

Abstract
Next‐generation sequencing (NGS) has been implemented in clinical oncology to analyze multiple genes and to guide therapy. In patients with advanced lung cancer, small biopsies such as computed tomography‐guided needle biopsy (CTNB), endobronchial ultrasound‐guided transbronchial needle aspiration (EBUS‐TBNA) and transbronchial biopsy (TBB) are less invasive and are preferable to resection to make a pathological diagnosis. However, the quality of DNA/RNA and NGS from small lung tumor biopsy samples is unknown. Between April 2017 and March 2018, 107 consecutive samples were obtained from thoracic tumors or metastatic sites for targeted NGS analysis. Fifteen samples were obtained through CTNB, 11 through EBUS‐TBNA, 11 through TBB and 70 through surgical resection. All samples were formalin‐fixed and paraffin‐embedded. DNA and RNA quality was measured using the ddCq method and the percentage of RNA fragments above 200 nucleotides (DV200), respectively. Our custommade probes were designed to capture exon sequences of 464 cancer‐related genes and transcripts of 463 genes. DNA and RNA yield from the 3 biopsy methods were similar, and less than the yield obtained from resected samples. The quality of DNA and RNA was similar across all methods. Overall, 12 of 15 CTNB samples (80%), all 11 EBUS‐TBNA samples, and 9 of 11 TBB samples (82%) underwent successful NGS assays from DNA. NGS analysis from RNA was successful in all 12 CTNB samples, 9 of 11 EBUS‐TBNA samples (82%), and 8 of 11 TBB samples (73%). CTNB, EBUS‐TBNA and TBB mostly resulted in adequate DNA and RNA quality and enabled high‐quality targeted NGS analysis.
Keywords: biopsy, bronchoscopy, CT‐guided needle biopsy, lung cancer, targeted next generation sequencing
1. INTRODUCTION
Next‐generation sequencing (NGS) was first used to analyze the biology of cancers.1 It has since been rapidly implemented in clinical oncology to guide therapy.2, 3 EGFR, ALK, ROS1 and BRAF mutations account for approximately 30% and 60% of adenocarcinomas in the United States and Japan, respectively, and treatment targeting these gene alterations has been approved globally.4, 5 In addition, expression levels of PD‐L1 and tumor mutation burden have been shown to predict response to immune checkpoint inhibitors.6, 7, 8 As the number of genes to analyze has increased, the need to simultaneously analyze multiple genes has grown.
Targeted sequencing is considered superior to whole genome or whole exome sequencing in the clinical setting because of higher accuracy and lower costs.2, 9, 10, 11 From the perspective of the sequencing laboratory, samples are ideally obtained through surgical resection to analyze sufficient amounts of tumor cells and correctly call mutations. However, when patients have advanced lung cancer, CT‐guided needle biopsy (CTNB), endobronchial ultrasound‐guided transbronchial needle aspiration (EBUS‐TBNA) or transbronchial biopsy (TBB) are less invasive and are preferable to resection to make a pathological diagnosis. These advanced cancer patients are also the ones likely to benefit most from NGS. It is unknown whether DNA and RNA of adequate quality can be extracted from these samples to allow high‐quality sequencing.
The aim of this study was to compare CTNB, EBUS‐TBNA and TBB with surgical resection and to determine whether samples obtained through these methods are feasible for clinically targeted NGS.
2. MATERIALS AND METHODS
One hundred and seven consecutive samples from 67 patients were obtained from thoracic tumors or metastatic sites between April 2017 and March 2018 at the Department of Respiratory Medicine and the Department of Thoracic Surgery of The University of Tokyo Hospital. Multiple samples were analyzed in 21 patients; no sample was obtained from the same lesion. Fifteen samples were obtained through CTNB, 11 samples through EBUS‐TBNA, 11 samples through TBB with or without the use of EBUS‐ guide sheath (GS), and 70 samples through surgical resection, including lobectomy, partial lung resection and resection of pleural tumors. Eighteen‐gauge needles were used for CTNB. Bronchoscopy was performed under local anesthesia and intravenous midazolam. We used one of the following bronchoscopes: BF‐1T260, BF‐260, BF‐P260F or BF‐UC260FW (Olympus Corporation). EBUS‐TBNA was performed using a ViziShot 22‐Gauge needle (Olympus). A small K‐201 Guide Sheath Kit (Olympus) was used in combination with a radial EBUS probe, UM‐S20‐17S (Olympus). FB‐15C, FB‐20C or FB‐21C forceps were used for TBB without the use of EBUS‐GS. Samples were fixed in 20% neutral buffered formalin solution and paraffin‐embedded (FFPE) between 6 and 24 hours. According to the Japanese Society of Pathology Guidelines, 10% and 20% neutral buffered formalin solution results in similar DNA yield after 1 day.12 Twenty percent allows complete fixation of surgical specimens after 1 day, and results in higher RNA yield after 1 day compared with 10%. Whenever possible, 10‐20 slides, each with 10‐μm thickness, were prepared; areas enriched in tumor cells were macrodissected by a board‐certified pathologist.
Details of DNA/RNA extraction and targeted sequencing have been described elsewhere.11 In brief, DNA was extracted from whole blood using a Maxwell RSC Blood DNA Kit (Promega), and DNA and RNA were extracted from FFPE samples using a GeneRead DNA FFPE Kit (Qiagen) and an RNeasy FFPE Kit (Qiagen), respectively. DNA yield was quantified by quantitative PCR and DNA quality was determined by the ddCq method using FFPE DNA QC Assay v2 (Thermo Fisher Scientific); this compares the amplification efficiency of short and long amplicons, which reflects the amount of DNA that has been degraded.13 RNA yield was quantified by Qubit4 (Thermo Fisher Scientific) and DV200 was measured using the 4200 TapeStation System (Agilent Technologies). Our custom‐made “Todai OncoPanel” consists of a DNA panel to capture exon sequences of 464 cancer‐related genes and an RNA panel to capture transcripts of 463 genes. Massively parallel sequencing of the isolated fragments was performed with the NextSeq 500 platform (Illumina). Unique reads were counted by removing PCR duplicates from total paired‐end mapped reads; the percentage on target was calculated as the number of reads mapped to our target region divided by the number of total paired‐end mapped reads. NGS analysis from DNA was considered successful if the average depth was over 200×; cutoffs for DNA yield, quality and sequencing were set so that the average depth would be 500× or higher. NGS analysis from RNA was considered successful if 100× coverage was achieved in more than 70% of housekeeping genes; cutoffs for RNA yield, quality and sequencing were set to achieve this goal.
As all analyzed data did not show normal distributions, Kruskal‐Wallis one‐way analysis of variance on ranks was used to compare multiple groups. Pairwise multiple comparisons were performed using Dunn's test. We confirmed that one‐way analysis of variance with Tukey's post‐hoc analysis gave similar results. Statistical analyses were performed using SigmaPlot 12.3 (Systat Software) and R (version 3.5.0).
3. RESULTS
A median of 2, 4 and 7 biopsies were taken from CTNB, EBUS‐TBNA and TBB, respectively. The diameter of biopsy samples taken from CTNB is approximately 1 mm; the median sum of the lengths was 3 mm. The median diameter of the samples taken through EBUS‐TBNA and TBB was 2 mm. Two samples were biopsied after EGFR tyrosine kinase inhibitor and one was biopsied after chemotherapy. DNA and RNA were extracted from samples within 1 year after biopsy in 33 of 37 samples (89%).
We first analyzed the quantity and quality of DNA and RNA. DNA yield from the 3 biopsy methods was similar and less than the yield obtained from resected samples (Figure 1A and Table 1, P < 0.001). Quality of DNA, as measured by ddCq, was similar across all 4 methods (Figure 1B). RNA yield from the 3 biopsy methods was also similar and less than the yield obtained from resected samples (Figure 1C, P < 0.001). DV200 was similar across all 4 methods (Figure 1D). In short, the yield was lower with biopsy compared to resection, but the quality was similar for both DNA and RNA.
Figure 1.
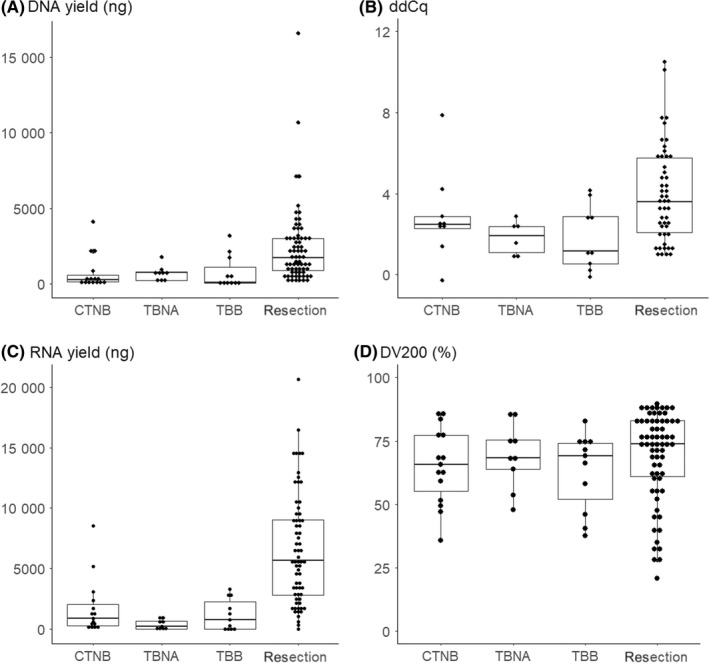
DNA and RNA yield and quality of each biopsy method. A, DNA yield. B, ddCq. C, RNA yield. D, DV200. CTNB, CT‐guided needle biopsy; EBUS‐TBNA, endobronchial ultrasound‐guided transbronchial needle aspiration; TBB, trans‐bronchial biopsy
Table 1.
Median DNA and RNA quality control values for each lung tumor biopsy method
| CTNB | TBNA | TBB | Resection | P‐value | |
|---|---|---|---|---|---|
| DNA QC measures | |||||
| DNA yield (ng) | 258 | 763 | 100 | 1738 | <.001 |
| ddCq (cycles) | 2.49 | 1.93 | 1.15 | 3.61 | >.05 |
| Total reads from DNA (×10⁶ reads) | 80.0 | 61.5 | 69.4 | 70.5 | >.05 |
| Unique reads (×10⁶ reads) | 32.5 | 33.1 | 26.8 | 30.9 | >.05 |
| Depth (×) | 866 | 891 | 645 | 760 | >.05 |
| % on target | 71% | 74% | 69% | 71% | >.05 |
| Tumor content | 36% | 33% | 24% | 32% | >.05 |
| Number of mutations | 5 | 12 | 3 | 5 | >.05 |
| RNA QC measures | |||||
| RNA yield (ng) | 894 | 256 | 788 | 5661 | <.001 |
| DV200 | 66% | 68% | 69% | 74% | >.05 |
| Total reads from RNA (×10⁶ reads) | 72.4 | 57.7 | 52.7 | 69.5 | >.05 |
| Housekeeping 100 × coverage | 84% | 83% | 83% | 83% | >.05 |
CTNB, CT‐guided needle biopsy; DV200, percentage of RNA fragments above 200 nucleotides; QC, quality control; TBB, transbronchial biopsy; TBNA, transbronchial needle aspiration.
We next analyzed the quality of sequencing. Total read number, number of unique reads and percentage of reads on target were all similar for DNA analysis (Table 2). The average depth was similar across all 4 methods (Figure 2A). RNA analysis also gave a similar number of total reads across the 4 methods, and the percentage of housekeeping genes with coverage of over 100× was also similar (Figure 2B).
Table 2.
Percentage of samples considered satisfactory for each lung tumor biopsy method
| CTNB (%) | TBNA (%) | TBB (%) | Resection (%) | |
|---|---|---|---|---|
| Success rates for DNA | ||||
| DNA yield > 50 ng | 80 | 100 | 82 | 99 |
| ddCq < 8.0 | 100 | 100 | 100 | 94 |
| Total reads > 40 000 000 | 93 | 100 | 100 | 97 |
| Unique reads > 12 000 000 | 87 | 100 | 82 | 97 |
| Average depth > 200 | 93 | 100 | 82 | 99 |
| % on target > 30% | 100 | 100 | 100 | 99 |
| Fully adequate analysis from DNA | 80 | 100 | 82 | 93 |
| Success rates for RNA | ||||
| RNA yield > 50 ng | 100 | 67 | 64 | 98 |
| DV200 > 30% | 100 | 100 | 100 | 95 |
| Total RNA reads > 20 000 000 | 100 | 82 | 73 | 98 |
| Housekeeping 100x coverage > 70% | 100 | 82 | 73 | 94 |
| Fully adequate analysis from RNA | 100 | 67 | 64 | 92 |
CTNB, CT‐guided needle biopsy; ddCq, delta delta quantitation cycle; DV200, percentage of RNA fragments above 200 nucleotides; TBB, transbronchial biopsy; TBNA, transbronchial needle aspiration.
Figure 2.
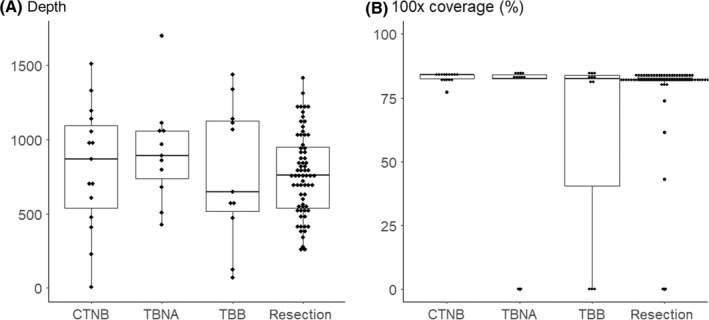
Quality control measures of targeted next generation sequencing for each biopsy method. A, Average depth. B, Estimated tumor content
To determine the rates of adequate DNA and NGS quality, cutoffs for DNA yield were set at 50 ng, ddCq at 8.0, total reads at 40 million, unique reads at 12 million, percentage on target at 30%, and depth at 200×. DNA yield was higher than 50 ng in 80% of CTNB samples, 100% of EBUS‐TBNA samples, 82% of TBB samples and 99% of resected samples (Table 2). Average depth was over 200× in 93% of CTNB samples, 100% of EBUS‐TBNA samples, 82% of TBB samples and 99% of resected samples. Overall, 12 of 15 CTNB samples (80%), all 11 EBUS‐TBNA samples, 9 of 11 TBB samples (82%) and 65 of 70 resected samples (93%) underwent fully adequate NGS assays from DNA.
Although estimated tumor content of samples is not directly related to the quality of DNA, low tumor content can lead to false‐negative results. Median tumor content was similar among the 4 biopsy methods. Tumor content was above 12% in 85% of CTNB samples, 91% of EBUS‐TBNA samples, 73% of TBB samples and 78% of resected samples. The number of mutations for each biopsy method can also be used as a surrogate for NGS quality because low mutation number can be due to false‐negative results. The median numbers of mutations were 5, 12, 3 and 5 for CTNB, EBUS‐TBNA, TBB and resection, respectively (P = 0.09).
Next‐generation sequencing analysis from RNA was successful in all 15 CTNB samples, 9 of 11 EBUS‐TBNA samples (82%), 8 of 11 TBB samples (73%) and 63 of 66 resected samples (95%). Seven samples had RNA yields below the detection limit. Of 7 samples, 4 also had DV200 of <50% and all 4 samples were not suitable for NGS analysis, while 2 of 3 samples with RNA yield below the detection limit but DV200 over 50% had successful NGS runs.
4. DISCUSSION
Precision medicine has been adopted in clinical oncology to maximize survival of patients with advanced cancer. NGS helps to characterize each cancer and is rapidly being implemented to guide therapy. However, surgical resection is too invasive in patients with advanced cancer, and small biopsy samples, which are a few millimeters in size, are taken from the tumor to reach a pathological diagnosis. We have shown that small biopsy samples mostly yield adequate quality DNA and RNA, enabling high‐quality NGS analysis.
Previous studies have relied on the number of gene mutations detected as a surrogate for NGS quality. A large study analyzed 7 genes in 500 samples obtained from biopsies and fine needle aspirations, and detected mutations in over 90% of samples.14 A smaller study analyzing 467 genes in 17 samples found that 16 samples “yielded a full report.”15 Another study analyzed 50 genes in 162 patients and detected mutations in 161 patients.16 However, the quality of the samples, extracted DNA and sequencing itself are unknown. In addition, to our knowledge, no previous study has performed clinical targeted NGS using RNA extracted from FFPE samples.
The most comprehensive study to date has focused on the feasibility of EBUS‐TBNA for NGS analysis.17 Of the 115 samples sent for NGS analysis, 16 (14%) were considered unsuccessful, defined as <10% proportion of tumor cells by visual estimate or <50 ng of extracted DNA. In addition, 17 (15%) were considered to have borderline tumor content, defined as the proportion of tumor cells between 10% and 20%, resulting in 82 (71%) samples which underwent fully adequate assays. Of the 16 samples with unsuccessful assays, 5 had low a DNA yield of <50 ng, 8 had insufficient tumor content, and 2 were sequence failures. Other biopsy methods were not analyzed, and RNA analysis was not performed.
We have shown that feasibility of CTNB, EBUS‐TBNA and TBB are comparable to resection. We have found that 89% of all small biopsy samples gave adequate DNA quantity and quality, which resulted in an 86% success rate of NGS analysis. Of the 4 samples with low DNA yield and low average depth, 2 were samples from more than 2 years ago and 1 was a biopsy taken after chemotherapy was performed. Avoiding these samples should lead to a higher percentage of samples being suitable for NGS analysis.
In addition to NGS analysis using DNA, RNA analysis allows robust detection of fusion transcripts and exon skipping.11 We have shown that small biopsy samples are feasible for targeted NGS analysis using RNA extracted from FFPE samples. RNA analysis was successfully performed in 30 of 37 biopsy samples. Of the 5 samples that failed, 4 had both low RNA yield and high RNA degradation, defined as RNA yield < 50 ng and DV200 < 50%. Of note, 3 out of 5 samples were biopsied by TBB only with the use of the guide sheath, and the same 3 samples also had low DNA yield of <100 ng. Using the guide sheath leads to higher cancer detection rate of TBB but smaller size of biopsied samples.18 To perform NGS analysis from RNA, samples should be biopsied using standard forceps.
The Japanese Society of Pathology Guidelines recommend 10% neutral buffered formalin to fix samples intended for NGS analysis.12 This is based on data showing higher DNA quality compared with 15% or 20% after 3 days.12 The same figure shows that 10% and 20% neutral buffered formalin solution result in similar DNA yield after 1 day. Twenty percent allows faster fixation, and also results in higher RNA yield after 1 day compared with 10%. All our samples were fixed using 20% neutral buffered formalin and gave excellent results. Pathologists and researchers need to keep in mind that samples should be fixed for 24 hours or less.
Limitations of our study include the retrospective nature of our analysis and the small number of samples. More samples are needed before concluding that older archival tissue and post–chemotherapy tissue are better avoided. More samples are also necessary to determine whether DV200 is superior to RNA yield to predict successful RNA analysis.
In conclusion, CTNB, EBUS‐TBNA and TBB mostly resulted in adequate DNA and RNA quality, enabling high‐quality targeted NGS analysis. Our results indicate that small biopsies may be feasible for targeted NGS in general.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Kage H, Kohsaka S, Shinozaki‐Ushiku A, et al. Small lung tumor biopsy samples are feasible for high quality targeted next generation sequencing. Cancer Sci. 2019;110:2652–2657. 10.1111/cas.14112
REFERENCES
- 1. Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607‐616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering‐integrated Mutation Profiling of Actionable Cancer Targets (MSK‐IMPACT): a hybridization capture‐based next‐generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inoue Y, Shiihara J, Miyazawa H, et al. A highly specific and sensitive massive parallel sequencer‐based test for somatic mutations in non‐small cell lung cancer. PLoS ONE. 2017;12:e0176525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kohno T, Nakaoku T, Tsuta K, et al. Beyond ALK‐RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 2015;4:156‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lindeman NI, Cagle PT, Aisner DL, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J Thorac Oncol. 2018;13:323‐358. [DOI] [PubMed] [Google Scholar]
- 6. Rizvi H, Sanchez‐Vega F, La K, et al. Molecular determinants of response to anti–programmed cell death (PD)‐1 and anti–programmed death‐ligand 1 (PD‐L1) blockade in patients with non‐small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol. 2018;36:633‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reck M, Rodriguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 8. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093‐2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kohsaka S, Tatsuno K, Ueno T, et al. Comprehensive assay for the molecular profiling of cancer by target enrichment from formalin‐fixed paraffin‐embedded specimens. Cancer Sci. 2019;110:1464‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kanai Y, Nishihara H, Miyagi Y, et al. The Japanese Society of Pathology Guidelines on the handling of pathological tissue samples for genomic research: standard operating procedures based on empirical analyses. Pathol Int. 2018;68:63‐90. [DOI] [PubMed] [Google Scholar]
- 13. Dang J, Mendez P, Lee S, et al. Development of a robust DNA quality and quantity assessment qPCR assay for targeted next‐generation sequencing library preparation. Int J Oncol. 2016;49:1755‐1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Illei PB, Belchis D, Tseng LH, et al. Clinical mutational profiling of 1006 lung cancers by next generation sequencing. Oncotarget. 2017;8:96684‐96696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DiBardino DM, Rawson DW, Saqi A, Heymann JJ, Pagan CA, Bulman WA. Next‐generation sequencing of non‐small cell lung cancer using a customized, targeted sequencing panel: emphasis on small biopsy and cytology. Cytojournal. 2017;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ku BM, Heo MH, Kim JH, et al. Molecular screening of small biopsy samples using next‐generation sequencing in Korean patients with advanced non‐small cell lung cancer: Korean Lung Cancer Consortium (KLCC‐13‐01). J Pathol Transl Med. 2018;52:148‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Turner SR, Buonocore D, Desmeules P, et al. Feasibility of endobronchial ultrasound transbronchial needle aspiration for massively parallel next‐generation sequencing in thoracic cancer patients. Lung Cancer. 2018;119:85‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Izumo T, Sasada S, Chavez C, Matsumoto Y, Hayama M, Tsuchida T. The diagnostic value of histology and cytology samples during endobronchial ultrasound with a guide sheath. Jpn J Clin Oncol. 2015;45:362‐366. [DOI] [PubMed] [Google Scholar]