

Abstract
The human prolyl isomerase PIN1, best known for its association with carcinogenesis, has recently been indicated in the disease of pancreatic ductal adenocarcinoma (PDAC). However, the functions of PIN1 and the feasibility of targeting PIN1 in PDAC remain elusive. For this purpose, we examined the expression of PIN1 in cancer, related paracarcinoma and metastatic cancer tissues by immunohistochemistry and analyzed the associations with the pathogenesis of PDAC in 173 patients. The functional roles of PIN1 in PDAC were explored in vitro and in vivo using both genetic and chemical PIN1 inhibition. We showed that PIN1 was upregulated in pancreatic cancer and metastatic tissues. High PIN1 expression is significantly association with poor clinicopathological features and shorter overall survival and disease‐free survival. Further stratified analysis showed that PIN1 phenotypes refined prognostication in PDAC. Inhibition of PIN1 expression with RNA interference or with all trans retinoic acid decreased not only the growth but also the migration and invasion of PDAC cells through regulating the key molecules of multiple cancer‐driving pathways, simultaneously resulting in cell cycle arrest and mesenchymal‐epithelial transition in vitro. Furthermore, genetic and chemical PIN1 ablation showed dramatic inhibition of the tumorigenesis and metastatic spread and then reduced the tumor burden in vivo. We provided further evidence for the use of PIN1 as a promising therapeutic target in PDAC. Genetic and chemical PIN1 ablation exerted potent antitumor effects through blocking multiple cancer‐driving pathways in PDAC. More potent and specific PIN1 targeted inhibitors could be exploited to treat this aggressive cancer.
Keywords: all‐trans retinoic acid, metastatic spread, pancreatic ductal adenocarcinoma, PIN1, tumorigenesis
1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease and is the fourth leading cause of cancer‐associated mortality.1 The difference between the incidence and the death rate is still small due to its early distant spread and the lack of efficacious current therapies.2, 3 Whole exome and genome sequencing have identified PDAC as a disease of high genetic variability including core gene alterations and general copy number variations.4, 5, 6 However, for personalized treatment strategies, efforts to date in targeting key oncogenic pathways have reported unsatisfied results.7, 8 A wide range of interactive and/or redundant pathways may be simultaneous activated when blocking a single pathway, resulting in less effect in solid tumors, especially for aggressive or drug‐resistant tumors.9, 10 Thus, targeting a variety of tumor‐driven gene therapies will become an important strategy for PDAC.
Recently, human unique prolyl isomerase PIN1 has been indicated to exhibit profound functions in pathogenesis of cancer disease by amplifying numerous oncoproteins and then disrupting its balance with tumor suppressors.11 PIN1 is a central controller in the regulation of proline‐directed phosphorylation, which is a common event relating to tumorigenesis.12, 13 Overactivation or upregulation of PIN1 contributes to carcinogenesis in the majority of human tumors, including PDAC,14, 15, 16, 17, 18, 19 whereas alteration of PIN1 single nucleotide polymorphisms (SNP) at the promoter region has inverse results.20 In addition, PIN1 is likely to be an indispensable orchestrator in the process of oncogene‐driven carcinogenesis.21, 22, 23, 24 Given that Pin1−/− homozygous mice grow normally and there are no general side effects on normal cells,25 PIN1 is a potential molecular target for cancer treatment. Notably, targeting of PIN1 has been achieving some promising results in animal studies on leukemia, triple negative breast cancer and hepatocellular carcinoma.26, 27, 28, 29 However, little evidence on the function of PIN1 in the progression of PDAC has been seen.
The PIN1 inhibitor has been explored for 2 decades. Clinical use has not been widespread because of the lack of required specificity and cell permeability, but this could change with the identification of all trans retinoic acid (ATRA) as a PIN1 inhibitor based on high‐throughput screen (HTS) technology.11, 26 ATRA can induce cell cycle arrest, apoptosis and epithelial cell differentiation of PDAC to inhibit tumor cell growth.30, 31 However, paradoxical results after ATRA treatment in different PDAC cells were observed.32, 33 Similar findings26 were obtained regarding ATRA for different breast cancer cells. However, cellular susceptibility against ATRA depends on the level of PIN1 expression and the degree of phosphorylation at S71 of PIN1.26 ATRA can bind to the catalytically active sites of PIN1 and subsequently inhibit its activity, resulting in PIN1 protein degradation. Furthermore, ATRA exerts potent antitumor activity against multiple refractory cancers through inhibiting and ablating PIN1 to inactivate multiple oncogenes or activate tumor suppressors simultaneously in vivo.26, 27, 28, 29 However, the role of ATRA and its targeting PIN1 in PDAC remain elusive.
Thus, in this present study, we examined the expression of PIN1 and explored the role of PIN1 in the pathogenesis of PDAC. The association between PIN1 and clinical pathological features was also examined. We showed that PIN1 was upregulated in pancreatic cancer and metastatic tissues. High PIN1 expression is significantly associated with shorter survival and stratified with outcomes in patients. Genetic and chemical PIN1 inhibition decreased not only the growth but also the invasiveness of PDAC cells through regulating the key molecules of multiple cancer‐driving signaling pathways simultaneously. Furthermore, such PIN1 ablation inhibited both the tumorigenesis and metastatic spread and reduced tumor burden in vivo. Thus, PIN1 may be a promising biomarker and a potential therapeutic target for PDAC disease.
2. MATERIALS AND METHODS
2.1. Patients and tissues
A total of 173 consecutive cases at the First Affiliated Hospital of Fujian Medical University were collected from the years 2006 to 2016. All original H&E slides were reviewed and patients’ clinical pathological parameters were retrieved from the medical records. Disease‐free survival (DFS) was defined as the duration from the date of initial diagnosis to the first detection of pancreatic cancer‐specific relapse or death. Overall survival (OS) was defined as the duration from the date of initial diagnosis to the date of death. None of the patients were dead within 1 month after surgery. Tissue microarray (TMA) was constructed for immunohistochemistry (IHC). For the TMA construction, 3 representative regions of each H&E‐stained slides were chosen, from the center, the edge of the tumor and the peri‐tumoral tissues, respectively; 2.5 mm cores at each corresponding area were taken and brought into “recipient” paraffin blocks by manual operation. Serial 4‐μm sections were cut and transferred to SuperfrostPlus glass slides (Matsunami Glass Industry, Japan). One section from each tissue array block was H&E stained to confirm the presence of representative tumors. IHC staining was performed with monoclonal antiPIN1 antibody provided by Dr Kun Ping Lu14 (Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA) using the Dako EnVision FLEX+ detection system (Dako, produktionsvej42, DK‐2600 Glostrup,Denmark). PIN1 staining was scored following the methods of Chen et al (2015)34 or analyzed by Image‐Pro Plus soft, Version 6.0. For PIN1 score, we used the average of immunoscore as a cutoff to define PIN1 low and high expression. The research project has been approved by the Institutional Research Ethics Committee of the First Affiliated Hospital of Fujian Medical University (2018055).
2.2. Cell culture, reagents and animals
Cell lines 293T, PANC1 and BXPC3, obtained from the Cell Bank of the Chinese Academy of Sciences, were cultured in DMEM (Hyclone, Xiamen, China) supplemented with 10% FBS (Gibco, Mexico) and 1% penicillin‐streptomycin (Hyclone). The reagents and the primary antibodies for the research are summarized in Table S1. For the present study, 4‐6‐week‐old female BALB/c nude mice (Shanghai SLAC laboratory Animal, Shanghai, China) were maintained in specific‐pathogen‐free conditions at the Laboratory Animal Center of Fujian Medical University. Animal care and experimental protocols were performed according to the Experimental Animal Ethics Committee of Fujian Medical University.
2.3. Stable PIN1 knockdown cell construction
Cells for PIN1 knockdown (KD) were infected with the lentiviruses expressing PIN1 shRNA or Scrambled, selected with puromycin (2 μg/mL). Briefly, the validated PIN1 shRNA (5′‐CCACCGTCACACAGTATTTAT‐3′)35, 36 was subcloned into the pKLO.1 lentiviral vector and generated, and was transfected into 293T cells with the helper plasmids (pVSVG, pMDL and pREV) for 48‐72 h. The supernatant containing virus was collected and stored. All the plasmids used were provided from Dr Kun Ping Lu as described previously.
2.4. Cell proliferation assays
Proliferation assays for cells with PIN1KD or ATRA (Innovative Research of America, Sarasota, FL, USA) treatments were determined by counting the number or using MTT assays. For colony formation assays, cells were seeded in each well of 6‐well plates (Corning, Kennebunk, USA) and cultured for 2 weeks. The number of colonies per well were counted after staining with crystal violet (Hyclone, Xiamen, China).
2.5. Migration and invasion assays
Migration and invasion assays for cells with PIN1KD or ATRA treatments were carried out with the Boyden Chamber system; 200 μL of 5 × 104 cells starved in media with 1% FBS were placed in the upper chamber and 500 mL of media with 10% PBS were placed in the lower chamber (Corning). The cells were allowed to migrate for 24 h or 48 h and stained with crystal violet. For invasion assays, cells were inserted into the upper chamber with a Matrigel coated membrane (Sigma, St.Louis, MO, USA). Migrated cells adhering to the underside of the inserts were photographed (AXIO Observer Z1, Carl Zeiss, Gottingen, Germany) and counted.
2.6. Western blotting analysis
Cells were lysed in ice cold RIPA buffer and quantified using a BCA Protein Assay Kit (Beyotime, Shanghai, China). Proteins were separated and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% milk and incubated with primary antibodies. After washing, the membranes were incubated with (HRP) conjugated secondary antibodies and enhanced with a chemiluminescence HRP substrate (Millipore, Burlington, MA, USA).
2.7. Animal studies
For xenograft experiments, 1‐2.0 × 106 PANC1 cells were inoculated subcutaneously into the mice. For ATRA treatments, the mice were grouped to receive different doses of ATRA‐releasing pellets (Innovative Research of America) when tumor growth was approximately 50 mm3. All tumor sizes were recorded and tumor volumes were calculated using the formula: length*width*width/2. For tail vein transport experiments, 1.5‐2.0 × 106 BXPC3 cells were injected into lateral tail veins of 6‐week‐old nude mice. Mice were killed at 50 days after tail vein injection for histopathologic evaluation. The number of metastatic tumor nodules were counted under a microscope. For ATRA treatments, the mice were grouped into receive ATRA‐releasing pellets after metastatic nodules were confirmed by histological evaluation. The time of death of the mice was recorded.
2.8. Statistical analysis
The findings were analyzed using the statistical software SPSS for Windows, Version 18. χ2 analysis or Fisher's exact test was used to test for the association of PIN1 expression with clinicopathological parameters. Survival data were evaluated with Kaplan–Meier analysis. Multivariate Cox regression analysis was performed. Student's t test was used in the in vitro and in vivo experiments. One‐way ANOVA were performed for comparing means of multiple groups. Statistical significance was established at P < 0.05.
3. RESULTS
3.1. PIN1 highly expression is associated with poor clinicopathological features and stratified with outcomes in pancreatic ductal adenocarcinoma
PIN1 is prevalently overexpressed in common human tumors.17 To explore the role of PIN1 in PDAC, we detected the expression of PIN1 protein in tumors and their paired paracarcinoma tissues by IHC. For paracarcinoma tissues, acinar and ductal epithelial cells both showed patchy weak or negative staining. For cancer tissues, PIN1 expression was observed in both the cytoplasm and nucleus or in the cytoplasm in 127/173 cases (Figure 1A). PIN1 expression in cancer tissues was significantly stronger than that in the paracarcinoma tissues (Figure 1B). To address the level of transcription and gene status of PIN1, we analyzed data in TCGA and Oncomine databases, respectively. The results showed that the transcriptional level of PIN1 in PDAC dramatically increased in comparison with normal pancreas tissues according to TCGA (Figure S1A, P = 1.76 × 10−5) and Pei datasets (Figure S1B, P = 0.002). However, the gene copy number of PIN1 was increased only in a small fraction of cases (Figure S1C) and no significant result was found in comparison with peripheral blood and pancreas tissues (Figure S1D, P = 0.293).
Figure 1.
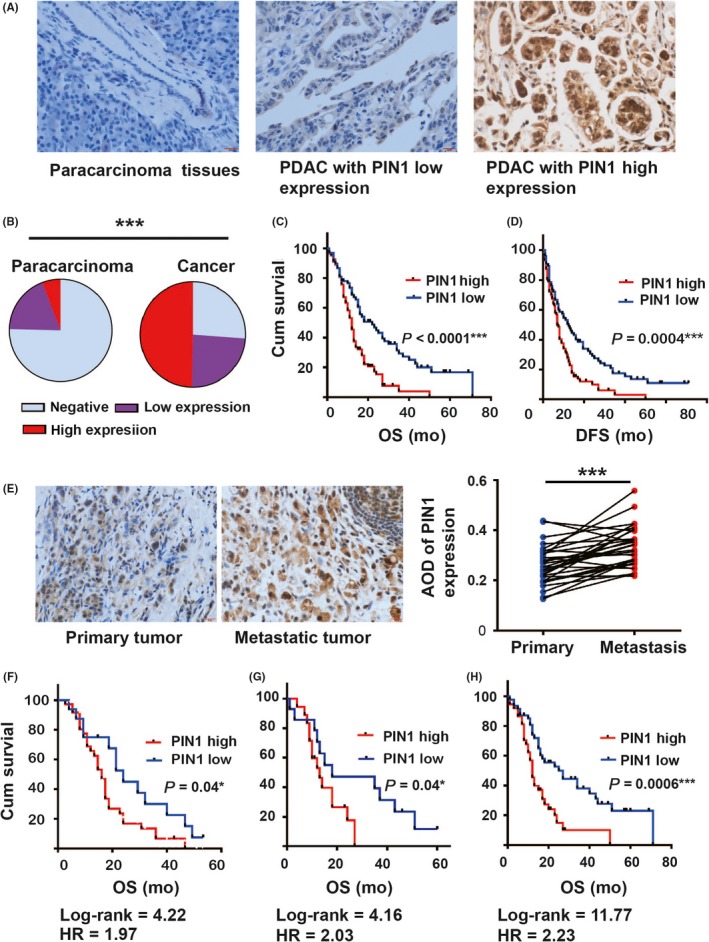
PIN1 expression in pancreatic ductal carcinoma (PDAC) and association with outcomes. A, B, Representative immunochemical staining of PIN1 expression in paracarcinoma and tumor tissues (A, scale bar = 20 μm) and analyzed (B, n = 173). C, D, Kaplan‐Meier analysis of overall survival (OS) (C) and disease‐free survival (DFS) (D) according to PIN1 expression in PDAC. E, Representative illustrations of PIN1 expression in primary tumor and its paired metastatic tissues and analyzed (scale bar = 20 μm, n = 30). F, G, H, Comparisons of OS between PIN1 expression in patients with TNM stage 2b (F), same moderate histological grade (G) and no lymph node involvement (H). (Fisher's exact test, the paired t test and the log‐rank test were used; *P < 0.05, **P < 0.01, ***P < 0.001)
Regarding the potential significance of PIN1 location in cancer tissues, we analyzed the relationship between the different locations and the clinicopathological features regardless of the intensity of PIN1 staining. PIN1 expression both in the cytoplasm and nucleus was significantly associated with histological grade (P = 0.001) and nerve invasion (P = 0.007), whereas no significant results were found between the groups that were PIN1 negative and cytoplasmic positive (Table S2). However, with dichotomization of PIN1 expression (average immunoscore = 97 as threshold) into high or low groups combining the percentage and intensity of PIN1 expression, high PIN1 expression was strongly significantly associated with tumor diameter (P = 0.05), histological grade (P = 0.001), lymph node involvement (P = 0.009) and nerve invasion (P = 0.006) in PDAC (Table 1). PIN1 low or high expression was shown to be an independent prognostic factor by multivariate Cox analysis for both DFS and OS in PDAC (Table 2). Patients with PIN1 high expression showed the worst OS (Figure 1C, Log‐rank = 16.77, HR = 2.001) and DFS (Figure 1D, Log‐rank = 12.57, HR = 1.759). Furthermore, the results showed that PIN1 expression was stronger in metastatic tissues than in the corresponding primary cancer (Figure 1E). Of note, stratified analysis revealed that PDAC with the same histological grade, no lymph node involvement and TNM 2b stage could be stratified into groups with different outcomes based on PIN1 expression. For TNM stage 2b (Figure 1F), cases with PIN1 low expression showed better OS. Similar trends were also shown in patients with the same moderate histological grade (Figure 1G) or with no lymph node involvement (Figure 1H).
Table 1.
Relationship between PIN1 expression and clinicopathological features
| Clinicopathological features | Cases (n = 173) | PIN1 expression | P‐value | |
|---|---|---|---|---|
| Low (n = 84) (%) | High (n = 89) (%) | |||
| Age | ||||
| <60 | 83 | 42 (50.0) | 41 (46.7) | 0.605 |
| ≥60 | 90 | 42 (50.0) | 48 (53.3) | |
| Gender | ||||
| Female | 67 | 28 (33.3) | 39 (43.8) | 0.164 |
| Male | 106 | 56 (66.7) | 50 (56.2) | |
| Tumor sites | ||||
| Head | 136 | 67 (79.8) | 69 (77.5) | 0.853 |
| Non‐head | 37 | 17 (20.2) | 20 (22.5) | |
| Histological grade | ||||
| Well+moderate | 87 | 53 (63.1) | 34 (38.2) | 0.001** |
| Poor | 86 | 31 (36.9) | 55 (61.8) | |
| Tumor diameters (cm) | ||||
| <2 | 25 | 17 (20.2) | 8 (9.0) | 0.05 |
| ≥2 | 148 | 67 (79.8) | 81 (91.0) | |
| Lymph node involvements | ||||
| Absence | 97 | 56 (66.7) | 41 (46.1) | 0.009** |
| Presence | 76 | 28 (33.3) | 48 (53.9) | |
| Nerve invasion | ||||
| Absence | 60 | 38 (45.2) | 22 (24.7) | 0.006** |
| Presence | 113 | 46 (54.8) | 67 (75.3) | |
Fisher's exact test, *P < 0.05; **P < 0.01.
Table 2.
Multivariate Cox regression analysis on overall survival (OS) and disease‐free survival (DFS) in patients with pancreatic ductal adenocarcinoma
| Parameters | OS | |||||
|---|---|---|---|---|---|---|
| Univariate analysis | Multivariate analysis | |||||
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Age | 1.291 | 0.899‐1.854 | 0.166 | |||
| Gender | 0.991 | 0.684‐1.434 | 0.960 | |||
| Tumor sites | 1.181 | 0.772‐1.806 | 0.442 | |||
| Nerve invasion | 1.089 | 0.745‐1.592 | 0.661 | |||
| Grade | 1.476 | 1.182‐1.843 | 0.001 | 1.293 | 1.021‐1.637 | 0.033 |
| pT | 1.345 | 1.042‐1.736 | 0.023 | 1.314 | 1.002‐1.723 | 0.048 |
| pN | 1.715 | 1.289‐2.283 | <0.0001 | 1.385 | 1.020‐1.882 | 0.037 |
| PIN1 | 2.165 | 1.469‐3.190 | <0.0001 | 1.621 | 1.062‐2.447 | 0.025 |
| Parameters | DFS | |||||
|---|---|---|---|---|---|---|
| Univariate analysis | Multivariate analysis | |||||
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Age | 1.17 | 0.838‐1.633 | 0.356 | |||
| Gender | 1.082 | 0.768‐1.523 | 0.653 | |||
| Tumor sites | 1.163 | 0.779‐1.736 | 0.460 | |||
| Nerve invasion | 1.177 | 0.825‐1.678 | 0.368 | |||
| Grade | 1.399 | 1.148‐1.704 | 0.001 | 1.282 | 1.045‐1.572 | 0.017 |
| pT | 1.45 | 1.140‐1.846 | 0.003 | 1.397 | 1.083‐1.802 | 0.01 |
| pN | 1.718 | 1.328‐2.222 | <0.0001 | 1.472 | 1.121‐1.933 | 0.005 |
| PIN1 | 1.841 | 1.299‐2.609 | 0.001 | 1.436 | 0.994‐2.073 | 0.05 |
Multivariate analysis: adjusted by histological grade, pT, pN and PIN1 expression.
3.2. Genetic PIN1 suppression inhibits both proliferation and invasiveness of pancreatic ductal adenocarcinoma cells in vitro
PIN1 was upregulated in pancreatic cancer tissues and several human pancreatic ductal cells.19 Next, we verify the functions of PIN1 in PDAC using PANC1 (poorly differentiated cells)37 and BXPC3 (highly metastatic cells)38 cells with RNA interference. Stable PIN1 KD cells showed a dramatic decrease in endogenous PIN1 by immunoblot analysis (Figure 2A). After genetic PIN1 suppression, PDAC cells showed a dramatic decrease in cell proliferation, colony formation, migration and invasion (Figure 2B‐F). Subsequently, cell cycle analysis (Doc S1) indicated that the population of PIN1KD cells increased at G0/G1 phase but decreasing at S phase (Figure S2A). Meanwhile, upregulation of the epithelial marker E‐cadherin (E‐cad) but downregulation of the mesenchymal markers Vimentin (Vim) were observed in PIN1KD cells by immunofluorescence staining (Doc S2, Figure S2B,C).
Figure 2.
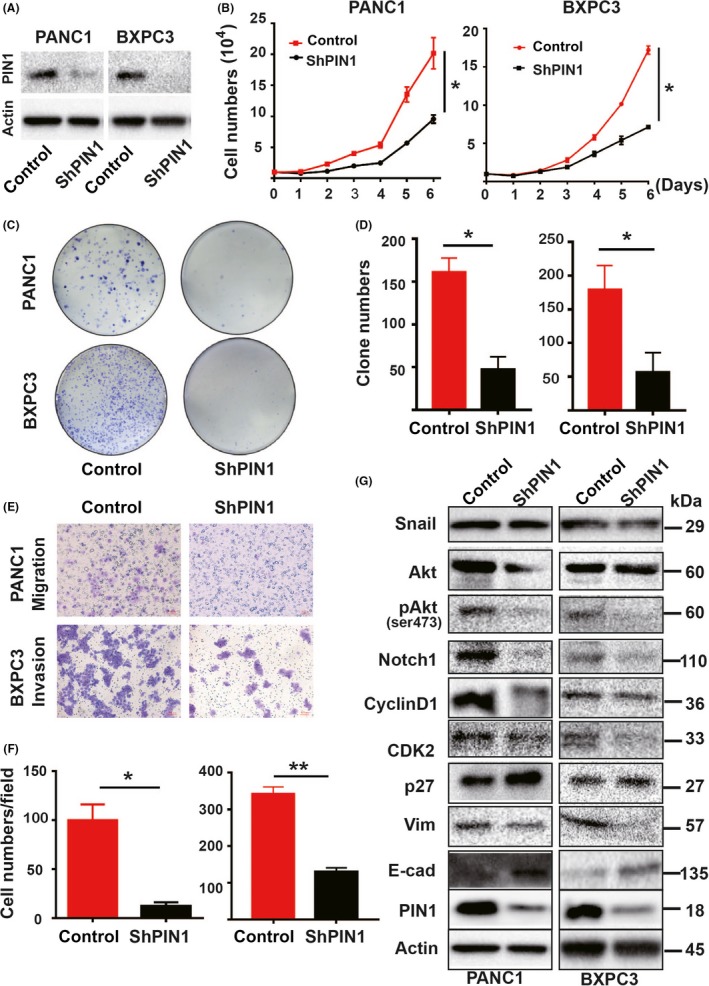
Genetic PIN1 suppression affected pancreatic ductal carcinoma (PDAC) cell functions in vitro. Stable PIN1 knockdown (KD) of PANC1 and BXPC3 cells with RNA interference and their controls were used, respectively. A, PIN1 KD cells resulted in marked reduction of PIN1 expression determined by immunoblot. B, PIN1 KD decreased in cell proliferation. C, D, PIN1 KD decreased cell colony formation. E, F, PIN1 KD reduced both migration and invasion. G, Cell lysates of PIN1 KD and controls were performed by immunoblot with specific antibodies. (All experiments were performed independently at least 3 times. Data are shown as mean ± SEM. One‐way ANOVA and Student's t test were used; *P < 0.05, **P < 0.01.)
PIN1 can activate more than 40 oncogenes whereas inactivate at least 20 tumor suppressing genes.11 To address the underlying mechanisms of PIN1‐mediated tumorigenesis, we detected downstream molecules that have been demonstrated to be substrates of PIN1. Monitors of G1/S phase transition, Notch1 signaling and AKT/mTOR pathway are the core signaling pathways in relation to pancreatic carcinogenesis,6 which could be regulated by PIN1.39, 40, 41, 42, 43, 44 As expected, PIN1KD cells decreased the expression of these oncoproteins, including Cyclin D1, CDK2, Notch1 and pser473Akt, and increased suppressor p27kip. Changes in EMT‐related protein were observed in PIN1KD cells. although notable downregulation of Vim was observed in PANC1 cells, there was notable upregulation of E‐cad in BXPC3 cells (Figure 2G).
3.3. Genetic PIN1 suppression inhibits tumor growth and metastatic spread for pancreatic ductal adenocarcinoma cells in vivo
To explore whether targeting PIN1 could affect tumorigenesis and metastatic spread for PDAC cells in vivo, we performed xenograft tumor and tail vein transport experiments in mice. For xenograft tumors, PANC1 PIN1KD and their paired controls were expanded and subcutaneously injected into the left and right flanks of the same mice, respectively. Tumor growth curves were established over time by measuring the tumor size every week until sacrifice criteria were met in the first mice. All the tumors were extracted and photographed and weighed. Compared to the controls, the PANC1 PIN1KD group showed significant growth inhibition, as illustrated by the tumor growth decrease (Figure 3A) and tumor weight lost (Figure 3B,C). Further analysis was undertaken by IHC for PIN1, Cyclin D1 and Ki67 to verify the function of PIN1 on tumor cell proliferation in vivo. As expected, the density and mitosis of tumor cells with PIN1KD obviously decreased, with dramatically lower expression of Cyclin D1 and Ki67 index (Figure 3D,E).
Figure 3.
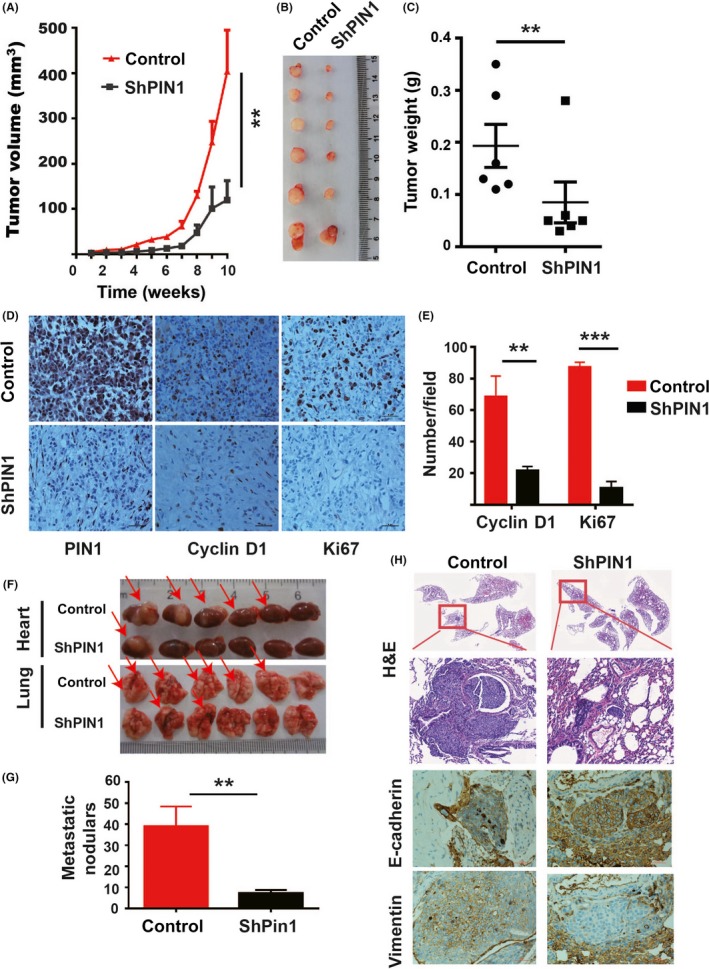
Genetic PIN1 suppression affected pancreatic ductal carcinoma (PDAC) cell functions in vivo. Xenograft tumor and tail vein transport models were constructed with PANC1 and BXPC3 PIN1KD and their control cells, respectively. A, PIN1 knockdown (KD) modulated tumor volume growth in vivo (n = 6). B, Photographic illustration of tumors collected from nude mice. C, PIN1 KD‐induced tumor weight lost in vivo (n = 6, measured at the end point). D, E, Representative illustrations of PIN1, Cyclin D1 and Ki67 expression in xenograft tumor tissues (D) and analyzed (E). F, Photographic illustrations of hearts and lungs collected from nude mice (red arrows indicate the tumor nodules). G, PIN1 KD decreased the capability of metastatic spread in vivo (n = 6). H, Representative histomorphology and expression of Vimentin and E‐cadherin in lung metastatic nodules of mice. (Data are shown as mean ± SEM. One‐way ANOVA and Student's t test were used; *P < 0.05, **P < 0.01, ***P < 0.001.)
For tail vein transport experiments, BXPC3 PIN1KD and their paired controls were expanded and injected into the lateral tail vein of mice. Mice were monitored by weight and vital signs until being killed at 50 days after tail vein injection. All the hearts and lungs of mice were extracted and photographed. The size and the number of metastatic tumor nodules of heart and lung tissues significantly decreased in the PIN1KD group (Figure 3F,G). To further verify the functions of PIN1 on EMT in vivo, we detected the protein expression of E‐cad and Vim in the lung tissues of mice. As results in vitro, both enhanced expression of E‐cad and reduced expression of Vim were observed in PIN1KD group (Figure 3H).
3.4. PIN1 chemical inhibitor all trans retinoic acid inhibits both proliferation and invasiveness of pancreatic ductal adenocarcinoma cells through PIN1 ablation in vitro
Given the oncogenic role of PIN1 in PDAC, we wondered whether targeting PIN1 with chemical inhibitor ATRA would show any therapeutic benefits. First, we treated PANC and BXPC3 cells with different doses of ATRA to detect PIN1 expression. The results showed that ATRA could induce PIN1 degradation in a dose‐dependent manner (Figure S3A). After chemical PIN1 suppression, PDAC cells exhibited similar changes as with genetic PIN1 suppression, with dramatic decreases in cell proliferation, colony formation, migration and invasion (Figure 4A‐F). However, the functions of ATRA on cell growth inhibition and invasiveness disappeared with PIN1 ablation in PDAC cells (Figure S3B‐E). Similarly, results of G0/G1 cell phase increasing with decreasing proportion in S phase and changes of MET were also found in cells with ATRA treatments (Figure S4A,B). Furthermore, for PIN1 downstream oncoproteins, changes in the core signaling pathways and EMT‐related proteins under ATRA treatments occurred, with similar tendencies to the results of PIN1KD (Figure 4G).
Figure 4.
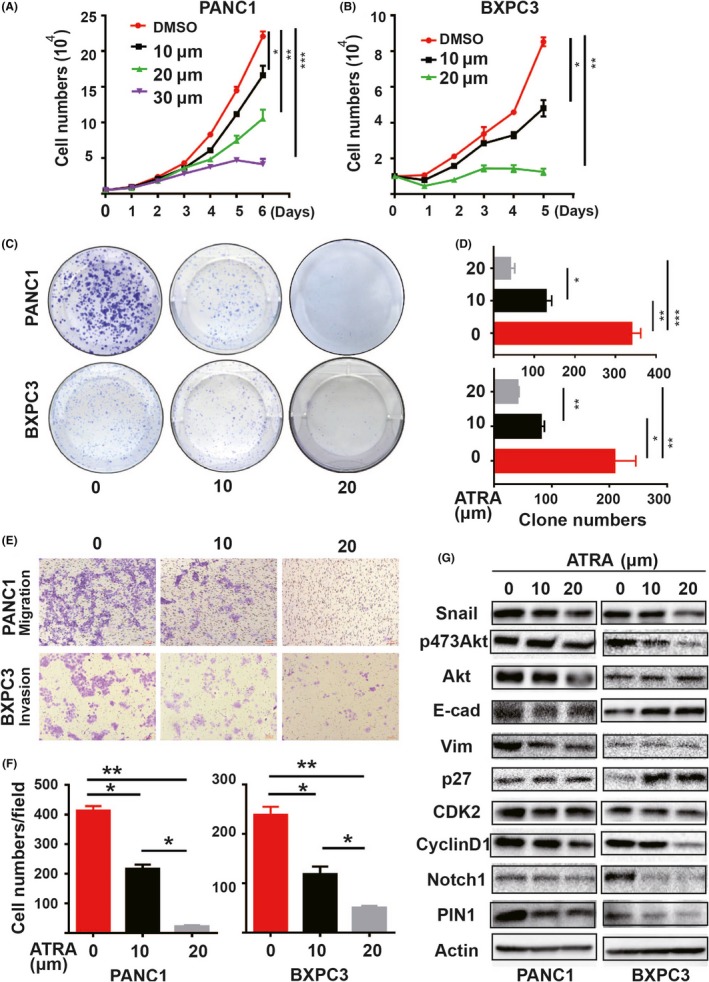
PIN1 chemical inhibitor all trans retinoic acid (ATRA) affected pancreatic ductal carcinoma (PDAC) cell functions in vitro. Both PANC1 and BXPC3 cells were performed with different doses of ATRA treatments. A, B, ATRA inhibited cell proliferation. C, D, ATRA decreased cell colony formation. E, F, ATRA reduced both migration and invasion. G, Cell lysates with ATRA treatments were performed by immunoblot with specific antibodies. (All experiments were performed independently at least 3 times. Data are shown as mean ± SEM. One‐way ANOVA and Student's t test were used; *P < 0.05, **P < 0.01,***P < 0.001.)
3.5. PIN1 chemical inhibitor all trans retinoic acid exerts potent anticancer activity against pancreatic ductal adenocarcinoma in vivo
Regarding targeting, PIN1 with chemical inhibitor ATRA has received noticeable results in vitro; we thus examined the functions of ATRA in vivo. Because regular ATRA is a light‐sensitive drug with a half‐life of 45 minutes in humans,45, 46 we used slow‐releasing ATRA to maintain a relatively stable drug level in blood for 21 days, as done previously.26 Mice were grouped to receive 5 or 10 mg ATRA or placebo pills implanted under the skin in the back of the neck, and tumor growth was then measured every 3 days. As revealed by tumor growth curves or final tumor weights, 5 and 10 mg ATRA both significantly inhibited PANC1 tumor growth (Figure 5A,B,D). To verify if ATRA could induce PIN1 degradation in vivo, total protein of tumor was obtained. The level of expression of PIN1 decreased with ATRA treatments, as indicated by PIN1 immunoblot (Figure 5C). Furthermore, the results of immunofluorescence staining indicated the lower expression of PIN1 and Ki67 under ATRA treatments (Figures 5E,F, S4C); results of proliferation inhibition were similar to those with PIN1 genetic inhibition.
Figure 5.
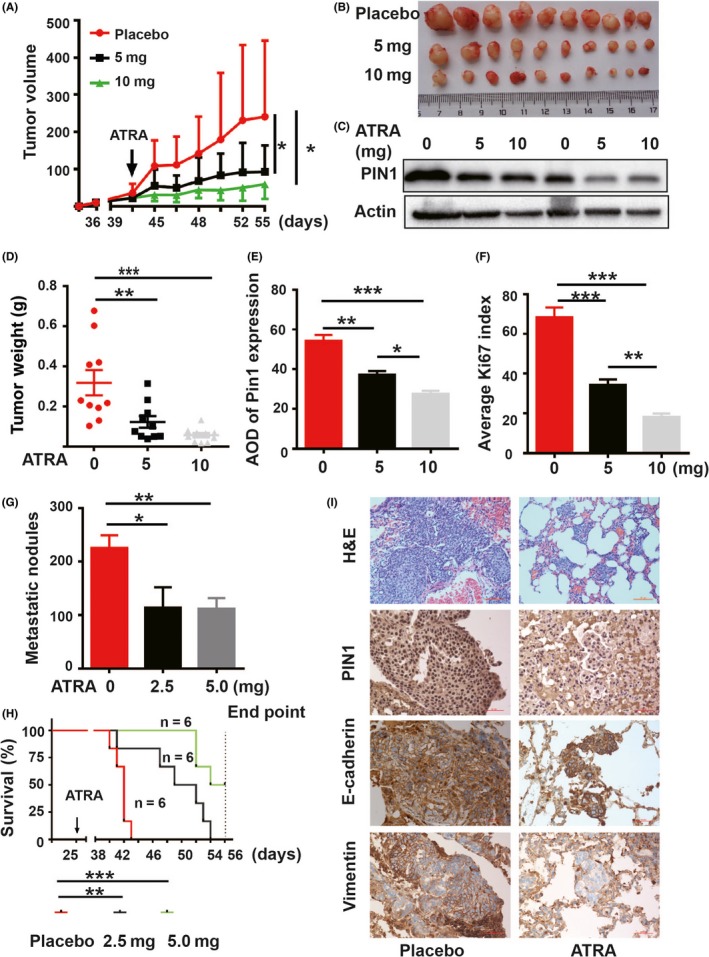
PIN1 chemical inhibitor all trans retinoic acid (ATRA) affected pancreatic ductal carcinoma (PDAC) cell functions in vivo. Xenograft tumor and tail vein transport models were constructed with PANC1 and BXPC3 cells, respectively. Different doses of slow‐releasing ATRA pellets were given for 21 d. A, ATRA decreased tumor volumes in vivo. B, Photographic illustration of tumors collected from nude mice. C, ATRA induced endogenous PIN1 degradation determined by immunoblot in xenograft tumor samples. D, ATRA induced tumor weight lost in vivo (measured at the end point). E, F, Statistical analysis on PIN1 (E) and Ki67 (F) expression in xenograft tumor tissues. G, ATRA reduced metastatic nodules in vivo. H, Kaplan‐Meier survival analysis for ATRA‐treated and control mice. I, Representative histomorphology and expression of Vimentin and E‐cadherin in lung metastatic nodules of mice. (Data shown as mean ± SEM. One‐way ANOVA, Student's t test and the log‐rank test were used, respectively; *P < 0.05, **P < 0.01,***P < 0.001.)
PDAC is a lethal disease with an overall 5‐year survival rate of less than 5% due to early distant spread.1 To determinate if chemical PIN1 suppression could inhibit metastatic spread of PDAC cells in vivo and prolong the OS of mice, we used BXPC3 cells to construct tail vein transport models for verification. To verify success in metastatic tumor models, a mouse was confirmed to have metastatic tumor nodules by histological evaluation at 20 days after tail vein injection. Then, mice were grouped to receive 2.5 or 5 mg slow‐releasing ATRA pellets or placebo pills and monitored by weight and vital signs until death occurred. Compared to the controls, the results showed that 2.5 mg and 5 mg ATRA both significantly reduced tumor burden (Figure 5G) and improved the OS of mice, as revealed by Kaplan‐Meier analysis (Figure 5H). Similar to the results in vitro, both enhanced expression of E‐cad and reduced expression of Vim were observed in tumor cells under ATRA treatments (Figure. 5I).
4. DISCUSSION
The search for an alternative means or a targeted agent for PDAC management has been ongoing for several years. Traditional prognostic tools and common therapeutic targets have not achieved satisfactory results in clinical studies of high intratumor heterogeneity in PDAC.7, 8 Targeting PIN1, to block multiple signaling pathways, has been demonstrated to exhibit potent antitumor effects in tumors with poor prognosis but limited therapeutic efficiency in clinical practice.26, 27, 28, 29, 47 As for the devastating disease of PDAC, our present study indicated that PIN1 may be an attractive predictive marker and targeting PIN1 could exert potent antitumor activity. Our results showed PIN1 overexpression in most PDAC without an increase in the gene copy number. PIN1 may be aberrantly activated by numerous mechanisms, including various signaling pathways, epigenetic regulation and post‐translational modification.11 High PIN1 expression showed significant association with poor outcomes and could further refine prognostication in PDAC. Especially for the heterogeneous category of TNM stage 2b, which comprises the vast majority of PDAC patients, the tools of PIN1 expression will greatly facilitate clinical management and prognostication. Not only that but genetic and chemical inhibition of PIN1 both significantly inhibited tumor growth and metastatic spread in vivo. Thus, PIN1 could be an important agent for targeting therapy in PDAC.
Pancreatic ductal adenocarcinoma is fundamentally a genetic disease with frequent genetic mutations.4, 5, 6 As Hong et al48 indicated, specific genetic disorders are strongly correlated with pathological and clinical findings in PDAC. PIN1 ablation has been demonstrated to induce cell apoptosis to inhibit tumor growth of KRAS‐mutated PDAC cells by mediating mitochondrial dysfunction.19 Similar findings of tumor proliferation inhibition were found after PIN1 suppression in our PDAC cells, which show less dependence on KRAS.37 Furthermore, PIN1 suppression could dramatically inhibit invasiveness and metastatic spread in our studies. PIN1 can induce EMT,36, 49 which is often considered to increase invasiveness and metastatic potential of cancer cells.9, 50 In addition, PIN1 inhibition could downregulate oncoproteins of Notch1, pSer473Akt, Cyclin D1 and CDK2, and upregulate suppressor p27kip, resulting in cell cycle arrest and phenotypic changes in EMT. The Notch1 signaling pathway is 1 of the 12 core signal pathways of aberration in PDAC, which is frequently upregulated in PDAC tissues and induces cell proliferation and EMT consistent with cancer stem phenotype in pancreatic cancer cells.4, 5, 51 Akt is a key molecule in PI3K/Akt/mTOR signaling pathways, which is a significant prognostic indicator for PDAC.52 Continuous activity of the Akt molecule is dependent on phosphorylation of Ser473, which is a downstream molecule of PIN1.43 Cyclin D1, CDK2 and p27kip are coordinated in the progression of G1‐S phase transition in the cell cycle, and the overexpression of Cyclin D1 contributes to tumorigenesis.53 Although the effects of the single client proteins followed by PIN1 inhibition were indicated to be only moderate, accumulative amplified effects of cancer‐related phenotypes depending on PIN1 function were observed. Therefore, the simultaneous impairment of Notch1, pser473Akt, Cyclin D1, CDK2 and p27kip resulted in a dramatic specifically for tumor invasiveness in our presented study after PIN1 ablation.
Targeting PIN1 using chemical inhibitor ATRA exhibits therapeutic benefits on PDAC, as shown in breast cancer and liver cancer.26, 28, 29 ATRA could dramatically inhibit tumor growth and metastatic spread in a dose‐independent manner by inducing PIN1 degradation and blocking multiple signaling pathways. In practice, ATRA has been used to treat APL for decades but with less efficiency against solid tumors, including PDAC,54, 55 for which the reason is partly the underlying molecular determinants. 26 Retinoic acid receptors (RAR) are as a major substrate of ATRA. However, more effective targeting drugs, including ATRA analogues and retinoid derivatives against RAR, have not been confirmed thus far for treatment of APL.56, 57 Recent series of studies have demonstrated that PIN1 degradation majorly influenes the effects of ATRA treatments.26, 27, 28, 29 For normal immortalized cells or hepatocelluar carcinoma cells with PIN1KD, ATRA treatments showed no effect on inhibition of cell proliferation,26, 29 which was also observed in our PIN1KD cells. Thus, PIN1 is a mainly target for ATRA treatment and PIN1 degradation is important for anticancer therapy in PDAC. When we used slow‐releasing ATRA pellets that can maintain ATRA serum concentrations in mice constantly enough to induce PIN1 inhibition,26 ATRA exhibited great potent anticancer effects in tumor models. This formulation of ATRA significantly reduced tumor burden and improved the OS of mice. Thus, it is urgent to develop a new longer‐acting ATRA or a more specific and potent PIN1 inhibitor to treat PDAC.
In summary, the results of this study support the role of PIN1 in PDAC progression. We found that PIN1 was highly expressed in most PDAC tissues and significantly correlated with the worst outcomes in patients. Here, we provided further evidence for the use of PIN1 as a promising therapeutic target in PDAC. Genetic and chemical PIN1 inhibition exerted potent antitumor activity through blocking multiple cancer‐driving pathways in PDAC. Longer half‐life ATRA or more potent and specific PIN1 targeting inhibitors could be exploited to treat this aggressive cancer.
DISCLOSURE
All authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We thank Mrs Cailing Yan (Service Centre, Public Technology, Fujian Medical University) for technical assistance.
Chen L, Xu X, Wen X, et al. Targeting PIN1 exerts potent antitumor activity in pancreatic ductal carcinoma via inhibiting tumor metastasis. Cancer Sci. 2019;110:2442‐2455. 10.1111/cas.14085
REFERENCES
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics 2018. CA Cancer J Clin. 2018;68:7‐30. [DOI] [PubMed] [Google Scholar]
- 2. Ren B, Liu X, Suriawinata AA. Pancreatic ductal adenocarcinoma and its precursor lesions: histopathology, cytopathology, and molecular pathology. Am J Pathol. 2019;189:9‐21. [DOI] [PubMed] [Google Scholar]
- 3. Esposito I, Konukiewitz B, Schlitter AM, Klöppel G. Pathology of pancreatic ductual adenocarcinoma: facts, challenges and future developments. World J Gastroenterol. 2014;20:13833‐13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waddell N, Pajic M, Patch AM, et al. Whole genome redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960‐1966. [DOI] [PubMed] [Google Scholar]
- 8. Van Cutsem E, van de Velde H, Karasek P, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430‐1438. [DOI] [PubMed] [Google Scholar]
- 9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 10. Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov. 2010;9:427‐428. [DOI] [PubMed] [Google Scholar]
- 11. Zhou XZ, Lu KP. The isomerase PIN1 controls numerous cancer‐driving pathways and is a unique drug target. Nat Rev Cancer. 2016;16:463‐478. [DOI] [PubMed] [Google Scholar]
- 12. Lu KP, Hanes SD, Hunter T. A human peptidyl‐prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544‐547. [DOI] [PubMed] [Google Scholar]
- 13. Lu PJ, Zhou XZ, Shen M, Lu KP. A function of WW domains as phosphoserine‐ or phosphothreonine‐binding modules. Science. 1999;283:1325‐1328. [DOI] [PubMed] [Google Scholar]
- 14. Ayala G, Wang D, Wulf G, et al. Pin1 is a novel prognostic marker in prostate cancer. Can Res. 2003;63:6244‐6251. [PubMed] [Google Scholar]
- 15. Shi M, Chen L, Ji J, et al. Pin1 is overexpressed and correlates with poor prognosis in gastric cancer. Cell Biochem Biophys. 2015;71:857‐864. [DOI] [PubMed] [Google Scholar]
- 16. Lin FC, Lee YC, Goan YG, et al. Pin1 positively affects tumorigenesis of esophageal squamous cell carcinoma and correlates with poor survival of patients. J Biomed Sci. 2014;21:75‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bao L, Kimzey A, Sauter G, et al. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pang R, Yuen J, Yuen MF, et al. PIN1 overexpression and beta‐catenin gene mutations are distinct oncogenic events in human hepatocellular carcinoma. Oncogene. 2004;23:4182‐4186. [DOI] [PubMed] [Google Scholar]
- 19. Liang C, Shi S, Liu M, et al. PIN1 maintains redox balance via the c‐Myc/NRF2 axis to counteract Kras‐induced mitochondrial respiratory injury in pancreatic cancer cells. Cancer Res. 2019;79:133‐145. [DOI] [PubMed] [Google Scholar]
