R491E and D581E mutants are able to create a dimer of EBNA1 dimers bound to DNA (Fig. 5); however, the EMSA data also indicate that these EBNA1-DNA complexes are less stable and structurally different than complexes formed by WT EBNA1. Since neither of these mutants could successfully bring the MCM3 complex to the oriP, it is possible that the three-dimensional structure induced by the EBNA1 dimer-dimer interface is key to recruiting certain cellular factors to the EBV episome and producing successful plasmid replication.
We predict that EBNA1 binding to the other half of DS (DS1 and DS2) would look similar to the structure reported here for DS34. The sequences of DS2 and DS1 differ only slightly from those of DS4 and DS3, and the 3-bp separation, CCC versus GTT, is likely to allow for similar flexibility at the interface (8). While these discrepancies could lead to minor differences between the two half-sites, it is likely that the dimer-dimer interface and overall structure would be highly similar. Full activity of the DS is reconstituted by combination of all four DS binding sites, as well as the nonomer repeats that bind telomere repeat factors (35). Our structure does not provide sufficient resolution at these nonamer sites to determine if they are structurally perturbed. Future studies with higher resolution, and cocrystals involving TRF1 and TRF2 in addition to EBNA1, would further illuminate how the DS controls replication of EBV episomes in cells.
The architecture of the FR region at the oriP is quite different than the DS. The FR is a series of 20 tandem copies of 30-bp repeats. Each repeat is an 18-bp EBNA1 site followed by an AT-rich 12-bp sequence. Seven sites are minimally needed for function, and this region is essential to plasmid maintenance (35, 36). EBNA1 has recently been crystallized as a trimer of dimers where the interface between dimers is distinct from the dimer-dimer interface crystallized in this study (37). Mutation of key residues in that interface did not affect episome replication but strongly impaired episome maintenance, suggesting that this multimer is important to EBNA1 assembly on the FR. Taken together with the structure reported here, those findings lead us to propose that the interfaces relevant to EBNA oligomerization at the DS and FR are distinct from each other.
Latent EBV infection is associated with several cancers, including nasopharyngeal carcinoma, which is endemic to Southeast Asia. Patients have a high probability of recurrence or distant metastases and low survival rates (38, 39). Currently, no therapeutic option exists for latent EBV infection, but the idea that EBNA1 function can be targeted by a small molecule is novel and has been explored with preliminary success (40–42). The dimer-dimer structure reported here identifies a necessary interface for EBV episome replication; when this interface is disrupted, replication of the plasmid is impaired. Over time, this phenomenon will result in cellular loss of the EBV episome and thus latent EBV infection. Recently, Gianti et al. computationally identified four druggable pockets on the EBNA1 DBD surface capable of binding a small-molecule ligand (43). One such pocket sits adjacent to this dimer-dimer interface and could be exploited for small molecule drug development against latent EBV infection.
MATERIALS AND METHODS
EBNA1 DBD expression and purification.
Residues 459 to 607 of B95.8 EBNA1 DBD were cloned into a modified pET DUET vector containing the SMT3 gene, which encodes the yeast small ubiquitin-like modifier (SUMO) protein. The hexahistidine and SUMO protein tag were fused to the N terminus of the EBNA1 protein. E. coli cells were transformed with plasmid, grown in the presence of 100 μg/ml of ampicillin, and induced via autoinduction (44). Cell pellets were resuspended in 20 mM Tris (pH 8.0), 1 M NaCl, 5 mM β-mercaptoethanol, 1 mM MgCl2, 5 mM imidazole, 1 mM phenylmethane sulfonyl fluoride (PMSF), and 100 μg/ml of lysozyme. Cells were lysed by sonication at 4°C, and the insoluble fraction was separated by centrifugation at 18,000 rpm (SS-24 Sorvall) for 30 min. The soluble lysate fraction was passed over preequilibrated nickel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen) and washed with 20 mM Tris (pH 8.0), 1 M NaCl, 5 mM β-mercaptoethanol, and 30 mM imidazole. Protein was batch eluted with 20 mM Tris (pH 8.0), 1 M NaCl, 5 mM β-mercaptoethanol, and 450 mM imidazole. Eluted protein was mixed with 500 μg of SUMO1 protease. The cleavage reaction was dialyzed overnight into 20 mM Tris (pH 8.0), 1 M NaCl, and 5 mM β-mercaptoethanol at 4°C. Purified EBNA was obtained by passing the cleavage reaction over preequilibrated Ni-NTA resin. The protein was further purified using a size exclusion column (HiLoad 26/60 Superdex 75; GE Life Sciences) equilibrated with 1 mM HEPES (pH 7.2), 500 mM NaCl, and 10 mM dithiothreitol (DTT). Fractions were run on a bis-Tris acrylamide gel to verify size and purity. Finally, protein was pooled, concentrated, aliquoted, and frozen at −80°C for long-term storage.
Site-directed mutagenesis.
Primers were designed to generate the mutants (R491E, R491A, D581E, D581H, and D581A) in the same modified pET DUET vector as previously described and the mammalian vector pRep10. A two-stage PCR protocol for site-directed mutagenesis was adapted from Stratagene (45). Following Dpn1 digestion and heat inactivation, PCR products were transformed into DH5α cells. Purified plasmids from colonies were sequenced to confirm the mutations. 6×His-SUMO fusion proteins were expressed and purified as previously described except that the 6×His-SUMO tag was not cleaved from the protein.
Crystallization, data collection, and analysis.
Purified WT EBNA1 from B95.8 concentrated at 10.5 mg/ml was incubated with a 1.5× molar excess of DS34 DNA (Fig. 1B). Crystals were grown for 24 to 36 h at room temperature in 200 mM sodium malonate (pH 6.75) and 24% polyethylene glycol 3350 (PEG 3350). Crystals were frozen in oil. Data were collected from a single crystal on the 19-DS-D beamline at the Advanced Photon Source of Argonne National Laboratory (Argonne, IL). Data were indexed, reduced, and scaled using HKL3000. The structure was solved by molecular replacement using PHASER integrated into PHENIX (46) with the structure of Bochkarev et al. (PDB code 1B3T) as a search model (12). Models were refined in PHENIX using simulated annealing, minimization, and individual B-factor refinement. Between refinement cycles, the model was manually rebuilt using the program Coot (47). Data collection and refinement statistics are summarized in Table 1.
DNA binding by Biacore.
DNA corresponding to DS4 (biotin-5′-CTAACCTAATTCAATAGCATATGTTACCCAAC-3′ and 5′-GTTGGGTAACATATGCTATTGAATTAGGGTTAG-3′) and DS34 (biotin-5′-TAACCCTAATTCGATAGCATATGCTTCCCGTTGGGTAACATATGCTATTGAATTAGGGTTAG-3′ and 5′-CTAACCCTAATTCAATAGCATATGTTACCCAACGGGAAGCATATGCTATCGAATTAGGGTTA-3′) were purchased from Integrated DNA Technologies. DNA was coupled to a Biacore T200-compatible streptavidin (SA) sensor chip (GE Healthcare Life Sciences). Binding between EBNA1 (0 nM to 200 nM) and these DNAs was performed in 20 mM Tris (pH 8.0), 250 mM NaCl, 5 mM β-mercaptoethanol, and 1 mM MgCl2. Resulting sensorgrams were used to obtain a Kd plot ([EBNA-DNA] versus [EBNA]). DS4 binding to EBNA1 (WT and mutants) were fit for Kd (nanomolar concentration), Rmax, and chi2 using the Biacore T200 evaluation software (GE Healthcare Life Sciences). Chi2 is less than 10% of Rmax for an acceptable data set. DS34 binding to EBNA1 (WT and mutants) were fit in GraphPad Prism using the nonlinear regression (curve fit) model “Specific Binding with Hill Slope.”
DNA binding by EMSA.
Both DNA strands corresponding to DS34 (as described above) were ordered with a 5′-IRDye700 tag from Integrated DNA Technologies. All reactions were done in 20 mM Tris (pH 8.0), 100 mM NaCl, 5 mM β-mercaptoethanol, 1 mM MgCl2, and 7.6% glycerol, with 1 nM DNA. Concentrations of 6×His-SUMO-EBNA1 WT and mutants varied from 0 nM to 100 nM. Six percent acrylamide-TBE gels were prerun for 30 min at 100 V. Reaction mixtures were incubated at room temperature for 10 min, loaded immediately, and run for 45 min at 100 V. Gels were visualized with the Odyssey Clx imaging system from Li-COR.
EBV episome replication assay, colony formation, and ChIP.
pREP10 plasmids with B95.8 EBNA1 WT, mutant, or empty vector were transfected into 1.5 million HEK 293 cells (ATCC) growing in a 10-cm plate with Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). At 48 h posttransfection, cells were replated into a 15-cm plate and allowed to grow for an additional 24 h. All cells were harvested from the 15-cm plate and washed with 1× phosphate-buffered saline (PBS; Gibco), and pellets were resuspended in 1 ml of 1× PBS. Twenty percent of the cells were lysed with radioimmunoprecipitation (RIPA) buffer, and protein concentrations were determined via bicinchoninic acid (BCA) assay (Pierce). Normalized samples were run on a bis-Tris acrylamide gel, transferred to a polyvinylidene difluoride (PVDF; Bio-Rad) membrane, blocked with 5% milk, and probed with anti-EBNA1 (in-house) and anti-β-actin-horseradish peroxidase (HRP) (Ab-Cam) to determine EBNA1 expression. Eighty percent of the cells were lysed with Hirt lysate buffer (0.6% SDS, 20 mM EDTA, and 10 mM Tris [pH 7.6]) to extract the plasmid DNA. Samples were treated with 200 μg of RNase (Invitrogen) and 200 μg of proteinase K (New England BioLabs), and then aqueous fractions were taken after treatment with phenol-chloroform (Sigma-Aldrich) and chloroform-isoamyl alcohol (Sigma-Aldrich). DNA was precipitated with isopropanol on dry ice for 2 h and washed with 70% ethanol, and pellets were dried overnight. DNA was resuspended in 50 μl of Tris-EDTA (TE) buffer and stored at −20°C until use.
Ninety percent of the extracted DNA was cut overnight at 37°C with DpnI (Fig. 6) or DpnI plus EcoRV (Fig. 7). The remaining 10% was subjected to a mock digestion (Fig. 6) or linearized with EcoRV (Fig. 7). DNA was purified following digestion by extraction with chloroform-isoamyl alcohol and precipitation by ethanol. Pellets were washed with 70% ethanol, dried overnight, and resuspended in TE buffer. Samples were loaded onto a 0.9% agarose gel and run at 50 V overnight. DNA within the gel was depurinated by soaking in 0.25 M HCl for 15 min. After two washes with water, the DNA was denatured by soaking in 0.5 M NaOH and 1 M NaCl for 30 min. After two washes, the gel was soaked in neutralizing buffer (0.5 M Tris [pH 7.4], 3 M NaCl) for 30 min. DNA was transferred to a nylon membrane (Millipore) and probed with 32P-labeled WT plasmid. Gels were exposed overnight and read with the Typhoon phosphoimager system (GE Healthcare).
Colony formation assay was done as described previously (28). 293T cells were cotransfected with 10 μg of OriP plasmids (REP10) expressing WT or EBNA1 mutants. The cells were split after 24 h and selected with 0.5 μg/ml of hygromycin. After 2 weeks of selection, the resulting hygromycin-resistant colonies were stained with toluidine blue (0.5 mg of toluidine blue in 2% sodium carbonate). Colonies that were at least 2 mm in size were scored as positive. Colonies were counted using a colony counting macro written with NIH Image.
Chromatin immunoprecipitation (ChIP) was performed using a procedure similar to that for the replication assay. pREP10 plasmids with B95.8 EBNA1 WT and the R491E and D581E mutants were transfected into 1.5 million HEK 293 cells (ATCC) growing in a 10-cm plate with Lipofectamine 2000 according to the manufacturer’s protocol (Invitrogen). At 48 h posttransfection, cells were replated into a 15-cm plate and allowed to grow for an additional 24 h. The remaining steps were performed as previously described (48).
Structure analysis.
Buried surface area was calculated by uploading the coordinates for chains A and C to the Protein Data Bank (in Europe) database PISA server v1.51 (22 September 2014). Surface conservation was mapped using the ConSurf Server (49). The multiple-sequence alignment file used to create Fig. 8 and the final coordinates of the dimer-dimer/DNA structure were input and the conservation score was placed in the B-factor column. The new coordinates were opened in PyMOL (50), and the protein surface was colored according to the B-factors. To model the mutant surfaces, individual amino acid changes were drawn with Coot within the full dimer-dimer/DNA complex. PDB files were then imported into Chimera and energy minimization was performed with standard settings. Mutant interactions of the energy-minimized structures were rendered using PyMOL.
Data availability.
The coordinates and structure factor files of EBNA1 dimer-dimer bound to the dyad symmetry half-site have been deposited in the Protein Data Bank under accession code 6PW2.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joel Cassell and the Wistar Core Facilities for Molecular Screening for their technical support.
This work was supported by NIH grants RO1 CA093606, RO1 DE017336, and P30 CA010815, a Wellcome Trust seeding drug discovery grant (WT096496), and a Wistar training grant to J.S.D. (T32 CA09171).
We declare a conflict of interest. Paul M. Lieberman has an ownership interest in Vironika, LLC. The Wistar Institute on behalf of the authors T.E.M. and P.M.L. has filed patents covering composition of matter and their use of EBNA1 inhibitors for the treatment of human cancer and other diseases (patent numbers WO2015073864, EBNA1 Inhibitors and Their Method of Use, and WO2016183534, EBNA1 Inhibitors and Methods using Same). Wistar, T.E.M., and P.M.L. have received financial benefit from the license to Cullinan Apollo Corp. and would receive additional financial benefit should the development of the EBNA1 inhibitor meet certain milestones, including commercialization.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00487-19.
REFERENCES
- 1.Young LS, Yap LF, Murray PG. 2016. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 16:789–802. doi: 10.1038/nrc.2016.92. [DOI] [PubMed] [Google Scholar]
- 2.Lieberman PM. 2015. Chromatin structure of Epstein-Barr virus latent episomes. Curr Top Microbiol Immunol 390:71–102. doi: 10.1007/978-3-319-22822-8_5. [DOI] [PubMed] [Google Scholar]
- 3.Tempera I, Lieberman PM. 2014. Epigenetic regulation of EBV persistence and oncogenesis. Semin Cancer Biol 26:22–29. doi: 10.1016/j.semcancer.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bashaw JM, Yates JL. 2001. Replication from oriP of Epstein-Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J Virol 75:10603–10611. doi: 10.1128/JVI.75.22.10603-10611.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yates JL, Warren N, Sugden B. 1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812–815. doi: 10.1038/313812a0. [DOI] [PubMed] [Google Scholar]
- 6.Lupton S, Levine AJ. 1985. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol Cell Biol 5:2533–2542. doi: 10.1128/mcb.5.10.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yates J, Warren N, Reisman D, Sugden B. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A 81:3806–3810. doi: 10.1073/pnas.81.12.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrison S, Fisenne K, Hearing J. 1994. Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J Virol 68:1913–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yates JL, Camiolo SM, Bashaw JM. 2000. The minimal replicator of Epstein-Barr virus oriP. J Virol 74:4512–4522. doi: 10.1128/jvi.74.10.4512-4522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bochkarev A, Barwell JA, Pfuetzner RA, William Furey J, Edwards AM, Frappier L. 1995. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA1. Cell 83:39–46. doi: 10.1016/0092-8674(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 11.Ambinder RF, Shah WA, Rawlins DR, Hayward GS, Hayward SD. 1990. Definition of the sequence requirements for binding of the EBNA-1 protein to its palindromic target sites in Epstein-Barr virus DNA. J Virol 64:2369–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bochkarev A, Bochkareva E, Frappier L, Edwards AM. 1998. The 2.2 A structure of a permanganate-sensitive DNA site bound by the Epstein-Barr virus origin binding protein, EBNA1. J Mol Biol 284:1273–1278. doi: 10.1006/jmbi.1998.2247. [DOI] [PubMed] [Google Scholar]
- 13.Frappier L, O’Donnell M. 1991. Epstein-Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc Natl Acad Sci U S A 88:10875–10879. doi: 10.1073/pnas.88.23.10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawlins DR, Milman G, Hayward SD, Hayward GS. 1985. Sequence-specific DNA binding of Epstein-Barr virus nuclear antigen (EBNA1) to clustered sites in the plasmid maintenance region. Cell 42:859–868. doi: 10.1016/0092-8674(85)90282-X. [DOI] [PubMed] [Google Scholar]
- 15.Jones CH, Hayward SD, Rawlins DR. 1989. Interaction of the lymphocyte-derived Epstein-Barr virus nuclear antigen EBNA-1 with its DNA-binding sites. J Virol 63:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frappier L, O’Donnell M. 1991. Overproduction, purification and characterization of EBNA1, the origin binding protein of Epstein-Barr virus. J Biological Chemistry 266:7819–7826. [PubMed] [Google Scholar]
- 17.Ambinder RF, Mullen MA, Chang YN, Hayward GS, Hayward SD. 1991. Functional domains of Epstein-Barr nuclear antigen EBNA-1. J Virol 65:1466–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah WA, Ambinder RF, Hayward GS, Hayward SD. 1992. Binding of EBNA-1 to DNA creates a protease-resistant domain that encompasses the DNA recognition and dimerization functions. J Virol 66:3355–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, Edwards AM. 1996. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84:791–800. doi: 10.1016/S0092-8674(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 20.Hedge RS, Grossman S, Laimins L, Sigler PB. 1992. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding protein bound to its DNA target. Nature 359:505–512. doi: 10.1038/359505a0. [DOI] [PubMed] [Google Scholar]
- 21.Correia B, Cerqueira SA, Beauchemin C, Pires de Miranda M, Li S, Ponnusamy R, Rodrigues L, Schneider TR, Carrondo MA, Kaye KM, Simas JP, McVey CE. 2013. Crystal structure of the gamma-2 herpesvirus LANA DNA binding domain identifies charged surface residues which impact viral latency. PLoS Pathog 9:e1003673. doi: 10.1371/journal.ppat.1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niller H, Glaser G, Knuchel R, Wolf H. 1995. Nucleoprotein complexes and DNA 5′-ends at oriP of Epstein-Barr virus. J Biol Chem 270:12864–12868. doi: 10.1074/jbc.270.21.12864. [DOI] [PubMed] [Google Scholar]
- 23.Hseih D-J, Camiolo SM, Yates JL. 1992. Constitutive binding of EBNA1 protein to the Epstein-Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J 12:4933–4944. doi: 10.1002/j.1460-2075.1993.tb06187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, Schepers A. 2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J Cell Sci 116:3971–3984. doi: 10.1242/jcs.00708. [DOI] [PubMed] [Google Scholar]
- 25.Koons M, Van Scoy S, Hearing J. 2001. The replicator of the Epstein-Barr virus latent cycle origin of DNA replication, oriP, is composed of multiple functional elements. J Virol 75:10582–10592. doi: 10.1128/JVI.75.22.10582-10592.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edwards AM, Bochkarev A, Frappier L. 1998. Origin DNA-binding proteins. Curr Opin Struct Biol 8:49–53. doi: 10.1016/S0959-440X(98)80009-2. [DOI] [PubMed] [Google Scholar]
- 27.Summers H, Barwell JA, Pfuetzner RA, Edwards AM, Frappier L. 1996. Cooperative assembly of EBNA1 on the Epstein-Barr virus latent origin of replication. J Virol 70:1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dheekollu J, Malecka K, Wiedmer A, Delecluse H-J, Chiang AKS, Altieri DC, Messick TE, Lieberman PM. 2017. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr virus episomal latency. Oncotarget 8:7248–7264. doi: 10.18632/oncotarget.14540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ceccarelli DF, Frappier L. 2000. Functional analyses of the EBNA1 origin DNA binding protein of Epstein-Barr virus. J Virol 74:4939–4948. doi: 10.1128/jvi.74.11.4939-4948.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schepers A, Ritzi M, Bousset K, Kremmer E, Yates JL, Harwood J, Diffley JF, Hammerschmidt W. 2001. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J 20:4588–4602. doi: 10.1093/emboj/20.16.4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frappier L, O'Donnell M. 1992. EBNA-1 distorts oriP, the Epstein-Barr virus latent replication origin. J Virol 66:1786–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hearing J, Mulhaupt Y, Harper S. 1992. Interaction of Epstein-Barr virus sequences that encode a nuclear antigen expressed in latently infected lymphocytes. J Virol 66:694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hellert J, Weidner-Glunde M, Krausze J, Lünsdorf H, Ritter C, Schulz TF, Lührs T. 2015. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc Natl Acad Sci U S A 112:6694–6699. doi: 10.1073/pnas.1421804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Leo A, Deng Z, Vladimirova O, Chen HS, Dheekollu J, Calderon A, Myers KA, Hayden J, Keeney F, Kaufer BB, Yuan Y, Robertson E, Lieberman PM. 2019. LANA oligomeric architecture is essential for KSHV nuclear body formation and viral genome maintenance during latency. PLoS Pathog 15:e1007489. doi: 10.1371/journal.ppat.1007489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frappier L. 2012. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr Opin Virol 2:733–739. doi: 10.1016/j.coviro.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 36.Hammerschmidt W, Sugden B. 2013. Replication of Epstein-Barr viral DNA. Cold Spring Harb Perspect Biol 5:a013029. doi: 10.1101/cshperspect.a013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deakyne JS, Malecka KA, Messick TE, Lieberman PM. 2017. Structural and functional basis for an EBNA1 hexameric ring in Epstein-Barr virus episome maintenance. J Virol 91:e01046-17. doi: 10.1128/JVI.01046-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsang J, Lee VH, Kwong DL. 2014. Novel therapy for nasopharyngeal carcinoma—where are we. Oral Oncol 50:798–801. doi: 10.1016/j.oraloncology.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 39.Sun X, Tong LP, Wang YT, Wu YX, Sheng HS, Lu LJ, Wang W. 2011. Can global variation of nasopharynx cancer be retrieved from the combined analyses of IARC Cancer Information (CIN) databases? PLoS One 6:e22039. doi: 10.1371/journal.pone.0022039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang M, Lee E, Soni V, Lewis T, Koehler A, Srinivasan V, Kieff E. 2011. Roscovitine inhibits EBNA1 serine 393 phosphorylation, nuclear localization, transcription, and episome maintenance. J Virol 85:2859–2868. doi: 10.1128/JVI.01628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yasuda A, Noguchi K, Minoshima M, Kashiwazaki G, Kanda T, Katayama K, Mitsuhashi J, Bando T, Sugiyama H, Sugimoto Y. 2011. DNA ligand designed to antagonize EBNA1 represses Epstein-Barr virus-induced immortalization. Cancer Sci 102:2221–2230. doi: 10.1111/j.1349-7006.2011.02098.x. [DOI] [PubMed] [Google Scholar]
- 42.Thompson S, Messick TE, Schultz D, Reichman M, Lieberman PM. 2010. Development of a high-throughput screen for inhibitors of Epstein-Barr virus EBNA1. J Biomol Screen 15:1107–1115. doi: 10.1177/1087057110379154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gianti E, Messick TE, Lieberman PM, Zauhar RJ. 2016. Computational analysis of EBNA1 “druggability” suggests novel insights for Epstein-Barr virus inhibitor design. J Comput Aided Mol Des 30:285–303. doi: 10.1007/s10822-016-9899-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Studier F. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, Malcolm B. 1999. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange site-directed mutagenesis. Biotechniques 26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- 46.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tempera I, Deng Z, Atanasiu C, Chen CJ, D'Erme M, Lieberman PM. 2010. Regulation of Epstein-Barr virus OriP replication by poly(ADP-ribose) polymerase 1. J Virol 84:4988–4997. doi: 10.1128/JVI.02333-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. 2016. ConSurf. 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeLano WL. 2002. The PyMOL molecular graphics system. Proteins Struct Funct Bioinformatics 30:442–454. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factor files of EBNA1 dimer-dimer bound to the dyad symmetry half-site have been deposited in the Protein Data Bank under accession code 6PW2.