

Abstract
Liquid biopsy of circulating tumor cells (CTC) and circulating tumor DNA (ctDNA) is gaining attention as a method for real‐time monitoring in cancer patients. Conventional methods based upon epithelial cell adhesion molecule (EpCAM) expression have a risk of missing the most aggressive CTC subpopulations due to epithelial‐mesenchymal transition and may, thus, underestimate the total number of actual CTC present in the bloodstream. Techniques utilizing a label‐free inertial microfluidics approach (LFIMA) enable efficient capture of CTC without the need for EpCAM expression. In this study, we optimized a method for analyzing genetic alterations using next‐generation sequencing (NGS) of extracted ctDNA and CTC enriched using an LFIMA as a first‐phase examination of 30 patients with head and neck cancer, esophageal cancer, gastric cancer and colorectal cancer (CRC). Seven patients with advanced CRC were enrolled in the second‐phase examination to monitor the emergence of alterations occurring during treatment with epidermal growth factor receptor (EGFR)‐specific antibodies. Using LFIMA, we effectively captured CTC (median number of CTC, 14.5 cells/mL) from several types of cancer and detected missense mutations via NGS of CTC and ctDNA. We also detected time‐dependent genetic alterations that appeared during anti–EGFR therapy in CTC and ctDNA from CRC patients. The results of NGS analyses indicated that alterations in the genomic profile revealed by the liquid biopsy could be expanded by using a combination of assays with CTC and ctDNA. The study was registered with the University Hospital Medical Information Network Clinical Trials Registry (ID: UMIN000014095).
Keywords: circulating tumor cell, circulating tumor DNA, gastrointestinal cancer, head and neck cancer, liquid biopsy
1. INTRODUCTION
Genetic and phenotypic variations occur between tumors involving different tissues and cell types as well as between individuals with the same tumor type.1, 2, 3 To enhance understanding of these phenomena, longitudinal tumor sampling approaches will be essential to elucidate the impact of tumor heterogeneity on cancer evolution.4 Although molecular profiling data obtained from tissue biopsy samples can facilitate delivery of precision medicine by enabling selection of the most effective chemotherapy approach for an individual patient, tissue biopsy is invasive, risky and painful. In addition, the biological behavior of tumor cells can change with each passing moment in response to selective pressures associated with cancer therapies. Therefore, is important to develop real‐time monitoring methods that are minimally invasive to profile the biological behavior of an individual tumor. Such methods could enhance understanding of the mechanisms underlying cancer diversity and drug resistance. Indeed, addressing cancer drug resistance was a key recommendation of the Blue Ribbon Panel that advised the Beau Biden Cancer Moonshot initiative.5
Liquid biopsy methods have gained increasing attention as tools for real‐time monitoring of cancer patients. Recent technological advances in the detection and characterization of circulating tumor cells (CTC) and circulating tumor DNA (ctDNA) could enable the examination of genomic alterations in minimally invasive examinations and facilitate tailoring of treatments based on real‐time monitoring of tumor evolution. A major advantage of CTC profiling is the ease of obtaining samples for monitoring tumor evolution and studying the mechanism of acquired drug resistance. Indeed, genomic analyses of CTC from non–small cell lung cancer patients identified the T790M gatekeeper mutation, which confers resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors.6, 7 Furthermore, next‐generation sequencing (NGS) of breast cancer CTC revealed significant interpatient heterogeneity that could be monitored over time.8, 9
Among current technologies for CTC detection, the only one cleared by the FDA for use in clinical settings is the CellSearch system. In this system, CTC are isolated with an antibody against the epithelial cell adhesion molecule (EpCAM) as a biomarker. However, conventional methods based on detection of EpCAM carry the risk of missing the most aggressive CTC subpopulations due to epithelial‐mesenchymal transition (EMT), potentially leading to underestimation of the total number of actual CTC present in the bloodstream. To overcome this drawback, a label‐free inertial microfluidics approach (LFIMA) was recently developed to enrich CTC from blood samples.10 This system enables efficient isolation of CTC without affinity purification of epithelial biomarkers, thereby avoiding underestimation of CTC subpopulations exhibiting downregulation of EpCAM expression.
Here, we established a method using LFIMA with NGS for the analysis of genomic alterations in CTC isolated from patients with head and neck cancer (HNC) or gastrointestinal (GI) cancer. In addition, we carried out blood‐based molecular profiling to identify actionable drug targets, monitor drug resistance, and track tumor dynamics using CTC and ctDNA from patients with metastatic colorectal cancer (CRC).
2. MATERIALS AND METHODS
2.1. Patients and peripheral blood samples
A total of 37 patients diagnosed at the National Cancer Center Hospital with HNC or GI cancer (esophageal cancer [OC], gastric cancer [GC] or CRC) between June 2013 and July 2016 were enrolled for this prospective study. All participants provided signed informed consent prior to sample collection. Peripheral blood was collected in 5‐mL EDTA vacutainers (TERUMO) and processed within 24 hours. The study was approved by the ethics committee of the National Cancer Center and registered with the University Hospital Medical Information Network Clinical Trials Registry (ID: UMIN000014095).
In the second‐phase examination focusing on CRC, patients received irinotecan plus panitumumab or cetuximab (anti–EGFR antibody selection was at the physician's discretion) until disease progression or unacceptable toxicity was noted. Cetuximab (Merck KGaA) was administered initially at a dose of 400 mg/m2, followed by weekly infusions of 250 mg/m2. Panitumumab (Takeda) was administered at a dose of 6 mg/kg every 2 weeks. The dose of irinotecan was selected by each physician according to the patient, based on prior symptoms of toxicity experienced with twice‐weekly irinotecan.
2.2. Circulating tumor cell enrichment
A ClearCell FX system (Biolidics [previously Clearbridge Biomedics], Singapore) compatible with the LFIMA was used to capture and enrich CTC from peripheral blood samples according to the manufacturer's protocol. A total of 5 mL of blood was mixed with 15 mL of red blood cell lysis buffer (G‐Biosciences) at room temperature for 10 minutes. After incubation, the samples were centrifuged at 500 g for 10 minutes followed by aspiration of the supernatant, with final addition of 4.3 mL of suspension reagent supplied by the manufacturer.10
2.3. Immunofluorescence cytochemistry
Circulating tumor cell slides were prepared using a cyto‐spin device and stored at −80°C. Cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature and permeabilized with 0.1% Triton X‐100 (Sigma‐Aldrich). The cells were then incubated with anti–pan CK rabbit polyclonal antibody (NICHIREI BIOSCIENCE), followed by incubation with anti–rabbit IgG Alexa Fluor 488 (Thermo Fisher Scientific).11
2.4. DNA extraction and quantification
DNA from CTC was prepared immediately after isolation, and whole‐genome amplification (WGA) was carried out using a REPLI‐g Single Cell Kit (Qiagen GmbH). The amplified DNA was purified using an Agencourt AMPure XP system (Beckman Coulter). Fragmentation of the output DNA of the WGA reaction was assessed using a TaqMan RNase P Detection Reagents Kit and FFPE DNA QC assay (Thermo Fisher Scientific).
A five‐point standard curve was prepared using human control genomic DNA (included in the TaqMan RNase P Detection Reagents Kit), and absolute DNA concentrations were determined against the standard curve using real‐time PCR. The degree of DNA fragmentation was estimated using the DNA ratio (relative quantification, RQ) of long amplicons (256 bp) to short amplicons (87 bp). ctDNA was extracted from 2 mL of plasma using a QIAmp Circulating Nucleic Acid Kit (Qiagen) or Maxwell RSC ccfDNA Plasma kit (Promega) according to the manufacturer's instructions. Human blood genomic DNA was purified from 250 to 1000 μL of buffy coat using a QIAmp DNA Mini Kit (Qiagen) or Maxwell RSC Buffy Coat DNA kit (Promega) to serve as a standard. The extracted ctDNA and human blood genomic DNA were purified using an Agencourt AMPure XP system (Beckman Coulter). DNA was quantified using a Qubit 2.0 fluorometer according to the manufacturer's instructions.
2.5. Targeted NGS
Library preparation was performed using 20 ng of CTC DNA, ctDNA, and 10 ng buffy coat DNA using an Ion AmpliSeqs Library Kit Plus and Ion AmpliSeq Cancer Hotspot Panel v2 (Thermo Fisher Scientific). The AmpliSeq Cancer Hotspot Panel was designed to amplify 207 amplicons covering approximately 2790 COSMIC mutations from 50 oncogenes and tumor suppressor genes (Table S1). Emulsion PCR was performed using the Ion 510 & Ion 520 & Ion 530 Kit – Chef and Ion Chef system (Thermo Fisher Scientific). Sequencing was performed on an Ion S5XL System using a 530 chip (Thermo Fisher Scientific).
2.6. Sequencing data analysis and variant calling
Sequencing data were assessed using Torrent Suite software, version 5.6. Variants were called using ion‐plugin‐coverageAnalysis, version 5.6.0.1, and ion‐plugin‐variantCaller, version 5.6.0.4. Single nucleotide variants, insertions and deletions were annotated using the Ion Reporter software, version 5.6 (Thermo Fisher Scientific). The allele frequency threshold was set to 5%, and minimum coverage per target amplicon was set to 250× to report de novo mutations.
3. RESULTS
3.1. Development of efficient analytical methods using DNA extracted from liquid biopsy samples from patients in the first‐phase examination
The study workflow is summarized in Figure 1A. To develop efficient methods for the analysis of mutations in CTC collected from patient blood samples, we optimized the CTC capture method using an LFIMA from the first examination phase, which consisted of 30 patients with HNC, OC, GC and CRC.
Figure 1.
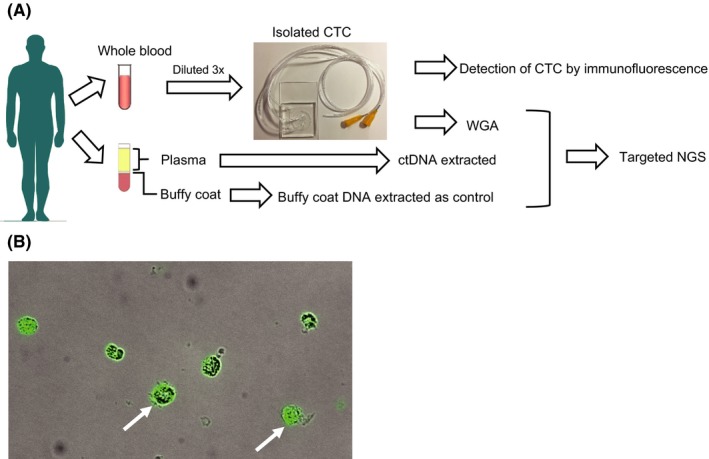
Flow diagram of the optimized protocol for detecting genomic alterations in circulating tumor cells (CTC) and ctDNA, and immunofluorescence cytochemistry of isolated CTC. A, Blood from patients (up to 2 × 5 mL of peripheral blood) was collected using EDTA vacutainers. One collection tube of hemolyzed whole blood was diluted 3‐fold. CTC were isolated from the blood using a label‐free inertial microfluidics approach (LFIMA). ctDNA and buffy coat DNA were isolated from the other collection tube. Targeted next‐generation sequencing was performed using the extracted DNA. B, Fluorescence image of isolated CTC stained for cytokeratin (green). NGS, next‐generation sequencing; WGA, whole‐genome amplification
Baseline characteristics of the 10 patients with HNC are shown in Table 1. The median age was 70 years (range, 42‐80 years). The tumor locations were as follows: 5 in the oral cavity (50%), 1 in the salivary glands (50%), 3 in the pharynx (30%) and 1 in the cervical esophagus (10%). Nine patients had squamous cell carcinoma, and 1 patient had adenoid cystic carcinoma. The number of patients at clinical stage II, III and IV was 3 (30%), 1 (10%) and 6 (60%), respectively.
Table 1.
Clinicopathologic characteristics of head and neck cancer patients in the first examination phase
| Female | Male | Total (%) | |
|---|---|---|---|
| Age, years, median (range) | 75 (59‐77) | 67 (42‐80) | 70 (42‐80) |
| Sex | 5 | 5 | 10 |
| Primary tumor site | |||
| Oral cavity | 3 | 2 | 5 (50) |
| Salivary gland | 1 | 0 | 1 (10) |
| Pharynx | 0 | 3 | 3 (30) |
| Cervical esophagus | 1 | 0 | 1 (10) |
| Histology | |||
| Squamous cell carcinoma | 4 | 5 | 9 (90) |
| Adenoid cystic carcinoma | 1 | 0 | 1 (10) |
| Stagea | |||
| II | 2 | 1 | 3 (30) |
| III | 0 | 1 | 1 (10) |
| IV | 3 | 3 | 6 (60) |
According to the International Union Against Cancer (UICC) TNM Classification of Malignant Tumours, 7th edition (2010).
Baseline characteristics of the 20 patients with advanced GI cancers, which consisted of 8 (40%) patients with OC, 1 with GC (5%) and 11 with CRC (55%), are shown in Table 2. The median age was 61.5 years (range, 46‐73 years). Eastern Cooperative Oncology Group performance status at consent of 0, 1 and 2 were 9 (45%), 10 (50%) and 1 (5%), respectively. Of these patients, 14 had recurrence. The number of patients with prior chemotherapy lines was 3 (15%) with 1 line, 5 (25%) with 2 lines and 3 (15%) with greater than 4 lines.
Table 2.
Clinicopathologic characteristics of gastrointestinal cancer patients in the first examination phase
| Female | Male | Total (%) | |
|---|---|---|---|
| Age, years, median (range) | 61.5 (63‐73) | 59.5 (46‐67) | 61.5 (46‐73) |
| Sex | 4 | 16 | 20 |
| Primary tumor site | |||
| Esophagus | 1 | 7 | 8 (40) |
| Stomach | 0 | 1 | 1 (5) |
| Colon and rectum | 3 | 8 | 11 (55) |
| ECOG performance status | |||
| 0 | 3 | 6 | 9 (45) |
| 1 | 0 | 10 | 10 (50) |
| 2 | 1 | 0 | 1 (5) |
| Disease statusa | |||
| Stage IV | 1 | 5 | 6 (30) |
| Recurrence | 3 | 11 | 14 (70) |
| Number of prior chemotherapy lines | |||
| 0 | 1 | 8 | 9 (45) |
| 1 | 1 | 2 | 3 (15) |
| 2 | 1 | 4 | 5 (25) |
| ≥4 | 1 | 2 | 3 (15) |
According to the International Union Against Cancer (UICC) TNM Classification of Malignant Tumours, 7th edition (2010).
3.2. Genomic profiling of circulating tumor cells and ctDNA using next‐generation sequencing in the first‐phase examination
To determine the number of CTC, we carried out immunofluorescent analyses with anti–pan keratin antibody (Figure 1B). Of the patients enrolled in the first‐phase examination, the number of CTC was determined for 27 patients (Figure 2A and S1). The median number of CTC was 14.5/mL (range, 3‐133/mL).
Figure 2.
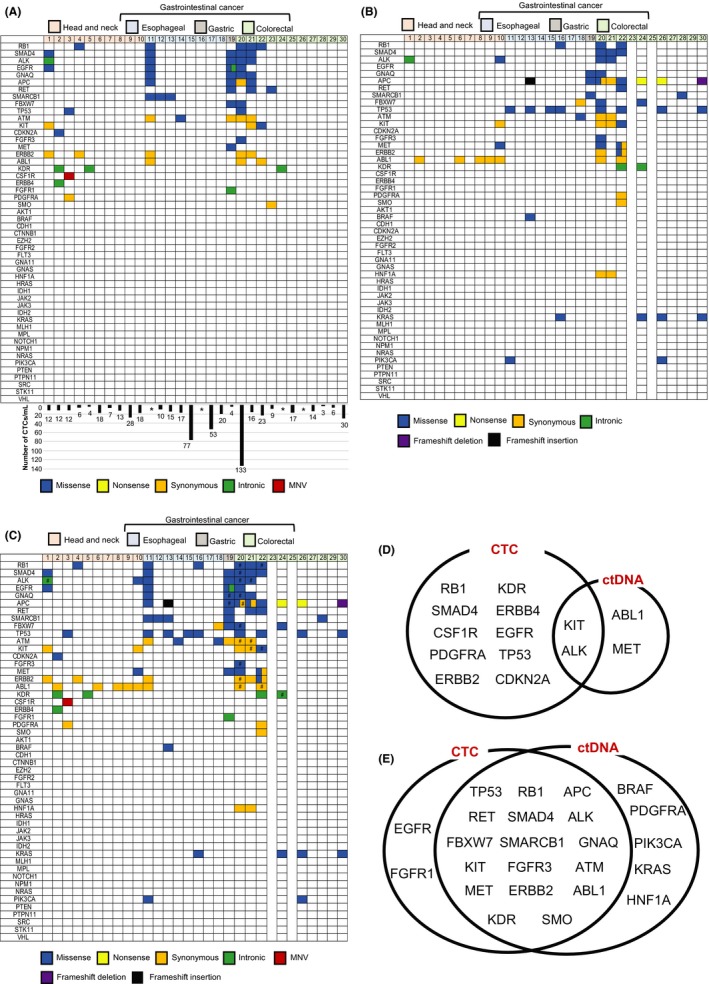
Targeted next‐generation sequencing and combination analysis of genomic alterations using circulating tumor cells (CTC) and ctDNA. A, Genomic alterations in CTC from patients with head and neck cancer (HNC), esophageal cancer (OC), gastric cancer (GC) and colorectal cancer (CRC). The number of CTC is shown in the columns. *The number of CTC could not be determined for 4 patients. B, Genomic alterations in ctDNA from patients with HNC, GC, and CRC. ctDNA could not be extracted from 2 patients with CRC. C, Genomic alterations in CTC and ctDNA in patients with HNC, GC and CRC. #The same amino acid changes were detected in both CTC and ctDNA. D, Combination analysis of genomic alterations using CTC and ctDNA from patients with HNC. E, Combination analysis of genomic alterations using CTC and ctDNA from patients with CRC
The results of genomic profiling by NGS of HNC CTC samples are shown in Figure 2A. Details regarding CTC mutation profiles, allele frequencies and coverages are shown in Table S2. Missense mutations were detected in 4 out of 10 (40%) HNC patients; these included mutations in EGFR and SMAD4 (n = 1), TP53 (n = 1), RB1 (n = 1) and CDKN2A (n = 1). The missense mutations in EGFR and SMAD4 were detected in the same case of HNC. In contrast, the missense mutations in TP53, RB1 and CDKN2A were detected in different cases.
The most frequent missense mutations in CTC from GI cancer were detected in ALK, GNAQ, RB1 and SMAD4, and these mutations occurred in 4 cases (20%). Moreover, missense mutations in APC, EGFR, RET and SMARCB1 were detected in 3 cases (15%). The most frequent missense mutations in cases of OC and CRC occurred in SMARCB1 (3/8, 37.5%) and RB1 (3/11, 27.3%), respectively. The missense mutations analyzed in this first‐phase examination of CTC occurred in 4 cases of OC (4/8, 50%), 1 case of GC (1/1, 100%) and 4 cases of CRC (4/11, 36.4%).
Of the 30 patients enrolled in first‐phase examination, ctDNA was obtained from 28 patients to confirm mutations in cells circulating in the plasma using NGS. Details regarding ctDNA mutation profiles, allele frequencies and coverages are shown in Table S2. The mutation profile is shown in Figure 2B. Missense mutations in ALK and MET were detected in 1 case of HNC; however, these missense mutations could not be detected in the remaining 9 cases of HNC. The most frequent missense mutations in GI cancers occurred in TP53, and these were detected in 8 cases of GI cancer (44.4%). The most frequent missense mutations in both OC and CRC occurred in TP53, and these were detected in 4 cases of OC (4/8, 50%) and 4 cases of CRC (4/9, 44.4%). Nonsense mutations in APC were detected in 2 cases of CRC. A frameshift deletion in APC was detected in 1 case of CRC; in addition, a frameshift insertion in APC was detected in a case of OC. No missense/nonsense or frameshift‐insertion/‐deletion mutations were detected in 3 cases of OC (3/8, 37.5%) and 2 cases of CRC (2/9, 22.2%), respectively.
3.3. Combination analysis of genomic mutation profiles obtained from circulating tumor cells and ctDNA
The results of genomic mutation profiling of CTC and ctDNA (Figure 2A,B) suggested that the genetic mutational concordance between profiles of CTC and ctDNA was not high. As tumor heterogeneity suggested that the CTC and ctDNA samples exhibited different profiles, we conducted a combination analysis (Figure 2C).
The combination analysis improved the rate of genomic alteration detection compared to the assays of CTC or ctDNA alone. The combination analysis detected missense mutations in 5 cases of HNC (5/10, 50%) and 15 cases of GI cancer (15/18, 83.3%). The same amino acid changes were detected in 6 of 28 cases in which both CTC and ctDNA were analyzed (Tables S2 and S3). Details of associations between genomic alterations detected in CTC and ctDNA are shown in Figure 2D (HNC) and E (GI cancer).
3.4. Second‐phase examination: Analysis of genomic alterations in circulating tumor cells and ctDNA over time during anti–epidermal growth factor receptor therapy for metastatic colorectal cancer
We evaluated the fragmentation of CTC DNA in the second‐phase examination. Representative RQ values for assessment of CTC DNA fragmentation are shown in Figure S2. CTC DNA was unfragmented. The mean amount of amplified and purified CTC DNA was 249 ng (range, 93‐444 ng).
The clinical management of individuals who develop resistance to anti–EGFR therapy through the emergence of gene mutations remains challenging. To address this issue, we monitored changes in genetic profiles of CTC and ctDNA over time in patients undergoing anti–EGFR therapy for metastatic CRC (Figure 3A). We examined blood samples from 7 patients with metastatic CRC who received anti–EGFR therapy (Table S4). These patients were monitored at 2 time points: before initiation of anti–EGFR therapy and at disease progression. Details regarding mutation profiles, allele frequencies and coverages are shown in Table S5. In patients 1 and 3, genomic alterations emerged in both CTC and ctDNA with disease progression. Patient 1 developed a new missense mutation in SMARCB1 (p.T72K) in CTC and missense mutations in FGFR3 (p.N644D), RB1 (p.I680T), RB1 (p.L670P) and SMAD4 (p.V354L), and an intronic mutation in EGFR in ctDNA. In patient 3, a missense mutation in SMARCB1 (p.T72K) in CTC and several other missense mutations were detected during irinotecan and cetuximab treatment.
Figure 3.
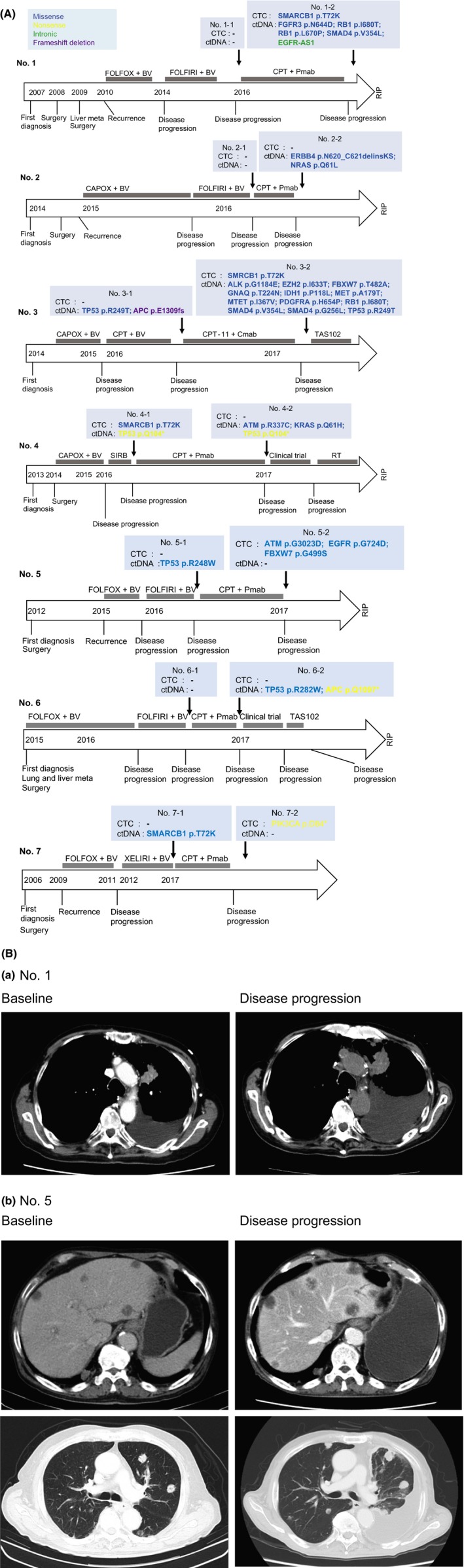
A, Clinical course in colorectal cancer patients who received anti–epidermal growth factor receptor (EGFR) therapy and genomic alterations in circulating tumor cells (CTC) and ctDNA. A, Monitoring genomic profiles of CTC and ctDNA during anti–EGFR therapy.BV, bevacizumab; CAPOX, capecitabine, oxaliplatin; Cmab, cetuximab; CPT, irinotecan; FOLFIRI, folinic acid, fluorouracil and irinotecan; FOLFOX, folinic acid, fluorouracil and oxaliplatin; Pmab, panitumumab; RT, radiation therapy; SIRB, tegafur/gimeracil/oteracil, irinotecan and bevacizumab; TAS102, trifluridine and tipiracil. B, Representative CT images. (a) Growth of lung metastases and increased pleural effusion were observed during disease progression in patient 1. (b) Growth of liver and lung metastases and increased pleural effusion were observed during disease progression in patient 5
In patients 2, 4 and 6, genomic alterations with disease progression were detected only in ctDNA. In patient 2, new missense mutations in ERBB4 (p.N620_C621 delinsKS) and NRAS (p.Q61L) were detected. Although patient 4 exhibited a missense in SMARCB1 (p.T72K) before beginning treatment with irinotecan and panitumumab, the mutation disappeared as the disease progressed. In ctDNA, new missense mutations in ATM (p.R337C) and KRAS (p.Q61H) were detected in addition to a nonsense mutation in TP53 present before treatment. Patient 6 developed a new missense mutation in TP53 (p.R282W) and a nonsense mutation in APC (p.Q1097*) as disease progressed.
In patient 7, a nonsense mutation in PIK3CA (p.D84*) was detected in the CTC analysis during progression after irinotecan and panitumumab therapy.
Interestingly, genomic alterations were detected in DNA extracted from CTC and/or ctDNA in all cases examined. Representative baseline and disease progression CT images are shown in Figure 3B.
4. DISCUSSION
In this prospective observational study, we evaluated whether CTC can be used to monitor molecular changes in real time throughout the clinical management of patients with advanced cancer. We effectively captured CTC from several types of cancers using LFIMA and performed targeted sequencing of CTC and ctDNA using NGS technology. Furthermore, we designed a strategy combining analyses of genomic mutation profiles of CTC and ctDNA to identify unique mutations that arise during anti–EGFR therapy in patients with metastatic CRC.
Circulating tumor cells released into the bloodstream from primary tumors and metastases may reflect current tumor status. Genomic alterations in CTC are of growing interest because their identification could aid in the development of targeted therapies. However, CTC are extremely rare in the blood, with 1‐100 CTC/mL among millions of white blood cells and billions of red blood cells.12 To overcome the challenge of isolating CTC, multiple platforms have been developed for enrichment and detection. We used the LFIMA to enrich CTC rather than the conventional method based on the identification of circulating cells expressing epithelial markers, most notably EpCAM.10 Using the LFIMA, we could detect a larger CTC pool (independent of EpCAM expression) to identify CTC subpopulations or EMT‐derived CTC. Enriched samples from at least 28 patients enrolled in the first‐phase examination of our study contained 3‐133 CTC/mL of blood. In previous reports describing isolation of CTC from HNC and CRC patients using the CellSearch system, only 10 of 80 cases (12.5%) of HNC13 and 7 of 20 cases (35%) of CRC14 harbored CTC in the peripheral blood. In our previous study, we compared the number of CTC in samples from metastatic CRC patients using the ClearCell FX and CellSearch systems. The median number of CTC was higher with the ClearCell FX system (14 cells [range 3‐26]/mL) compared with the CellSearch system (0 cells/7.5 mL).15
Because the number of CTC in the bloodstream is extremely low in comparison with other normal hematopoietic cells, WGA methods are generally required for analyzing NGS data for DNA obtained from CTC. WGA strategies are known to introduce artifacts and errors in variation detection studies.16 Recently, the results of NGS analyses of CTC using WGA methods have been reported. Multiple displacement amplification technology was developed for analyzing genetic alterations from extremely small amounts of DNA extracted from single cells. The accuracy of multiple displacement amplification technology has been evaluated in terms of its reliability in WGA using CTC.12, 17 To confirm the reliability of WGA, we assessed the fragmentation of amplified CTC DNA. No fragmentation of CTC DNA was detected. Thus, experiments involving WGA were deemed reliable because the method was the same for all CTC analyses. We optimized the efficiency of a method for NGS analysis of CTC DNA using patient samples with multiple displacement amplification technology.
Molecular and cellular heterogeneity are hallmarks of cancer that have important impacts on the diagnosis and treatment of tumors.18 A previous study showed that the degree of intratumor heterogeneity can be highly variable, with 0 to >8000 coding mutations found to be heterogeneous within primary tumors or between primary and metastatic or recurrent sites.19 Liquid biopsy is an essential tool for non‐invasive real‐time monitoring of cancer and also enables characterization of tumor heterogeneity because blood carries DNA derived from cancer cells located at distinct metastatic sites, in contrast to single‐tissue biopsies.20 CTC and ctDNA may represent a molecular proxy of the overall disease. In this study, when comparing mutations detected in CTC and ctDNA from patients with HNC and CRC, we found that in some blood samples, CTC exhibited mutations that were not detected in ctDNA, whereas in others, ctDNA exhibited mutations that were not detected in CTC. This suggests that CTC and ctDNA exhibit heterogeneity, and therefore, both must be evaluated in the clinical setting to enable optimal surveillance of disease progression and treatment selection. In this study, NGS data revealed that the same genetic alteration could be detected in data obtained from CTC and ctDNA using multiple displacement amplification technology. However, we found that the genetic alteration profiles were not perfectly correlated between CTC and ctDNA. These data suggest that CTC and ctDNA exhibit unique genetic alteration profiles. In other words, using a combination assay involving CTC and ctDNA could increase the sensitivity of detecting genetic alterations without decreasing the specificity, thus contributing to the establishment of precision medicine for cancer.
Considerable recent attention has focused on the biological heterogeneity of CTC. However, in this study, we did not assess the heterogeneity of CTC because the technique utilizing a label‐free inertial microfluidics approach enriched CTC in bulk according to cell size, and we carried out WGA immediately after isolation. In addition, to predict the sensitivity of tumors to molecular therapies, the mutation status of a majority of the tumors must be known. NGS analysis of the genome of bulk CTC could facilitate better predictions of the efficacy of personalized molecular targeted therapies compared with analyses of single CTC.
Anti–EGFR therapy, either alone or in combination with chemotherapy, is the standard treatment for patients with RAS wild‐type metastatic CRC.21, 22, 23, 24, 25 It has become apparent that RAS mutations are correlated with resistance to anti–EGFR therapy, and the presence of RAS mutations accounts for approximately 50%‐60% of patients with metastatic CRC refractory to anti–EGFR therapy.26 In addition to mutations in RAS, mutations in BRAF and PIK3CA can induce constitutive activation of the EGFR and subsequent intracellular signaling, ultimately leading to drug resistance.27, 28, 29 Several studies have detected these mutations in CTC and ctDNA isolated from patients with CRC.30, 31, 32 We also detected genomic alterations in KRAS, NRAS, and PIK3CA in CTC and ctDNA using targeted NGS in patients resistant to anti–EGFR therapy, as described in previous reports. In liquid biopsy of CTC and ctDNA, codon 61 mutations in KRAS and NRAS that were detected in our study are more frequently observed after CRC patients have acquired resistance to anti–EGFR therapy than before starting the anti–EGFR therapy.20
More than 80% of mutations detected in PIK3CA have been reported in 2 hotspots, the helicase domain of exon 9 and the kinase domain in exon 20, suggesting that these mutations can be used to predict response to treatment with anti–EGFR therapy.27 The exon 1 mutation we detected in CTC was very infrequent, and its importance and function remain incompletely understood. As for other genes, mutations in FBXW7 and SMAD4 were frequently detected, and these mutations, which are located in the same domain as the mutation we reported in this study, are involved in acquired resistance to anti–EGFR therapy.33, 34 Our data, which were obtained from a relatively small number of samples, showed that further study is needed to determine whether genetic information for CRC cells obtained from liquid biopsy of patients resistant to anti–EGFR therapy could enhance efforts to overcome drug resistance.
However, this study has some limitations. First, our study consisted of a small number of samples. The results thus need to be validated in a prospective manner with a sufficient sample size. Second, CTC were defined as cytokeratin‐positive in this study. Recently, CTC have been distinguished from other cells present in the blood based on being: (i) nuclear positive; (ii) cytokeratin positive; and (iii) CD45 negative.35 We previously stained cells for these markers to identify CTC; however, it is now possible to distinguish CTC morphologically. Indeed, we reported the untargeted molecular profiling of single CTC obtained from patients with gastric cancer and colorectal cancer using live single‐cell mass spectrometry integrated with microfluidics‐based cell enrichment techniques.36 In that study, we detected CTC based on CD45 staining and morphology and demonstrated clear differences in the metabolomics profiles of CTC and leucocytes. The present study was not a single‐cell analysis of CTC but rather a mutational analysis of CTC. Therefore, we employed immunostaining with an anti–cytokeratin antibody rather than anti–CD45 antibody. Third, we did not assess the concordance of gene mutations between primary tumor tissues and liquid biopsy sample because almost all cases were advanced stages or recurrences, and the acquisition of biopsy specimens from patients was difficult in the clinical setting at the timely manner. However, whether the tumor mutation profile obtained from tumor biopsy samples truly reflects tumor heterogeneity is unclear,37, 38 and in the case of surgical specimens, it may be difficult to compare the gene mutation status of liquid biopsy samples with that of surgical specimens, given that the biological behavior of tumor cells can change moment to moment in response to selective pressures associated with cancer therapies, and the clonal revolution occurred in primary tumors.39 Therefore, by comparing the gene mutation status of liquid biopsy samples and responsiveness to specific cancer therapies, we aim to establish biomarkers that predict responsiveness based on the gene mutation profile obtained from liquid biopsy samples without being influenced by the mutation profile of the primary tumors.
In summary, we optimized the efficiency of a platform for capturing CTC using an LFIMA and revealed the importance of both CTC and ctDNA as diagnostic tools. In addition, our data suggest that both CTC and ctDNA can be used to closely monitor the emergence of molecular changes in patients with metastatic CRC.
DISCLOSURE
The authors have no conflicts of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank all of the participants, physicians, nurses and staff members involved in this study.
Onidani K, Shoji H, Kakizaki T, et al. Monitoring of cancer patients via next‐generation sequencing of patient‐derived circulating tumor cells and tumor DNA. Cancer Sci. 2019;110:2590–2599. 10.1111/cas.14092
Onidani and Shoji contributed equally to this work.
REFERENCES
- 1. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature. 2013;499:214‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jamal‐Hanjani M, Wilson GA, McGranahan N, et al. Tracking the evolution of non‐small‐cell lung cancer. N Engl J Med. 2017;376:2109‐2121. [DOI] [PubMed] [Google Scholar]
- 4. Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501:338‐345. [DOI] [PubMed] [Google Scholar]
- 5. Jacks T, Jaffee E, Singer D. Cancer Moonshot Blue Ribbon Panel Report 2016. (National Cancer Advisory Board)).
- 6. Sundaresan TK, Sequist LV, Heymach JV, et al. Detection of T790M, the acquired resistance EGFR mutation, by tumor biopsy versus noninvasive blood‐based analyses. Clin Cancer Res. 2016;22:1103‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung‐cancer cells. N Engl J Med. 2008;359:366‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu M, Bardia A, Aceto N, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Luca F, Rotunno G, Salvianti F, et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget. 2016;7:26107‐26119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hou HW, Warkiani ME, Khoo BL, et al. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci Rep. 2013;3:1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miura N, Kamita M, Kakuya T, et al. Efficacy of adjuvant chemotherapy for non‐small cell lung cancer assessed by metastatic potential associated with ACTN4. Oncotarget. 2016;7:33165‐33178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu HE, Triboulet M, Zia A, et al. Workflow optimization of whole genome amplification and targeted panel sequencing for CTC mutation detection. NPJ Genom Med. 2017;2:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grobe A, Blessmann M, Hanken H, et al. Prognostic relevance of circulating tumor cells in blood and disseminated tumor cells in bone marrow of patients with squamous cell carcinoma of the oral cavity. Clin Cancer Res. 2014;20:425‐433. [DOI] [PubMed] [Google Scholar]
- 14. Germano G, Mauri G, Siravegna G, et al. Parallel evaluation of circulating tumor DNA and circulating tumor cells in metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17:80‐83. [DOI] [PubMed] [Google Scholar]
- 15. Kato K, Shoji H, Kakizaki F, et al. Next generation sequencing of circulating tumor cells isolated from the peripheral blood of patients with gastrointestinal cancer. Circle‐1 trial. Ann Oncol. 2014;25(suppl_4):iv558. [Google Scholar]
- 16. Macaulay IC, Voet T. Single cell genomics: advances and future perspectives. PLoS Genet. 2014;10:e1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hou Y, Wu K, Shi X, et al. Comparison of variations detection between whole‐genome amplification methods used in single‐cell resequencing. GigaScience. 2015;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Johnson BE, Mazor T, Hong C, et al. Mutational analysis reveals the origin and therapy‐driven evolution of recurrent glioma. Science. 2014;343:189‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX‐4 as first‐line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535‐1546. [DOI] [PubMed] [Google Scholar]
- 22. Van Cutsem E, Peeters M, Siena S, et al. Open‐label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy‐refractory metastatic colorectal cancer. J Clin Oncol. 2007;25:1658‐1664. [DOI] [PubMed] [Google Scholar]
- 23. Heinemann V, von Weikersthal LF, Decker T, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment for patients with metastatic colorectal cancer (FIRE‐3): a randomised, open‐label, phase 3 trial. Lancet Oncol. 2014;15:1065‐1075. [DOI] [PubMed] [Google Scholar]
- 24. Venook AP, Niedzwiecki D, Lenz HJ, et al. Effect of first‐line chemotherapy combined with cetuximab or bevacizumab on overall survival in patients with KRAS wild‐type advanced or metastatic colorectal cancer: a randomized clinical trial. JAMA. 2017;317:2392‐2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Price T, Kim TW, Li J, et al. Final results and outcomes by prior bevacizumab exposure, skin toxicity, and hypomagnesaemia from ASPECCT: randomized phase 3 non‐inferiority study of panitumumab versus cetuximab in chemorefractory wild‐type KRAS exon 2 metastatic colorectal cancer. Eur J Cancer. 2016;68:51‐59. [DOI] [PubMed] [Google Scholar]
- 26. Misale S, Di Nicolantonio F, Sartore‐Bianchi A, Siena S, Bardelli A. Resistance to anti‐EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 2014;4:1269‐1280. [DOI] [PubMed] [Google Scholar]
- 27. De Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy‐refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753‐762. [DOI] [PubMed] [Google Scholar]
- 28. Sanz‐Garcia E, Argiles G, Elez E, Tabernero J. BRAF mutant colorectal cancer: prognosis, treatment, and new perspectives. Ann Oncol. 2017;28:2648‐2657. [DOI] [PubMed] [Google Scholar]
- 29. Therkildsen C, Bergmann TK, Henrichsen‐Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti‐EGFR treatment in metastatic colorectal cancer: a systematic review and meta‐analysis. Acta Oncol. 2014;53:852‐864. [DOI] [PubMed] [Google Scholar]
- 30. Gasch C, Bauernhofer T, Pichler M, et al. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin Chem. 2013;59:252‐260. [DOI] [PubMed] [Google Scholar]
- 31. Heitzer E, Auer M, Gasch C, et al. Complex tumor genomes inferred from single circulating tumor cells by array‐CGH and next‐generation sequencing. Can Res. 2013;73:2965‐2975. [DOI] [PubMed] [Google Scholar]
- 32. Xu JM, Wang Y, Wang YL, et al. PIK3CA mutations contribute to acquired cetuximab resistance in patients with metastatic colorectal cancer. Clin Cancer Res. 2017;23:4602‐4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lupini L, Bassi C, Mlcochova J, et al. Prediction of response to anti‐EGFR antibody‐based therapies by multigene sequencing in colorectal cancer patients. BMC Cancer. 2015;15:808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mehrvarz Sarshekeh A, Advani S, Overman MJ, et al. Association of SMAD4 mutation with patient demographics, tumor characteristics, and clinical outcomes in colorectal cancer. PLoS ONE. 2017;12:e0173345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yagi S, Koh Y, Akamatsu H, et al. Development of an automated size‐based filtration system for isolation of circulating tumor cells in lung cancer patients. PLoS ONE. 2017;12:e0179744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abouleila Y, Onidani K, Ali A, et al. Live single cell mass spectrometry reveals cancer‐specific metabolic profiles of circulating tumor cells. Cancer Sci. 2019;110:697‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Normanno N, Rachiglio AM, Lambiase M, et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann Oncol. 2015;26:1710‐1714. [DOI] [PubMed] [Google Scholar]
- 38. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials