

With the onset of the 2015 ZIKV outbreak, ZIKV pathogenesis has been of extreme global public health interest, and a better understanding of interactions with the host would provide insight into molecular mechanisms driving the severe neurological outcomes of ZIKV disease. Here is the initial report on the relationship between ZIKV and the host oligoadenylate synthetase-RNase L (OAS-RNase L) system, a potent antiviral pathway effective at restricting replication of diverse viruses. Our study elucidated a unique mechanism whereby ZIKV production is impervious to antiviral RNase L activity, through a mechanism of viral RNA protection that is not mimicked during infection with numerous other RNase L-activating viruses, thus identifying a distinct replication strategy potentially important for ZIKV pathogenesis.
KEYWORDS: RNA, RNase L, Sindbis virus, Zika virus, dengue virus, innate immunity, replication factories, viral genome
SUMMARY
There is currently no knowledge of how the emerging human pathogen Zika virus (ZIKV) interacts with the antiviral endoribonuclease L (RNase L) pathway during infection. Since activation of RNase L during infection typically limits virus production dramatically, we used CRISPR-Cas9 gene editing technology to knockout (KO) targeted host genes involved in the RNase L pathway to evaluate the effects of RNase L on ZIKV infection in human A549 cells. RNase L was activated in response to ZIKV infection, which degraded ZIKV genomic RNA. Surprisingly, despite viral genome reduction, RNase L activity did not reduce ZIKV infectious titers. In contrast, both the flavivirus dengue virus and the alphavirus Sindbis virus replicated to significantly higher titers in RNase L KO cells compared to wild-type (WT) cells. Using MAVS/RNase L double KO cells, we demonstrated that the absence of increased ZIKV production in RNase L KO cells was not due to compensation by enhanced type I interferon transcripts to thus inhibit virus production. Finally, when synthetic double-stranded RNA was detected by OAS3 to induce RNase L antiviral activity prior to ZIKV infection, we observed reduced ZIKV replication factory formation, as well as a 42-fold reduction in virus yield in WT but not RNase L KO cells. This study proposes that ZIKV evades RNase L antiviral activity by generating a viral genome reservoir protected from RNase L cleavage during early infection, allowing for sufficient virus production before RNase L activation is detectable.
IMPORTANCE With the onset of the 2015 ZIKV outbreak, ZIKV pathogenesis has been of extreme global public health interest, and a better understanding of interactions with the host would provide insight into molecular mechanisms driving the severe neurological outcomes of ZIKV disease. Here is the initial report on the relationship between ZIKV and the host oligoadenylate synthetase-RNase L (OAS-RNase L) system, a potent antiviral pathway effective at restricting replication of diverse viruses. Our study elucidated a unique mechanism whereby ZIKV production is impervious to antiviral RNase L activity, through a mechanism of viral RNA protection that is not mimicked during infection with numerous other RNase L-activating viruses, thus identifying a distinct replication strategy potentially important for ZIKV pathogenesis.
INTRODUCTION
The introduction of Zika virus (ZIKV) into the Western hemisphere in 2015 triggered an epidemic spanning the Americas (1). There were several remarkable characteristics of this outbreak that had not previously been linked to ZIKV, notably sexual and vertical transmission of the virus, the latter of which can cause severe neurological sequelae in the developing fetus (2–4). While many studies have compared characteristics of recent outbreak strains from the Asian lineage with those of older ZIKV strains of the African lineage (5), the molecular determinants involved in pathogenesis and cellular tropism to confer transplacental migration or sexual transmission have yet to be mapped. An improved understanding of ZIKV interaction with the host on a molecular level will provide essential information for development of antiviral strategies not only against ZIKV infection but also to combat other important human-pathogenic flaviviruses.
In addition to ZIKV, the Flaviviridae family is comprised of other prominent mosquito-borne human viruses, including West Nile virus, yellow fever virus, Japanese encephalitis virus, and dengue virus (DENV). After entering the cell by endocytosis, the viral polyprotein is translated from the positive-sense single-stranded RNA (ssRNA) genome containing a single open reading frame (ORF), and cleaved into 10 proteins. The negative-sense antigenome is synthesized from the RNA genome and used as a template for generation of positive-strand progeny genome (6) in a process that involves generation of double-stranded RNA (dsRNA) intermediates. Flavivirus replication occurs within invaginations of the endoplasmic reticulum (ER), deemed replication factories (RFs), which result from extensive virus-induced ER membrane and cytoskeletal remodeling (7–9). After virus particle assembly, also within ER vesicles, virus buds into the ER lumen and exploits the host secretory pathway for release from the plasma membrane. Polyprotein translation and processing, and assembly of progeny virus all occur in close proximity to RFs, for spatial coordination, as well as simultaneous evasion of host detection throughout the virus replication cycle (10).
Flavivirus subversion of the type I interferon (IFN) pathway is well described (11, 12) and includes reports of degradation of STAT2 and subsequent reduction of IFN signaling during ZIKV infection of human cells (13–16). The relationship between ZIKV and the oligoadenylate synthetase-RNase L (OAS-RNase L) pathway has yet to be investigated, although there are reports of other flaviviruses sensitive to this pathway (17, 18). The OAS-RNase L pathway is initiated by detection of the viral replication intermediate dsRNA by OAS sensors (Fig. 1). While there are three human OAS isoforms with enzymatic function, we have previously shown that the OAS3 isoform is the predominant mediator of antiviral effects during infections of human cells with a variety of viruses (19). OAS3 generates 2′,5′-phosphodiester-linked oligoadenylates (2-5A) from ATP, which bind RNase L to trigger homodimerization and nucleolytic activation (20). This results in a milieu of antiviral effects, including protein translation inhibition as a result of cellular rRNA (rRNA) and mRNA degradation and inhibition of viral replication and translation due to viral ssRNA cleavage, as well as the induction of inflammation (21) and apoptosis (22–24). This pathway is highly effective in inhibiting replication of RNA and DNA viruses, and previous findings from our lab revealed that virus resistance to RNase L activity can dictate cellular tropism (25, 26), valuable information in the context of ZIKV.
FIG 1.
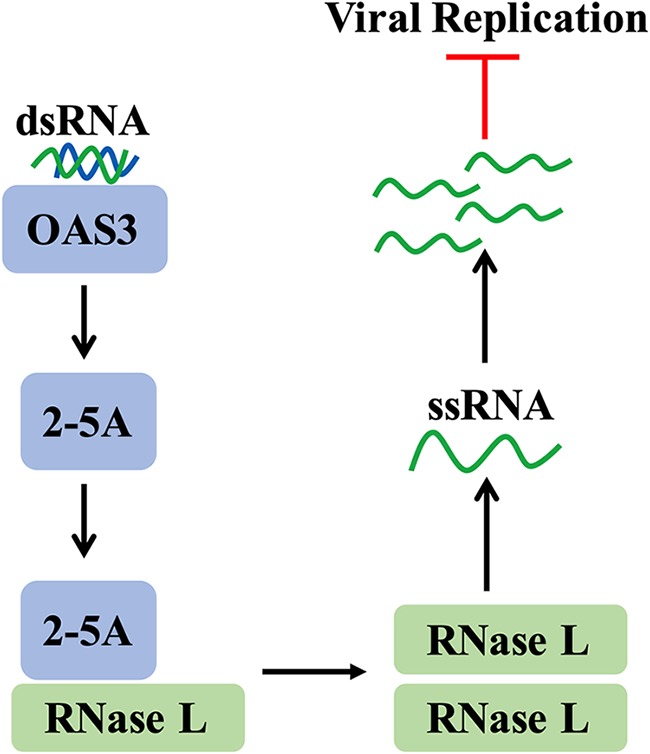
OAS-RNase L pathway. Viral replication intermediate dsRNA is detected in the cytoplasm of an infected cell by host OAS3 receptors, which synthesize the small molecule 2-5A. The molecule 2-5A binds and activates RNase L, which homodimerizes and targets host and viral ssRNA for cleavage, resulting in the inhibition of viral replication.
Our present study demonstrates the atypical occurrence of RNase L-mediated degradation of ZIKV genome in the absence of any restriction of ZIKV production. We reveal that only under conditions of artificial RNase L activation prior to ZIKV infection is RNase L capable of facilitating potent antiviral effects on ZIKV production. To emphasize the uniqueness of ZIKV circumvention of activated RNase L function, we employ the related flavivirus DENV and the alphavirus Sindbis virus (SINV) as controls known to exhibit susceptibility to RNase L antiviral activity (17, 19, 27).
RESULTS
ZIKV infection activates the OAS-RNase L pathway.
We first assessed the relevance of the host OAS-RNase L pathway (Fig. 1) to ZIKV infection using A549 cells, which are human lung epithelial cells easily modified using clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 gene editing technology to knockout (KO) specific target genes in this pathway. To determine whether the ZIKV replication intermediate dsRNA was detected by OASs to activate RNase L, we infected wild-type (WT) A549 cells with ZIKV and at various times postinfection examined rRNA cleavage, a hallmark of RNase L activation. We initially assessed both MR766 and Puerto Rico 2015 (PR) strains to determine whether there were any discrepancies in activation of this pathway between the African and Asian lineages, using SINV as a control for strong RNase L activation (19). At the earlier time point of 16 h postinfection (hpi) rRNA cleavage was not yet detected; however, at 24 and 40 hpi robust degradation of 28S and 18S rRNA was observed (Fig. 2A), indicating that ZIKV activated RNase L between 16 and 24 hpi. We did not observe any differences between strains and thus proceeded with the ZIKV PR strain for all future infections. To confirm rRNA cleavage was a direct result of RNase L activation, we showed that rRNA remained intact in ZIKV-infected A549 cells with either OAS3 or RNase L CRISPR-Cas9-mediated gene knockouts (OAS3 KO or RNase L KO), which we also verified using SINV infection as a control (Fig. 2B). In addition, rRNA cleavage was maintained during ZIKV or SINV infection of A549 cells with STAT1 or STAT2 gene knockouts (STAT1 KO or STAT2 KO) (Fig. 2B), demonstrating RNase L activation by both viruses in the absence of type I IFN signaling. Cleavage of rRNA was also observed in ZIKV-infected MAVS KO cells, but not in RNase L KO or MAVS/RNase L double KO (DKO) cells, further establishing RNase L catalytic activity as independent of type I IFN responses during ZIKV infection.
FIG 2.

ZIKV infection activates the OAS-RNase L pathway. (A) A549 cells were mock infected or infected with ZIKV MR766, ZIKV PR, or SINV at an MOI of 1, and RNA was harvested at the indicated times postinfection and analyzed by using an Agilent bioanalyzer for rRNA degradation. (B) A549 cells (WT or KO) were infected with ZIKV PR or SINV at an MOI of 1, and at 24 hpi RNA was harvested and analyzed by using an Agilent bioanalyzer for rRNA degradation. DKO, MAVS/RNase L double KO. Two different RNase L KO clones were assessed (representative data are shown). The data shown are representative of two to three experiments.
Activated RNase L reduces ZIKV genome levels.
In addition to targeting cellular RNA, RNase L restricts viral replication by cleaving viral ssRNA; therefore, we investigated the effect of activated RNase L on ZIKV genomic RNA levels by Northern blotting for ZIKV genome. To thoroughly ascertain RNA status, we designed probes that bind distinct NS2B, NS4a, or NS5 regions of the genome but which should each detect the 10.8-kb full-length genome. With each probe and at each time postinfection, genomic RNA levels were reduced in WT cells compared to RNase L KO cells (Fig. 3A). We compared effects on ZIKV genome levels to those of DENV, another related flavivirus, and SINV, both of which are sensitive to RNase L antiviral activity. Examining viral genome over time, we first detected ZIKV RNA at low levels in WT cells at 18 hpi, shortly before we observed robust RNase L activation during ZIKV infection (Fig. 2A), after which genome diminished and was barely visible by 32 hpi (Fig. 3B). In RNase L KO cells, genome abundance at 18 hpi increased throughout 32 hpi, consistent with RNase L cleavage of ZIKV genome in the WT cells, resembling that of SINV and DENV at later time points (Fig. 3B). In addition, quantitative reverse transcription-PCR (qRT-PCR) analysis demonstrated significant increases in RNA levels of ZIKV, SINV, and DENV in RNase L KO cells compared to WT at 24 and 30 hpi and as early as 18 hpi in SINV-infected cells (Fig. 3C). To visually confirm effects of RNase L on the ZIKV genome, we used fluorescence in situ hybridization (FISH) to detect ZIKV genome in infected WT and RNase L KO cells and immunofluorescent staining of the ZIKV NS3 protease as a marker for sites of replication. At 16 hpi before RNase L activation, similar levels of genome were detected during ZIKV infection in WT and RNase L KO cells; however, at 20 and 24 hpi, after which RNase L is activated in WT cells, genomic RNA accumulated throughout infected RNase L KO cells but not in WT cells (Fig. 3D). Due to poor sensitivity of RNA detection by Northern blotting compared to qRT-PCR and FISH, infections for Northern blotting were conducted at a higher MOI (an MOI of 10 for Northern blotting in Fig. 3A and B versus an MOI of 1 in qRT-PCR and FISH assays in Fig. 3C and D), potentially resulting in variations in RNA temporal expression between the different assays. However, the pattern of genome expression over time between WT and RNase L KO cells was consistent among the different assays for each virus evaluated.
FIG 3.

Activated RNase L reduces ZIKV genome levels. (A) A549 cells were mock infected or infected with ZIKV at an MOI of 10, and RNA was harvested at 24 or 30 hpi and analyzed by Northern blotting with probes designed to detect the indicated coding regions of the ZIKV genome. (B) A549 cells were infected with ZIKV, SINV, or DENV at an MOI of 10, and RNA was harvested at the indicated times postinfection for analysis by Northern blotting with virus genome-specific probes. (C) A549 cells were infected with ZIKV, SINV, or DENV at an MOI of 1, and RNA was harvested at the indicated times postinfection for qRT-PCR for viral RNA expression. The cycle threshold (CT) values were normalized to 18S rRNA to generate ΔCT values (ΔCT = CT gene of interest – CT 18S rRNA). Technical triplicates were averaged and displayed using the equation 2–ΔCT. Statistical significance was determined by two-way ANOVA; the means of three replicates ± the SD are shown (**, P < 0.01; ****, P < 0.0001). (D) A549 cells were infected with ZIKV at an MOI of 1, and at the indicated time points the cells were fixed and stained with a FISH probe detecting positive-strand ZIKV genome (red) before immunofluorescence staining with anti-ZIKV NS3 (green) primary antibody and anti-mouse Alexa Fluor 488 secondary antibody and DAPI (blue) for nuclei. Scale bar, 50 μm. Two different RNase L KO clones were assessed; representative images are shown. The data shown are representative of two experiments.
RNase L activation does not impair ZIKV replication.
We next assessed how RNase L activation and its cleavage of cellular and viral RNAs during ZIKV infection would affect ZIKV infectious particle production over time by measuring viral titers from supernatants of A549 cells infected with ZIKV at a low MOI to permit examination of multiple rounds of replication over a 72-h period. Surprisingly, despite a reduction in ZIKV genome levels in WT cells compared to RNase L KO (Fig. 3), viral titers were not increased in RNase L KO cells. Instead, ZIKV titers were significantly decreased, albeit modestly, in RNase L KO cells compared to WT (Fig. 4A). These results were consistently observed at different MOIs as well as in two clones of RNase L KO cells (representative data for one clone shown). Due to the remarkably contradictory effect of activated RNase L on ZIKV replication levels in comparison to any other RNase L-activating virus we have evaluated (19, 25), we confirmed the lack of RNase L-mediated inhibition of ZIKV replication in HT1080 cells, another human cell line, and again observed a failure of RNase L to limit ZIKV replication (Fig. 4B). In contrast to ZIKV infection, SINV and DENV titers were significantly increased in RNase L KO cells compared to WT by 8 or 48 hpi, respectively (Fig. 4C and D), consistent with previous findings (17, 19).
FIG 4.

RNase L activation does not impair ZIKV replication. Supernatants were collected from infected cells at the indicated times postinfection and titrated on Vero cells for determination of the viral titer in PFU/ml over time postinfection. (A) A549 cells were infected with ZIKV PR at MOIs of 0.01 and 0.1. Two different RNase L KO clones were assessed, and representative titers from one clone are shown. (B) HT1080 cells infected with ZIKV PR at an MOI of 0.1. (C) A549 cells infected with DENV at MOIs of 0.01 and 0.1. (D) A549 cells infected with SINV at an MOI of 0.1. Statistical significance was determined by two-way ANOVA; the means of three replicates ± the SD are shown (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001). The data shown are representative of two to three experiments.
Since activated RNase L targets host mRNAs for cleavage in addition to viral RNA, we considered that lower than expected ZIKV titers in RNase L KO cells may be the result of increased type I IFN transcript levels in the absence of RNase L cleavage (28). Increased type I IFN responses could subsequently dampen any increases in ZIKV titers that KO of RNase L may provide. However, we found that in A549 cells with targeted KO of key components of the type I IFN production or signaling pathways, namely STAT1, STAT2, or MAVS, as well as in MAVS/RNase L DKO cells, ZIKV titers were not enhanced over those of WT cells (Fig. 5), demonstrating that type I IFN is effectively antagonized by ZIKV and not responsible for lagging viral titers in RNase L KO cells.
FIG 5.

Type I IFN production and signaling does not impair ZIKV replication. Supernatants were collected from infected cells at the indicated times postinfection and titrated on Vero cells for determination of viral titer in PFU/ml over time postinfection. (A) A549 WT, STAT1 KO, or STAT2 KO cells infected with ZIKV PR at an MOI of 0.1. (B) A549 WT, MAVS KO, RNase L KO, or MAVS/RNase L DKO infected with ZIKV PR at an MOI of 0.1. The statistical significance was determined by two-way ANOVA; the means of three replicates ± the SD are shown. No significance found from 24 to 72 hpi between titers from WT and any KO cells is displayed. The data shown are representative of two experiments.
RNase L activation does not inhibit ZIKV protein synthesis.
Since degradation of host and viral RNA had no effect on ZIKV infectious particle production, we investigated protein translation arrest, another consequence of RNase L activation, as potentially antagonized by ZIKV to block RNase L antiviral activity. Degradation of cellular RNA by RNase L inhibits global protein translation to limit viral infection, although many viruses that induce cellular protein translation arrest have found ways to concurrently maintain viral protein synthesis (29, 30). Inhibition of protein translation during ZIKV infection has been previously described (29, 31); we therefore investigated whether translation arrest was mediated by RNase L, and, if so, whether ZIKV nascent protein synthesis was affected by activation of RNase L. We infected WT or RNase L KO A549 cells with ZIKV, and at different time points postinfection treated cells with puromycin, which is incorporated into nascent protein, before harvesting whole-cell lysate. Coomassie blue staining for preexisting total cellular and viral protein, as well as Western blotting for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) protein, from both WT and RNase L KO cells over time confirmed equal amounts of total protein abundance in each sample (Fig. 6) (32, 33). Western blotting for puromycin expression revealed a decline in de novo cellular protein over time in ZIKV-infected WT and RNase L KO cells (Fig. 6). However, expression of the viral protease NS3, a component of the viral replication complex, increased over time in both cell types, indicating that protein translation arrest during ZIKV infection is independent of RNase L activity and has no effect on viral protein expression.
FIG 6.
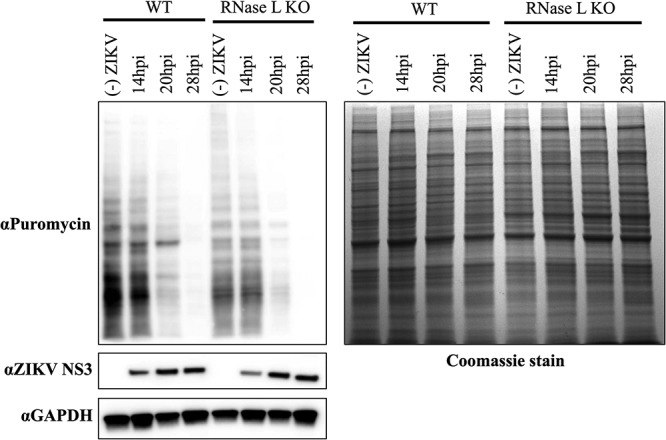
RNase L activation does not inhibit ZIKV protein synthesis. A549 cells were seeded onto six-well plates and mock infected or infected with ZIKV at an MOI of 5 for 14, 20, or 28 h; 10 μg/ml puromycin was then added to the cells for incorporation into de novo-synthesized protein for 10 min before whole protein lysates were harvested. Next, 30 μg of protein was loaded onto SDS-PAGE gels for Western blotting, followed by probing with anti-puromycin at 1:6,000 (Millipore, MABE342) for cellular and viral nascent protein and anti-ZIKV NS3 1:1,000 (GeneTex, GTX133309) for changes in viral protein expression. Loading controls were provided by Coomassie blue staining for preexisting total protein, as well as by staining for anti-GAPDH (1:1,000; Thermo Fisher MA5-15738). The data shown are representative of two experiments.
Activation of RNase L prior to ZIKV infection impairs viral replication and infectious particle production.
ZIKV genome is synthesized within RFs at perinuclear ER membrane invaginations, which are proximal to virus assembly sites also within ER folds. We sought to determine whether RFs protect genome from cleavage by activated RNase L. To evaluate the importance of RF formation in resistance to RNase L activation, we transfected cells with the synthetic dsRNA poly(rI)-poly(rC) [pIC] for 2 h to induce RNase L activation prior to infection with ZIKV. At the indicated times postinfection, we compared RF assembly and viral titers in pIC-treated cells to those without pIC treatment, the latter of which activate RNase L by 22 hpi as measured by rRNA degradation (Fig. 7A and B). We were unable to detect any transfected pIC with the anti-dsRNA J2 antibody (Scicons) by immunofluorescent staining, likely due to a combination of the low concentration of pIC used (250 ng/ml) and the length of time posttransfection of pIC at which we were evaluating dsRNA expression. Using ZIKV dsRNA as a marker for RFs and protein disulfide isomerase (PDI) to label the ER, at 16 hpi dsRNA ER localization characteristic of ZIKV RFs was detectable in both WT and RNase L KO cells without pIC treatment, whereas in pIC-treated cells RFs were detectable in the RNase L KO cells but not in WT cells (Fig. 7C). By 24 hpi, after which RNase L is activated by ZIKV alone, we again detected dsRNA in both WT and RNase L KO cells without pIC; however, once more only in the RNase L KO cells could RF assembly overcome pIC pretreatment, indicating that impaired RF formation at both time points was a result of early RNase L activation and not due to RNase L-independent host responses induced by pIC (Fig. 7C). This correlates with the absence of any antiviral effects of type I IFN production and signaling on ZIKV infectious particle production (Fig. 5A and B). In addition, dsRNA expression was only absent in WT cells where RNase L was activated prior to ZIKV infection and not in WT cells with RNase L activation induced naturally by virus infection, indicating that RF assembly is required to occur prior to RNase L activation during ZIKV infection to preserve genome replication (Fig. 7C). In agreement with dsRNA staining, viral titers in WT cells pretreated with pIC were significantly reduced (42-fold at 28 hpi) compared to those in WT cells without pIC (Fig. 7D). ZIKV titers in RNase L KO cells were unaffected by pIC treatment and only displayed the slight attenuation consistently observed between RNase L KO cells and WT cells independent of pIC pretreatment (Fig. 7D). To further ascertain absence of any type I IFN-mediated reduction in viral titers in the WT cells, pIC treatment of MAVS KO cells before ZIKV infection significantly decreased ZIKV production, whereas pIC treatment of MAVS/RNase L DKO cells had no effect on ZIKV titer, confirming RNase L as the sole mediator of ZIKV restriction (Fig. 7E).
FIG 7.

Activation of RNase L prior to ZIKV infection impairs viral replication and infectious particle production. (A) Overview of panels B, C, D, and E. A549 WT and RNase L KO cells and (in panel E) MAVS KO and RNase L/MAVS DKO cells were transfected with or without pIC for 2 h before infection at an MOI of 1 with ZIKV PR or mock infected. Downstream assays included rRNA cleavage assay, immunofluorescence staining, and analysis of viral replication kinetics by plaque assay, all at the indicated times postinfection. (B) RNA was harvested at 2, 16, and 22 hpi for rRNA cleavage analysis by using an Agilent bioanalyzer. (C) Cells were fixed at either 16 or 24 hpi for staining with anti-dsRNA (green) and anti-PDI ER marker (red) primary antibodies and then anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 594 secondary antibodies and DAPI (blue) for nuclei. Scale bar, 50 μm. (D) Supernatants were titrated on Vero cells to determine viral titers in PFU/ml over time postinfection; the statistical significance displayed was determined by two-way ANOVA comparing WT with or without pIC (**, P < 0.01; ****, P < 0.0001) or RNase L KO with or without pIC (ns). The means of three replicates ± the SD are shown. (Results for 28 hpi, WT without pIC versus RNase L KO without pIC [****, P < 0.001] and WT with pIC versus RNase L KO with pIC [**, P < 0.01] are not displayed.) (E) Supernatants from 28 hpi were titrated on Vero cells to determine viral titers in PFU/ml over time postinfection; the statistical significance displayed was determined by using a Student t test (**, P < 0.01). The means of three replicates ± the SD are shown. DKO, MAVS/RNase L double KO. The data shown are representative of two to three experiments.
DISCUSSION
Numerous studies with diverse viruses have established the paradigm that activated RNase L degrades viral ssRNA, as well as host rRNA and mRNA, leading to restriction of viral replication and spread (34). Indeed, as demonstrated here, SINV and DENV undergo RNase L-mediated genome cleavage and are significantly limited in replication. We detected decreased ZIKV genome in infected WT cells compared to RNase L KO cells by Northern blotting, FISH staining, and qRT-PCR analysis for ZIKV RNA (Fig. 3). We were therefore surprised to discover that infectious ZIKV release into the supernatant of infected WT cells was unrestricted despite RNase L-mediated cleavage of viral genome. Instead, WT cells produced slightly higher levels of virus compared to RNase L KO cells (Fig. 4A and B), suggesting that ZIKV assembly and exit from the cell were not only unaffected by RNase L activation, but even slightly defective without RNase L expressed. Our data suggest that ZIKV genome synthesized within perinuclear RFs was protected from cleavage by activated RNase L (Fig. 3D), thereby permitting the efficient virion production exhibited by viral replication curves (Fig. 4), while extraneous ZIKV RNA outside RFs is accessible to activated RNase L and thus degraded (Fig. 3). Interestingly, the related flavivirus DENV also establishes RFs at ER invaginations upon infection (7). However, our data suggest that these RFs may not provide equal protection as those in ZIKV infection, since we observe a reduction of replication during DENV infection of WT cells compared to RNase L KO cells (Fig. 4C), indicative of sensitivity to RNase L activity and suggestive of a unique mechanism of ZIKV tolerance to RNase L antiviral activity.
All positive-strand RNA viruses form RFs for the basic purpose of maximizing a confined concentration of viral proteins and genome to complete the replication cycle efficiently. ZIKV and other flaviviruses likely form RFs not only for the productive coupling of replication, translation, and assembly at the ER and eventual ease of secretory pathway appropriating for trafficking to the plasma membrane but also as a protective compartment for all aforementioned activities to transpire undetected by host antiviral sensors (10). Recent structural characterization of ZIKV RFs identified narrow pores in the RF open to the ER lumen (8). This provides explanation for the eventual availability of dsRNA in the cytoplasm where it must at some point be detected by OAS3 to activate RNase L, as well as how excess ZIKV genome “escapes” into RNase L-accessible cytoplasmic compartments for subsequent cleavage (Fig. 3, WT versus RNase L KO). The aforementioned study also described differences between the composition of cytoskeletal proteins remodeled to generate RFs during infection with ZIKV and DENV, in particular in the perinuclear region, offering explanation for discrepancies between RFs formed by ZIKV that shield genome from RNase L cleavage and those formed by DENV that are not as protective from RNase L (8). Indeed, only after pIC treatment to activate RNase L before ZIKV infection did we observe the expected difference in ZIKV titers between WT and RNase L KO cells (Fig. 7D, dotted lines), indicating ZIKV evasion of early RNase L activation as crucial for productive infection in WT cells. It is likely that RF formation and associated localization of dsRNA and viral genome throughout different stages of the virus replication cycle are determinants for how effective the OAS/RNase L pathway is at limiting virus infection. Future studies will focus on the variation in RF composition and formation between ZIKV and other flaviviruses to enable protection of dsRNA and genome from OAS3 and RNase L, respectively, to allow for productive virus assembly.
Many viruses have evolved specific mechanisms and in some cases specific proteins (coronaviruses, rotaviruses, and Theiler’s virus) to evade the antiviral effects of RNase L (35–39), which we suggest is at least in part because RNase L can be activated even while IFN signaling is antagonized (Fig. 2B) (40). Indeed, many diverse RNA (influenza virus, coronaviruses, and hepatitis C virus [HCV]) and DNA (vaccinia virus and herpesvirus) viruses antagonize the OAS-RNase L system (41), the majority of viruses targeting upstream of RNase L homodimerization and activation as effects on genome downstream of that are typically devastating to virus production. In the present study, we detected protein translation arrest during ZIKV infection and, although it was independent of RNase L, there was no inhibition of viral protein translation (Fig. 6). Some HCV strains have evolved resistance to activated RNase L by removal of RNase L cleavage sites from the viral genome (42–44). In addition, one study suggests that some reovirus strains replicate more efficiently in the presence of RNase L due to RNase L-mediated inhibition of host protein translation to provide a more suitable environment for reovirus replication (45), although further studies are required to assess RNase L activation and function in reovirus-infected cells (46). Thus, to our knowledge, ZIKV is the only virus to date that robustly activates RNase L during infection but subsequently avoids its antiviral effects at the protein level despite degradation of genome.
We identified OAS3 as the required isoform for mediating RNase L activation during ZIKV infection (Fig. 2B), which is in agreement with our preceding study establishing the importance of the OAS3 isoform in particular in detection of viral dsRNA from diverse virus families (19). While OAS proteins can be upregulated by type I IFN production and signaling, we established here that RNase L activation during ZIKV infection occurs independent of type I IFN production and signaling, revealed by the presence of rRNA degradation in infected STAT1 KO, STAT2 KO, and MAVS KO cells (Fig. 2B), which is consistent with our previous findings in primary murine cells (40). KO of STAT1, STAT2, or MAVS from A549 cells also had no effect on ZIKV replication compared to WT cells (Fig. 5A and B), and pIC-induced IFN responses caused only a transient delay in ZIKV replication in RNase L KO cells (Fig. 7D), all of which combined with previously published studies indicate strong antagonism of the type I IFN pathway by ZIKV (13–16). Thus, these findings suggest that basal OAS3 levels in ZIKV-infected A549 cells are sufficient for detection of ZIKV dsRNA and subsequent activation of RNase L even when the type I IFN pathway is antagonized during viral infection.
In addition to targeting viral RNA and host machinery utilized by the virus to limit replication, RNase L has also been shown to cleave abundant host mRNAs, which can include type I IFN transcript, therefore KO of RNase L may increase the abundance of these transcripts to thereby boost activation of innate immune pathways (28). Despite the strong evidence for ZIKV antagonism of type I IFN responses, we considered the possibility that in the absence of RNase L cleavage of RNA, increases in type I IFN mRNA to subsequently enhance type I IFN production and downstream antiviral responses could potentially reduce ZIKV titers in RNase L KO cells to those of WT cells. In particular, we were interested determining the cause of the consistently observed minor, yet significant, decrease in ZIKV titers in the absence of RNase L compared to in WT cells, which contradicted the canonical outcome of RNase L activation on virus production. However, using both MAVS KO and MAVS/RNase L DKO cells, we confirmed that MAVS signaling had no detectable effect on ZIKV titers in the presence or absence of RNase L activity (Fig. 5B), correlating with the aforementioned data indicating ZIKV evasion of type I IFN responses. These findings are also in agreement with a recently published study indicating that production of type I, as well as type III, IFNs are spared from RNase L-mediated translation inhibition, providing additional explanation for the lack of difference in ZIKV titers between infected RNase L KO and MAVS/RNase L DKO cells (47).
A mechanism for a possible proviral role of RNase L during ZIKV infection is therefore focus of ongoing investigation, specifically its involvement in assembly of RFs during early infection, prior to its catalytic activation around 20 hpi (Fig. 2A). While RNase L antiviral nuclease activity is well understood, fewer efforts have focused on RNase L noncatalytic function, although there have been several accounts of RNase L interaction with the cytoskeleton. Latent RNase L interaction with the actin cytoskeleton, namely the scaffolding protein Filamin A, inhibits viral entry, and this interaction is disrupted by activation of RNase L (48). In the context of ZIKV infection, a potential latent RNase L-cytoskeletal interaction could improve replication by aiding in RF formation rather than prevent it, consistent with other viruses that engage cytoskeletal components to enable viral life cycle events (49, 50).
This study reveals a striking inconsistency between intracellular ZIKV genome levels and infectious virus production in the context of RNase L activity. Our data suggest that during ZIKV infection, the formation of replication factories is essential for replication and optimal virus production. In addition to providing a center for coordinated RNA synthesis, translation, and virus assembly, RFs establish a physical barrier shielding viral products from host innate immune sensors, thereby limiting antiviral activity. This presently unique feature of ZIKV permits efficient virus assembly and release from infected cells despite RNase L activation and cleavage of viral genome. It is likely that RNase L cleaves accessible, excess viral genome disseminated throughout the cytoplasm, while an essential genome reservoir is protected within the perinuclear replication and assembly sites. Further efforts will evaluate the importance of ZIKV assembly of RFs to protect against activated RNase L and other host RNA sensors in primary human cells.
MATERIALS AND METHODS
Cell culture.
A549 cells were grown in Roswell Park Memorial Institute medium 1640 (Gibco, 11875) supplemented with l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (Gibco), and 10% (vol/vol) fetal bovine serum (FBS; HyClone). Vero cells were grown in Dulbecco modified Eagle medium (DMEM; Gibco, 11965) supplemented with 4.5 g/liter d-glucose, 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 50 μg/ml gentamicin (Gibco). HEK 293T cells were cultured in DMEM supplemented with 10% FBS and 1 mM sodium pyruvate. HT1080 cells were cultured in DMEM supplemented with 10% FBS.
Viruses.
Zika virus isolate MR766 (GenBank accession no. KX377335) was obtained from Robert Tesh, University of Texas Medical Branch, Galveston, TX, and propagated in Vero cells. Zika virus 2015 Puerto Rico isolate PRVABC59 (KX377337) and Dengue virus type 2 (DENV2; New Guinea C, BEI Resources) were obtained from Scott Hensley, University of Pennsylvania, Philadelphia, and were propagated in Vero cells. Sindbis virus Girdwood (G100) was obtained from Mark Heise, University of North Carolina, Chapel Hill, and was prepared as previously described (51).
rRNA cleavage assay.
A549 cells were infected with the indicated virus at a multiplicity of infection (MOI) of 1 or 5 for the indicated number of hours. Cells were harvested in RLT buffer (RNeasy minikit; Qiagen) and lysed through a QIAshredder (Qiagen). Total RNA was extracted and analyzed on RNA microchips using an Agilent 2100 BioAnalyzer (25, 52).
qRT-PCR.
A549 cells were seeded into 12-well plates and the next day infected with the indicated virus at an MOI of 1 for the indicated number of hours. Cells were harvested in RLT buffer (RNeasy Plus minikit; Qiagen), lysed through a QIAshredder (Qiagen), and DNase treated. Total RNA was extracted, and 200 ng of RNA was reverse transcribed into cDNA with SuperScript III 4 (Invitrogen). cDNA was amplified using virus-specific qRT-PCR primers, iQ SYBR Green Supermix (Bio-Rad), and the QuantStudio 3 PCR system (Thermo Fisher). The ZIKV NS5 gene was amplified with the forward (5′–3′) primer ACTTGGTGGTGCAGCTTATC and the reverse (3′–5′) primer CACTTTCTCTGGCTTCCTCAA. The SINV nsP4 gene was amplified with the forward primer CACTGAGCGACTGCTTTCAGG and the reverse primer TAGTTCGCCGGTACACTGCTGG. The DENV NS5 gene was amplified with the forward primer GTCCTCTGCGTGACTCAAGTGTTG and the reverse primer AAACCTCCCTGGATTTCCTTCCCAC. The 18S rRNA gene was amplified with the forward primer TTCGATGGTAGTCGCTGTGC and the reverse primer CTGCTGCCTTCCTTGAATGTGGTA. Cycle threshold (CT) values were normalized to 18S rRNA to generate ΔCT values (ΔCT = CT gene of interest – CT 18S rRNA). Technical triplicates were averaged and displayed using equation 2–ΔCT.
Northern blotting.
A549 cells were seeded into six-well plates and the next day infected with the indicated virus at an MOI of 10 for the indicated number of hours. Cells were harvested in RLT buffer (RNeasy minikit; Qiagen) and lysed through a QIAshredder (Qiagen). Total RNA was extracted, and 5 to 12 μg of RNA was resolved on a formaldehyde gel. Before transfer, the gel was stained with ethidium bromide (EtBr) to visualize 28S and 18S rRNA bands using UV light in a Bio-Rad Gel Doc to ensure equal loading of all RNA samples. The gel was then incubated in 50 mM NaOH–10 mM NaCl for 20 min at room temperature and transferred to a nylon membrane (Hybond-N+; Amersham) using capillary transfer overnight. Probes were generated against viral RNA of interest and horseradish peroxidase (HRP)-conjugated with an enhanced chemiluminescence (ECL) direct nucleic acid labeling and detection system (GE, RPN 3001). RNA was UV cross-linked to the nylon membrane and hybridized with the indicated probes overnight at 40°C. Probes for detection of ZIKV were generated by labeling restriction enzyme-digested plasmids expressing ZIKV protein NS2b, NS4a, or NS5. These plasmids—pLV_Zika_NS2B_Flag, pLV_Zika_Flag_NS4a, and pLV_Zika_NS5_Flag—were gifts from Vaithi Arumugaswami (Addgene, catalog numbers 79637, 79636, and 79639, respectively). Probes for detection of SINV and DENV were made by labeling PCR product after gel isolation. The SINV nsP4 gene was amplified with the forward (5′–3′) primer CATGATCGATCTCGACACGTCGAAAGAG and the reverse (3′–5′) primer TAAGCATCGTACTCGTCGCTCGAGTATC. The DENV NS5 gene was amplified with the forward primer ATGCTCCTAGTCCTCTGCGTGAC and the reverse primer AGTATGACCAGCCTCCTCTGCC. The hybridized RNA was visualized by chemiluminescence using the ECL direct nucleic acid labeling and detection system and an Amersham Imager (GE). RNA band of interest size was determined by alignment of the chemiluminescent image of the nylon membrane with a UV light image of the same nylon membrane to visualize prior EtBr staining of the RNA transferred from the gel, using the known sizes of the 28S and 18S rRNAs as size markers.
RNA fluorescence in situ hybridization.
A549 cells were seeded onto eight-well chamber slides (EMD Millipore) and the next day infected with ZIKV PR at an MOI of 1 for the indicated number of hours. Cells were fixed with 10% formalin for 30 min at room temperature, dehydrated, rehydrated, treated with protease, and stained with fluorescent labels specific to positive-strand ZIKV genome (catalog no. 521511) using an RNAscope fluorescent multiplex reagent kit (Advanced Cell Diagnostics, Inc.). After FISH staining, the slides were rinsed with water and then phosphate-buffered saline (PBS) before blocking with bovine serum albumin (BSA) for immunofluorescent staining (see “Immunofluorescence Assays” below). ZIKV genomic RNA was imaged with the Amp 4 Alt B fluorescence option with excitation at 550 nm and emission at 580 nm at ×60 magnification on a Nikon Ti2E fluorescence microscope.
Generation of KO cell lines using CRISPR-Cas9 gene editing.
Single guide RNAs (sgRNA) were generated by designing oligonucleotide sequences (IDT) targeting human STAT1 or STAT2 genes. The STAT1 forward (5′–3′) primer sequence was CACCGGCAGCTTGACTCAAAATTCC, and the reverse (3′–5′) primer was AAACGGAATTTTGAGTCAAGCTGCC, targeting exon 3. The STAT2 forward primer was CACCGGGTGCAGCTGATCCTGAAAG, and the reverse primer was AAACCTTTCAGGATCAGCTGCACCC, targeting exon 2 (53). Oligonucleotides were annealed, phosphorylated, and inserted into BsmBI-digested pLenti-CRISPR v1 (Addgene) (53). pLenti-CRISPR-sgSTAT1 and pLenti-CRISPR-sgSTAT2 plasmids were confirmed by sequencing. Pseudolentiviruses were packaged in HEK 293T cells using psPAX2 and pCMV-VSV-G plasmids, as previously described (19). Supernatants harvested 48 h later were used to generate STAT1 and STAT2 KO single cell clones by limited dilution in A549 cells. Knockout of STAT1 or STAT2 protein expression was confirmed by Western blotting and DNA sequencing. STAT1 sequencing from WT cells generated sequence for the 82.72-kDa full-length amino acid product with sequence “…QLDSKFLEQVHQL…,” while the STAT1 KO product was 2.64 kDa with the amino acid sequence “…QLDSKFPGAGSPAL” before termination, the bolded letters indicating the altered sequence. The STAT2 sequencing from WT cells generated the 93.17-kDa full-length product with amino acid sequence “MAQWEMLQNLDSPFQDQLHQLYSHSLLPVDIRQYLAVWIEDQNWQEAALGSDDSKATMLF…,” whereas sequencing from STAT2 KO cells generated the product “MAQWEMLQNLDSPSFTRTASCLWTFDSTWLSGLKTRTGRKLHLGVMIPRLPCYSSTSWIS” of only 6.6 kDa before early termination. OAS3 KO, RNase L KO (two clones), and MAVS/RNase L DKO A549 cells, as well as HT1080 KO cells, were generated as previously described (19, 22).
Virus replication curves.
Cells were seeded into 24-well plates and infected the next day at the indicated MOI in triplicate. After 1 h of incubation of cells with virus at 37°C, virus was removed, and cells were washed three times with PBS before the addition of 1 ml of complete medium. At the indicated times postinfection, 150 μl of the supernatant was harvested and stored at –80°C until titration, and 150 μl of complete medium was replaced in each well so that all time points from one replicate contained supernatant from the same infected well throughout the time course.
Plaque assays.
Virus supernatant from infected cells was serially diluted in DMEM supplemented with 2% FBS and added to Vero cell monolayers in six-well plates. Plates were incubated for 1 h at 37°C before overlaying infected monolayers with DMEM containing 3% FBS, 8% NaCO3, 10 mM HEPES, 1× l-glutamine, 250 μg/ml amphotericin B, and 1.4% agarose. Plaques were stained with neutral red after 2 days (SINV) or 4 days (ZIKV, DENV) for 16 to 18 h before plaques were counted. Viral titers for each triplicate were calculated as PFU per ml of supernatant and as the mean of duplicates. All viral titers are displayed in log10 scale as the means ± the standard deviations (SD).
Immunofluorescence assays.
Cells were seeded onto eight-well glass chamber slides (EMD Millipore) and the next day mock infected or infected with ZIKV PR at an MOI of 1. Cells were fixed with 10% formalin at the indicated time point postinfection, permeabilized with 0.1% Triton X-100 in PBS for 10 min, and blocked with fresh 2% BSA for 1 h. Slides were incubated in primary then secondary antibody diluted in BSA for 1 h at room temperature, with three PBS washes after each antibody. Mouse monoclonal J2 anti-dsRNA antibody diluted 1:500 was purchased from Scicons. Rabbit polyclonal anti-Zika virus NS3 antibody diluted 1:1,000 was purchased from GeneTex (catalog no. GTX133320). Rabbit monoclonal anti-protein disulfide isomerase (PDI) 3501 was purchased from Cell Signaling (C81H6). Goat anti-mouse Alexa Fluor 488 (Invitrogen, catalog no. A11029) and goat anti-rabbit Alexa Fluor 594 (Invitrogen, catalog no. A11037) secondary antibodies were used at 1:400. Coverslips were cured onto slides using ProLong gold antifade mountant (Life Technologies) overnight before cells were imaged using a Nikon Ti2E fluorescence microscope at ×60 magnification. For immunofluorescence assay (IFA) staining in combination with FISH staining, standard FISH staining preceded a water rinse and a PBS rinse, before BSA blocking for IFA staining, with DAPI (4′,6′-diamidino-2-phenylindole) staining only after IFA staining. All images were processed in ImageJ, applying equivalent, linear brightness/contrast alterations to all images in their entirety.
pIC transfections.
Cells were seeded in 24-well plates, or in eight-well glass chamber slides (EMD Millipore) if for IFAs, and the next day transfected with or without 250 ng/ml pIC using Lipofectamine 2000 (Invitrogen) for 2 h prior to infection with ZIKV PR at the indicated MOI. Cells not treated with pIC were transfected with Lipofectamine only. Cells were either fixed (IFA) or supernatant (replication curves) and RNA (rRNA cleavage assay) were harvested at the indicated time points postinfection for downstream analysis.
Western blotting.
Whole-cell lysate was boiled for 10 min at 95°C, loaded onto precast 10% polyacrylamide gels (Bio-Rad), migrated through the gel by SDS-PAGE at 110 V, transferred onto polyvinylidene difluoride membrane (EMD Millipore) for 3 h at 150 mA, blocked in 5% milk overnight, incubated in primary and HRP-conjugated secondary antibodies in 5% milk for 1 h each incubation, and imaged using chemiluminescent HRP substrate (Thermo Scientific SuperSignal West Pico). For evaluation of de novo protein synthesis, A549 cells were seeded onto six-well plates, mock infected or infected with ZIKV at an MOI of 5 for 14, 20, or 28 h, after which 10 μg/ml puromycin was added to cells for incorporation into de novo-synthesized protein for 10 min before whole protein lysates were harvested. Then, 30 μg of protein was loaded onto SDS-PAGE gels, probing with anti-puromycin at 1:6,000 (Millipore MABE342) for cellular and viral nascent protein, anti-ZIKV NS3 1:1,000 (GeneTex, GTX133309) for changes in viral protein expression, and anti-GAPDH (1:1,000, Thermo Fisher MA5-15738) for loading control. For total protein and an additional loading control, gel was stained with Coomassie dye for 4 h before being destained for 2 h with a solution containing 7% methanol and 5% acetic acid.
Statistical analysis.
All analyses were performed using GraphPad Prism version 8.1.1. qRT-PCR data were analyzed by two-way analysis of variance (ANOVA). Plaque assay data obtained from replication curves compared multiple cell lines across several time points by two-way ANOVA, while comparison of a single time point in one cell line with or without pIC was done using a Student t test (ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
ACKNOWLEDGMENTS
We thank Ruth Elliott, Stephen Goldstein, and Shannon Christensen for performing preliminary experiments for this study; Judith Phillips and Paul Bates for their technical expertise; and Sebastien Felt for confirming the STAT1 and STAT2 KO cell lines by sequencing.
This study was supported by NIH grants R21NS100182 (to S.R.W.) and R01AI104887 (to S.R.W. and R.H.S.). J.N.W. was supported in part by T32NS007180.
J.N.W., Y.L., R.H.S., and S.R.W. designed the experiments. J.N.W. and S.R.W. wrote the manuscript. J.N.W. performed the experiments. J.N.W., Y.L., R.H.S., and S.R.W. edited the manuscript. S.R.W. supervised all research.
The authors declare no competing interests.
REFERENCES
- 1.Depoux A, Philibert A, Rabier S, Philippe HJ, Fontanet A, Flahault A. 2018. A multi-faceted pandemic: a review of the state of knowledge on the Zika virus. Public Health Rev 39:10. doi: 10.1186/s40985-018-0087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miner JJ, Diamond MS. 2017. Zika virus pathogenesis and tissue tropism. Cell Host Microbe 21:134–142. doi: 10.1016/j.chom.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brasil P, Pereira JP, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, Rabello RS, Valderramos SG, Halai U-A, Salles TS, Zin AA, Horovitz D, Daltro P, Boechat M, Raja Gabaglia C, Carvalho de Sequeira P, Pilotto JH, Medialdea-Carrera R, Cotrim da Cunha D, Abreu de Carvalho LM, Pone M, Machado Siqueira A, Calvet GA, Rodrigues Baião AE, Neves ES, Nassar de Carvalho PR, Hasue RH, Marschik PB, Einspieler C, Janzen C, Cherry JD, Bispo de Filippis AM, Nielsen-Saines K. 2016. Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med 375:2321–2334. doi: 10.1056/NEJMoa1602412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoen B, Schaub B, Funk AL, Ardillon V, Boullard M, Cabié A, Callier C, Carles G, Cassadou S, Césaire R, Douine M, Herrmann-Storck C, Kadhel P, Laouénan C, Madec Y, Monthieux A, Nacher M, Najioullah F, Rousset D, Ryan C, Schepers K, Stegmann-Planchard S, Tressières B, Voluménie J-L, Yassinguezo S, Janky E, Fontanet A. 2018. Pregnancy outcomes after ZIKV infection in French territories in the Americas. N Engl J Med 378:985–994. doi: 10.1056/NEJMoa1709481. [DOI] [PubMed] [Google Scholar]
- 5.Beaver JT, Lelutiu N, Habib R, Skountzou I. 2018. Evolution of two major Zika virus lineages: implications for pathology, immune response, and vaccine development. Front Immunol 9:1640. doi: 10.3389/fimmu.2018.01640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Selisko B, Wang C, Harris E, Canard B. 2014. Regulation of flavivirus RNA synthesis and replication. Curr Opin Virol 9:74–83. doi: 10.1016/j.coviro.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. 2009. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortese M, Goellner S, Acosta EG, Neufeldt CJ, Oleksiuk O, Lampe M, Haselmann U, Funaya C, Schieber N, Ronchi P, Schorb M, Pruunsild P, Schwab Y, Chatel-Chaix L, Ruggieri A, Bartenschlager R. 2017. Ultrastructural characterization of Zika virus replication factories. Cell Rep 18:2113–2123. doi: 10.1016/j.celrep.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neufeldt CJ, Cortese M, Acosta EG, Bartenschlager R. 2018. Rewiring cellular networks by members of the Flaviviridae family. Nat Rev Microbiol 16:125–142. doi: 10.1038/nrmicro.2017.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paul D, Bartenschlager R. 2013. Architecture and biogenesis of plus-strand RNA virus replication factories. World J Virol 2:32–48. doi: 10.5501/wjv.v2.i2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Best SM. 2017. The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J Virol 91:e01970-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miorin L, Maestre AM, Fernandez-Sesma A, García-Sastre A. 2017. Antagonism of type I interferon by flaviviruses. Biochem Biophys Res Commun 492:587–596. doi: 10.1016/j.bbrc.2017.05.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, García-Sastre A. 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Liu Q, Zhou J, Xie W, Chen C, Wang Z, Yang H, Cui J. 2017. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov 3:17006. doi: 10.1038/celldisc.2017.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS. 2017. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PLoS Pathog 13:e1006164. doi: 10.1371/journal.ppat.1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC. 2016. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17:1766–1775. doi: 10.15252/embr.201642627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin RJ, Yu HP, Chang BL, Tang WC, Liao CL, Lin YL. 2009. Distinct antiviral roles for human 2′,5′-oligoadenylate synthetase family members against dengue virus infection. J Immunol 183:8035–8043. doi: 10.4049/jimmunol.0902728. [DOI] [PubMed] [Google Scholar]
- 18.Scherbik SV, Paranjape JM, Stockman BM, Silverman RH, Brinton MA. 2006. RNase L plays a role in the antiviral response to West Nile virus. J Virol 80:2987–2999. doi: 10.1128/JVI.80.6.2987-2999.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci U S A 113:2241–2246. doi: 10.1073/pnas.1519657113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong B, Silverman RH. 1995. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem 270:4133–4137. doi: 10.1074/jbc.270.8.4133. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarti A, Banerjee S, Franchi L, Loo YM, Gale M Jr, Nunez G, Silverman RH. 2015. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17:466–477. doi: 10.1016/j.chom.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, Weiss SR. 2017. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 6:e25687. doi: 10.7554/eLife.25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. 1997. Interferon action and apoptosis are defective in mice devoid of 2′,5′-oligoadenylate-dependent RNase L. EMBO J 16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castelli JC, Hassel BA, Wood KA, Li XL, Amemiya K, Dalakas MC, Torrence PF, Youle RJ. 1997. A study of the interferon antiviral mechanism: apoptosis activation by the 2-5A system. J Exp Med 186:967–972. doi: 10.1084/jem.186.6.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao L, Jha BK, Wu A, Elliott R, Ziebuhr J, Gorbalenya AE, Silverman RH, Weiss SR. 2012. Antagonism of the interferon-induced OAS-RNase L pathway by murine coronavirus ns2 protein is required for virus replication and liver pathology. Cell Host Microbe 11:607–616. doi: 10.1016/j.chom.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Weiss SR. 2016. Antagonism of RNase L is required for murine coronavirus replication in Kupffer cells and liver sinusoidal endothelial cells but not in hepatocytes. J Virol 90:9826–9832. doi: 10.1128/JVI.01423-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girardi E, Chane-Woon-Ming B, Messmer M, Kaukinen P, Pfeffer S. 2013. Identification of RNase L-dependent, 3′-end-modified, viral small RNAs in Sindbis virus-infected mammalian cells. mBio 4:e00698. doi: 10.1128/mBio.00698-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee S, Chakrabarti A, Jha BK, Weiss SR, Silverman RH. 2014. Cell-type-specific effects of RNase L on viral induction of beta interferon. mBio 5:e00856. doi: 10.1128/mBio.00856-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth H, Magg V, Uch F, Mutz P, Klein P, Haneke K, Lohmann V, Bartenschlager R, Fackler OT, Locker N, Stoecklin G, Ruggieri A. 2017. Flavivirus infection uncouples translation suppression from cellular stress responses. mBio 8:e02150-16. doi: 10.1128/mBio.02150-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walsh D, Mathews MB, Mohr I. 2013. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol 5:a012351. doi: 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hou S, Kumar A, Xu Z, Airo AM, Stryapunina I, Wong CP, Branton W, Tchesnokov E, Gotte M, Power C, Hobman TC. 2017. Zika virus hijacks stress granule proteins and modulates the host stress response. J Virol 91:e00474-17. doi: 10.1128/JVI.00474-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Donovan J, Rath S, Kolet-Mandrikov D, Korennykh A. 2017. Rapid RNase L-driven arrest of protein synthesis in the dsRNA response without degradation of translation machinery. RNA 23:1660–1671. doi: 10.1261/rna.062000.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aulas A, Fay MM, Lyons SM, Achorn CA, Kedersha N, Anderson P, Ivanov P. 2017. Stress-specific differences in assembly and composition of stress granules and related foci. J Cell Sci 130:927–937. doi: 10.1242/jcs.199240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman RH. 2007. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J Virol 81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silverman RH, Weiss SR. 2014. Viral phosphodiesterases that antagonize double-stranded RNA signaling to RNase L by degrading 2-5A. J Interferon Cytokine Res 34:455–463. doi: 10.1089/jir.2014.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drappier M, Jha BK, Stone S, Elliott R, Zhang R, Vertommen D, Weiss SR, Silverman RH, Michiels T. 2018. A novel mechanism of RNase L inhibition: Theiler’s virus L* protein prevents 2-5A from binding to RNase L. PLoS Pathog 14:e1006989. doi: 10.1371/journal.ppat.1006989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sánchez-Tacuba L, Rojas M, Arias CF, López S. 2015. Rotavirus controls activation of the 2′-5′-oligoadenylate synthetase/RNase L pathway using at least two distinct mechanisms. J Virol 89:12145–12153. doi: 10.1128/JVI.01874-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, Patton JT, Silverman RH, Weiss SR. 2013. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci U S A 110:13114–13119. doi: 10.1073/pnas.1306917110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein SA, Thornbrough JM, Zhang R, Jha BK, Li Y, Elliott R, Quiroz-Figueroa K, Chen AI, Silverman RH, Weiss SR. 2017. Lineage A betacoronavirus NS2 proteins and the homologous torovirus Berne pp1a carboxy-terminal domain are phosphodiesterases that antagonize activation of RNase L. J Virol 91:e02201-16. doi: 10.1128/JVI.02201-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Birdwell LD, Zalinger ZB, Li Y, Wright PW, Elliott R, Rose KM, Silverman RH, Weiss SR. 2016. Activation of RNase L by murine coronavirus in myeloid cells is dependent on basal oas gene expression and independent of virus-induced interferon. J Virol 90:3160–3172. doi: 10.1128/JVI.03036-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drappier M, Michiels T. 2015. Inhibition of the OAS/RNase L pathway by viruses. Curr Opin Virol 15:19–26. doi: 10.1016/j.coviro.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han JQ, Wroblewski G, Xu Z, Silverman RH, Barton DJ. 2004. Sensitivity of hepatitis C virus RNA to the antiviral enzyme ribonuclease L is determined by a subset of efficient cleavage sites. J Interferon Cytokine Res 24:664–676. doi: 10.1089/1079990042475698. [DOI] [PubMed] [Google Scholar]
- 43.Washenberger CL, Han JQ, Kechris KJ, Jha BK, Silverman RH, Barton DJ. 2007. Hepatitis C virus RNA: dinucleotide frequencies and cleavage by RNase L. Virus Res 130:85–95. doi: 10.1016/j.virusres.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han JQ, Barton DJ. 2002. Activation and evasion of the antiviral 2′,5′-oligoadenylate synthetase/ribonuclease L pathway by hepatitis C virus mRNA. RNA 8:512–525. doi: 10.1017/S1355838202020617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith JA, Schmechel SC, Williams BR, Silverman RH, Schiff LA. 2005. Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J Virol 79:2240–2250. doi: 10.1128/JVI.79.4.2240-2250.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baglioni C, De Benedetti A, Williams GJ. 1984. Cleavage of nascent reovirus mRNA by localized activation of the 2′,5′-oligoadenylate-dependent endoribonuclease. J Virol 52:865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chitrakar A, Rath S, Donovan J, Demarest K, Li Y, Sridhar RR, Weiss SR, Kotenko SV, Wingreen NS, Korennykh A. 2019. Real-time 2-5A kinetics suggest that interferons beta and lambda evade global arrest of translation by RNase L. Proc Natl Acad Sci U S A 116:2103–2111. doi: 10.1073/pnas.1818363116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malathi K, Siddiqui MA, Dayal S, Naji M, Ezelle HJ, Zeng C, Zhou A, Hassel BA. 2014. RNase L interacts with Filamin A to regulate actin dynamics and barrier function for viral entry. mBio 5:e02012. doi: 10.1128/mBio.02012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foo KY, Chee HY. 2015. Interaction between flavivirus and cytoskeleton during virus replication. Biomed Res Int 2015:427814. doi: 10.1155/2015/427814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walsh D, Naghavi MH. 2018. Exploitation of cytoskeletal networks during early viral infection. Trends Microbiol 27:39–50. doi: 10.1016/j.tim.2018.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suthar MS, Shabman R, Madric K, Lambeth C, Heise MT. 2005. Identification of adult mouse neurovirulence determinants of the Sindbis virus strain AR86. J Virol 79:4219–4228. doi: 10.1128/JVI.79.7.4219-4228.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiang Y, Wang Z, Murakami J, Plummer S, Klein EA, Carpten JD, Trent JM, Isaacs WB, Casey G, Silverman RH. 2003. Effects of RNase L mutations associated with prostate cancer on apoptosis induced by 2′,5′-oligoadenylates. Cancer Res 63:6795–6801. [PubMed] [Google Scholar]
- 53.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]