- 20. Li Q, Dong Z, Lin Y, et al. The rs2233678 polymorphism in PIN1 promoter region reduced cancer risk: a meta‐analysis. PLoS ONE. 2013;8:e68148‐e68155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wulf G, Garg P, Liou YC, et al. Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 2004;23:3397‐3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Girardini JE, Napoli M, Piazza S, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell. 2011;20:79‐91. [DOI] [PubMed] [Google Scholar]
- 23. D'Artista L, Bisso A, Piontini A, et al. Pin1 is required for sustained B cell proliferation upon oncogenic activation of Myc. Oncotarget. 2016;7:21786‐21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Franciosa G, Diluvio G, Gaudio FD, et al. Prolyl‐isomerase Pin1 controls Notch3 protein expression and regulates T‐ALL progression. Oncogene. 2016;35:4741‐4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujimori F, Takahashi K, Uchida C, Uchida T. Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest. Biochem Biophys Res Comm. 1999;265:658‐663. [DOI] [PubMed] [Google Scholar]
- 26. Wei S, Kozono S, Kats L, et al. Active Pin1 is a key target of all‐trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med. 2015;21:457‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lian X, Lin YM, Kozono S, et al. Pin1 inhibition exerts potent activity against acute myeloidleukemia through blocking multiplecancer‐driving pathways. J Hematol Oncol. 2018;11:73‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao XH, Zhang AL, Zheng M, et al. Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer‐driving pathways. Sci Rep. 2017;7:43639‐43649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang D, Luo W, Wang J, et al. A novel controlled release formulation of the Pin1 inhibitor ATRA to improve liver cancer therapy by simultaneously blocking multiple cancer pathways. J Control Release. 2018;269:405‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li J, Orr B, White K, et al. Chmp 1A is a mediator of the anti‐proliferative effects of all‐trans retinoic acid in human pancreatic cancer cells. Mol Cancer. 2009;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo JM, Xiao BX, Lou YR, et al. The effects of all‐trans‐retinoic acid on cell cycle and alkaline phosphatase activity in pancreatic cancer cells. Med Chem. 2006;2:457‐461. [DOI] [PubMed] [Google Scholar]
- 32. Leelawat K, Ohuchida K, Mizumoto K, et al. All‐trans retinoic acid inhibits the cell proliferation but enhances the cell invasion through up‐regulation of c‐met in pancreatic cancer cells. Cancer Lett. 2005;224:303‐310. [DOI] [PubMed] [Google Scholar]
- 33. Gupta S, Pramanik D, Mukherjee R, et al. Molecular determinants of retinoic acid sensitivity in pancreatic cancer. Clin Cancer Res. 2012;18:280‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen LY, Tsang JY, Ni YB, et al. Bcl2 and Ki67 refine prognostication in luminal breast cancers. Breast Cancer Res Treat. 2015;149:631‐643. [DOI] [PubMed] [Google Scholar]
- 35. Nakamura K, Greenwood A, Binder L, et al. Proline isomer‐specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell. 2012;149:232‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo ML, Gong C, Chen CH, et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem‐like cell traits in breast cancer. Cancer Res. 2014;74:3603‐3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang JJ, Zhu Y, Xie KL, et al. Yin Yang‐1 suppresses invasion and metastasis of pancreatic ductal adenocarcinoma by downregulating MMP10 in a MUC4/ErbB2/p38/MEF2C‐dependent mechanism. Mol Cancer. 2014;13:130‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liou YC, Ryo R, Huang HK, et al. Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1‐null phenotypes. Proc Natl Acad Sci. 2002;99:1335‐1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wulf GM, Ryo A, Wulf GG, et al. Pin1 is overexpressed in breast cancer and potentiates the transcriptional activity of phosphorylated c‐Jun towards the cyclin D1 gene. EMBO J. 2001;20:3459‐3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng CW, Leong KW, Ng YM, et al. The peptidyl‐prolyl isomerase PIN1 relieves cyclin‐dependent kinase 2 (CDK2) inhibition by the CDK inhibitor p27. J Biol Chem. 2017;292:21431‐21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou W, Yang Q, Low CB, et al. Pin1 catalyzes conformational changes of Thr‐187 in p27Kip1 and mediates its stability through a polyubiquitination process. J Biol Chem. 2009;284:23980‐23988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liao Y, Wei Y, Zhou X, et al. Peptidyl‐prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene. 2009;28:2436‐2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rustighi A, Tiberi L, Soldano A, et al. The prolyl‐isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol. 2009;11:133‐142. [DOI] [PubMed] [Google Scholar]
- 45. Muindi J, Frankel SR, Miller WH Jr, et al. Continuous treatment with all‐trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood. 1992;79:299‐303. [PubMed] [Google Scholar]
- 46. Smith MA, Adamson PC, Balis FM, et al. Phase I and pharmacokinetic evaluation of all‐trans‐retinoic acid in pediatric patients with cancer. J Clin Oncol 1992;10:1666‐1673. [DOI] [PubMed] [Google Scholar]
- 47. Campaner E, Rustighi A, Zannini A, et al. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat Commun. 2017;8:15772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hong SM, Park JY, Hruban RH, et al. Molecular signatures of pancreatic cancer. Arch Pathol Lab Med. 2011;135:716‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim MR, Choi HK, Cho KB, Kim HS, Kang KW. Involvement of Pin1 induction in epithelial‐mesenchymal transition of tamoxifen‐resistant breast cancer cells. Cancer Sci. 2009;100:1834‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319‐8326. [DOI] [PubMed] [Google Scholar]
- 51. Bao B, Wang Z, Ali S, et al. Notch‐1 induces epithelial‐mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011;307:26‐36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52. Yamamoto S, Tomita Y, Hoshida Y, et al. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin Cancer Res. 2004;10:2846‐2850. [DOI] [PubMed] [Google Scholar]
- 53. Tashiro E, Tsuchiya A, Imoto M. Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression. Cancer Sci. 2007;98:629‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brembeck FH, Schoppmeyer K, Leupold U, et al. A phase II pilot trial of 13‐cis retinoic acid and interferon‐alpha in patients with advanced pancreatic carcinoma. Cancer. 1998;83:2317‐2323. [DOI] [PubMed] [Google Scholar]
- 55. Moore DF Jr, Pazdur R, Sugarman S, Jones D 3rd, et al. Pilot phase II trial of 13‐cis‐retinoic‐acid and interferon‐alpha combination therapy for advanced pancreatic adenocarcinoma. Am J Clin Oncol. 1995;18:525‐527. [DOI] [PubMed] [Google Scholar]
- 56. Ramlau R, Zatloukal P, Jassem J, et al. Randomized phase III trial comparing bexarotene (L1069‐49)/cisplatin/vinorelbine with cisplatin/vinorelbine in chemotherapy‐naive patients with advanced or metastatic non‐small‐cell lung cancer: SPIRIT I. J Clin Oncol. 2008;26:1886‐1892. [DOI] [PubMed] [Google Scholar]
- 57. Connolly RM, Nguyen NK, Sukumar S. Molecular pathways: current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin Cancer Res. 2013;19:1651‐1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials