

Summary
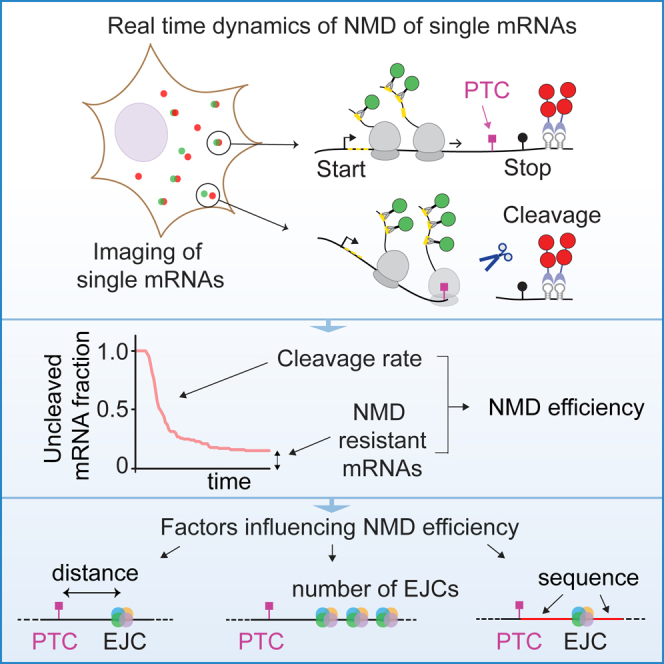
Nonsense-mediated decay (NMD) is a surveillance system that degrades mRNAs containing a premature termination codon (PTC) and plays important roles in protein homeostasis and disease. The efficiency of NMD is variable, impacting the clinical outcome of genetic mutations. However, limited resolution of bulk analyses has hampered the study of NMD efficiency. Here, we develop an assay to visualize NMD of individual mRNA molecules in real time. We find that NMD occurs with equal probability during each round of translation of an mRNA molecule. However, this probability is variable and depends on the exon sequence downstream of the PTC, the PTC-to-intron distance, and the number of introns both upstream and downstream of the PTC. Additionally, a subpopulation of mRNAs can escape NMD, further contributing to variation in NMD efficiency. Our study uncovers real-time dynamics of NMD, reveals key mechanisms that influence NMD efficiency, and provides a powerful method to study NMD.
Keywords: nonsense-mediated mRNA decay, NMD, SunTag, single-molecule imaging, live-cell imaging, translation, pioneer round of translation, XRN1
Graphical Abstract
Highlights
-
•
Nonsense-mediated decay (NMD) of single mRNA molecules is visualized in live cells
-
•
Each terminating ribosome has an equal probability of inducing NMD
-
•
NMD efficiency is affected by the number and position of introns
-
•
Kinetics of XRN1-dependent decay of the 3′ mRNA cleavage fragment are uncovered
Hoek et al. develop a method to visualize nonsense-mediated mRNA decay (NMD) of individual mRNA molecules in live cells. Using this method, they uncover the precise timing of NMD, identify multiple parameters that affect NMD efficiency, and determine the kinetics of decay of the 3′ mRNA cleavage fragments by the exonuclease XRN1.
Introduction
Nonsense mutations (i.e., point mutations that create a premature termination codon [PTC]) are responsible for ∼20% of all disease-associated single-base pair substitutions (Mort et al., 2008). In addition to genetic mutations, a PTC can also be introduced into an mRNA molecule stochastically, through errors in transcription or splicing. Understanding the fate of mRNAs containing a nonsense mutation is critical to understand the phenotypic outcome of such mutations. Transcripts harboring a PTC are rapidly degraded by a process called nonsense-mediated mRNA decay (NMD), which prevents the synthesis of truncated, and potentially toxic, proteins (Kurosaki and Maquat, 2016, Karousis et al., 2016, Lykke-Andersen and Jensen, 2015, He and Jacobson, 2015).
Pre-mRNA splicing is a critical regulator of NMD. Most mammalian genes contain multiple introns, which are spliced out before nuclear export of the mRNA (Sakharkar et al., 2004). Concomitant with intron splicing, the exon junction complex (EJC) is loaded onto the mRNA 20–24 nt upstream of the exon-exon junction (Le Hir et al., 2000, Singh et al., 2012, Saulière et al., 2012). Since stop codons are generally located in the last exon of a gene, all EJCs will usually be deposited in the coding sequence of the mRNA. During translation, these EJCs are removed by the first ribosome translating the mRNA (Dostie and Dreyfuss, 2002, Sato and Maquat, 2009, Lejeune et al., 2002), so translation termination occurs in the absence of EJCs bound to the mRNA. In contrast, PTCs are frequently located upstream of one or more introns, and translation termination on PTC-containing transcripts can thus occur while one or more EJCs are still bound to the mRNA. These mRNA-bound EJCs are thought to communicate with the ribosome during translation termination through the NMD factor UPF1 and the translation termination factors eRF1/3 (Kashima et al., 2006). EJC-ribosome communication triggers degradation of the PTC-containing mRNA, which occurs either through endonucleolytic cleavage of the mRNA by SMG6 followed by exonucleolytic decay of the cleavage fragments (Huntzinger et al., 2008, Eberle et al., 2009, Gatfield and Izaurralde, 2004), or through deadenylation, decapping, and exonucleolytic decay stimulated by the SMG5/7 complex (Unterholzner and Izaurralde, 2004, Loh et al., 2013).
For many NMD substrates, residual levels of PTC-containing transcripts can be detected in steady-state measurements (Cheng and Maquat, 1993, Cheng et al., 1994, Belgrader et al., 1994, Trcek et al., 2013, Thermann et al., 1998, Boehm et al., 2014, Lindeboom et al., 2016), suggesting that mRNA molecules with the same sequence display heterogeneity in timing of decay and/or susceptibility to NMD. Interestingly, the amount of residual mRNA for an NMD substrate appears to vary depending on the gene and the position of the PTC (Lindeboom et al., 2016, Thermann et al., 1998, Cheng et al., 1994). Uncovering the underlying cause of this variability, as well as the factors that influence it, would provide important insights into the mechanisms that control NMD efficiency and would provide a better understanding of the clinical outcome of disease-associated genetic mutations. However, current technologies such as northern blot analysis or qRT-PCR only report the average mRNA levels of an NMD target in a population of cells and thus preclude insight into differences within mRNA populations. Differences in levels of NMD targets could arise not only from differences in the rate of NMD-dependent mRNA decay but also from differences in the fraction of mRNA molecules that is susceptible to NMD or even differences in NMD efficiency between subpopulations of cells. Furthermore, as NMD requires translation of the target mRNA in the cytoplasm, steady-state mRNA levels are likely also affected by differences in nuclear export rates and differences in the onset and efficiency of translation of different mRNAs. Finally, NMD has been proposed to occur preferentially during a “pioneer round” of translation on mRNAs bound to the nuclear cap-binding complex (CBC) (Ishigaki et al., 2001, Maquat et al., 2010), which is replaced by the cytoplasmic cap-binding protein eIF4E after nuclear export, although this model is debated (Durand and Lykke-Andersen, 2013, Rufener and Mühlemann, 2013). The inability to precisely determine the timing of both NMD and CBC replacement by eIF4E has hampered the understanding of the effect of CBC replacement on NMD.
To overcome the technical challenges associated with bulk and “snapshot” analyses, we have developed an imaging method that allows real-time visualization of both mRNA translation and NMD of single mRNA molecules in living cells. Using this system, we precisely define the timing of NMD, describe a subpopulation of mRNA molecules of variable size that is resistant to NMD, uncover the key parameters that control NMD efficiency, and provide the first real-time kinetic measurements of degradation of the 3′ mRNA cleavage fragment generated by NMD. Taken together, this single-molecule imaging approach reveals key determinants of NMD variability and efficiency and provides a powerful assay to study NMD.
Results
An Assay to Visualize NMD of Single mRNA Molecules in Real Time
To analyze NMD of single mRNA molecules in real time, we modified the SunTag fluorescence labeling approach that we (Tanenbaum et al., 2014, Yan et al., 2016) and others (Wang et al., 2016, Morisaki et al., 2016, Wu et al., 2016, Pichon et al., 2016) have recently developed for studying translation of single mRNA molecules. Briefly, this system uses a reporter mRNA that encodes (1) an array of 24 SunTag peptides near the 5′ end of the coding sequence to monitor translation, and (2) 24 binding sites for the PP7 bacteriophage coat protein (PCP) within the 3′ UTR to monitor the mRNA itself (Chao et al., 2008) (Figure 1A). Upon translation, the SunTag peptides recruit GFP-tagged antibodies (scFv-GFP, stably expressed in the cell) that fluorescently label the nascent protein (Figures 1A–1C). The reporter mRNA is fluorescently labeled and tethered to the plasma membrane via PCP-mCherry-CAAX (Figures 1A–1C); we previously showed that tethering improves visualization without affecting translation (Yan et al., 2016).
Figure 1.

An Assay for Real-Time Visualization of NMD of Single mRNA Molecules
(A) Schematic of NMD single-molecule imaging assay before (top) or after (bottom) NMD induction. Green and red spots (insets) show nascent proteins and reporter mRNA, respectively, as observed through the microscope.
(B) Schematic of the NMD reporter constructs. e, exon; in, intron. PTC160 and PTC1 indicate PTC at amino acids 160 and 1, respectively.
(C–J) U2OS cells expressing scFv-sfGFP and either PCP-mCherry-CAAX (C–G, I, and J) or PCP-HaloTag (H) were transfected with indicated reporter constructs (C–J) and siRNAs (G and J) and were analyzed by time-lapse microscopy.
(C) Representative images of mRNA molecules of indicated reporters are shown. Scale bar, 1 μm. Time is shown in min:s.
(D–J) The time from first detection of translation until separation of red and green foci (i.e., mRNA cleavage) (D–H) or the time from mRNA cleavage until disappearance of the 3′ cleavage fragment (red spot) (I and J) was quantified.
Solid lines and corresponding shaded regions in (D)–(J) represent mean ± SEM. Dotted lines in (H) indicate that the data are replotted from an earlier figure panel for comparison. Number of measurements for each experiment are listed in Table S1. See also Figures S1 and S2 and Videos S1, S2, S3, and S4.
To generate NMD reporter mRNAs, we introduced the sequence encoding the model NMD target Triose Phosphate Isomerase (TPI) in the reporter (Boehm et al., 2014, Belgrader et al., 1994), either with the native stop codon (TPIWT), or with a PTC at amino acid 160 (TPIPTC160), or at amino acid 1 (TPIPTC1) (Figure 1B). Furthermore, we included a sequence encoding BFP upstream of TPI to confirm expression of the reporter and placed the reporter mRNA under control of a doxycycline-inducible promoter (Figure 1B). Human U2OS cells expressing TPIPTC160 or TPIPTC1 displayed a 4- or 12-fold lower level of mRNA expression, respectively, than cells expressing TPIWT, as determined by qRT-PCR (Figure S1A). TPIPTC160 and TPIPTC1 abundance increased 3- to 6-fold upon depletion of the key NMD factor UPF1, consistent with NMD of TPIPTC160 and TPIPTC1 (Figures S1B and S1C). Degradation of NMD reporter mRNAs was not affected by expression of scFv-GFP and PCP-mCherry-CAAX (Figure S1A), demonstrating that fluorescence labeling of mRNA and nascent polypeptides or mRNA tethering do not interfere with NMD.
To observe NMD of single mRNA molecules in real time, human U2OS cells expressing TPIWT or TPIPTC160 reporter mRNAs were followed using time-lapse spinning disk confocal microscopy with a 30 s time interval. Cells expressing the TPIWT reporter displayed red mRNA foci in the cytoplasm within 15–30 min of transcription induction by doxycycline addition. The majority of mRNAs (86% ± 1.1%, mean ± SEM) initially appeared without associated green translation signal but rapidly initiated translation (after 2.3 ± 0.2 min, mean ± SEM). Translation was generally maintained for the remainder of the experiment (∼30–60 min) (Figures 1C, top panel, S1D–S1F; Video S1) (see STAR Methods). Quantitative analysis revealed that the initial GFP appearance on mRNAs represented the first round of translation for almost all mRNAs (>99%) (see STAR Methods).
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIWT. Images were acquired every 30 s (movie duration is 28 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state and then undergo translation. Note that these mRNAs do not undergo cleavage. Movie field of view is 6.21 × 6.21 μm.
Kinetics of transcription and of translation initiation of TPIPTC160 mRNAs were similar to those of TPIWT mRNAs (Figures S1D–S1F). However, the green and red foci associated with individual translating TPIPTC160 mRNAs often separated rapidly after translation had initiated (79% ± 3% in 20 min, mean ± SEM) (Figure 1C, bottom panel; Video S2). In contrast, only 13% ± 3% of TPIWT mRNAs showed foci separation during this time period (Figure 1D). We also observed rapid foci separation for TPIPTC1 reporter mRNAs (90% ± 3%, Video S3), or when TPI was replaced by another NMD model substrate, β-globinPTC39 (84% ± 3%, compared to 8% ± 1% for the β-globinWT reporter) (Figure 1E). Separation of red and green foci likely represents endonucleolytic cleavage of the mRNA, rather than translation shutdown, because multiple ribosomes (5–30) associate with a single mRNA molecule (Figure S1G) and the entire GFP spot (i.e., all 5–30 ribosomes) separates from the mRNA signal in a single step (Figures S1H and S1I; see STAR Methods). The following observations support that the observed mRNA cleavage is induced by NMD; first, cleavage only occurred after the first ribosome reached the stop codon, consistent with an essential role of translation termination at the PTC in NMD induction (Figures 1D and S1J; see STAR Methods). Second, very little cleavage was observed in the absence of a PTC (Figures 1D and 1E). Third, mRNA cleavage required the presence of introns in the mRNA (Figure 1F). Finally, cleavage was strongly reduced after depletion of the important NMD factor UPF1 and the endonuclease SMG6 (9% and 28% cleavage in 20 min, respectively, compared to 78% in mock-treated cells, Figure 1G). Surprisingly, the small amount of cleavage observed with the TPIWT reporter was also reduced upon UPF1 depletion (Figure S1K), suggesting that a small fraction of mRNA molecules are targeted for NMD even in the absence of a PTC, possibly due to stochastic errors in transcription or splicing of TPIWT mRNAs. Together, these results show that red and green foci separation is an accurate readout of NMD induction of single mRNA molecules.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIPTC60. Images were acquired every 30 s (movie duration is 15 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo the pioneer round of translation and get cleaved. Large, bright red structures in the top of the field of view are lysosomes, which can be distinguished from mRNA based on size, fluorescence intensity, and speed of movement. Movie field of view is 8.32 × 8.32 μm.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIPTC1. Images were acquired every 30 s (movie duration is 29 min and 30 s) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo the pioneer round of translation and get cleaved. Large, bright red structures in the top-left of the field of view are lysosomes, which can be distinguished from mRNA based on size, fluorescence intensity, and speed of movement. Movie field of view is 25.52 × 25.52 μm.
Interestingly, for all NMD reporters, a small subset of mRNAs molecules appeared resistant to cleavage (approximately 20%, 10%, and 10% for TPIPTC160, TPIPTC1, and β-globinPTC39 reporters, respectively, Figures 1D and 1E), consistent with heterogeneous NMD kinetics for different subpopulations of mRNAs (Trcek et al., 2013). This cleavage-resistant population could not be explained by heterogeneity in NMD efficiency among different cells (Figures S2A–S2D; see STAR Methods). The cleavage-resistant population was also not a result of differences in translation efficiency between NMD-sensitive and resistant mRNAs (Figure S2E). Therefore, these data suggest that different mRNA molecules are heterogeneous with respect to NMD.
Long 3′ UTRs can stimulate NMD under certain conditions (Singh et al., 2008, Boehm et al., 2014, Bühler et al., 2006), so we tested whether the relatively long 3′ UTR in our reporter mRNA (1,986 nt, including the 24x PP7 binding sites) might affect NMD induction. However, reducing the length of the TPIPTC160 3′ UTR length to 765 nt (containing 5x PP7 binding sites) did not alter NMD kinetics (Figures S2F and S2G; see STAR Methods). Tethering the reporter mRNAs to the plasma membrane also did not detectably alter the kinetics of NMD (Figure 1H; Video S4). Furthermore, we observed similar cleavage kinetics upon transient transfection or stable integration of the reporter gene (Figures S2H and S2I). Therefore, to facilitate the experimental setup and analyses, we used transiently transfected reporters, which contained 24x PP7 binding sites and were tethered to the plasma membrane, unless indicated otherwise.
A U2OS cell expressing scFv-sfGFP (green) and PCP-Halo (red) was transfected with the NMD reporter TPIPTC1. Cells were incubated with 50nM Halo-TMR for 30 min before imaging. Images were acquired every 15 s (movie duration is 20 min) on a spinning disk confocal microscope. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo the pioneer round of translation and get cleaved. Movie field of view is 7.29 × 7.29 μm.
Finally, we examined the fate of the two mRNA fragments that are produced by cleavage of the reporter mRNA. The 5′ fragment rapidly diffused out of the plane of observation after endonucleolytic cleavage, precluding analysis of its degradation. The 3′ fragment, which remains tethered to the membrane, disappeared rapidly after mRNA cleavage (Figures 1C and 1I). Small interfering RNA (siRNA)-mediated depletion of the 5′-to-3′ exonuclease XRN1, but not UPF1, reduced the rate by which red foci disappeared by over 10-fold (Figures 1J and S2J), indicating that red foci disappearance represents XRN1-mediated decay of the 3′ cleavage fragment. In conclusion, our imaging approach allows us to monitor NMD of individual mRNA molecules from start to finish in real time.
Each Ribosome that Terminates Translation at the PTC Has an Equal Probability of Inducing NMD
Earlier studies have suggested that NMD occurs preferentially during the pioneer round of translation, which is generally defined as the first, or first few ribosomes that translate an mRNA, while the mRNA is bound by CBC (Maquat et al., 2010), while other studies provided evidence that NMD can occur during any round of translation (Durand and Lykke-Andersen, 2013, Rufener and Mühlemann, 2013). Since our method allows precise measurements of the timing of the first round of translation and NMD, we wished to determine which ribosome induced NMD.
When examining the cleavage kinetics of TPIPTC1 in more detail, we noticed three distinct phases in the distribution of cleavage times (Figure S3A); in the first phase (0–3 min after GFP appearance), which represents the time it takes for the first ribosome to translate the coding sequence and reach the PTC, very little cleavage occurred (Figure S3A). In the second phase (from 3 to 10 min for TPIPTC1), the curve showed a very steep downward slope, indicating that most mRNA molecules were cleaved during this phase. Finally, during the third phase (>10 min) only a few cleavage events occurred, indicating that the mRNAs that had not yet been cleaved (∼5%–10% of molecules for TPIPTC1) were largely resistant to NMD. Similar phases were observed for other reporters as well (Figures 1D and 1E). The first ribosome was predicted to arrive at the PTC of TPIPTC1 after ∼3 min, yet cleavage was observed over a period of 3–10 min. Two possible models could explain the observed variability in timing of cleavage; first, it is possible that the first ribosome translating an mRNA always induces NMD, and that the variability is caused by variation in arrival time of the first ribosome at the PTC. Alternatively, the first ribosome may arrive at the PTC around 3 min on all mRNAs, and the variability in cleavage time is caused by variation in which ribosome induces NMD (i.e., the first ribosomes or one of the following ribosomes). To distinguish between these two models, we precisely determined the average time as well as the variation in time it takes ribosomes to reach the PTC, which we found to be 2.6 ± 0.8 min (mean ± SD) (Figure S3B; see STAR Methods). We then performed stochastic simulations to determine whether the experimentally determined variation in rate of ribosome translocation could explain the observed cleavage time distribution. In brief, we developed a model to describe the cleavage time distribution using two parameters: (1) the time of arrival of the first ribosome, for which we used our experimentally determined values (Figure S3B), and (2) the fraction of NMD-resistant mRNAs (see STAR Methods). We found that the observed cleavage kinetics of the TPIPTC1 reporter were poorly described by a model in which NMD is induced by the first ribosome (Figure 2A, cf. blue and black lines).
Figure 2.

NMD Occurs with Equal Probability during Each Round of Translation
(A) Experimentally determined cleavage time distribution of TPIPTC1 is shown (black dotted line). Predicted cleavage time distributions based on stochastic simulations are shown for indicated models.
(B) Schematic of indicated reporters.
(C–F) U2OS cells expressing scFv-sfGFP and PCP-mCherry-CAAX were transfected with indicated reporter plasmids (C–F) and siRNAs (E) and were analyzed by time-lapse microscopy.
(C) Representative images of a single HP21-TPIPTC1 mRNA molecule. Scale bar, 1 μm. Time is shown in min:s. Graph shows GFP fluorescence intensity of the mRNA over time. Red filled areas represent peaks that were called as translation events. Yellow numbers indicate the number of ribosomes that contributed to the peak.
(D) Quantification of the number of ribosomes that translated HP21-TPIPTC1 mRNAs before cleavage occurred.
(E and F) The time from first detection of translation until mRNA cleavage was quantified. Black lines indicate the best fit from simulations.
Dotted lines in (A) and (F) indicate that the data are replotted from an earlier figure panel for comparison. Solid lines and corresponding shaded regions in (D)–(F) represent mean ± SEM. Numbers of measurements for each experiment are listed in Table S1. See also Figure S3 and Video S5.
We therefore tested an alternative model, in which the cleavage rate reflects both the time of arrival of the first ribosome and the probability that a ribosome will induce NMD upon translation termination. To this end, we added a third parameter to the model, a probability for each terminating ribosome of inducing NMD. This parameter requires knowledge of the frequency of ribosome termination events, which we calculated to be 3.2 ribosomes per minute based on the translation elongation rate and ribosome occupancy of TPIPTC1 (Figures S1G and S3B; see STAR Methods). The second model resulted in a very good fit with the data (Figure 2A, cf. red and black lines, Akaike information criterion (AIC) = −639 compared to AIC = −308 for the “first ribosome” model described above; see STAR Methods) and revealed that 90% ± 3% (mean ± SEM) of TPIPTC1 mRNAs were degraded in the rapidly degrading population, and that each terminating ribosome induced NMD with a probability of 0.11 ± 0.01 (i.e., a 11% chance of inducing NMD per ribosome) (see STAR Methods). Together, these results strongly suggest that each terminating ribosome has an equal probability of inducing NMD.
To unambiguously determine which ribosome induced NMD of each mRNA, we engineered a TPIPTC1 reporter with a strongly reduced initiation rate by introducing a 21-nucleotide hairpin in the 5′ UTR (which reduced translation by ∼30-fold [Figure S3C]) that allowed us to count individual ribosomes translating the reporter mRNA (HP21-TPIPTC1, Figure 2B). When tracking GFP intensity over time on HP21-TPIPTC1 mRNAs, we observed clear peaks of GFP signal that lasted several minutes, interspersed by periods lacking detectable GFP signal, and we could assign the precise number of ribosomes that made up each peak (Figure 2C; Figure S3E; Video S5; see STAR Methods).
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter HP21-TPIPTC1. Images were acquired every 15 s (movie duration is 17 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo several translation events and finally disappear, indicating mRNA cleavage and 3′ fragment degradation. Movie field of view is 5.94 × 5.94 μm.
When mRNAs are translated by a single ribosome, nascent chain release during translation termination and mRNA cleavage both result in complete separation of the GFP and mCherry foci and are thus indistinguishable. Therefore, to monitor NMD of the HP21-TPIPTC1 mRNAs we defined mRNA cleavage as red and green foci separation rapidly followed by the disappearance of the red foci (i.e., decay of the 3′ cleavage fragment), which was a reliable readout for NMD of HP21-TPIPTC1 mRNAs (Figures S3F and S3G). When counting individual ribosomes translating HP21-TPIPTC1 reporter mRNAs, we found that 8 ribosomes (median) typically translated the HP21-TPIPTC1 reporter mRNA before NMD was triggered (Figure 2D). This corresponds to a probability of 0.10 ± 0.03 (mean ± SEM) of inducing NMD for each terminating ribosome (see STAR Methods), which is in good agreement with the probability of 0.11 per ribosome determined through our stochastic simulation approach for the TPIPTC1 reporter lacking the hairpin sequence (Figure 2A).
NMD Does Not Occur Preferentially on CBC-Bound mRNAs
If NMD occurs preferentially on CBC-bound mRNAs, a progressively slower NMD decay rate over time should be observed due to gradual replacement of CBC by eIF4E within the population of mRNAs. However, the cleavage rate of TPIPTC1, TPIPTC160, and β-globinPTC39 was constant over time (i.e., fit an exponential decay distribution; excluding the subset of mRNAs that are resistant to NMD) (Figure 2A). Decay of these reporters was, however, very rapid (<10 min), so it is possible that all of these mRNAs are degraded while the mRNA is still bound to CBC because the CBC-eIF4E exchange occurs at a time-scale of >10 min. To determine whether the NMD decay rate remains constant over longer time periods, we reduced the NMD efficiency to extend the time window during which the decay rate can be analyzed. First, we performed partial depletion of UPF1 in cells expressing either TPIPTC160 or β-globinPTC39, which revealed that the decay rate remained largely constant over the entire 40 min experiment, as evident from a good fit of the data with an exponential decay distribution (Figures 2E and S3H). Second, we examined NMD efficiency of an mRNA with a lower translation initiation rate to reduce the speed of NMD. For this, we re-plotted the cleavage kinetics of the HP21-TPIPTC1 reporter and found that it also showed a similar rate of decay over the entire experiment (Figure 2F). Together, these results show that NMD occurs at a constant rate over at least 40 min (∼100 rounds of translation).
To definitively determine the efficiency of NMD of both CBC- and eIF4E-bound mRNAs, we wished to determine the exact moment of CBC-to-eIF4E replacement. For this, we made use of a specific inhibitor of eIF4E-dependent translation; a hyperactive, non-phosphorylatable, non-degradable variant of the protein 4E-BP1 (ha4E-BP1) (Yanagiya et al., 2012, Durand and Lykke-Andersen, 2013). As CBC does not bind to 4E-BP1, we reasoned that translation initiation rates of single mRNAs would be unaffected by overexpression of ha4E-BP1 as long as mRNAs were bound to CBC (Durand and Lykke-Andersen, 2013). However, at some point in time, translation rates of single mRNAs would decrease in ha4E-BP1-overexpressing cells compared to control cells, and this time point would represent the moment of replacement of CBC by eIF4E. In the absence of ha4E-BP1 expression, the majority of newly transcribed mRNAs rapidly initiated translation, as evident by the appearance of a green fluorescence signal and continued translating for the remainder of the movie (20–45 min) (Figures 3A, 3B, and S4A). In cells overexpressing ha4E-BP1, initial appearance of green fluorescence occurred with similar kinetics as in control cells (Figures 3C and S4B), suggesting that initial translation is likely driven by CBC on most mRNAs. However, in ha4E-BP1-expressing cells, translation was rapidly shut down on the majority of mRNAs within minutes of initial translation initiation (Figures 3A, 3B, and S4A), indicative of the CBC-to-eIF4E switch.
Figure 3.

NMD Does Not Occur Preferentially on CBC-Bound mRNAs
(A–G) U2OS cells expressing scFv-sfGFP, PCP-mCherry-CAAX, and the translation reporter shown in (A) were transfected with ha4E-BP1 or mock transfected and analyzed by time-lapse microscopy.
(A) (top) Schematic of standardized translation reporter. Representative images of a single mRNA molecule of either mock (upper image panel) or ha4E-BP1 (lower image panel) transfected cells are shown. Scale bar, 1 μm. Time is shown in min:s.
(B–E) GFP fluorescence intensity over time of a representative mRNA (B, D, and E) or of the average of all mRNAs (C). Blue lines (D and E) indicate the best fit from simulations. Blue triangles indicate translation initiation events.
(F) Quantification of the mean translation initiation rate determined by the fitting approach illustrated in (D) and (E).
(G) Quantification of the number of ribosomes that initiated in the first burst of translation. Data were fit with a single exponential decay distribution (blue line).
(H) The calculated fraction of mRNAs that is targeted for NMD while CBC is bound to the mRNA cap (mean ± SEM).
Dotted lines in (D) and (E) indicate that the data are replotted from an earlier figure panel for comparison. Solid lines and corresponding shaded regions in (C), (F), and (G) represent mean ± SEM. Number of measurements for each experiment are listed in Table S1. See also Figure S4.
To determine the precise moment of translation initiation of each ribosome, we made use of a fluorescence fitting algorithm, RiboFitter, which we recently developed (Boersma et al., 2018). In control cells, we found an average translation initiation rate of 2–3 ribosomes per minute, which remained mostly constant over time (Figures 3D and 3F). In contrast, in ha4E-BP1-expressing cells most mRNAs showed a brief burst of translation initiation during which the initiation rate was similar as in control cells, followed by a period without initiation (Figures 3E and 3F). Note that after the period with no translation initiation events, additional bursts of initiation were often observed, which might represent removal of ha4E-BP1 from the cap or binding of a new eIF4E molecule that was not bound to ha4E-BP1 to the mRNA cap (Figures 3E and S4C). The first burst of translation initiation in ha4E-BP1-expressing cells likely reflects CBC-bound translation, and the end of the translation initiation burst thus reflects the transition of CBC-to-eIF4E and binding of ha4E-BP1 to eIF4E. Analysis of the size of the first translation initiation burst in ha4E-BP1-expressing cells revealed that the median number of ribosomes that initiated on a CBC-bound mRNA before CBC was replaced by eIF4E was 8.7 (average of 3 experiments, Figure 3G). As the NMD cleavage rate remained constant for >40 min (∼100 rounds of translation, much longer than the duration of CBC-dependent translation), it follows that NMD efficiency is similar on CBC- and eIF4E-bound mRNAs.
Using the quantitative data on the kinetics of the CBC-to-eIF4E switch, we developed a model to calculate the fraction of mRNAs on which NMD was induced while CBC was bound to the mRNA cap. This fraction not only depends on the efficiency of NMD induction, but also on the length of the open reading frame (ORF); ribosomes require more time to reach the stop codon of long ORFs, which increases the time during which CBC-to-eIF4E replacement could occur. Our modeling approach suggests that for highly efficient NMD substrates with a short ORF, such as the endogenous β-globin mRNA with a PTC at position 39 (ORF of 117 nt), 56% of mRNAs would be targeted for NMD while CBC is bound to the cap (Figure 3H). However, on mRNAs that are less rapidly targeted for NMD, such as weaker NMD substrates (e.g., TPIPTC160), mRNAs with a lower translation initiation rate (e.g., HP21-TPIPTC1), or substrates with a longer ORF (e.g., endogenous mRNAs with a long ORF, or reporters mRNAs containing the SunTag sequence), NMD will be induced more frequently when eIF4E is bound to the cap. For example, our modeling suggests that only 13% of cleavage events of the HP21-TPIPTC1 reporter mRNA (ORF length of 2,517 nt) occur while the mRNA is bound to CBC (Figure 3H).
To confirm that a subset of TPIPTC160 or TPIPTC1 mRNAs are bound by eIF4E at the moment of NMD induction, we also analyzed their cleavage rate in ha4E-BP1-expressing cells. As expected, NMD was delayed in these cells compared to control cells, and the magnitude of this delay was close to the magnitude predicted by our modeling (Figures S4D and S4E). Taken together, these data demonstrate that NMD occurs with equal probability on CBC- and eIF4E-bound mRNAs and that the fraction of mRNAs undergoing NMD while bound to CBC depends on the NMD efficiency, translation initiation rate, and ORF length.
Exon Sequence Downstream of the PTC Influences NMD Efficiency
Our real-time imaging approach revealed that both the cleavage rate and the fraction of NMD-resistant mRNAs can vary between different reporter mRNAs (e.g., Figure 1D). This assay therefore provides a unique opportunity to determine the mechanisms regulating these parameters, and thus NMD efficiency.
First, we focused on the role of gene-specific nucleotide sequences on NMD efficiency. We found that the sequence of the PTC sequence itself did not affect NMD efficiency (Figure S5A). Next, we examined the effect of nucleotide sequences downstream of the PTC. To directly compare different sequences, we generated a standardized reporter that contains a constant coding (Kif18b) and PTC (TAA) sequence but variable sequences downstream of the PTC. We then inserted 14 randomly selected single introns with their native flanking exons (“exon-intron-exon” referred to as “intron cassettes”) in the standardized reporter downstream of the PTC (Figure 4A). First, we assessed splicing efficiency of the introns in each of these reporters, either using a two-color fluorescence splicing reporter (Figure S5B; see STAR Methods), or by qPCR (Figures S5C and S5D). As expected, reporters that were efficiently spliced generally showed NMD, while unspliced reporters did not (Figure S5E) and hence were excluded from further analysis. Two reporters, ERAL1 and MITD1, showed moderate to strong splicing in the fluorescence-activated cell sorting (FACS)-based reporter but no splicing by qPCR, yet both reporters showed cleavage in the NMD assay. The observed cleavage was dependent on splicing of the intron, as cleavage was eliminated upon removal of the intron or mutation of the splice sites (Figures S5G and S5H), indicating that they are undergoing splicing-dependent NMD as well.
Figure 4.

The NMD Cleavage Rate Is Variable and Depends on Exon Sequences Downstream of the PTC
(A) Schematic of indicated reporter.
(B–G) U2OS cells expressing scFv-sfGFP and PCP-mCherry-CAAX were transfected with indicated reporter plasmids (B–G) and UPF1 siRNA (D) and analyzed by time-lapse microscopy. The time from first detection of translation until mRNA cleavage was determined (B–F).
(B) Reporters were designed as in (A) and encoded an intron cassette of indicated genes.
(C) Cleavage rates for each reporter shown in (B).
(E) Schematic of indicated reporters.
(G) Quantification of the duration that individual mRNAs that were not cleaved could be observed over time.
Solid lines and corresponding shaded regions in (B), (D), (F), and (G) represent mean ± SEM. Dotted lines in (D) and (F) indicate that the data are replotted from an earlier figure panel for comparison. Error bars in (C) represent SEM. Number of measurements for each experiment are listed in Table S1. See also Figure S5.
Among well-spliced reporters, we observed striking differences in the cleavage rates, with the probability of inducing NMD for each terminating ribosome ranging from 0.008 to 0.14 (for MITD1 and TPI, respectively, Figures 4B, 4C, and S5F). For three reporters with varying cleavage rates, we confirmed that cleavage was dependent on UPF1 and on splicing (Figures 4D, S5G, and S5H), confirming that the observed cleavage is caused by NMD. The differences in the observed cleavage efficiencies were not caused by differences in intron sequences, as swapping introns did not affect NMD efficiency (Figures 4E and 4F), suggesting that the exon sequences downstream of the PTC determine the NMD cleavage rate. For reporters that showed a relatively slow rate of cleavage (ERAL1, MITD1, DNAL4, and NUBP2), we examined whether the mRNA molecules that were not cleaved were instead degraded through exonucleolytic decay but found no evidence for exonucleolytic decay within the time frame of our experiments (Figure 4G). These results show that NMD cleavage rates can vary substantially depending on the mRNA sequence downstream of the PTC.
The PTC-to-Intron Distance Affects Both the Cleavage Rate and Fraction of NMD-Resistant mRNAs
Genome-wide studies revealed that in long exons, a large PTC-to-intron distance can result in a reduced NMD efficiency (Lindeboom et al., 2016). However, if a PTC is close (<50–55 nt) to the last intron, NMD efficiency is also reduced, likely because EJCs are displaced from the mRNA by the translocating ribosome (Dostie and Dreyfuss, 2002, Lejeune et al., 2002). Using our assay, we can distinguish whether PTC-to-intron distance affects the cleavage rate or fraction of NMD-resistant mRNAs, which could shed new light on the mechanisms underlying these observations. We introduced linker sequences of different lengths (100, 175, 250, and 1,000 nt) between the PTC and downstream intron of the Kif18bPTC-TPI-intron 6 reporter, in which the PTC is located 91 nt upstream of intron (Figure 5A). Increasing the linker length decreased the cleavage rate up to 7-fold (Figures 5B–5D), suggesting that the probability that a terminating ribosome will induce NMD depends on its proximity to the downstream EJC. The dependence of the cleavage rate on the PTC-to-intron distance was also observed with other linker sequences and reporters (Figures S6A–S6E). Interestingly, although larger PTC-to-intron distances resulted in reduced cleavage rates, the fraction of NMD-resistant mRNAs was not substantially unaffected (Figures 5C and 5D).
Figure 5.

PTC-to-Intron Distance Affects Both the Decay Rate and Fraction of NMD-Resistant mRNA Molecules
(A) Schematic of indicated reporters.
(B–D and F) U2OS cells expressing scFv-sfGFP and PCP-mCherry-CAAX were transfected with indicated reporter plasmids and analyzed by time-lapse microscopy.
(B) The time from first detection of translation until mRNA cleavage was determined.
(C and D) Cleavage rates (C) and fraction of mRNAs sensitive to NMD (D) for each reporter shown in (B).
(E) Schematic of indicated reporters.
(F) The time from first detection of translation until mRNA cleavage was determined.
Dots represent experiments; lines show the mean of all experiments. Solid lines and corresponding shaded regions in (B) and (F) represent mean ± SEM. Dotted line in (B) indicates that the data are replotted from an earlier figure panel for comparison. Number of measurements for each experiment are listed in Table S1. See also Figure S6.
To explore the effects of very short PTC-to-intron distances on NMD, we introduced new PTCs in the TPI gene (Figure 5E). Decreasing the distance between the PTC and the downstream intron to 73, 52, or 40 nt (TPIPTC185, TPIPTC192, and TPIPTC196, respectively) led to a strong reduction in NMD (Figure 5F), as expected. Interestingly, the reduction in NMD efficiency was mostly caused by a substantial increase in the fraction of NMD-resistant mRNAs (Figure 5F), consistent with a model in which the EJC can be displaced from the mRNA by the translating ribosome if it is too close to the PTC, thereby preventing NMD during future rounds of translation. Surprisingly, we found that a fraction of mRNAs was still susceptible to NMD in all 3 reporters (TPIPTC185/192/196) (Figure 5F), suggesting that the EJC was not displaced by translating ribosomes from all mRNAs, even when it is positioned very closely to the PTC.
The Number of Introns Both Upstream and Downstream of the PTC Affects the NMD Cleavage Rate and the Fraction of NMD-Resistant mRNAs
Next, we examined the effect of altering the number of introns in an mRNA on the NMD decay rate and the fraction of NMD-resistant mRNAs. We added an extra copy of a single intron cassette to our standardized reporter (Kif18bPTC-CD63) to create the Kif18bPTC-2xCD63 reporter (Figure 6A). These reporters have an identical PTC-to-intron distance, and the same nucleotide sequence immediately downstream of the PTC, but a different number of introns. Introduction of a second intron downstream of the PTC resulted in a faster decay rate and also a reduced fraction of NMD-resistant mRNA molecules (Figure 6B), indicating that multiple downstream introns enhance NMD through two parallel mechanisms. Similar results were obtained with a second set of reporters (Figures S7A and S7B). To support these findings, we performed analysis of the effects of nonsense mutations on mRNA levels in a large cohort of previously sequenced cancer samples (Lindeboom et al., 2016). Genome-wide analysis revealed that PTCs with only a single EJC downstream of the PTC had a significantly lower NMD efficiency than PTCs with multiple (>3) downstream EJCs (p = 4.9 ∗ 10−5, Mann-Whitney U test, Figure 6C).
Figure 6.

The Number and Position of Introns Relative to the PTC Affects the NMD Decay Rate and Fraction NMD-Resistant mRNA Molecules
(A) Schematic of indicated reporters.
(B and E) U2OS cells expressing scFv-sfGFP and PCP-mCherry-CAAX were transfected with indicated reporter plasmids and analyzed by time-lapse microscopy.
(B) The time from first detection of translation until mRNA cleavage was determined.
(C and F) Genome-wide analysis of the effect of the number of downstream (C) or upstream (F) EJCs on NMD efficiency in a large cohort of cancer samples. Only PTCs with a single downstream EJC were included in (F).
(D) Schematic of indicated reporters.
(E) The time from first detection of translation until mRNA cleavage was determined.
Solid lines and corresponding shaded regions in (B) and (E) represent mean ± SEM. Dotted lines in (B) and (E) indicate that the data are replotted from an earlier figure panel for comparison. In boxplots of (C) and (F), the boxes represent the interquartile range with the central line depicting the median, and the whiskers extend to the extreme values after removing outliers. p values are indicated as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by two-tailed Mann-Whitney U tests. Number of measurements for each experiment are listed in Table S1. See also Figure S7.
We considered two possible models to explain the enhanced NMD cleavage rate of mRNAs containing multiple introns. First, deposition of multiple EJCs downstream of the PTC could increase the probability that a terminating ribosome interacts with an EJC and induces NMD. Alternatively, multiple introns could enhance NMD through altered mRNA processing, for example, by enhancing the loading of a protein factor onto the mRNA that stimulates NMD. In the first model, NMD efficiency is only enhanced when introns are placed downstream of a PTC, while, in the second model, NMD efficiency could also be affected when introns are inserted upstream of a PTC. To distinguish between these models, we placed additional introns either upstream or downstream of the PTC in the Kif18bPTC-NUBP2 reporter (Figure 6D) (which had a moderate cleavage rate and thus allows detection of both increases and decreases in the cleavage rate). Interestingly, inserting 4 additional intron cassettes upstream of the PTC enhanced NMD efficiency (Figure 6E). The observed cleavage was still mostly dependent on the presence of the PTC, suggesting that upstream introns did not trigger NMD through generation of new PTCs due to splicing errors (Figure 6E). Inserting 4 intron cassettes downstream of the PTC stimulated the NMD decay rate even further (Figure 6E), suggesting that the presence of multiple EJCs downstream of the PTC also enhances NMD induction during translation termination. Similar results were obtained when using multiple different sets of reporters (Figures S7C–S7F). Enhancement of NMD by upstream introns was also observed in the genome-wide cancer dataset; when the PTC was located in the penultimate exon, we observed a significantly lower NMD efficiency when 0 or 1 upstream introns were present than when 4 or more upstream introns were present (p = 0.04 and p = 0.001, respectively, Mann-Whitney U test, Figure 6F). Upstream introns did not further increase NMD efficiency when two or more introns were present downstream of the PTC (Figure S7G). Together, these results suggest that the presence of multiple introns in an mRNA enhances NMD through two distinct mechanisms.
Decay Kinetics of the 3′ Fragment after mRNA Cleavage
After NMD-dependent mRNA cleavage, the 3′ cleavage fragment is degraded by XRN1 (Gatfield and Izaurralde, 2004; Figure 1J). While in vitro studies have shown that XRN1 displays high processivity (Jinek et al., 2011, Chang et al., 2011, Stevens, 1980), very little is known about the speed and processivity by which XRN1 degrades mRNAs in vivo. To examine the kinetics of XRN1-mediated mRNA degradation in vivo, we imaged TPIPTC160 mRNAs with high temporal resolution (5 s interval) (Figures 7A and 7B) and quantified the decrease in red fluorescence intensity of individual mRNAs over time (Figures 7B and 7C). This analysis revealed an exonucleolytic decay speed of 38 nt/s (median, see STAR Methods and Figure 7C). To examine processivity of XRN1, we determined how partial depletion of XRN1 affects the onset and rate of decay; if XRN1 is highly processive, reduced levels of XRN1 would be expected to slow the onset but not the rate of decay. In contrast, if XRN1 is non- or weakly processive, decay should initiate at approximately the same time but show a decreased rate (Figure 7D). We found that the onset of decay was delayed by XRN1 depletion (160 s versus 50 s in XRN1-depleted versus control cells, Figures 7D and 7E) but that the decay rate was similar (median rate 38 versus 31 nucleotides/s; Figure 7F; see STAR Methods), suggesting that most TPIPTC160 3′ cleavage fragments are degraded by a single, processive XRN1 molecule.
Figure 7.

mRNA Decay Kinetics of XRN1
(A) Schematics of indicated reporters.
(B, C, and E–K) U2OS cells expressing scFv-sfGFP and PCP-Cherry-CAAX were transfected with either TPIPTC160 (B, C, E, and F) or with TPIPTC160-48xPP7 (G–K) reporter plasmids and with indicated siRNAs (C, E, F, and I-K). Cells were analyzed by time-lapse microscopy at 5 s (C, E, and F) or 15 s (H–K) time interval. (B and H) Representative images of single mRNA molecules are shown. Scale bar, 1 μm. Time is shown in min:s. (B, C, H, and I) mCherry fluorescence intensity over time of representative example mRNAs. Dashed blue lines in (C) indicate best fit from simulations (see STAR Methods).
(D) Schematic illustrating the expected fluorescence intensities over time upon XRN1 depletion.
(E, F, J, and K) Quantification of the time between cleavage and onset of 3′ fragment degradation (E and J) and the time between onset of degradation and complete disappearance of the 3′ cleavage fragment (F) and of the duration of the plateau phase (K). 3 experiments in (J) were grouped because of the low number of mRNAs that could be analyzed. (G) mCherry fluorescence intensities of individual mRNA foci of indicated reporters were measured at the time point immediately before the mRNA was cleaved. Dots in (E) and (F) represent single mRNA molecules; lines show mean ± SEM. Solid lines and corresponding shaded regions in (K) represent mean ± SEM. Number of measurements for each experiment are listed in Table S1.
See also Video S6.
To analyze XRN1 processivity more precisely, we generated a new reporter with a second 24x PP7 array followed by a 2.6 kb linker sequence upstream of the original 24x PP7 array (TPIPTC160-48xPP7), which showed an approximately 2-fold increased mCherry fluorescence, as expected (Figures 7A and 7G). The TPIPTC160-48xPP7 reporter has a much longer 3′ UTR than TPIPTC160 (5 versus 1.5 kb), increasing the likelihood of XRN1 dissociation from the 3′ cleavage fragment, thus providing a more sensitive readout for XRN1 processivity. Decay of the first 24x PP7 array results in an ∼50% decrease in fluorescence, after which the fluorescence intensity remains constant during the decay of the 2.6 kb linker sequence (referred to as the plateau phase), followed by another decrease in fluorescence when the second 24x PP7 array is degraded (Figures 7H and 7I; Video S6). Interestingly, when analyzing many mRNA molecules, the duration of the plateau phase showed a clear bimodal distribution; ∼60% of molecules displayed rapid degradation of the linker (median time 0.8 min, Figure 7J, black line), which is in good agreement to the predicted decay time of ∼1.1 min for the linker sequence based on our measured XRN1 decay speed. The remaining molecules displayed much slower decay of the linker (median time 8.5 min). The rapidly degrading mRNA population likely represents processive degradation of the linker via a single XRN1 binding event, whereas the slowly degrading mRNAs could represent degradation that requires two or more XRN1 binding events. Consistent with this hypothesis, depletion of XRN1 did not substantially affect the fraction of rapidly degraded mRNA molecules (∼50%) but dramatically increased the decay time of the slowly degrading mRNAs (Figure 7J). Furthermore, a strong delay in the initial recruitment of XRN1 was observed by XRN1 depletion, and this delay was similar to the delay in the degradation time of the linker (Figures 7J and 7K; see STAR Methods). This further suggests that the slowly decaying population of mRNAs represent mRNAs in which XRN1 fell off the mRNA before completing degradation, and that the increased time required for degradation of the linker in XRN1-depleted cells is caused by slower recruitment of a new XRN1 molecule when cellular XRN1 levels are low. Together, these results show that XRN1 is a fast and highly processive enzyme in vivo, but many mRNAs nonetheless require two or more XRN1 binding events for complete mRNA degradation.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIPTC60-48xPP7. Images were acquired every 15 s (movie duration is 10 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. After cleavage of the mRNA, the 3′ cleavage fragment (red) becomes progressively dimmer as XRN1 degrades the mRNA. Movie field of view is 3.6 × 3.6 μm.
Discussion
SunTag Translation Imaging—A Method to Study NMD
A major strength of our single-molecule imaging NMD assay is that it accounts for many variable factors that can influence the steady-state mRNA levels of an NMD target. For example, delayed nuclear export or delayed translation initiation would extend the lifetime of mRNAs, making them appear less sensitive to NMD in bulk mRNA decay measurements, while our assay distinguishes between these possibilities. In addition, single-molecule measurements can uncover different mRNA subpopulations, for instance, with distinct sensitivities to NMD. Although exogenous mRNA sequence elements are required for the assay (i.e., SunTag and PP7 binding sites), we find that our NMD reporter faithfully recapitulates key aspects of NMD, including the dependence on a PTC and an EJC downstream of the PTC, a requirement for the key NMD factors UPF1 and SMG6, and exonucleolytic decay of the 3′ cleavage fragment by XRN1. We note that a small percentage of mRNAs (∼5%) is cleaved even in the absence of a PTC or EJCs, so it is possible that these mRNAs are cleaved through EJC-independent NMD or through a mechanism other than NMD. Long 3′ UTRs can stimulate NMD by increasing the distance between a PTC and the poly(A) tail (Bühler et al., 2006, Singh et al., 2008, Eberle et al., 2008). However, for the reporters tested here, the length of the 3′ UTR did not play a major role in inducing NMD (Figures 1F, S2F, S2G, S5G, and S5H). Thus, our single-molecule imaging method faithfully recapitulates most, if not all, aspects of NMD and therefore adds a unique tool to study NMD timing, kinetics, and heterogeneity. The mRNA cleavage and exonucleolytic decay assays developed here may also be adapted to study other forms of mRNA quality control and more generally other aspects of RNA biology involving mRNA translation and decay.
Probability that a Ribosome Induces NMD during Termination
We provide multiple lines of evidence that NMD occurs with equal probability during each round of translation (Figure 2) and that NMD efficiency is not preferentially induced on CBC-bound mRNAs (Figure 3). Nonetheless, we find that on efficient NMD substrates up to 56% of mRNA molecules are targeted for NMD, while the mRNA is still bound to the CBC. Together, these findings could reconcile the apparently contradictory observations that NMD is preferentially induced during the “pioneer” round of translation (Ishigaki et al., 2001) and observations that NMD can be efficiently induced on eIF4E-bound mRNAs (Durand and Lykke-Andersen, 2013, Rufener and Mühlemann, 2013), thus potentially providing a unifying model for NMD induction.
The observation that only a relatively small subset of termination events results in NMD may also explain why translation of upstream open reading frames (uORFs) does not result in substantial NMD (Calvo et al., 2009). If a ribosome translates a uORF before the main ORF has been translated, it would terminate on the uORF stop codon, while EJCs are still associated with the main ORF, potentially triggering NMD. Since only a few termination events would occur on the uORF stop codon before the main ORF is translated and EJCs are removed, a low probability of inducing NMD for each translation termination event would largely prevent NMD on uORF-containing mRNAs, while allowing rapid NMD of mRNAs containing a bona fide PTC.
An NMD-Resistant Subpopulation of mRNA Molecules
Most mRNA molecules are efficiently degraded by NMD, but a subpopulation of mRNA molecules, generally ranging from 5%–30%, is resistant to NMD-dependent mRNA cleavage, consistent with a previous report (Trcek et al., 2013). One possibility is that NMD-resistant mRNA molecules arise from a (stochastic) failure in splicing or EJC deposition on a subset of mRNA molecules. Consistent with this, our results show that insertion of additional introns downstream of a PTC reduces the fraction of NMD-resistant mRNAs (Figure 6B). The presence of multiple introns would increase the fraction of mRNAs that contains at least one EJC complex downstream of the PTC. In this study, all mRNAs were expressed from a single promoter, so an interesting open question is to what extent the promoter and chromatin context affects the fidelity of splicing and EJC loading, and thus potentially the fraction of NMD-resistant mRNAs. While variability in splicing and/or EJC deposition may explain part of the NMD-resistant mRNAs, a small fraction of mRNAs escapes NMD even in reporter mRNAs that contain multiple introns located downstream of the PTC, suggesting that an additional mechanism may also contribute to NMD escape.
XRN1 Speed and Processivity
We found that XRN1 degrades 3′ cleavage fragments with high speed and processivity but occasionally dissociates from the mRNA. The observed XRN1 degradation speed (38–55 nt/s) is substantially higher than the translocation speed of ribosomes (9–15 nt/s), which explains why XRN1 trails ribosomes during co-translational mRNA decay (Pelechano et al., 2015). We also found that human XRN1 efficiently degrades structured RNAs with tightly bound RBPs (i.e., PP7 binding site with bound PCP), consistent with a previous study, but contrasting results obtained with yeast XRN1 (Garcia and Parker, 2015, Horvathova et al., 2017, Kim et al., 2019). Finally, a recent study found that degradation of PTC-containing mRNAs in zebrafish can lead to transcriptional adaptation, a process in which genes with sequence similarity to the degraded mRNA are upregulated, and the study showed that transcriptional adaptation was dependent on XRN1-mediated decay of NMD substrates (El-Brolosy et al., 2019). Upregulation of these genes was sequence specific, and the authors speculated that mRNA decay intermediates may play a role. Since we found that XRN1 occasionally dissociates from an mRNA during degradation, which results in production of decay intermediates, XRN1 dissociation from mRNAs during decay could be important for this process of transcriptional adaptation.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | GIBCO | Cat# 31966021 |
| Leibovitz’s L15 medium | GIBCO | Cat# 21083-027 |
| Penicillin-Streptomycin | GIBCO | Cat# 15140-122 |
| Fetal Bovine Serum (FBS) | Sigma-Aldrich | Cat# F7524 |
| Doxycycline | Sigma-Aldrich | Cat# D9891-1G |
| Opti-MEM | Sigma-Aldrich | Cat# 11058-021 |
| FuGENE 6 Transfection Reagent | Promega | Cat# E231A |
| Lipofectamine RNAi-MAX | Invitrogen | Cat# 13778-075 |
| Polyethylenimine | Polysciences Inc | Cat# 23966 |
| Zeocin | Invitrogen | Cat# R25001 |
| Trimethoprim | Sigma-Aldrich | Cat# T7883-5G |
| TRIsure | Bioline | Cat# Bio-38033 |
| Bioscript Reverse Transcriptase | Bioline | Cat# Bio-27036 |
| Halo-TMR ligand | Promega | Cat# G8252 |
| RNase Inhibitor | New England Biolabs (NEB) | Cat# M0307L |
| Polybrene | Santa Cruz Biotechnology, Inc | Cat# sc-134220 |
| Critical Commercial Assays | ||
| iQ SYBR Green SuperMix | Bio-Rad | Cat# 1708885 |
| Experimental Models: Cell Lines | ||
| Human U2OS cells | Tanenbaum lab | Cat# HTB-96 |
| HEK293T cells | Tanenbaum lab | Cat# CRL-3216 |
| Oligonucleotides | ||
| siRNA targeting sequence-UPF1:GCAGUUCCGCUCCAUUUUGAU | Dharmacon | N/A |
| siRNA targeting sequence-XRN1: AGAUGAACUUACCGUAGAAUU | Dharmacon | N/A |
| siRNA targeting sequence-SMG6: GGGUCACAGUGCUGAAGUAUU | Dharmacon | N/A |
| See Table S1 for all primers used for RT-qPCRs. | This study | N/A |
| Recombinant DNA | ||
| See Table S1 for all plasmids used in the paper | This study | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Graphpad Prism 7 | GraphPad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| MATLAB R2012b | The Mathworks, Inc. | https://nl.mathworks.com/products/matlab.html |
| Micromanager for microscope control | Micro-Manager 1.4.22 | https://micro-manager.org |
| NIS elements 5.11.01 | Nikon | https://www.microscope.healthcare.nikon.com/en_EU/products/software |
| TransTrack (MATLAB) | Boersma et al., 2018 | https://github.com/TanenbaumLab |
| RiboFitter (R) | Boersma et al., 2018 | https://github.com/TanenbaumLab |
| Other | ||
| 96-well glass bottom imaging plates-(Matriplates) | Brooks Life Science Systems | Cat# MGB096-1-2-LG-L |
| Deposited Data | ||
| Raw data of imaging experiments | Mendeley data | https://doi.org/10.17632/bw255hcw7h.1 |
Contact for Reagents and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Marvin Tanenbaum (m.tanenbaum@hubrecht.eu)
Experimental Model and Subject Details
U2OS and HEK293T cell culture
Human U2OS cells and HEK293T cells (ATCC) were grown in DMEM (4.5g/L glucose, GIBCO) containing 5% fetal bovine serum (Sigma-Aldrich) and 1% penicillin/streptomycin (GIBCO). Cells were grown at 37°C and with 5% CO2.
Method Details
Cells, Plasmids, Transfections and Lentiviral infections
Plasmids
The complete list and sequence of all plasmids used in this study is provided in Table S1.
Plasmid and siRNA transfections
For imaging experiments, plasmid transfection of U2OS cells was performed in 96-well glass-bottom imaging plates 24 hr before imaging, using 0.5 μl FuGENE 6 (Promega) and 100-200 ng DNA per well. In experiments in which ha4E-BP1 was overexpressed, cells were transfected with ha4E-BP1 plasmid 16h before the start of imaging to reduce toxicity associated with overexpression of this protein. The transfection mix was prepared in OptiMEM (Sigma-Aldrich) and added to the cells in a total volume 150-200 μL of medium. Transfections in 24-well plates were performed using 1 μL FuGENE and 200-400 ng DNA per well in a total volume of 300 μl.
For experiments in which siRNA transfections and plasmid transfections were combined, U2OS cells were first reverse transfected with siRNAs at a final concentration of 10 nM (unless stated otherwise) using Lipofectamine RNAiMAX (Invitrogen) and seeded in plastic 24-well plates. After 24hr, the cells were trypsinized, transfected with a second dose of 10 nM siRNA and re-plated in 96-well glass-bottom imaging plates. 48 hr after the first siRNA transfection, cells were transfected with plasmid DNA, as described above. 24 hr after DNA transfection, cells were analyzed by time-lapse microscopy. The sequences of the siRNAs used in this study are listed in the Key Resource table.
For generation of cells stably expressing reporter mRNAs, U2OS cells were transfected with indicated reporter plasmids. 24 hr after transfection, selection for stable integration was performed using 0.4mg/ml Zeocin (Invitrogen) for 10 days.
Lentivirus production and infection
For lentivirus production, HEK293T cells were transfected with the lentiviral vector along with lentiviral packaging plasmids pMD2.g and pspax2 using Polyethylenimine (PEI) (Polysciences Inc). The medium was replaced the day after transfection with fresh culture medium, and 72 hr after transfection, viral supernatant was collected. For lentiviral infections, cells were seeded in a 6-well plate at about 70% confluency. Viral supernatant was added to the cells along with Polybrene (10μg/ml) (Santa Cruz Biotechnology Inc) and the cells were spun at 2000 rpm for 90 min at 22°C (Spin-infection). After the spin-infection, the culture medium was replaced with fresh medium, and cells were incubated for at least 48 hr before further analysis.
Microscopy
Unless stated otherwise, all live-cell imaging experiments were performed using U2OS cells expressing TetR, scFv-sfGFP and PCP-mCherry-CAAX (Yan et al., 2016, Ruijtenberg et al., 2018). Cells were seeded 48h before imaging in 96-well glass bottom dishes (Matriplates, Brooks Life Science Systems) at 20%–25% confluency. Cells were transfected with reporter plasmid DNA 24h before imaging. Thirty minutes before imaging, the cell culture medium was replaced with pre-warmed CO2-independent Leibovitz’s-15 medium (GIBCO) and transcription of the reporters was induced by addition of doxycycline (1 μg/ml) (Sigma-Aldrich). Images were acquired using a Nikon TI inverted microscope with perfect focus system equipped with a Yokagawa CSU-X1 spinning disc, a 100x 1.49 NA objective and an iXon Ultra 897 EM-CCD camera (Andor) using Micro-Manager software (Edelstein et al., 2010) and NIS software (Nikon). During the experiment, cells were maintained at a constant temperature of 37°C. Unless stated otherwise, single Z-plane images were acquired, with the bottom of the cell in the focal plane. Camera exposure times of 500 ms were used for both GFP and mCherry, and images were acquired with an interval of 30 s, unless stated otherwise. Of note, mCherry-positive lysosomes were visible in most cells, but these could easily be distinguished from mRNAs based on fluorescence intensity and diffusion kinetics.
In experiments in which untethered mRNAs were tracked, U2OS cells expressing PCP-Halo (instead of PCP-mCherry-CAAX) were used. Cells were labeled with Halo-TMR ligand (Promega) (50nM concentration for 2 h) before imaging. Single Z-plane images were acquired, with the region just below the nucleus of the cell in the focal plane. Images were taken with an interval of 15 s and camera exposure times of 500 ms were used for both GFP and TMR.
In experiments in which the intensity of green spots was measured in 3D before and after cleavage, Z stacks were acquired for GFP. We acquired 11 slices with an inter-slice distance of 1 μm each, and used a 100 ms exposure time. For mCherry, a single Z-plane was imaged with 500 ms exposure time. Images were acquired at a 10 s time interval.
In experiments in which the intensity of red spots was measured after mRNA cleavage, low laser power (∼8x lower than used for other imaging) and high exposure times (1500 ms) were used for mCherry to reduce photobleaching and increase signal-to-noise. This enabled accurate detection and measurement of mCherry foci for > 300 time points.
Quantitative RT-PCR
U2OS cells stably expressing TetR or TetR, scFv-sfGFP, PCP-mCherry-CAAX (Yan et al., 2016) were seeded in 24-well plastic bottom plates at ∼10% confluency 72h before harvesting of cells. When the effect of UPF1 siRNA on reporter expression was assessed, a reverse transfection with 10 nM siRNA against UPF1 was performed during seeding, and another siRNA transfection was performed 24h after seeding. 48h after seeding, equal amounts of TPIWT, TPIPTC160 or TPIPTC1 reporter constructs were co-transfected with a control plasmid, and doxycycline was added for 24h to induce transcription of the reporters. 72hr after cell seeding, RNA was isolated using RNeasy plus mini kit (QIAGEN) according to manufacturer’s guidelines, and cDNA was generated using Bioscript reverse transcriptase (Bioline) and Oligo-d(T) primers. qPCRs were performed using SYBR-Green Supermix (Bio-Rad) on a Bio-Rad Real time PCR machines (CFX Connect Real-Time PCR Detection System). RNA abundance of reporter mRNAs was measured using two different primer sets that amplified a ∼200 nt regions upstream or downstream of the PTC. Reporter mRNA abundance was normalized to the expression of the control plasmid that was co-transfected to control for differences in transfection efficiency. The average of the two primer sets was then used as the final value for mRNA abundance.
For checking efficiencies of UPF1 and XRN1 siRNAs by qPCR, U2OS cells expressing TetR, scFv-sfGFP and PCP-mCherry-CAAX (Yan et al., 2016) were seeded in 24-wells plates. Respective siRNAs (10mM) were transfected during cell plating using reverse translation and cells were harvested 3 days after transfection. RNA isolation was performed using TRIsure (Bioline) according to manufacturer’s protocol. cDNA synthesis and qPCRs were performed as described above, except that GAPDH mRNA levels were used to normalize mRNA abundance.
For determining splicing efficiency of NMD reporters, U2OS cells expressing TetR were seeded in 24-wells plates at 20%–25% confluency 48h before harvesting. After 24h cells were transfected with either a kif18b reporter that contained an intron or a matched reporter in which the intron sequence was removed from the plasmid. 3h before harvesting of cells, doxycycline was added to the cell culture medium to induce transcription, and 200 μg/ml cycloheximide was added to prevent degradation of spliced transcripts by NMD. RNA was isolated using RNeasy plus mini kit (QIAGEN) with on-column DNase treatment (RNase-free DNase set, QIAGEN) according to manufacturer’s protocol. cDNA synthesis and qPCR were performed as described above. To assess splicing efficiency, we used a primer set that amplified the reporter mRNA independent of its splicing status (total transcript) and a primer set for which one primer binds at the exon-exon junction, which only generates a PCR product when the transcript is spliced. The no-intron control reporter has the same mRNA sequence as the spliced transcript, and should therefore be amplified by both primer sets. We compared the ratio in abundance of the two amplicons (e.g., ‘total’ and ‘spliced’), and normalized this ratio to the ratio of total and spliced amplicons obtained with the no-intron control reporter.
To ensure that the ‘spliced’ mRNA-specific primer set is indeed specific to spliced mRNAs, we tested the spliced mRNA-specific primer set on plasmid DNA. Amplification was tested on plasmid DNA in which the intron was present, which resembles unspliced mRNA and should thus not be amplified, and plasmid DNA in which the intron was not present, which resembles spliced mRNA and should be efficiently amplified. Each spliced mRNA-specific primer set amplified plasmid DNA lacking an intron > 500 fold more efficiently than plasmid DNA containing an intron, confirming the specificity of these primer sets.
Flow Cytometry
For analysis of splicing by flow cytometry using fluorescence splicing reporters, U2OS cells were seeded in 24-well plates at 20%–25% density. 24 hr after seeding, cells were transfected with plasmids encoding the splicing reporters and 1μg/ml doxycycline was added to induce expression of the reporter. 48 hr after seeding, cells were harvested and analyzed for GFP and BFP expression by flow cytometry using a Cytoflex analyzer (Beckman Coulter). The ratio of BFP-to-GFP signal intensity was then determined for each cell and the average BFP/GFP ratio for the cell population was calculated for each reporter. The BFP/GFP ratio of intron-containing reporters was then normalized to the average ratio of BFP/GFP of 6 no-intron control reporters (which represent fully spliced mRNAs) to determine the splicing efficiency.
Quantitation and Statistical Analysis
Quantification of mRNA degradation
mRNA cleavage
To calculate the precise moment of cleavage of reporter mRNA molecules relative to the start of translation, we determined for each mRNA at which moment we could first visually observe GFP signal, and at which moment the red and the green signals separated from each other (cleavage). For mRNAs for which we did not observe cleavage, we determined the total time that the mRNA was tracked. An mRNA track was ended either when: 1) the end of the time lapse was reached, 2) two mRNAs crossed each other’s paths, 3) when translation of the mRNA could no longer be observed, 4) when the mRNA moved out of the field of view, or 5) when the mRNA detached from the plasma membrane. To ensure we only analyzed newly transcribed mRNAs, we excluded mRNAs that were already present at the membrane at the start of the time-lapse experiment, mRNAs that were already associated with a green fluorescent signal in the frame that they appeared in the field of view, or mRNAs on which we never observed a green fluorescence signal during the time lapse (∼50% of mRNAs). For each mRNA, we calculated the time from GFP appearance until cleavage or until the last time point in which the mRNA could be tracked, and we plotted the fraction of uncleaved mRNAs using a Kaplan-Meier plot, which takes into account both the track length of the cleaved and uncleaved mRNAs.
Cleavage of mRNAs with 5xPP7 binding sites
Reporter mRNAs containing a 5x PP7 binding array did not recruit sufficient mCherry molecules to detect the mRNA molecule over the background fluorescence. Therefore, mRNA cleavage could not be defined as physical separation of GFP and mCherry foci. As an alternative approach to define the time from translation initiation until mRNA cleavage, we analyzed the fluorescence intensity and diffusion of translation sites; translation initiation on a newly transcribed mRNA molecule was determined by the gradual appearance of a GFP spot that diffused slowly (indicative of membrane tethering). Cleavage was determined as either the rapid disappearance of the translation site, or a sudden large increase in the diffusion speed of the translation site, both of which occur because the 5′ cleavage product, which contains all the ribosomes and is thus GFP-labeled, is no longer physically connected to the PP7 binding sites in the 3′UTR after cleavage, and has thus lost its membrane tethering. An issue with determining the moment of cleavage based on the GFP signal alone is that cleavage cannot be distinguished from detachment of the entire mRNA from the plasma membrane. Membrane detachment may be especially prevalent when analyzing mRNAs containing only 5x PP7 binding sites, as they are connected to the membrane by fewer PP7 molecules. Therefore, for each reporter the rate of mRNA membrane detachment was determined by analyzing the rate of translation site disappearance for reporters that did not include a PTC. Cleavage times were then corrected for mRNA detachment by dividing the fraction of uncleaved mRNAs in the reporters containing a PTC by the fraction of remaining mRNAs (i.e., not detached) of the control reporter lacking a PTC for each time point.
Cleavage of HP21 reporters
Because reporters with reduced initiation rates are frequently translated by a single ribosome at a time, cleavage cannot be distinguished from translation termination as they both result in the separation of a single ribosome/SunTag array from the mRNA. Since mRNA cleavage, but not translation termination, results in rapid disappearance of the 3′ cleavage fragment, we classified disappearance of a green fluorescent signal as mRNA cleavage when the mRNA signal disappeared after GFP disappearance.
Although the mRNA foci can also disappear through mechanisms other than NMD cleavage (e.g., mRNA detachment from the membrane), mRNAs on which no NMD occurred rarely disappeared from the membrane, indicating that mRNA disappearance after GFP disappearance mostly represents NMD.
Cleavage of untethered mRNAs
To observe cleavage of mRNA molecules that were not tethered to the membrane, cells were imaged in a single focal plane just below the nucleus of the cell. This region was selected to allow observation of mRNAs immediately after nuclear export, and because mRNAs could be easily tracked in the region below the nucleus. Similar to analysis on tethered mRNAs, we excluded from our analysis mRNAs which were already present at the start of the time-lapse video, or first appeared in the field of view in a translating state. For mRNAs that were not cleaved for the duration of the video, the time was noted at which we could no longer accurately track the mRNA and/or GFP signal (e.g., when mRNAs get out of focus or cross each other). The fraction of cleaved mRNAs was plotted as Kaplan-Meier plots.
Estimating SMG5/7-dependent NMD
NMD-dependent mRNA decay through SMG5/7 does not result in cleavage, but rather in exonucleolytic decay (Unterholzner and Izaurralde, 2004, Loh et al., 2013). Exonucleolytic decay of an mRNA would result in disappearance of both the GFP and mCherry signals. Therefore, we tested whether reporter mRNAs that were not degraded by endonucleolytic decay were subject to exonucleolytic decay. To do this, we determined the duration that mRNA and translation foci that did not get cleaved could be observed.
Analysis of decay of 3′ cleavage fragments
To determine how quickly 3′ decay intermediates were degraded after mRNA cleavage (Figure 1I), we precisely determined the moment of mRNA cleavage (as described above) and the moment of mRNA disappearance. For mRNAs that did not disappear during the video, we determined the last frame in which we could track the mRNA. An mRNA track could be lost, because the mRNA spatially overlapped with another mRNA, moved out of the field of view, or because the end of the video was reached. We then calculated for each individual mRNA the time between cleavage and mRNA disappearance and determined the fraction of remaining mRNAs over time using a Kaplan-Meier plot.
Observing the first round of translation
It is possible that a minor fraction of the mRNAs that initially appeared in the field of view in the untranslated state had already undergone the first round of translation previously followed by translation shutdown. To determine the likelihood that an mRNA that appears without an associated GFP signal had previously been translated, we analyzed which fraction of their lifetime TPIWT mRNAs spend in a state of temporary translational shutdown. The duration of a temporary shutdown event was defined as the number of frames during which no GFP signal could be detected on an mRNA before translation reinitiated. We determined the total time that an mRNA was translated as the duration from first GFP appearance until the last frame in which GFP was observed. We then calculated which fraction of their lifetime TPIWT mRNA spends in a state of temporary shutdown, which was 4% ± 2%. If all newly appearing mRNAs would already have been translated, 96% of the newly appearing mRNAs should be associated with a green signal, while 4% should not have an associated green signal. However, 86% of the newly appearing mRNAs did not have an associated green signal, suggesting that the majority of these mRNAs had not yet initiated translation. The 14% of mRNAs that appeared with a GFP signal represent 96% of the mRNAs that had already initiated translation. Therefore, the 4% of mRNAs in a state of temporary shutdown should represent only 4/96 ∗ 14% = 0.6% of all mRNAs, meaning that we observe the first round of translation on > 99% for the mRNAs that appear without an associated GFP signal.
Measuring fluorescence intensities
Fluorescence intensities of mRNAs and translation sites
To determine the fluorescence intensity of individual translation or mRNA foci over time, mean spot intensities were measured in ImageJ in a region of interest (ROI) 4x4 pixels in size (0.54 × 0.54 μm). For each spot, local background fluorescence intensity was measured in a 10x10 pixel ROI directly next to the spot of interest, and mean background fluorescence intensities was subtracted from the mean spot intensity. To correct for photobleaching during the time-lapse video, the mean fluorescence intensity of the entire cell (for GFP fluorescence) or of mRNAs that remained present for the entire duration of the video (mCherry fluorescence) was determined at each time point in the video and the decrease in fluorescence over time was fit with a single exponential decay distribution, from which a bleaching rate was determined. All spot and background fluorescence intensity measurements were then corrected for the bleaching.
Tracking and intensity measurements of translation sites
For mRNA tracking and fluorescence intensity measurements of single mRNAs described in Figure 3, we used a previously described software package called ‘TransTrack’ (Boersma et al., 2018). Using TransTrack, we measured the GFP fluorescence intensity and performed background subtraction and bleach correction. All traces were manually curated.
Ribosome initiation and elongation rates
Calculating the number of ribosomes present on an mRNA
For TPIPTC1, calculation of the number of ribosomes present on individual mRNA molecules was performed as described previously (Yan et al., 2016). In brief, mRNAs were imaged with short (30 ms) exposure time and high laser intensity (∼20x higher than used for normal translation imaging). With this short exposure time, mature SunTag proteins that have completed translation and are freely diffusing in the cytoplasm and are not co-localizing with an mRNA molecule can be observed as distinct foci. In contrast, translation sites are visible as brighter foci that co-localized with an mRNA molecule. We compared the background-subtracted fluorescence intensity of translation sites with the mean fluorescence intensity of mature proteins in the same cell. Since some ribosomes (that have not yet translated the entire SunTag sequence) will recruit fewer scFv-sfGFP molecules, a mature protein produces more fluorescence intensity than an average ribosome translating an mRNA. Therefore, we corrected for the average fluorescence intensity associated with a translating ribosome to obtain the final ribosome occupancy on mRNA molecules, as described previously (Yan et al., 2016)).
For other reporters, we determined the ribosome occupancy by comparing the fluorescence intensity of translation sites of each of these reporters with the fluorescence intensity of TPIPTC1 translation sites, for which the ribosome number had been calculated, as described above.
Determining ribosome elongation speeds
To determine the time a ribosome takes to translate the entire coding sequence of a reporter mRNA, the 5′UTR sequence of the Emi1 gene (Yan et al., 2016, Tanenbaum et al., 2015) was introduced into the reporter, which severely reduces the translation initiation rate, frequently limiting the number of ribosomes per mRNA molecule to one. To determine the time taken by the ribosomes to translate the entire reporter mRNA coding sequence, we measured the time between appearance and disappearance of GFP signal. To ensure that only translation events by single ribosomes were analyzed, only events with low GFP signal were included in this analysis. In addition, only translation events on which the time between GFP appearance and disappearance was < 4.5 min were included, as longer events are more likely to represent either translation events by multiple ribosomes or stalled ribosomes. The duration of all analyzed translation events was then plotted, and fit with a Gaussian distribution. The mean of this Gaussian distribution (2.38 min) was used as a mean time from the moment of GFP appearance to GFP disappearance.
As GFP cannot be detected until ∼8 SunTag peptides have been synthesized due to limited fluorescence intensity, GFP will only be detected after the first ∼735 nucleotides have been translated (Yan et al., 2016). Therefore, the translation time of 2.38 min represents the time required for translation of the mRNA reporter from nt 735 until the end of the coding sequence (nt 2517), resulting in an effective coding sequence length of 2517 – 735 = 1782 nucleotides, which results in an elongation speed of 12.45 nucleotides/s (4.15 codons/s).
When translation initiation is not reduced (e.g., for TPIPTC1), multiple ribosomes will simultaneously translate the mRNA, resulting in a brighter fluorescent signal than when the mRNA is translated by a single ribosome. Therefore, the GFP signal can already be detected when the first ribosome has translated fewer than 8 SunTag peptides, resulting in a longer effective coding sequence length. With the determined ribosome occupancy of 10.85 ribosomes on TPIPTC1, the average distance between ribosomes will be 232 nucleotides, corresponding to 3.2 SunTag peptides. When the first ribosome has on average translated 5.6 SunTag peptides, or 562 nucleotides, the next ribosome trailing the first at a distance of 232 nucleotides will have translated 2.4 peptides, resulting in a total fluorescence of 8 SunTag peptides of the two ribosome combined, which we determined to be our detection limit (see above). Translation of the remaining 1955 nucleotides (2517 minus the 562 undetected nucleotides), which corresponds to the time required for the first ribosome to reach the stop codon after GFP appearance, should therefore require on average 1955/12.5 = 2.62 min.
In Figure 1D, we state that the timing of the first mRNAs undergoing NMD coincides with the expected time of arrival of the first ribosome at the stop codon. This statement is based on elongation rates measured in a previous study, in which we found elongation rates of 9-15 nt/s for different reporters ((Yan et al., 2016)). TPIPTC1 and TPIPTC160 have effective coding sequence lengths (see above) of 1955 and 2429 nt respectively. Assuming elongation rates of 9-15 nt/s, translation of TPIPTC1 should take between 1955/15 = 130 and 1955/9 = 217 s (2.1-3.6 min), which corresponds to 2.1-3.6 min. Translation of TPIPTC160 should take between 2.7-4.4 min.
Calculation of translation initiation rates
Calculation of the translation initiation rate was performed as described previously (Yan et al., 2016). In brief, it requires both knowledge of the translation elongation rate (4.15 codons/s, see ‘Determining ribosome elongation speeds’) and the inter-ribosome distance. The inter-ribosome distance was calculated based on the coding sequence length (2517 nucleotides for TPIPTC1) and the number of ribosomes present on the mRNA (10.85 for TPIPTC1, see ‘Calculating the number of ribosomes present on an mRNA’). The inter-ribosome distance for TPIPTC1 was 2517/10.85 = 232 nucleotides. In steady state, ribosomes need to initiate as frequently as they terminate. With an inter-ribosome distance of 232 nucleotides, and an elongation speed of 4.15 codons/s, a ribosome terminates and initiates every 232/12.45 = 18.6 s, corresponding to an initiation rate of 3.2 ribosomes per minute.
Cell-to-cell heterogeneity in NMD efficiency
Since we only analyze a relatively small number of mRNAs in each cell, cleavage distributions in individual cells are expected to show substantial variation, even when they have the same NMD efficiency. In order to determine if NMD efficiency was variable among different cells, it is therefore essential to determine the extent of heterogeneity that is expected between different cells based on random chance, assuming that each cells has an equal NMD efficiency. To determine how much heterogeneity is expected when all cells have equal NMD efficiency, we performed stochastic simulations for TPIWT, TPIPTC160 and TPIPTC1 reporters to determine the expected ranges in cleavage time distributions. The number of cells and the number of mRNAs per cell in each simulation were kept equal to the number of cells and mRNAs per cell in the corresponding experiment. For each simulated mRNA, the moment of cleavage was determined by randomly picking a value from the experimentally observed cleavage time distribution. Then, the cleavage time distribution for each simulated cells was plotted using a Kaplan-Meier plot.
Next, we determined whether the cell-to-cell variation in the experimental cleavage time distributions exceeded the variation in the simulated cleavage time distribution (which would indicate that different cells have distinct NMD efficiencies). For simulated data, we compared the cleavage time distributions of single cells with the experimentally observed average cleavage distribution of all cells, and calculated the Summed Squared Error (SSE) for each simulated cell individually. We then summed the SSE’s of all individual cells to get a measure of the total variation. These simulations were performed 10.000 times to generate a 95% confidence interval of the SSE that is expected if all cells have an equal NMD efficiency. Similarly, for experimental data, we compared the cleavage time distributions of single cells with the experimentally observed average cleavage distribution of all cells to get a measure of the total variation in the experiment. Finally, we compared the experimental variation with the variation in the simulations to determine if the experimental data showed significantly greater heterogeneity in NMD efficiency among different cells than the simulated data.
Counting ribosomes that translate HP21 mRNAs
Determining how many ribosomes have translated a HP21-TPIPTC1 reporter mRNA molecule, requires knowledge of the GFP fluorescence produced over time by a single ribosome translating the mRNA, and of the GFP fluorescence associated with an mRNA during each time point.
Distinguishing translation events from noise
To generate GFP intensity time traces, we measured the GFP fluorescence intensity of an mRNA at every time point in which the mRNA was present. GFP intensity time traces showed peaks of high fluorescence intensity that lasted for multiple consecutive time points, consistent with the expected fluorescence intensity produced by a translation event of one or more ribosomes. In order to count how many ribosomes had translated an mRNA, we first distinguished translation events (originating from one or multiple ribosomes translating the mRNA) from noise. Fluorescence intensity peaks were defined as translation events when the mean pixel intensity was either above 0.3 (arbitrary units) for 4 consecutive time points, or above 0.3 for 5 out of 6 consecutive time points. When a fluorescence intensity peak was categorized as a translation event, GFP intensity of earlier and later time points were also determined to obtain the entire fluorescence intensity peak associated with that translation event. Earlier time points were included until a time point was encountered in which the mean pixel intensity was < 0.05, or when a local minimum with an intensity of < 0.15 was encountered. Later time points were included until the first time point was encountered with a pixel intensity of < 0.2. The total fluorescence intensity of each translation event was then calculated as the sum of the fluorescence intensities at each time point of the translation event, i.e., the integrated fluorescence intensity of the peak.
Fluorescence intensity of a single ribosome
As the observed translation events (i.e., GFP peaks in the GFP intensity time traces) can originate from one or multiple ribosomes, interpretation of fluorescence intensities requires knowledge of the fluorescence intensity produced by single ribosome translation events. We reasoned that a single ribosome translation event can be distinguished from multi-ribosome translation events based one: 1) the duration of the translation event, and 2) the fluorescence intensity; translation events that include multiple ribosomes will, on average, last longer and have a higher fluorescence intensity. Translation events with a duration of 2.5 min or less always had a low maximum intensity, whereas translation events with a duration of more than 2.5 min occasionally had higher maximum intensities (Figure S2G). Therefore, we reasoned that all translation events with a duration of 2.5 min or less are highly enriched from translation events originating from a single ribosome.
We measured the maximum pixel intensity for all peaks of 2.5 min or less, and found that the average maximum intensity of GFP for those mRNAs was 0.76. To acquire the integrated GFP fluorescence intensity of a single ribosome translation HP21-TPIPTC1, we used our previously described model to correct for the lower fluorescence intensity when ribosomes have not yet completed translation of all 24 SunTag peptides (Yan et al., 2016). This correction takes into account the delay in fluorescence buildup caused by sequential production of SunTag peptides and the delay in binding of scFv-sfGFP to a newly translated SunTag peptide when the SunTag peptide has not left the ribosomal exit tunnel. Using this correction we estimated the mean fluorescence intensity of the entire translation event to be 0.43. Finally, the integrated fluorescence intensity was calculated as the mean fluorescence intensity multiplied by the average duration of a single-ribosome translation event, which we determined to be 2.38 min (in Emi1-TPIPTC1) or 9.5 frames, resulting in an integrated fluorescence intensity of 4.1 for single ribosomes.
Counting ribosomes translating an HP21-PTC1 mRNA
To calculate the total number of ribosomes that had translated an mRNA, we calculated how many ribosomes contributed to the fluorescence intensity of each translation event. The number was rounded, so that translation events with a total fluorescence intensity between 0.5 and 1.5 ribosomes (e.g., between 2.05 and 6.15 arbitrary intensity units) were scored as originating from 1 ribosome, translation events with intensities of 1.5-2.5 ribosomes as 2 ribosomes, etc. We then summed the number of ribosomes from all translation events observed on an mRNA to get the total number of ribosomes that had translated an mRNA.
Probability per ribosome to induce NMD of HP21-TPIPTC1 mRNAs
After counting how many ribosomes translated each HP21-TPIPTC1 mRNA before NMD was induced, we calculated the chance per ribosome by fitting the cumulative decay graph with a 2-component exponential decay distribution (containing a fast and slow decaying population, similar to the fitting approach described in ’Modeling of NMD kinetics’) in GraphPad Prism. Since 10% of TPIPTC1 mRNAs appeared to be insensitive to NMD (Figure 1D), we used a slow component that represented 10% of the mRNAs. The decay rate for this slow decaying population was set to the same value as the slow decay rate for our modeling of reporters without reduced translation initiation. The decay rate of the fast component was than fitted by GraphPad Prism. We then converted the decay rate that was determined by prism to a probability per ribosome to induce NMD. An exponential decay distribution is described the by the equation:
In this equation, R represents the number of ribosomes that have translated the mRNA, while λ is the decay rate (in ribosomes-1. Therefore, after 1 ribosome has translated an mRNA, the fraction of mRNAs that has not yet been degraded is given by:
The probability per ribosome to induce NMD is then given by:
Analysis of the first burst of translation
Fitting initiation events based on the fluorescence intensities
To determine the timing of individual translation initiation events based on fluorescence intensity time traces of single mRNAs, we used a previously described algorithm to fit initiation events to the fluorescence intensity trace of translation sites (Boersma et al., 2018). This algorithm calculates the fluorescence intensity trace of a single ribosome translating the mRNA, and fits multiple intensity traces of single ribosomes along the intensity trace of a translation site. The fit in which the combined traces of single ribosomes best fit the trace of the translation site is determined by minimizing the root mean squared error between the fit and the experimentially determine intensity trace. In the best fit, the timing of translation initiation events is recorded. Generating the intensity trace of a single ribosome requires knowledge of 1) the fluorescence intensity of a ribosome when all SunTag peptides are occupied by scFv-GFP, and 2) the duration of translation of the SunTag peptides and of the mRNA downstream of the SunTag peptides. To obtain these values, we measured the intensity of a single ribosome as described above for TPIPTC1 (‘Calculating the number of ribosomes present on an mRNA‘), which we found to be 0.155 a.u.. We calculated the time of translation of the SunTag peptides and the downstream sequence based on the previously published translation elongation rate of 3.3 codons/s for the Kif18b reporter (Yan et al., 2016).
Defining the first burst of translation
In cells overexpression ha4E-BP1, newly transcribed mRNAs frequently showed a high translation initiation rate initially (referred to as the “first burst of translation”), followed by a period in which no or very few new ribosomes initiated translation (For example, see Figure 3E). To distinguish initiation events that occurred during this first burst of initiation from events that happened during later bursts of translation, we defined the end of the first burst of translation as the first moment in which no translation initiation events had occurred for 2 consecutive minutes. In addition, we also called the end of a burst when only a very low initiation rate was observe for 3 consecutive minutes (initiation rate lower than 1 ribosome per minute). Finally, we plotted the duration of the first bursts of translation as a cumulative distribution, and fit this distribution with an exponential decay distribution to determine the average and median duration of the first burst.
XRN1 decay kinetics
Measurement of fluorescence intensities of mRNAs
To analyze changes in mCherry fluorescence caused by XRN1-mediated degradation of the PP7 binding site array, images were acquired with a time interval of 5 (TPIPTC160) or 15 (TPIPTC160-48xPP7) seconds. The mCherry fluorescence intensity of 3′ cleavage fragments was measured starting 10 frames before cleavage until the moment the mRNA could no longer be detected or the track was lost, and intensities were normalized to the average mCherry fluorescence intensity in the 10 frames before cleavage.
Calculating XRN1 decay kinetics
mCherry fluorescence intensity profiles during the decay of the 3′ cleavage fragments for the TPIPTC160 reporter (containing a 24x PP7 array) were characterized by an initial delay phase, during which no changes in fluorescence intensity were observed, followed by a degradation phase, during which the fluorescence intensity decreased linearly until the mRNA could no longer be detected. A fraction of mRNAs did not show a degradation phase for the duration of the experiment and were excluded from further analysis. To calculate the duration of the delay and the degradation phase, we determined the onset of the degradation phase by fitting each intensity trace with a model consisting of a plateau followed by a linear decrease. To determine onset of degradation, the quality of fit was determined for different initial moments of linear decrease, ranging from immediately after cleavage to the moment of degradation. The initial moment of linear decrease that best fit the data was then used for further calculations. The duration of the delay phase was calculated as the time from cleavage until the onset of degradation, while the duration of the degradation phase was calculated as the time from the onset of degradation till the last moment the mRNA signal could be detected.
Calculating mRNA decay kinetics using the TPIPTC160-48xPP7 reporter
Degradation of TPIPTC160-48xPP7 occurred in 4 phases: mCherry fluorescence initially remained constant during a delay phase, after which it decreased to ∼50%, followed by a plateau phase during which mCherry intensity again remained constant, and finally complete disappearance of the mCherry signal was observed. Because these traces showed a complex profile, we manually inspected all traces to identify the different phases. The duration of the initial delay phase was calculated as the time between mRNA cleavage and the first moment of a steep decline in fluorescence intensity to < 75% of the initial intensity. The duration of the plateau phase was defined by the number of frames during which fluorescence intensity remained constant over time while having a fluorescence intensity of 15%–75% of the initial intensity. For some traces, we could not observe all 4 phases because mRNAs could not be tracked sufficiently long; for these traces we noted the last frame in which the delay phase or plateau phase could be observed. We finally plotted the duration of delay phase and plateau phase using a Kaplan-Meier plot.
Calculation of the XRN1 degradation speed
To calculate the degradation speed of XRN1 degrading a 24x PP7 binding site array, we used the degradation time which was determined as described above. We estimated that we could no longer track the mRNA when 19 of 24 PP7 binding sites were degraded (i.e., when 5 binding sites were remaining), as we generally observed a steep decrease in fluorescence intensity when ∼1/5th of the fluorescence intensity was remaining, suggesting that we could no longer accurately identify the mRNA spot at that point and were instead measuring background fluorescence. Therefore, the degradation time represents the time required for degradation of 19/24th of the 24xPP7 array, or 1153 of the total 1456 nucleotides. Using this value, we calculated the degradation speed by dividing the 1153 nucleotides by the median degradation time (30 s for TPIPTC160) to obtain a degradation speed of 38 nucleotides/s.
To calculate the degradation speed of XRN1 degrading the 2642 nucleotide linker sequence of TPIPTC160-48xPP7, we assumed that plateau phases with durations of 2 or less min were degraded by a single XRN1 binding event, and that plateau phases that lasted longer were degraded by 2 or more XRN1 binding events (see main text ‘Decay kinetics of the 3′ fragment after mRNA cleavage’). The degradation speed was then calculated by dividing the linker length of 2642 nucleotides by the median degradation time (48 s for TPIPTC160-48x PP7) to obtain a degradation speed of 55 nucleotides/s.
Modeling of NMD kinetics
General information
We used MATLAB to perform stochastic simulations that describe NMD cleavage kinetics with quantitative parameters. Each simulation was run using four parameters: 1) the moment the ribosome reaches the stop codon, which was experimentally determined and was assumed to be constant for all reporters with the same coding sequence. 2) The fraction of mRNAs that is sensitive to NMD; this parameter was variable and the value that best described the data was determined by the modeling. 3) The probability that a terminating ribosomes induces NMD of an NMD-sensitive mRNA; this parameter was variable and the value that best described the data was determined by the modeling. 4) The probability that a terminating ribosomes induces NMD of an NMD-insensitive mRNA; this probability was constant and set to the decay rate of an NMD reporter without introns (TPIPTC1-no introns). Since 2 parameters were kept constant, decay kinetics were determined by the 2 variable parameters (fraction NMD-sensitive mRNAs and decay rate of NMD-sensitive mRNAs). Decay kinetics were simulated using a wide range of input values for the two variable parameters, and the best fitting values were determined based on similarity between data and simulation using least square fitting.
Running a single simulation
Each simulation was run for 100.000 mRNA molecules. The moment of cleavage was determined for each mRNA molecule and depended on the values of the four parameters.
First, the 100.000 mRNAs were divided into either NMD-sensitive or NMD-insensitive mRNAs based on the value of the parameter that describes the fraction of NMD-sensitive mRNAs
Next, the time from first detection of translation until induction of NMD (tdegraded) was calculated for each mRNA based on two parameters: 1) the time from first detection of translation until arrival of the first ribosome at the stop codon (t1), and 2) the time from arrival of the first ribosome at the stop codon until the moment of induction of NMD (t2), which depends on the probability that a terminating ribosome induces NMD:
A value for t1 was randomly selected from a Gaussian distribution; the mean and standard deviation of this Gaussian distribution were experimentally determined using the Emi-TPIPTC1 reporter (2.38 ± 0.79 min). For other reporters, the mean was scaled linearly based on the effective coding sequence length (See ‘Determining ribosome elongation speeds’), while the standard deviation was scaled with the square root of the effective coding sequence length (e.g., a reporter with a 2x longer effective coding sequence length will have a 1.41x higher standard deviation). Since a Gaussian distribution was used, ribosomes could occasionally be predicted to reach the stop codon in less than 0 s (< 0.1% of the cases); therefore a lower limit of 0 s was used for the time at which ribosomes reached the stop codon.
A value for t2 was randomly selected from an exponential decay distribution; the decay rate of this exponential decay distribution depended on the probability that a terminating ribosome induces NMD. For the NMD-sensitive population (NNMD sensitive), we used a fitting approach to determine which decay rate best described the experimental data (see below, ‘Comparing simulations and data’).
The decay rate was then converted to a probability per ribosome to induce NMD using the equation:
In which λ is the decay rate (in min-1), and t is the time (in min). Since 3.2 ribosomes terminate per minute, t can be converted to ribosomes (R):
After a single ribosome has translated the mRNA (R = 1), the fraction of mRNA that has not been degraded is given by:
Thus, the probability per ribosome to induce NMD is given by
In the ‘first ribosome’ model, degradation of the NMD-sensitive population was based purely on the moment the ribosome reached the stop codon (Figure 2A), and decay was therefore modeled to be induced immediately after the ribosome had reached the stop codon (t2 = 0).
For the NMD-insensitive population of mRNAs, the time required to induce NMD after the first ribosome had reached the stop codon was also randomly selected from an exponential decay distribution. The decay rate of this exponential distribution was set at a fixed value for all simulations, and was similar to the decay rate of TPIPTC1-no introns (decay rate = 0.0029 min-1)
After we determined the moment of degradation for all 100.000 molecules, a cumulative distribution function was made that describes the fraction of uncleaved mRNAs over time, similar to the Kaplan-Meier plots used to describe cleavage data. We then determined how well the simulation fit the data by calculating the Summed Square Error (SSE): at each time point (e.g., every 30 s for most experiments), we calculated the squared difference in fraction of remaining mRNAs in simulation and data, and then summed all these values to get the total SSE. Simulations that describe the data well will have a small SSE, while simulations that do not fit well will have a large SSE.
Comparing simulations and data
To find which values for the two variable parameters best described the data, we first ran simulations with a wide range of initial NMD decay rates and fractions of NMD-sensitive mRNAs. The initial values for (1/decay rate) ranged from 0-400 min with steps of 1 min, whereas the fraction of cleaved mRNAs range from 10 to 100% with steps of 1%. The lower limit of 10% prevented fitting very high decay rates in situations where a single mRNA molecule was cleaved early on but very little cleavage occurred during the rest of the experiment. We calculated the SSE for each simulation, and determined which parameter values resulted in the lowest SSE and thus best described the data. We then ran the simulation with a new, smaller range of parameter values centered on the values that were found to describe the data most accurately in the previous round. This was repeated for several rounds with increasingly small parameter ranges until the change in the parameter 1/decay rate was less than 0.01 min.
The entire fitting process was repeated 5 times, and the mean of best fitting parameter values of the 5 replicates were used as the final value that was used to describe the data. In general, the 5 replicate fitting processes had differences of < 1%, indicating that stochasticity does not have a major effect on the outcome of the simulation.
Comparing quality of fit of different models
We have used two different models to describe the cleavage kinetics of TPIPTC1 based on either one variable parameter (‘first ribosome’ model) or two variable parameters (‘any ribosome’ model). As models with two parameters generally produce better fits, we calculated Akaike information criterion (AIC) for both models, which corrects for the number of variable parameters through the following equation:
In this equation, SSE is the summed squared error of the fit and the data, n is the number of time points, which was 81 (i.e., 40 min), and K is the number of variable parameters in the model. The model that best describes the data will have the lowest (most negative) AIC, which we found to be the model with two variable components.
Predicting NMD decay rates in cells expressing ha4E-BP1
To predict the effect of ha4E-BP1 overexpression on kinetics of NMD of TPIPTC160 and TPIPTC1 reporters, we assumed that ha4E-BP1 did not affect the NMD decay rate while mRNAs were bound to CBC, but reduced the decay rate while mRNAs were bound to eIF4E by decreasing the number of ribosomes that initiate translation and could induce NMD. To calculate the predicted NMD decay rate in ha4E-BP1 overexpressing cells, two parameters are required. 1) The number of ribosomes that initiated on CBC-bound mRNAs. 2) The fold decrease in the NMD decay rate while eIF4E is bound to mRNAs for cells overexpressing ha4E-BP1, which is directly related to the initiation rate on eIF4E-bound mRNAs in the presence and absence of ha4E-BP1 overexpression. The number of ribosomes that initiated on CBC-bound mRNAs was described by an exponential decay distribution with an half-live of 8.7 ribosomes (see Figure 3G). Translation initiation rates on eIF4E-bound mRNAs were on average 2.4-fold lower in ha4E-BP1 overexpressing cells, thus we used a NMD decay rate that was also reduced by 2.4-fold upon ha4E-BP1 overexpression. We then adjusted the stochastic simulation described above (See ‘Running a single simulation‘) to predict how ha4E-BP1 overexpression affects the NMD decay rate.
For each mRNA, we simulated 3 values:
-
1)
t1, the time from first detection of translation until arrival of the first ribosome at the stop codon (see above, ‘running a single simulation’).
-
2)
t2, the time from arrival of the first ribosome at the stop codon until the moment of induction of NMD (in a situation without ha4E-BP1 overexpression,see above, ‘running a single simulation’).
-
3)
t3, the duration of the first burst of translation (in min), which was randomly selected from the exponential decay distribution that described the duration of the first burst of translation (and has a half-live of 8.7 ribosomes (See Figure 3G)).
We then determined for each simulated mRNA whether it was degraded by ribosomes that initiated in the first burst of translation, or by ribosomes that initiated after the first burst of translation. mRNAs were considered to be degraded in the first burst of translation when t3 > t2, as this indicated that the time required for degradation was shorter than the duration of the first burst of translation, and thus that NMD was induced by a ribosome that initiated in the first burst of translation. For these mRNAs, the moment of degradation was defined as above (‘running a single simulation’):
For mRNAs that were not degraded in the first burst of translation (t3 < t2), we assumed that the initiation rate (and thus NMD decay rate) was unaffected during the first burst of translation, and reduced by 2.4-fold after the first burst of translation in the presence of ha4E-BP1 (see Figure 3F). The time from the end of the first burst of translation until degradation (during which decay is delayed by 2.4-fold) is defined as:
Therefore, the moment of degradation for mRNAs not degraded during the first burst of translation is defined as:
After we determined the moment of degradation for all molecules in the simulation, a cumulative distribution function was made that describes the fraction of uncleaved mRNAs over time.
Fraction of mRNAs degraded while CBC-bound
To predict which fraction of mRNAs are degraded while still bound to CBC, we need to determine when replacement of CBC by eIF4E occurs relative to the moment of NMD induction for each reporter. We assumed that the CBC-to-eIF4E switch kinetics was similar for all reporters. Therefore, the fraction of mRNAs that are degraded while still bound to CBC could be calculated based on the NMD decay rate for each reporter. We assumed that the end of the first burst of translation (see above, ’Predicting NMD decay rates in cells expressing ha4E-BP1’) indicates the moment of CBC-replacement by eIF4E. For each mRNA, we again simulated the three values t1, t2 and t3. Next, we determined the moment of degradation (tdegraded) for each mRNA as described above in ‘Running a single simulation’. An mRNA was considered to be degraded while CBC-bound if degradation happened before CBC-replacement (i.e.. tdegraded < t3). We then determined for each mRNA whether degradation happened before (i.e., tdegraded < t3) or after (i.e., tdegraded > t3) CBC-replacement to calculate the fraction of mRNAs that is degraded while bound to CBC. Of note, this fraction was calculated for the population of mRNAs that was cleaved efficiently; we did not include the fraction uncleaved mRNAs in this calculation.
Analysis of genome-wide data on NMD efficiency
Filtering the TCGA dataset
To determine the effect of the number and position of upstream and downstream introns on NMD efficiency, we used the previously published TCGA dataset (Lindeboom et al., 2016). This dataset contains expression levels of PTC-containing transcripts for 2840 high-confidence nonsense mutations, compared to expression levels of their wild-type counterparts. These expression levels were filtered for non-random noise by principal component analysis (see (Lindeboom et al., 2016) for details), and the ratio of abundances of nonsense and wild-type transcripts is used as a measure for NMD efficiency. For our analyses, we removed all transcripts with an intron in their 3′ UTR (98 nonsense mutations removed), as they are likely to behave differently regarding the effect of the number of upstream and downstream introns on NMD efficiency. We also removed transcripts in which the PTC was located within 250 nucleotides of the start codon (413 nonsense mutations removed), as these PTCs have been shown to induce NMD less efficiently and would therefore skew analyses independently of the effect we are analyzing.
Effect of the number of downstream EJCs on NMD efficiency
To determine the effect of the number of downstream EJCs on NMD efficiency, we assumed that an EJC will be present on the mRNA when the intron is located at least 50 nucleotides downstream of the PTC, while the EJC will be displaced by translating ribosomes if the PTC is within 50 nucleotides of a downstream intron (refs). We grouped transcripts based on the number of EJCs downstream of the PTC, and compared NMD efficiency for transcripts with 0, 1, 2, 3, or more than 3 EJCs downstream of the PTC. Statistical significance was tested using a two-tailed Mann-Whitney U test.
Effect of the number of upstream introns on NMD efficiency
To determine the effect of the number of upstream EJCs on NMD efficiency, we grouped transcripts based on the number of introns upstream of the PTC, and compared NMD efficiency for transcripts with 0, 1, 2, 3, or more than 3 upstream introns. We performed this analysis independently for either transcripts containing a PTC with 1 downstream EJC, or containing a PTC with 2 or more downstream introns. Statistical significance was tested using a two-tailed Mann-Whitney U test.
Data and software availability
Raw imaging data of key experiments related to Figures 1, 2, 3, and 7 (including related supplemental figures) can be accessed on Mendeley data. Microscopy data reported in this paper can be found at Mendeley Data: https://doi.org/10.17632/bw255hcw7h1.
Acknowledgments
We thank Niels Gehring for sharing TPI plasmids (Addgene 65801 and Addgene 65804), and we would like to thank members of the Tanenbaum lab for helpful discussions. We would also like to thank Suzan Ruijtenberg, Matilde Galli, Lotte van Rijnberk, and Angela Andersen from Life Science Editors for critical reading of the manuscript. This work was financially supported by the European Research Council (ERC) through an ERC starting grant (ERC-STG 677936-RNAREG), two grants from the Netherlands Organization for Scientific Research (NWO) (ALWOP.290 and NWO/016.VIDI.189.005), and the Howard Hughes Medical Institute through an International Research Scholar grant to M.E.T. (HHMI/IRS 55008747). All authors are supported by the Oncode Institute that is partly funded by the Dutch Cancer Society (KWF). M.V. also acknowledges support by the gravitation program CancerGenomiCs.nl from the Netherlands Organization for Scientific Research (NWO). T.A.H. was supported by a PhD fellowship from the Boehringer Ingelheim Fonds.
Author Contributions
M.E.T. and T.A.H. conceived of the project. T.A.H. and D.K. performed all the experiments. R.G.H.L. performed bioinformatics analysis under supervision of M.V. S.S. and B.M.P.V. developed computational tools. S.B. provided reagents. T.A.H., D.K., and M.E.T. analyzed the data, prepared the figures, and wrote the manuscript. Equally contributing authors are listed in alphabetical order.
Declaration of Interests
The authors declare no competing interests.
Published: May 30, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.05.008.
Supplemental Information
References
- Belgrader P., Cheng J., Zhou X., Stephenson L.S., Maquat L.E. Mammalian nonsense codons can be cis effectors of nuclear mRNA half-life. Mol. Cell. Biol. 1994;14:8219–8228. doi: 10.1128/mcb.14.12.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm V., Haberman N., Ottens F., Ule J., Gehring N.H. 3′ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep. 2014;9:555–568. doi: 10.1016/j.celrep.2014.09.012. [DOI] [PubMed] [Google Scholar]
- Boersma S., Khuperkar D., Verhagen B.M.P., Sonneveld S., Grimm J.B., Lavis L.D., Tanenbaum M.E. Multi-color Single-Molecule Imaging Uncovers Extensive Heterogeneity in mRNA Decoding. Cell. 2018 doi: 10.1016/j.cell.2019.05.001. Published online June 6, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler M., Steiner S., Mohn F., Paillusson A., Mühlemann O. EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3′ UTR length. Nat. Struct. Mol. Biol. 2006;13:462–464. doi: 10.1038/nsmb1081. [DOI] [PubMed] [Google Scholar]
- Calvo S.E., Pagliarini D.J., Mootha V.K. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.H., Xiang S., Xiang K., Manley J.L., Tong L. Structural and biochemical studies of the 5′→3′ exoribonuclease Xrn1. Nat. Struct. Mol. Biol. 2011;18:270–276. doi: 10.1038/nsmb.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J.A., Patskovsky Y., Almo S.C., Singer R.H. Structural basis for the coevolution of a viral RNA-protein complex. Nat. Struct. Mol. Biol. 2008;15:103–105. doi: 10.1038/nsmb1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Maquat L.E. Nonsense codons can reduce the abundance of nuclear mRNA without affecting the abundance of pre-mRNA or the half-life of cytoplasmic mRNA. Mol. Cell. Biol. 1993;13:1892–1902. doi: 10.1128/mcb.13.3.1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J., Belgrader P., Zhou X., Maquat L.E. Introns are cis effectors of the nonsense-codon-mediated reduction in nuclear mRNA abundance. Mol. Cell. Biol. 1994;14:6317–6325. doi: 10.1128/mcb.14.9.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J., Dreyfuss G. Translation is required to remove Y14 from mRNAs in the cytoplasm. Curr. Biol. 2002;12:1060–1067. doi: 10.1016/s0960-9822(02)00902-8. [DOI] [PubMed] [Google Scholar]
- Durand S., Lykke-Andersen J. Nonsense-mediated mRNA decay occurs during eIF4F-dependent translation in human cells. Nat. Struct. Mol. Biol. 2013;20:702–709. doi: 10.1038/nsmb.2575. [DOI] [PubMed] [Google Scholar]
- Eberle A.B., Stalder L., Mathys H., Orozco R.Z., Mühlemann O. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol. 2008;6:e92. doi: 10.1371/journal.pbio.0060092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle A.B., Lykke-Andersen S., Mühlemann O., Jensen T.H. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009;16:49–55. doi: 10.1038/nsmb.1530. [DOI] [PubMed] [Google Scholar]
- Edelstein A., Amodaj N., Hoover K., Vale R., Stuurman N. Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. 2010 doi: 10.1002/0471142727.mb1420s92. Published online October 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy M.A., Kontarakis Z., Rossi A., Kuenne C., Günther S., Fukuda N., Kikhi K., Boezio G.L.M., Takacs C.M., Lai S.L. Genetic compensation triggered by mutant mRNA degradation. Nature. 2019;568:193–197. doi: 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J.F., Parker R. MS2 coat proteins bound to yeast mRNAs block 5′ to 3′ degradation and trap mRNA decay products: implications for the localization of mRNAs by MS2-MCP system. RNA. 2015;21:1393–1395. doi: 10.1261/rna.051797.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatfield D., Izaurralde E. Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature. 2004;429:575–578. doi: 10.1038/nature02559. [DOI] [PubMed] [Google Scholar]
- He F., Jacobson A. Nonsense-mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu. Rev. Genet. 2015;49:339–366. doi: 10.1146/annurev-genet-112414-054639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvathova I., Voigt F., Kotrys A.V., Zhan Y., Artus-Revel C.G., Eglinger J., Stadler M.B., Giorgetti L., Chao J.A. The dynamics of mRNA turnover revealed by single-molecule imaging in single cells. Mol. Cell. 2017;68:615–625. doi: 10.1016/j.molcel.2017.09.030. [DOI] [PubMed] [Google Scholar]
- Huntzinger E., Kashima I., Fauser M., Saulière J., Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA. 2008;14:2609–2617. doi: 10.1261/rna.1386208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y., Li X., Serin G., Maquat L.E. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell. 2001;106:607–617. doi: 10.1016/s0092-8674(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Jinek M., Coyle S.M., Doudna J.A. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol. Cell. 2011;41:600–608. doi: 10.1016/j.molcel.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis E.D., Nasif S., Mühlemann O. Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip. Rev. RNA. 2016;7:661–682. doi: 10.1002/wrna.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I., Yamashita A., Izumi N., Kataoka N., Morishita R., Hoshino S., Ohno M., Dreyfuss G., Ohno S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006;20:355–367. doi: 10.1101/gad.1389006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Vieira M., Kim H.J., Kesawat M.S., Park H.Y. MS2 Labeling of Endogenous Beta-Actin mRNA Does Not Result in Stabilization of Degradation Intermediates. Mol. Cells. 2019 doi: 10.14348/molcells.2019.2398. Published online March 28, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T., Maquat L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016;129:461–467. doi: 10.1242/jcs.181008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Hir H., Izaurralde E., Maquat L.E., Moore M.J. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F., Ishigaki Y., Li X., Maquat L.E. The exon junction complex is detected on CBP80-bound but not eIF4E-bound mRNA in mammalian cells: dynamics of mRNP remodeling. EMBO J. 2002;21:3536–3545. doi: 10.1093/emboj/cdf345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom R.G., Supek F., Lehner B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016;48:1112–1118. doi: 10.1038/ng.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh B., Jonas S., Izaurralde E. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013;27:2125–2138. doi: 10.1101/gad.226951.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S., Jensen T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- Maquat L.E., Tarn W.-Y., Isken O. The pioneer round of translation: features and functions. Cell. 2010;142:368–374. doi: 10.1016/j.cell.2010.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T., Lyon K., DeLuca K.F., DeLuca J.G., English B.P., Zhang Z., Lavis L.D., Grimm J.B., Viswanathan S., Looger L.L. Real-time quantification of single RNA translation dynamics in living cells. Science. 2016;352:1425–1429. doi: 10.1126/science.aaf0899. [DOI] [PubMed] [Google Scholar]
- Mort M., Ivanov D., Cooper D.N., Chuzhanova N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008;29:1037–1047. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- Pelechano V., Wei W., Steinmetz L.M. Widespread co-translational RNA decay reveals ribosome dynamics. Cell. 2015;161:1400–1412. doi: 10.1016/j.cell.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon X., Bastide A., Safieddine A., Chouaib R., Samacoits A., Basyuk E., Peter M., Mueller F., Bertrand E. Visualization of single endogenous polysomes reveals the dynamics of translation in live human cells. J. Cell Biol. 2016;214:769–781. doi: 10.1083/jcb.201605024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufener S.C., Mühlemann O. eIF4E-bound mRNPs are substrates for nonsense-mediated mRNA decay in mammalian cells. Nat. Struct. Mol. Biol. 2013;20:710–717. doi: 10.1038/nsmb.2576. [DOI] [PubMed] [Google Scholar]
- Ruijtenberg S., Hoek T.A., Yan X., Tanenbaum M.E. Imaging translation dynamics of single mRNA molecules in live cells. Methods Mol. Biol. 2018;1649:385–404. doi: 10.1007/978-1-4939-7213-5_26. [DOI] [PubMed] [Google Scholar]
- Sakharkar M.K., Chow V.T., Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. (Gedrukt) 2004;4:387–393. [PubMed] [Google Scholar]
- Sato H., Maquat L.E. Remodeling of the pioneer translation initiation complex involves translation and the karyopherin importin β. Genes Dev. 2009;23:2537–2550. doi: 10.1101/gad.1817109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulière J., Murigneux V., Wang Z., Marquenet E., Barbosa I., Le Tonquèze O., Audic Y., Paillard L., Roest Crollius H., Le Hir H. CLIP-seq of eIF4AIII reveals transcriptome-wide mapping of the human exon junction complex. Nat. Struct. Mol. Biol. 2012;19:1124–1131. doi: 10.1038/nsmb.2420. [DOI] [PubMed] [Google Scholar]
- Singh G., Rebbapragada I., Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G., Kucukural A., Cenik C., Leszyk J.D., Shaffer S.A., Weng Z., Moore M.J. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell. 2012;151:750–764. doi: 10.1016/j.cell.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A. Purification and characterization of a Saccharomyces cerevisiae exoribonuclease which yields 5′-mononucleotides by a 5′ leads to 3′ mode of hydrolysis. J. Biol. Chem. 1980;255:3080–3085. [PubMed] [Google Scholar]
- Tanenbaum M.E., Gilbert L.A., Qi L.S., Weissman J.S., Vale R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum M.E., Stern-Ginossar N., Weissman J.S., Vale R.D. Regulation of mRNA translation during mitosis. eLife. 2015;4 doi: 10.7554/eLife.07957. Published online August 25, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermann R., Neu-Yilik G., Deters A., Frede U., Wehr K., Hagemeier C., Hentze M.W., Kulozik A.E. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998;17:3484–3494. doi: 10.1093/emboj/17.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trcek T., Sato H., Singer R.H., Maquat L.E. Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 2013;27:541–551. doi: 10.1101/gad.209635.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L., Izaurralde E. SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell. 2004;16:587–596. doi: 10.1016/j.molcel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Wang C., Han B., Zhou R., Zhuang X. Real-time imaging of translation on single mRNA transcripts in live cells. Cell. 2016;165:990–1001. doi: 10.1016/j.cell.2016.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B., Eliscovich C., Yoon Y.J., Singer R.H. Translation dynamics of single mRNAs in live cells and neurons. Science. 2016;352:1430–1435. doi: 10.1126/science.aaf1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X., Hoek T.A., Vale R.D., Tanenbaum M.E. Dynamics of translation of single mRNA molecules in vivo. Cell. 2016;165:976–989. doi: 10.1016/j.cell.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagiya A., Suyama E., Adachi H., Svitkin Y.V., Aza-Blanc P., Imataka H., Mikami S., Martineau Y., Ronai Z.A., Sonenberg N. Translational homeostasis via the mRNA cap-binding protein, eIF4E. Mol. Cell. 2012;46:847–858. doi: 10.1016/j.molcel.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIWT. Images were acquired every 30 s (movie duration is 28 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state and then undergo translation. Note that these mRNAs do not undergo cleavage. Movie field of view is 6.21 × 6.21 μm.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIPTC60. Images were acquired every 30 s (movie duration is 15 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo the pioneer round of translation and get cleaved. Large, bright red structures in the top of the field of view are lysosomes, which can be distinguished from mRNA based on size, fluorescence intensity, and speed of movement. Movie field of view is 8.32 × 8.32 μm.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIPTC1. Images were acquired every 30 s (movie duration is 29 min and 30 s) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo the pioneer round of translation and get cleaved. Large, bright red structures in the top-left of the field of view are lysosomes, which can be distinguished from mRNA based on size, fluorescence intensity, and speed of movement. Movie field of view is 25.52 × 25.52 μm.
A U2OS cell expressing scFv-sfGFP (green) and PCP-Halo (red) was transfected with the NMD reporter TPIPTC1. Cells were incubated with 50nM Halo-TMR for 30 min before imaging. Images were acquired every 15 s (movie duration is 20 min) on a spinning disk confocal microscope. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo the pioneer round of translation and get cleaved. Movie field of view is 7.29 × 7.29 μm.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter HP21-TPIPTC1. Images were acquired every 15 s (movie duration is 17 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. Multiple mRNAs can be observed that appear in an untranslated state, after which they undergo several translation events and finally disappear, indicating mRNA cleavage and 3′ fragment degradation. Movie field of view is 5.94 × 5.94 μm.
A U2OS cell expressing scFv-sfGFP (green), PCP-mCherry-CAAX (red) and the NMD reporter TPIPTC60-48xPP7. Images were acquired every 15 s (movie duration is 10 min) on a spinning disk confocal microscope focusing near the bottom plasma membrane of the cell. After cleavage of the mRNA, the 3′ cleavage fragment (red) becomes progressively dimmer as XRN1 degrades the mRNA. Movie field of view is 3.6 × 3.6 μm.
Data Availability Statement
Raw imaging data of key experiments related to Figures 1, 2, 3, and 7 (including related supplemental figures) can be accessed on Mendeley data. Microscopy data reported in this paper can be found at Mendeley Data: https://doi.org/10.17632/bw255hcw7h1.