

Abstract
Multigenerational adverse effects from the environment such as nutrition and chemicals are among important concerns in environmental health issues. Previously, we have found that arsenite exposure of only F0 females during their pregnancy increases hepatic tumors in the F2 males in C3H mice. In the current study, we investigated the association of DNA methylation with the hepatic tumor increase in the F2 males of the arsenite group. Reduced‐representation bisulfite sequencing analysis newly identified that DNA methylation levels of regions around the transcriptional start sites of Tmem54 and Cd74 were decreased and the expression of these genes were significantly increased in the hepatic tumors of F2 males of the arsenite group. The associations between DNA methylation in these regions and gene expression changes were confirmed by treatment of murine hepatoma cell lines and hepatic stellate cell line with 5‐aza‐2′‐deoxycytidine. Overexpression of Cd74 in Hepa1c1c7 cells increased Trib3 expression and suppressed the expression of tumor suppressor genes Id3 and Atoh8. Human database analysis using the Cancer Genome Atlas indicated that TMEM54, CD74, and TRIB3 were significantly increased and that ATOH8 was decreased in hepatocellular carcinoma. The data also showed that high expression of TMEM54 and TRIB3 and low expression of ATOH8 were associated with poor survival. These results suggested that an increase in Tmem54 and Cd74 expression via DNA methylation reduction was involved in the tumor increase in the F2 male offspring by gestational arsenite exposure of F0 females. This study also suggested that genes downstream of Cd74 were involved in tumorigenesis.
Keywords: arsenite, DNA methylation, hepatic tumor, multigenerational effect, next‐generation sequencing
Abbreviations
- CCN, CCTn, CCTt:
normal liver tissues (N) and the nontumor (Tn) and tumor tissues (Tt) of tumor‐bearing livers of F2 males obtained by crossing control sires and control dams
- AAN, AATn, AATt:
normal liver tissues (N) and the nontumor (Tn) and tumor tissues (Tt) of tumor‐bearing livers of F2 males obtained by gestationally arsenite‐exposed F1 sires and gestationally arsenite‐exposed F1 dams
1. INTRODUCTION
Multigenerational adverse effects from environmental factors are one of the important concerns for health.1 In pregnant females (designated as the filial (F)0 generation), chemical exposure interacts not only with their offspring (F1 generation) but also the germ cells, the origin of their grandchildren (F2 generation), developing within the F1 animal.2 Exposure of pregnant F0 mice to various environmental factors has been reported to cause harmful health effects, such as metabolic disorders and cancers, in the F2 as well as the F1 generations.3, 4, 5
Epidemiological studies have reported an increased risk of carcinogenesis in adulthood by gestational arsenic exposure.6, 7, 8 Previous studies have reported that gestational arsenite exposure increased hepatic tumor incidence in F1 males in C3H mice,9, 10 whose males tend to develop spontaneous hepatic tumors.11 In the same experimental model,9, 10 we found that hepatic tumor incidence was increased even in the F2 males born to the F1 males gestationally exposed to arsenite compared with F2 males of control group, irrespective of exposure of F1 females.12 However, the mechanisms of hepatic tumor increase in the F2 by gestational arsenic exposure have not yet been clarified.
Epigenetic changes are inheritable and are suspected to be associated with multigenerational and transgenerational effects.13 Epigenetics is a regulation system of gene expression without changing DNA sequences. DNA methylation, histone modifications, and noncoding RNAs are known as major epigenetic modifications.14 The most abundant epigenetic mark of DNA is methylation of carbon 5 of cytosines, which creates 5‐methylcytosine (5mC). In addition, 5‐hydroxymethylcytosine (5hmC), an intermediate metabolite of 5mC in the active demethylation process, is another epigenetically modified base.15 Those methylation changes are related to adverse health effects including cancer.16, 17 Total levels of 5mC and 5hmC are known to be reduced in cancer tissues, and can compromise genomic integrity and lead to tumor growth.18, 19 In tumor tissues, site‐specific hypermethylation of cytosines have also been observed in the promoter regions of suppressor genes and are considered to contribute to tumor formation.20
In the current study, we investigated DNA methylation changes in the liver tissues of F2 C3H male mice that was induced by gestational arsenite exposure of F0 females. In particular, we measured 5mC and 5hmC content of in the genome using liquid chromatography tandem mass spectrometry (LC‐MS/MS) and detected genome‐wide site‐specific DNA methylation using a reduced‐representation bisulfite sequencing (RRBS) method.21 The results of this study propose novel DNA methylation‐dependent pathways that increase hepatic tumors in the F2 males following gestational arsenite exposure.
2. MATERIALS AND METHODS
2.1. Animal and arsenite treatment
Animal experiments were performed as previously described.12 Briefly, pregnant C3H/HeN mice (F0) were purchased from CLEA Japan (Tokyo, Japan) and given free access to a standard diet (CA‐1; CLEA Japan) and tap water with or without 85 ppm sodium arsenite (Sigma, St.Louis, MI, USA) from d 8 to d 18 of gestation.12 Throughout the experiments, arsenite was only given to F0 pregnant mice and not to F1 or F2 mice. F1 male and female mice were mated at 10 wk of age. The F2 males were reared until around 74 wk of age and used for the assessment. Hepatic tumors were visually separated from the livers. All animal procedures were approved by the Animal Care and Use Committee of the National Institute for Environmental Studies (NIES), Japan. All animals were treated humanely in accordance with the NIES guideline for animal experiments.
2.2. Measurement of 5mC and 5hmC amounts
Five genomic DNA (gDNA) samples from 5 mice born to different dams were used per group. gDNA (1 μg) was hydrolyzed as described previously.22 Standard materials 5‐hydroxymethyl‐2′‐deoxycytidine and 5‐hydroxymethyl‐2′‐deoxycytidine‐d3 were purchased from Toronto Research Chemicals Inc (North York, Canada). Internal standards, ie 100 ng 13C9,15N3‐2′‐deoxycytidine, 10 ng 13C10,15N2‐5‐methyl‐2′‐deoxycytidine,23 and 1 ng 5‐hydroxymethyl‐2′‐deoxycytidine‐d3 were added to 5 μL of the hydrolyzed sample, and the mixture was diluted with H2O. LC‐MS/MS analyses were performed using an LC‐MS‐8040 mass spectrometer and an electrospray ionization (ESI) probe (Shimadzu, Kyoto, Japan) in multiple reaction monitoring mode. Deoxycytidine, 5‐methyldeoxycytidine and 5‐hydroxymethyl‐2′‐deoxycytidine were separated using a reversed‐phase column (Atlantis T3; Waters, Milford, MA, USA) in gradient elution mode. Eluent A was a 10 mmol/L aqueous ammonium acetate solution, and eluent B was methanol. The following gradient eluting system was used at a flow rate of .2 mL/min: 2% B for 5 min, 2%‐8% B over .1 min, hold 3.9 min, 8%‐30% B over 4 min, and hold 2.5 min at a flow rate of .2 mL/min.
2.3. RRBS analysis and identification of differentially methylated cytosines (DMCs) and differentially methylated regions (DMRs)
Reduced‐representation bisulfite sequencing analysis was performed as previously described.24 Briefly, gDNA was prepared from 3 mice from different litters per group by phenol‐chloroform extraction and 100 ng of gDNA was used per sample for RRBS library preparation. The libraries were sequenced on an Illumina HiSeq2500. The sequencing reads were mapped on the mouse reference genome (mm10) with the use of the Bismark program25 and adapter trimming and quality control was performed using Trim Galore software (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The methylation status of each CpG was calculated using the read.Bismark function of methylKit package (v. 0.9.4)26 in R. DMCs and DMRs were selected using the methylKit package and eDMR package27 in R, respectively. DMC was defined as the CpG site with >20% methylation difference between the 2 groups, <.01 q‐value by the logistic regression method, and >10 reads in coverage in both groups. DMR was defined as the region that contained >3 CpGs and one DMC and whose methylation level was different over 20% with a statistically significant difference between the 2 groups. The distance of DMRs from the transcription start sites (TSSs) of the closest RefSeq genes were extracted using bedtools 2.25.0 (bedtools.readthedocs.io). Sequence data are publicly available at Gene Expression Omnibus (GEO), accession number GSE111420 for CCN and CCTt, and GSE123749 for the other 4 groups (CCTn, AAN, AATn, and AATt).
2.4. RNA isolation, microarray analysis, and quantitative reverse‐transcription polymerase chain reaction
Total RNA was extracted from tissues or cultured cells using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Qualities of RNA samples were checked using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Total RNA was hybridized to the SurePrint G3 Mouse GE 8x60K Microarray (Agilent Technologies) according to the manufacturer's protocol. Signal intensities were normalized to the 75th percentile of all measurement using Agilent Gene‐Spring GX software. The expression data flagged “glsSaturated = 0,” “glsFeatPopnOL = 0,” “gls Feat NonUnifOL = 0” and “glsWellaboveBG = 1” were utilized for analysis of liver tissues. The genes with a ≥2‐ fold increase or decrease in mean expression values (n = 3) were judged to be changed. In the experiment for Hepa1c1c7 cells with or without overexpression of Cd74 or Tmem54, genes with raw expression values of >50, and genes with an at least 2‐fold increase or decrease were utilized for further analysis. Array data are publicly available at GEO, accession number GSE104627 for CCN and CCTt groups and GSE123737 for CCTn, AAN, AATn, and AATt. Array data for Hepa1c1c7 cells are available at GEO, accession number GSE123718.
cDNA synthesis and RT‐qPCR analysis were performed as described previously.28 The primer sequences and annealing temperatures are shown in Table S1.
2.5. Cell culture and treatment
Murine hepatic stellate cell line GRX cells (0094) were purchased from the Rio de Janeiro Cell Bank (Rio de Janeiro, Brazil). Murine hepatoma cell line Hepa1c1c7 cells, Hepa1‐6 cells, and GRX cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Sigma, D5796) with 1% penicillin/streptomycin and10% fetal bovine serum (FBS). 5‐Aza‐2‐deoxycytidine (5‐aza‐dC) was obtained from Santa Cruz Biotechnology. Cells were seeded into 6‐well culture plates at a density of 3‐5 × 105 cells per well. After 24 h, cells were treated in culture medium with 1‐100 μmol/L 5‐aza‐dC for 72 h and subjected to bisulfite sequencing analysis. Mouse ORF clones Tmem54 (MC203978), Cd74 (MC200462), and pCMV6‐Kan/Neo vectors were purchased from Origene Inc (Rockville, MD, USA). Cells were transfected with these vectors using Amaxa nucleofector II (Lonza, Basel, Switzerland) according to the manufacturer's instructions. Briefly, 1 × 106 of Hepa1c1c7 cells were transfected with 2 μg of each vector using the Nucleofector Kit V (program X‐005). After 24 h, cells were collected and subjected to real‐time PCR and microarray analysis.
2.6. Bisulfite sequencing analysis and pyrosequencing
Five gDNA samples were isolated from 5 mice obtained from different litters per group. Bisulfite sequencing analysis was performed as previously described.29 The bisulfite‐treated gDNA were amplified with the primers shown in Table S1. PCR products were purified and cloned into the pGEM‐T Easy Vector (Promega, Madison, WI, USA). Obtained clones were cycle sequenced using M13RV primers and the BigDye Terminator version 3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA), and analyzed using an Applied Biosystems 3730 DNA analyzer. Pyrosequencing was performed as described previously.12 Briefly, bisulfite‐modified gDNAs were amplified by PCR. Primers are shown in Table S1. We performed sequencing of the products using the PryoMark Q96 ID System (Qiagen) according to the manufacturer's protocol.
2.7. Gene expression analysis of human hepatocellular carcinoma
Human hepatocellular carcinoma (HCC) data were acquired from the TCGA database (http://cancergenome.nih.gov/) as previously described.24 Fifty paired RNA‐seq datasets of normal livers and HCC tissues were downloaded and used for gene expression analysis.
2.8. Survival analysis
We analyzed the clinical data of liver hepatocellular carcinoma (LIHC) using Oncolnc (http://www.oncolnc.org/), which can link TCGA survival data to mRNA expression levels. The survival data were separated to 2 groups, a high expression group (top 33%) and a low expression group (bottom 33%), and were assessed for the correlation between survival and gene expressions.
2.9. Statistical analysis
Statistical significance was determined using Student's t test or the Tukey‐Kramer test. P‐values < .05 were considered significant. For survival analysis, the log‐rank test was performed and P‐values < .05 were considered significant.
3. RESULTS
3.1. Contents for genomic 5mC and 5hmC decreased in tumor tissues but were not changed between F2 males of the control group and the arsenite group
Levels of genomic 5mC and 5hmC are substantially reduced in cancer tissues. These changes are implicated in compromised genomic integrity and tumorigenesis.18, 19, 20 Therefore, we investigated whether reductions in 5mC and 5hmC levels were involved in tumor increase in F2 males following gestational arsenic exposure of F0 females. To determine the content of 5mC and 5hmC in the liver genome, we made accurate measurements of these in hepatic tissues of the F2 males by performing LC‐MS/MS with stable isotope‐labeled surrogates as the internal standards. We defined F2 males obtained by crossing control F1 males and females as control (CC), and those obtained by crossing gestationally arsenite‐exposed F1 males and females as arsenite (AA). The normal liver tissues (N) and the nontumor (Tn) and tumor tissues (Tt) of tumor‐bearing livers were designated CCN, CCTn, and CCTt for the control group and AAN, AATn, and AATt for the arsenite group, respectively. We observed that the content of genomic 5mC was significantly decreased in tumor tissues compared with normal tissues both in the control and arsenite groups (Table 1). As with the reduction in 5mC, content of genomic 5hmC was also significantly decreased in tumor tissues compared with normal tissues. However, there were not any significant changes in the content of genomic 5mC and 5hmC between the control and arsenite groups. These results indicated that reductions in total levels of 5mC and 5hmC were not involved in tumor increase in the F2 males from the arsenite group.
Table 1.
The contents of genomic 5mC and 5hmC in the liver tissues of F2 males
| F2 | Control | Arsenite | ||||
|---|---|---|---|---|---|---|
| CCN | CCTn | CCTt | AAN | AATn | AATt | |
| 5mC | % in total cytosine | % in total cytosine | ||||
| 5.66 ± .05 | 5.63 ± .04 | 5.30 ± .19a , b | 5.64 ± .04 | 5.77 ± .07a | 5.28 ± .13a , b | |
| 5hmC | % of (5mC + 5hmC) | % of (5mC + 5hmC) | ||||
| 10.27 ± .34 | 10.40 ± .38 | 3.50 ± .93a , b | 9.92 ± .31 | 10.21 ± .60 | 3.46 ± 1.43a , b | |
Significantly changed compared with a control tissues b nontumor tissues.
3.2. Genome‐wide DNA methylation profiles in the normal and tumor liver tissues of F2 males analyzed using the RRBS method
Next, to detect site‐specific changes in DNA methylation, we performed RRBS analysis. We obtained 1 975 828 CpG sites with at least 10× coverage in all samples. The average DNA methylation levels of the 3 types of liver tissues in the control group (CCN, CCTn and CCTt) and those in the arsenite group (AAN, AATn and AATt) are shown in Figure 1A. The average methylation levels ranged from 30.5 (AATt) to 31.5% (CCN) (Figure S1A). Upstream regions far from TSSs were highly DNA methylated, and the methylation levels gradually decreased toward TSSs, where they troughed at approximately 3%. The regions downstream of TSSs were again highly methylated except around the immediate vicinity of transcription end sites (TESs), where methylation levels dropped to approximately 20% (Figure 1A). The numbers of CpG sites were highly enriched in TSS regions and modestly in TES regions (Figure S1B).
Figure 1.
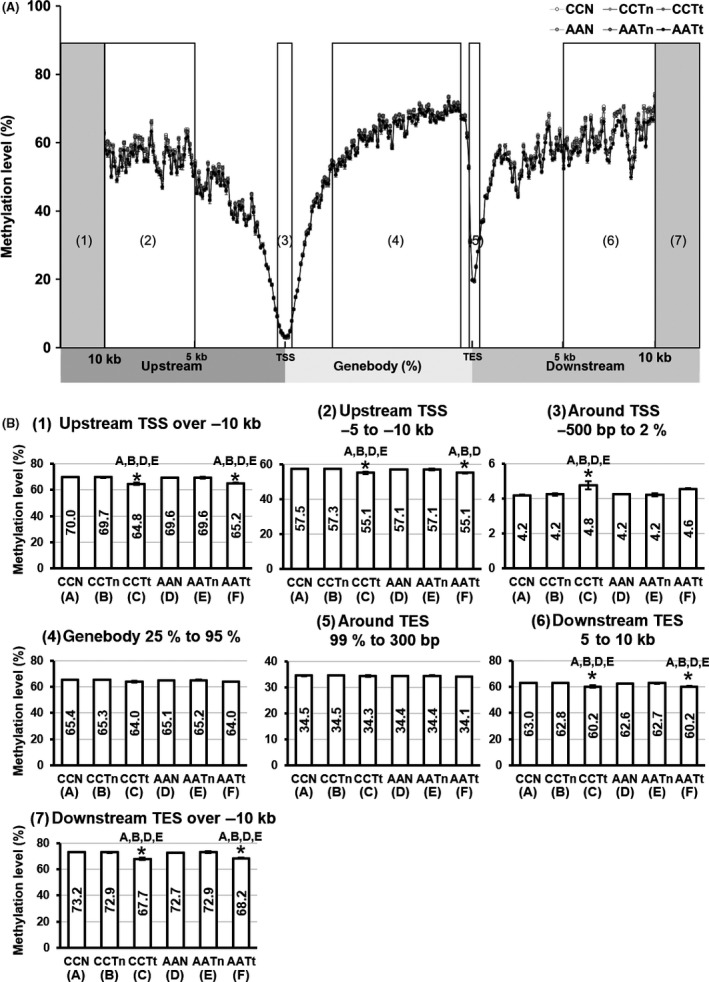
Average DNA methylation levels in F2 livers determined by reduced‐representation bisulfite sequencing analysis. A, DNA methylation levels in relation to distance from the transcriptional start site (TSS). We plotted DNA methylation levels in each 100 bp at intergenic regions and plotted in each 1% at intragenic region. B, Region‐specific changes of DNA methylation levels
Comparison of methylation levels in the 6 groups showed that the intergenic regions far from the TSSs and TESs (Figure 1B[1][2][6][7]) were significantly DNA hypomethylated in tumor tissues compared with normal and nontumor tissues. Gene body regions (Figure 1B[4]) were also slightly hypomethylated in tumor tissues, although not significantly. In contrast, the regions around TSSs were significantly hypermethylated in CCTt (Figure 1B[3]). The regions around TSSs were also slightly hypermethylated in AATt compared with normal tissues and nontumor tissues, although not significantly. DNA methylation levels around TES regions (Figure 1B[5]) were not changed among the groups. We did not detect any significant changes in the average DNA methylation levels between the control groups and arsenite groups.
3.3. Detection of region‐specific DNA methylation changes in the F2 livers associated with gestational arsenite exposure of F0 mice
To determine specific DNA methylation changes between groups, we detected DMCs and DMRs using methylKit and eDMR packages in R. We have reported a comparison between CCN and CCTt elsewhere.24 To confirm the success of the analysis, we compared the differences between the AAN group and AATt group. As a result, 24 495 hyper‐DMCs and 60 646 hypo‐DMCs were detected in AATt compared with AAN (Figure S2A[1]). The number of hypo‐DMCs was higher than that of hyper‐DMCs in almost all chromosomes (Figure S2A[2],[3]). The distribution of DMCs in genomic regions showed that approximately 40% of hyper‐DMCs were detected in the promoter and exon regions, whereas over 80% of hypo‐DMCs were detected outside the promoter and exon regions (Figure S2A[4]). These results were successfully consistent with the tumor features previously reported.30
Next, to detect DMCs and DMRs caused by gestational arsenite exposure, we compared RRBS data between the control and arsenite groups. The numbers of DMCs and DMRs were determined by comparisons between the control and arsenite groups in normal tissues, nontumor tissues, and tumor tissues, respectively. We detected 107 DMRs between CCN and AAN, 57 DMRs between CCTn and AATn, and 1547 DMRs between CCTt and AATt, respectively. Among these, we extracted DMRs at ±2000 bp of the TSS (promoter DMR) to seek DMRs that are closely associated with gene expression (Table S2). Then we checked these DMRs in relation to gene expression using microarray data. We selected the candidate genes that might have been affected by gestational arsenite exposure as follows: (a) DNA methylation changes of DMRs and gene expression changes were negatively correlated; (b) at least 2‐fold expression changes were detected; and (c) raw microarray signals were not lower than 1000. In CCTt vs AATt, 2 genes, Tmem54 and Cd74, met all the criteria, while no candidate genes were detected by comparison of CCN vs AAN or CCTn vs AATn.
3.4. Validation of gene expression and DNA methylation of Tmem54 and Cd74 in the hepatic tumors of F2 males
We focused on Tmem54 (transmembrane protein 54) and Cd74 (CD74 antigen). TMEM54 was identified as a component of the ubiquitin ligase complex including a cullin region, which is also conserved in mice orthologs.31 Figure 2A shows the DMR region displayed using the Integrative Genomics Viewer (IGV). Figure 2B shows the methylation levels of promoter DMR of Tmem54 obtained by RRBS analysis for normal tissues, nontumor tissues, and tumor tissues in the control and arsenite groups. The data showed statistically significant hypomethylation in AATt compared with CCTt. We confirmed DNA methylation levels of cytosines in the DMR for CCTt and AATt by bisulfite sequencing analysis. The analyzed region is shown in Figure 2A (marked as Bisulfite‐seq.) and the sequencing results of the region in 5 CCTt samples and 5 AATt samples are shown in Figure 2C,D. Among the 38 CpG sites analyzed, methylation levels of 8 sites were significantly decreased in AATt compared with CCTt (Figure 2D). The total methylation levels of the region were also significantly reduced in AATt (Figure 2D). Figure 2E shows the Tmem54 expression in the 6 groups. The data show that Tmem54 expression is specifically upregulated in the tumors and is significantly upregulated in AATt compared with CCTt. These results suggested that expression of Tmem54 is upregulated by some factors in tumor tissues (CCTt and AATt) and reduction of DNA methylation in the DMR by gestational arsenic exposure of F1 males is involved in the upregulation of Tmem54 in AATt compared with the expression in CCTt.
Figure 2.
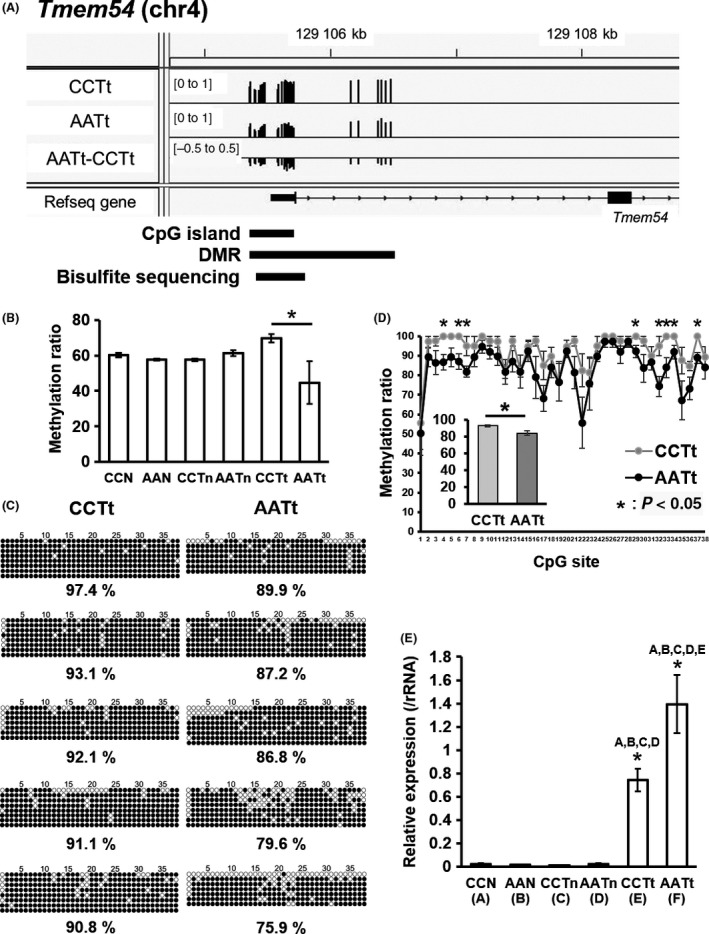
Validation of the promoter differentially methylated region (DMR) and the gene expression of Tmem54 in AATt and CCTt. A, Visualizing reduced‐representation bisulfite sequencing (RRBS) data for DNA methylation levels around the transcriptional start site (TSS) of Tmem54 by Integrative Genomics Viewer (IGV). B, DNA methylation levels of Tmem54 DMR obtained by RRBS analysis. C, Bisulfite sequencing analysis of Tmem54 DMR. ○, Unmethylated cytosine; ●, methylated cytosine. D, DNA methylation levels of Tmem54 DMR calculated from the data of bisulfite sequencing shown in C. E, Expression of Tmem54 measured by real‐time polymerase chain reaction (PCR)
CD74 is also a transmembrane protein and a transcription regulator functioning by interacting with the transcription factors RUNX and NF‐κB.32 The DMR region of Cd74 is shown in Figure 3A. The methylation levels of Cd74 DMR obtained by RRBS analysis in normal tissues, nontumor tissues, and tumor tissues in the control and arsenite group are shown in Figure 3B. The Cd74 DMR in AATt is significantly hypomethylated compared with CCTt, and also compared with CCN, AAN, and CCTn. The methylation level of DMC in Cd74 DMR was validated for CCTt and AATt using pyrosequencing (Figure 3C). Cd74 expression was confirmed to be significantly increased in AATt in comparison with CCTt, CCN, AAN, and CCTn using real‐time PCR (Figure 3D). These results demonstrated that the DNA methylation levels of DMR and gene expression were inversely correlated in Cd74, suggesting the involvement of DNA methylation in gene expression regulation.
Figure 3.
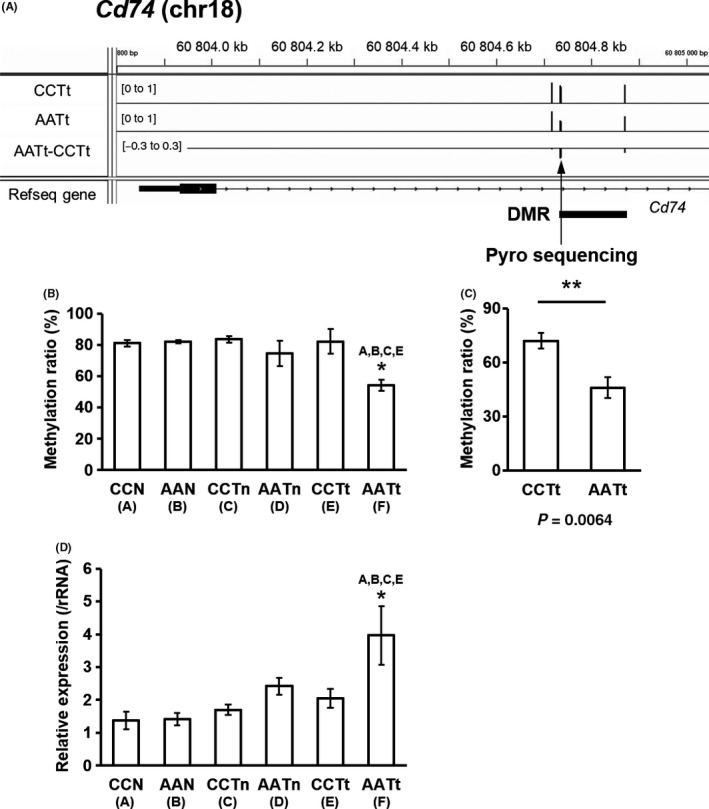
Validation of the differentially methylated cytosine (DMC) and the gene expression of Cd74 in AATt and CCTt. A, Visualizing reduced‐representation bisulfite sequencing (RRBS) data for DNA methylation levels around TSS region of Cd74 by Integrative Genomics Viewer (IGV). Arrowhead shows the DMC that was assessed by pyrosequencing. B, DNA methylation levels of Cd74 DMR obtained by RRBS analysis. C, Pyrosequencing analysis of the DMC of Cd74. D, mRNA expression of Cd74 measured by real‐time polymerase chain reaction (PCR)
3.5. Expression of Tmem54 and Cd74 is upregulated by 5‐aza‐dC treatment in hepatoma cells
To determine whether DNA methylation changes in the DMRs of Tmem54 and Cd74 are involved in the expression regulation of these genes, we confirmed the relationship using the DNA methylation inhibitor 5‐aza‐dC in the culture of murine hepatoma cell line Hepa1c1c7 cells and hepatic stellate cell line GRX cells. 5‐Aza‐dC treatment of Hepe1c1c7 cells showed upregulation of Tmem54 in a dose‐dependent manner (Figure 4A). DNA methylation of the same 38 CpG sites in the DMR region of Tmem54, as shown in Figure 2A, were confirmed to be reduced by 5‐aza‐dC treatment (Figures 4B, S3A). Significant upregulation of Tmem54 (Figure 4C) and hypomethylation of CpGs in the DMR (Figures 4D, S3B) was also confirmed by 5‐aza‐dC treatment of GRX cells.
Figure 4.
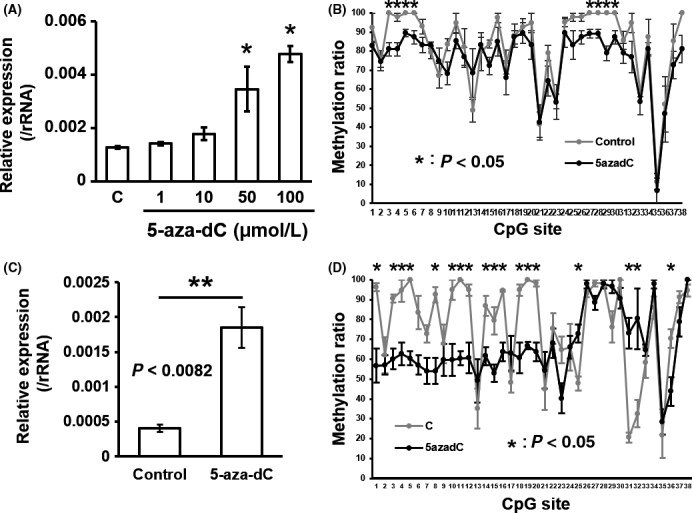
5‐Aza‐dC treatment inhibits DNA methylation of Tmem54 DMR and upregulates mRNA expression in Hepa1c1c7 cells and GRX cells. Expression of Tmem54 was measured in Hepa1c1c7 cells treated with different doses of 5‐aza‐dC (A) or GRX cells treated with 50 μmol/L of 5‐aza‐dC (C) by real‐time PCR. The methylation level of each CpG site in the DMR was validated in Hepa1c1c7 cells (B) and GRX cells (D) by bisulfite sequencing
Likewise, Cd74 expression was upregulated (Figure 5A,C,E) and DMC in the DMR was hypomethylated (Figure 5B,D,F) by 5‐aza‐dC treatment of Hepa1c1c7 cells, Hepa1‐6 cells, and GRX cells. These data support the notion that DNA methylation changes in Tmem54 and Cd74 are involved in gene expression.
Figure 5.
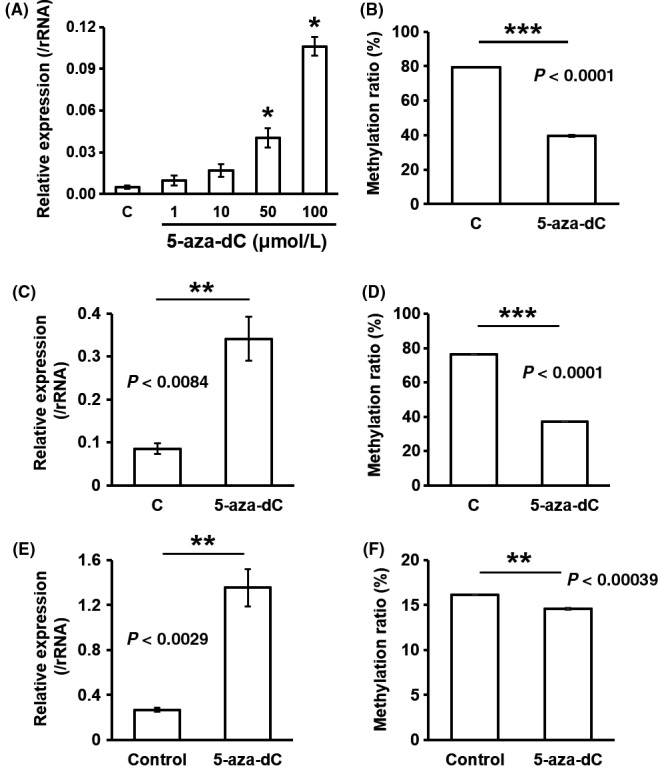
5‐Aza‐dC treatment inhibits DNA methylation of Cd74 DMR and upregulates mRNA expression in murine hepatic cells. mRNA expression of Cd74 was measured in Hepa1c1c7 cells treated with different doses of 5‐aza‐dC (A), Hepa1‐6 cells treated with 1 μmol/L of 5‐aza‐dC (C), or GRX cells treated with 50 μmol/L of 5‐aza‐dC (E) by real‐time polymerase chain reaction (PCR). Methylation level of Cd74 DMC was measured in Hepa1c1c7 cells (B), Hepa1‐6 cells (D) or GRX cells (F) by pyrosequencing
3.6. Overexpression of Cd74 suppressed tumor suppressor genes Atoh8 and Id3, and increased Trib3 in hepatoma cells
To explore the involvement of upregulation of Tmem54 and Cd74 in hepatic tumor increase, we performed overexpression experiments with Hepa1c1c7 cells. We confirmed that both Cd74 and Tmem54 mRNA were strikingly induced by each transfection (Figure 6A). Then, we performed microarray analysis to identify genes whose expressions were affected by overexpression of Tmem54 or Cd74. We selected candidate genes downstream of Cd74 or Tmem54 that might be related to tumor formation as follows: (a) the concordant directions of expression changes were observed in AATt compared with AAN in the microarray data of F2 males; and (b) they are reported as tumor‐related genes in PubMed. We validated mRNA expression of the candidate genes in Hepa1c1c7 cells and in tumor tissues of the F2 males using real‐time PCR and confirmed by real‐time PCR that the expression of Id3 and Atoh8 was decreased and that of Trib3 was increased by overexpression of Cd74 in Hepa1c1c7 cells (Figure 6B) and in AATt compared with AAN (Figure S4). We also measured the expression of these genes in CCN and CCTt in the F2 males (Figure S4). The results indicated that the expression of Id3 and Atoh8 was decreased and that of Trib3 was increased in tumor tissues both in the control and arsenite groups to a similar extent. The findings implied that those genes were regulated in a tumor‐specific manner by some factors in the hepatic tumors and Cd74 was not involved in the regulation, at least around the stage investigated. We discuss the regulatory mechanism of Cd74 in Discussion section. Conversely, we did not detect any significant mRNA changes by Tmem54 overexpression (data not shown).
Figure 6.
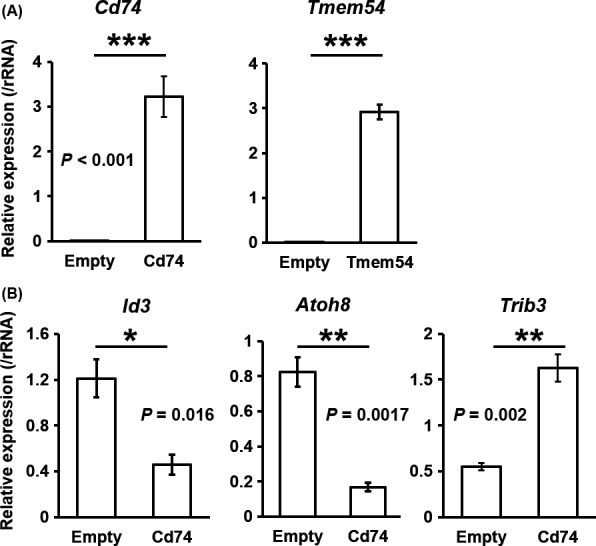
Overexpression of Cd74 in Hepa1c1c7 cells induces expression changes in cancer‐related genes. A, Validation of overexpression of Cd74 or Tmem54 in Hepa1c1c7 cells transfected with each vector. B, Real‐time polymerase chain reaction (PCR) validation of mRNA expression changes in Id3, Atoh8, and Trib3 by Cd74 overexpression
3.7. High expression of TMEM54, CD74, and TRIB3 and low expression of ATOH8 were associated with human liver hepatocellular carcinoma
To seek information on the association of Tmem54 with a hepatic tumor increase, we analyzed the expression of TMEM54 in HCC using the TCGA database. Expression of TMEM54 was found to be increased in human HCC compared with nontumor tissues (Figure 7A). In addition, prognosis was poorer in HCC with high TMEM54 expression than in those with low expression (Figure 7B). Likewise, expression of CD74 and TRIB3 was increased and ATOH8 was decreased in human HCC, as observed in AATt, in comparison with CCTt in the F2 males (Figure 6A). High expression of TRIB3 and low expression of ATOH8 predicted a poor prognosis (Figure 6B).
Figure 7.
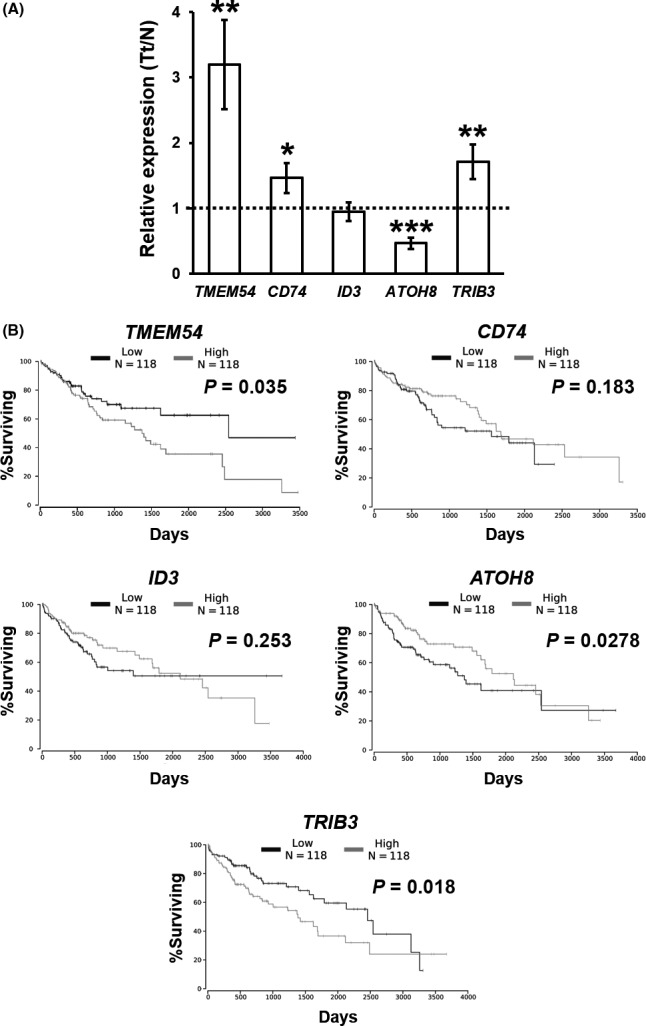
Expression of TMEM54, CD74 and CD74‐downstream genes in human hepatocellular carcinoma (HCC) and correlation with survival. A, Relative expression of genes in HCC compared with pair‐matched noncancer tissues. B, Kaplan‐Meier survival curves of patients with HCC categorized by gene expression levels
4. DISCUSSION
In this study, to identify DNA methylation changes involved in a hepatic tumor increase in F2 males by gestational arsenite exposure, we measured the contents of genomic 5mC and 5hmC by LC‐MS/MS and performed RRBS analysis that could detect genome‐wide DNA methylation levels of each CpG site in the 3 types of liver tissues (normal tissues, and nontumor and tumor tissues of tumor‐bearing livers) in a control group and arsenite group.
The contents of genomic 5mC and 5hmC were reduced in tumor tissues compared with normal tissues both in the control and arsenite groups by LC‐MS/MS analysis (Table 1). This result is consistent with previous reports that the contents of 5mC and 5hmC were reduced in human HCC.33, 34 Conversely, this difference was not detected in the contents of 5mC or 5hmC in normal tissues, nontumor tissues, and tumor tissues between the control group and arsenite group (Table 1).
Next, we compared the RRBS data between the control group and arsenite group. We could not detect any significant changes in DNA methylation levels in individual regions classified by TSS distance between the control group and arsenite group (Figure 1B). Conversely, a precise analysis of DMR revealed that the promoter DMR of Cd74 and Tmem54 were significantly hypomethylated in AATt compared with CCTt (Figures 2, 3). Furthermore, the DNA methylation changes in these DMRs were shown to be related to regulation of gene expression by treatment with 5‐aza‐dC in murine hepatoma cell lines and an hepatic stellate cell line (Figures 4, 5). The DMRs of Tmem54 and Cd74 represented arsenite‐induced tumor‐specific changes as they were not detected from comparison of CCN vs AAN or CCTn vs AATn. The 2 DMRs were also not detected from comparison of CCN vs CCTt reported in our previous study.24
In contrast with the broad DMR of Tmem54, we only detected 3 CpG sites with one DMC in the DMR of Cd74. Gene expression regulation by single‐site methylation has been reported in previous studies.35, 36 A single 5‐methylation of cytosine affects transcription factor binding through changes such as long‐range allosteric effects in DNA37 and transcription factor binding.38 These mechanisms might be implicated in regulation of Cd74 expression by its DMC. On the other hand, there were 29 CpG sites on one strand around the Cd74 TSS ± 2000 bp in addition to the CpGs we identified. However, as the coverage of these CpGs was <10 or not read at all in the present RRBS analysis, they were not subjected to analysis. Therefore, we could not exclude the possibility that DNA methylation of other CpG sites in the promoter region in addition to the DMR identified in the present study were involved in the regulation of Cd74 expression.
Although CD74 is not commonly detected in hepatocytes in normal livers, its expression was reported in the hepatocytes of severe acute autoimmune hepatitis patients and Ikkβ‐deleted mouse hepatocytes.39, 40 CD74 is also reported to be expressed in many types of cancers, however the regulatory mechanisms in individual cancers are largely unknown.41 A recent study reported that CD74 is among the regulated intramembrane proteolysis (RIP) proteins that stay dormant and are activated in response to a variety of stimuli and act as a transcription regulator.32 As expected, we newly identified downregulation of Id3 and Atoh8 and upregulation of Trib3 by overexpression of Cd74 in Hepa1c1c7 cells (Figure 6). Id3 has been reported to induce caspase3‐ and 9‐dependent apoptosis in human keratinocytes,42 and TRIB3 has been also reported to have an antiapoptotic role in gastric cancer cells exposed to anticancer drugs.43 Decreased Id3 and Atoh8, and increased Trib3 were reported to be involved in tumor progression.44, 45, 46 Moreover, the analysis of human HCC data from TCGA disclosed that the increased expression of CD74 and TRIB3 and decreased expression of ATOH8 were also observed in human HCC, and the expression changes were related to poor prognosis (Figure 7). Therefore, CD74 activation and subsequent expression changes in the downstream genes, such as TRIB3 and ATOH8, might contribute to human HCC. Consistently, the present study detected downregulation of Id3 and Atoh8 and upregulation of Trib3 in the hepatic tumors in the control and arsenic groups (Figure S4). However, the expression of these genes was not changed between CCTt and AATt, although Cd74 was upregulated in AATt compared with CCTt. These findings indicated that Cd74 was not a major regulator of CCTt and AATt in the hepatic tumors of C3H mice around 74 wk of age, although it might be involved in regulation of downstream genes at some point during tumor development in response to tumorigenic stimuli.
In experiments using human tissues and cell lines, TMEM54 (also known as CAC1) is shown to have a pivotal role in activating cyclin‐dependent kinase 2 and progression of the cell cycle.31 TMEM54 is also expressed in some cancers and the cell cycle associated function is implicated in carcinogenesis.31 Our analysis using the human database from TCGA revealed that the expression of TMEM54 was increased in HCC compared with paired noncancer tissues. Furthermore, an increase in TMEM54 expression was related to poor prognosis (Figure 7). These results supported the idea that TMEM54 was related to HCC, suggesting that Tmem54 might have an important role in tumor increase in the F2 males following earlier arsenite exposure.
We examined DNA methylation patterns around the TSS of TMEM54 and CD74 using the TCGA database to assess whether DNA methylation is also involved in expression regulation of these genes in human HCC. We utilized DNA methylation data from 377 HCC tissues and 50 normal hepatic tissues. We found that the methylation data from one CpG site (chr1:33 366 802) in the CpG island of TMEM54. Mann‐Whitney U test demonstrated that there was a statistically significant decrease in methylation in tumor tissues (P < .0001, data not shown). Likewise, methylation data were available for 7 CpGs at ±300 bp from the CD74 TSS. Results of the Mann‐Whitney U test showed that one site upstream of the TSS was significantly hypermethylated and that 6 sites downstream of the TSS were hypomethylated in tumors compared with normal tissues (data not shown). These inverse relationship between DNA methylation and gene expression may implicate DNA methylation regulation of expression of these genes.
Our previous study showed that the tumor augmenting effects following gestational arsenite exposure of F0 mice from gestational day (GD) 8‐18 were transmitted through F1 males, ie via sperm to F2 mice.12 This period is pivotal for epigenetic reprogramming of primordial germ cells (PGCs), the origin of future F2 mice, in the fetal F1 mice. Perturbation of reprogramming by environmental factors, such as nutrition and chemicals, can give rise to adverse health effects in subsequent generations.47 As an example, offspring of male mice fed a low‐protein diet exhibited DNA methylation changes associated with expression changes in lipid metabolism‐related genes in the liver,48 and compositional changes in the transfer RNA fragments in the sire's sperm were implicated.49 Further studies on epigenetic changes in F1 sperm will be needed to identify the mechanism that causes DNA methylation changes associated with F2 mice tumor increase observed in the present study.
Overall, our study proposes novel mechanisms of multigenerational effects of arsenite exposure in that expression of Cd74 and Tmem54 was regulated by DNA methylation that involved a tumor increase in F2 males following earlier gestational arsenite exposure.
DISCLOSURE
The authors declare that they have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We would like to thank Dr. Y Horibe (NRICHD) for his support for these experiments. We also thank J Matsushita, H Murai, Dr. A Furuyama (NIES), and M Ohashi and Dr. H Ogata (NRICHD) for their discussions and technical support, and Ms. Hayakawa for her secretarial assistance. The authors also wish to thank the staff of the Animal Care Company for their excellent assistance in the maintenance of mice. This study was partly supported by NIES (1115AA082; 1315AT001; 1620AA041, KN), and Grant‐in‐Aid for Scientific Research (26293154, 15K15246, KN) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Okamura K, Nakabayashi K, Kawai T, et al. DNA methylation changes involved in the tumor increase in F2 males born to gestationally arsenite exposed F1 male mice. Cancer Sci. 2019;110:2629–2642. 10.1111/cas.14104
REFERENCES
- 1. Schmidt CW. Uncertain inheritance transgenerational effects of environmental exposures. Environ Health Perspect. 2013;121:A298‐A303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Xin F, Susiarjo M, Bartolomei MS. Multigenerational and transgenerational effects of endocrine disrupting chemicals: a role for altered epigenetic regulation? Semin Cell Dev Biol. 2015;43:66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Radford EJ, Ito M, Shi H, et al. In utero effects. In utero undernourishment perturbs the adult sperm methylome and intergenerational metabolism. Science. 2014;345:1255903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Skinner MK. Environmental stress and epigenetic transgenerational inheritance. BMC Med. 2014;12:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yamasaki H, Loktionov A, Tomatis L. Perinatal and multigenerational effect of carcinogens: possible contribution to determination of cancer susceptibility. Environ Health Perspect. 1992;98:39‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hughes MF, Beck BD, Chen Y, Lewis AS, Thomas DJ. Arsenic exposure and toxicology: a historical perspective. Toxicol Sci. 2011;123:305‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith AH, Marshall G, Yuan Y, Ferreccio C, Steinmaus C. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ Health Perspect. 2006;114:1293‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuan Y, Marshall G, Ferreccio C, et al. Kidney cancer mortality: fifty‐year latency patterns related to arsenic exposure. Epidemiology. 2010;21:103‐108. [DOI] [PubMed] [Google Scholar]
- 9. Waalkes MP, Ward JM, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol Appl Pharmacol. 2003;186:7‐17. [DOI] [PubMed] [Google Scholar]
- 10. Waalkes MP, Liu J, Chen H, et al. Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J Natl Cancer Inst. 2004;96:466‐474. [DOI] [PubMed] [Google Scholar]
- 11. Maronpot RR, Fox T, Malarkey DE, Goldsworthy TL. Mutations in the ras proto‐oncogene: clues to etiology and molecular pathogenesis of mouse liver tumors. Toxicology. 1995;101:125‐156. [DOI] [PubMed] [Google Scholar]
- 12. Nohara K, Okamura K, Suzuki T, et al. Augmenting effects of gestational arsenite exposure of C3H mice on the hepatic tumors of the F2 male offspring via the F1 male offspring. J Appl Toxicol. 2016;36:105‐112. [DOI] [PubMed] [Google Scholar]
- 13. Klosin A, Casas E, Hidalgo‐Carcedo C, Vavouri T, Lehner B. Transgenerational transmission of environmental information in C. elegans. Science. 2017;356:320‐323. [DOI] [PubMed] [Google Scholar]
- 14. Choi SW, Friso S. Epigenetics: a new bridge between nutrition and health. Adv Nutr. 2010;1:8‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guibert S, Weber M. Functions of DNA methylation and hydroxymethylation in mammalian development. Curr Top Dev Biol. 2013;104:47‐83. [DOI] [PubMed] [Google Scholar]
- 16. Liyanage VR, Jarmasz JS, Murugeshan N, Del Bigio MR, Rastegar M, Davie JR. DNA modifications: function and applications in normal and disease States. Biology (Basel). 2014;3:670‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang J, Tang J, Lai M, Zhang H. 5‐Hydroxymethylcytosine and disease. Mutat Res, Rev Mutat Res. 2014;762:167‐175. [DOI] [PubMed] [Google Scholar]
- 18. Pfeifer GP, Xiong W, Hahn MA, Jin SG. The role of 5‐hydroxymethylcytosine in human cancer. Cell Tissue Res. 2014;356:631‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cimmino L, Aifantis I. Alternative roles for oxidized mCs and TETs. Curr Opin Genet Dev. 2017;42:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Esteller M. Aberrant DNA methylation as a cancer‐inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629‐656. [DOI] [PubMed] [Google Scholar]
- 21. Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high‐resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868‐5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nohara K, Tateishi Y, Suzuki T, et al. Late‐onset increases in oxidative stress and other tumorigenic activities and tumors with a Ha‐ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicol Sci. 2012;129:293‐304. [DOI] [PubMed] [Google Scholar]
- 23. Nohara K, Baba T, Murai H, et al. Global DNA methylation in the mouse liver is affected by methyl deficiency and arsenic in a sex‐dependent manner. Arch Toxicol. 2011;85:653‐661. [DOI] [PubMed] [Google Scholar]
- 24. Matsushita J, Okamura K, Nakabayashi K, et al. The DNA methylation profile of liver tumors in C3H mice and identification of differentially methylated regions involved in the regulation of tumorigenic genes. BMC Cancer. 2018;18:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics. 2011;27:1571‐1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Akalin A, Kormaksson M, Li S, et al. methylkit: a comprehensive R package for the analysis of genome‐wide DNA methylation profiles. Genome Biol. 2012;13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li S, Garrett‐Bakelman FE, Akalin A, et al. An optimized algorithm for detecting and annotating regional differential methylation. BMC Bioinformatics. 2013;14(suppl 5):S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nohara K, Ao K, Miyamoto Y, et al. Comparison of the 2,3,7,8‐tetrachlorodibenzo‐p‐dioxin (TCDD)‐induced CYP1A1 gene expression profile in lymphocytes from mice, rats, and humans: most potent induction in humans. Toxicology. 2006;225:204‐213. [DOI] [PubMed] [Google Scholar]
- 29. Takumi S, Aoki Y, Sano T, Suzuki T, Nohmi T, Nohara K. In vivo mutagenicity of arsenite in the livers of gpt delta transgenic mice. Mutat Res, Genet Toxicol Environ Mutagen. 2014;760:42‐47. [DOI] [PubMed] [Google Scholar]
- 30. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009;1:239‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kong Y, Nan K, Yin Y. Identification and characterization of CAC1 as a novel CDK2‐associated cullin. Cell Cycle. 2009;8:3552‐3561. [DOI] [PubMed] [Google Scholar]
- 32. Gil‐Yarom N, Radomir L, Sever L, et al. CD74 is a novel transcription regulator. Proc Natl Acad Sci USA. 2017;114:562‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lin CH, Hsieh SY, Sheen IS, et al. Genome‐wide hypomethylation in hepatocellular carcinogenesis. Cancer Res. 2001;61:4238‐4243. [PubMed] [Google Scholar]
- 34. Chen ML, Shen F, Huang W, et al. Quantification of 5‐methylcytosine and 5‐hydroxymethylcytosine in genomic DNA from hepatocellular carcinoma tissues by capillary hydrophilic‐interaction liquid chromatography/quadrupole TOF mass spectrometry. Clin Chem. 2013;59:824‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pogribny IP, Pogribna M, Christman JK, James SJ. Single‐site methylation within the p53 promoter region reduces gene expression in a reporter gene construct: possible in vivo relevance during tumorigenesis. Cancer Res. 2000;60:588‐594. [PubMed] [Google Scholar]
- 36. Zhang X, Wu M, Xiao H, et al. Methylation of a single intronic CpG mediates expression silencing of the PMP24 gene in prostate cancer. Prostate. 2010;70:765‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bie L, Du L, Yuan Q, Gao J. How a single 5‐methylation of cytosine regulates the recognition of C/EBPβ transcription factor: a molecular dynamic simulation study. J Mol Model. 2018;24:159. [DOI] [PubMed] [Google Scholar]
- 38. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Assis DN, Leng L, Du X, et al. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology. 2014;59:580‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koch KS, Leffert HL. Ectopic expression of CD74 in Ikkbeta‐deleted mouse hepatocytes. Acta Histochem. 2011;113:428‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schröder B. The multifaceted roles of the invariant chain CD74–More than just a chaperone. Biochim Biophys Acta. 2016;1863:1269‐1281. [DOI] [PubMed] [Google Scholar]
- 42. Simbulan‐Rosenthal CM, Daher A, Trabosh V, et al. Id3 induces a caspase‐3‐ and ‐9‐dependent apoptosis and mediates UVB sensitization of HPV16 E6/7 immortalized human keratinocytes. Oncogene. 2006;25:3649‐3660. [DOI] [PubMed] [Google Scholar]
- 43. Wu IJ, Lin RJ, Wang HC, Yuan TM, Chuang SM. TRIB3 downregulation enhances doxorubicin‐induced cytotoxicity in gastric cancer cells. Arch Biochem Biophys. 2017;622:26‐35. [DOI] [PubMed] [Google Scholar]
- 44. Song Y, Pan G, Chen L, et al. Loss of ATOH8 increases stem cell features of hepatocellular carcinoma cells. Gastroenterology. 2015;149:1068‐1081. e1065. [DOI] [PubMed] [Google Scholar]
- 45. Li J, Roy S, Kim YM, et al. Id2 collaborates with Id3 to suppress invariant NKT and innate‐like tumors. J Immunol. 2017;198:3136‐3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li K, Wang F, Cao WB, et al. TRIB3 promotes APL progression through stabilization of the oncoprotein PML‐RARalpha and Inhibition of p53‐Mediated Senescence. Cancer Cell. 2017;31:697‐710. e697. [DOI] [PubMed] [Google Scholar]
- 47. Barouki R, Melen E, Herceg Z, et al. Epigenetics as a mechanism linking developmental exposures to long‐term toxicity. Environ Int. 2018;114:77‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carone BR, Fauquier L, Habib N, et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084‐1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharma U, Conine CC, Shea JM, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351:391‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials