

Abstract
The t(11;14)/CCND1‐IGH, t(4;14)/NSD2(MMSET)‐IGH, and t(14;16)/IGH‐MAF gene rearrangements detected by fluorescence in situ hybridization (FISH) are used for risk stratification in patients with multiple myeloma (MM). Compared with conventional FISH techniques using fresh cells, immunohistochemistry (IHC) is much more cost‐ and time‐efficient, and can be readily applied to routinely prepared formalin‐fixed, paraffin‐embedded (FFPE) materials. In this study, we performed tissue FISH and IHC employing FFPE specimens, and examined the usefulness of IHC as a tool for detecting CCND1,NSD2, and MAF gene rearrangements. CD138 signals were used to identify plasma cells in tissue FISH and IHC analyses. With cohort 1 (n = 70), we performed tissue FISH and subsequently IHC, and determined IHC cut‐off points. In this cohort, the sensitivity and specificity for the 3 molecules were ≥.90 and ≥.96, respectively. With cohort 2, using MM cases with an unknown gene status (n = 120), we performed IHC, and the gene status was estimated using the cut‐off points determined with cohort 1. The subsequent FISH analysis showed that the sensitivity and specificity for the 3 molecules were ≥.92 and ≥.98, respectively. CCND1, NSD2, and MAF gene rearrangements were estimated accurately by IHC, suggesting that conventional FISH assays can be replaced by IHC.
Keywords: FFPE tissue sections, gene rearrangements, immunohistochemistry, multiple myeloma, tissue fluorescence in situ hybridization
1. INTRODUCTION
Multiple myeloma (MM) is an incurable plasma cell neoplasm, which develops through a long‐term series of genetic events.1, 2 Biological and clinical features of MM are associated with genetic aberrations such as gene rearrangements involving the immunoglobulin heavy chain gene (IGH) locus as well as chromosomal deletions, somatic gene mutations, and chromosomal hyperdiploidy involving odd number chromosomes.3 These chromosomal abnormalities provide important information for the treatment and management of MM patients, and have been included in the Revised International Staging System published in 2015.4 t(11;14)(q13;q32) involving the CCND1 locus is found in 15%‐25% of newly diagnosed MM cases, and is associated with a lymphoplasmacytic morphology, frequent CD20 expression, an indolent clinical course, and a relatively favorable outcome when no additional cytogenetic abnormalities are present.5 In contrast, t(4;14)(p16.3;q32) involving the NSD2/MMSET/FGFR3 locus is found in 10%‐15% of MM cases, and is associated with a frequent deletion of chromosome 13q, a common IgA subtype, and a relatively unfavorable outcome even in patients receiving high‐dose therapy with autologous stem cell transplantation.6, 7 The overall prognosis of MM patients harboring t(4;14) may improve after the introduction of proteasome inhibitors (PIs) such as bortezomib. However, the overall survival rate is <50% at 5 y even with bortezomib treatment.8 Another important chromosomal aberration observed in c. 5% of MM cases is t(14;16)(q32;q23) involving the MAF locus.1, 9, 10, 11 Many studies have suggested that MM cases carrying t(14;16) are associated with hypercalcemia and an unfavorable outcome as well as a lower frequency of extramedullary tumor formation even in the era of clinical innovations such as the use of PIs and immunomodulatory drugs (IMiDs).8, 12
Due to difficulties in obtaining mitotic figures, cytogenetic analysis is not an optimal method for detecting gene rearrangements. Detection is usually carried out using a fluorescent in situ hybridization (FISH) technique performed using fresh tumor samples fixed in Carnoy's solution soon after obtaining the tissue. Although this procedure usually provides excellent results, there are some disadvantages in a clinical setting. The FISH assay requires (a) fresh tumor cells, (b) a rapid sample fixation in Carnoy's solution, and (c) a fluorescence microscope equipped to detect gene rearrangements.
Formalin‐fixed, paraffin‐embedded (FFPE) tissue specimens are routinely prepared from bone marrow aspirates and biopsy samples in an ordinary clinical setting. The advantages of being able to use routinely prepared FFPE samples far outweigh any drawbacks. With recent technical advances, FISH assays can be carried out using FFPE materials. Nevertheless, compared with FISH, immunohistochemistry (IHC) is much more cost‐ and time‐efficient. There have been some reports describing the usefulness of IHC for the estimation of gene rearrangements.10, 13 However, FISH and IHC assays were performed separately using different tumor samples (eg, fresh tumor cells and FFPE sections), and concordance between FISH and IHC has not been well verified in a large‐scale tumor cohort. The aim of this study was to reach a comprehensive clarification of whether CCND1, NSD2, and MAF gene rearrangements can be detected successfully employing IHC for CCND1, NSD2, and MAF using FISH‐confirmed FFPE tumor samples.
2. MATERIALS AND METHODS
2.1. Study cohorts
We retrieved archival MM and non‐neoplastic cases with bone marrow specimens from the files of the Department of Pathology and Molecular Diagnostics and Department of Hematology and Oncology, Nagoya City University Graduate School of Medical Sciences. This study was approved by the Nagoya City University Internal Review Board. The cases were pathologically evaluated and the diagnosis was confirmed by expert hematopathologists (AM and HI) according to the criteria of the WHO classification of MM.14 All cases were within the morphologic boundaries of MM, were positive for MM‐associated antigens, and were restricted to either kappa or lambda light chain. All specimens used in this study were obtained by bone marrow aspiration, fixed in formalin, and embedded in paraffin. In the preliminary analysis, DNA extracted from FFPE sections of MM cases was subjected to amplification of a fragment of the PLZF gene (300 bp) by the polymerase chain reaction to confirm DNA quality.15 Cases negative for the amplification were excluded. The specimens were then separated into cohorts 1 and 2.
2.1.1. Cohort 1
Using data of the previously performed karyotyping, FISH for fresh tumor cells, and/or quantification for the CCND1, NSD2, and MAF transcripts, we retrieved 72 MM cases; 52 cases were highly suspected of having the gene rearrangements and the remaining 20 cases were thought to be silent for these genes. These 72 cases were subjected to tissue FISH analysis for CCND1, NSD2, and MAF genes to identify tissue FISH‐confirmed positive and negative controls. Two cases were excluded because their FISH signals were highly complicated. Consequently, cohort 1 consisted of 70 MM cases (20 cases positive for CCND1‐FISH, 20 cases positive for NSD2‐FISH, 10 cases positive for MAF‐FISH, and 20 MM cases negative for these 3 gene rearrangements). We then performed IHC for CCND1, NSD2, and MAF to develop IHC cut‐off points to determine the presence or the absence of respective rearrangements.
2.1.2. Cohort 2
Cohort 2 consisted of 120 MM cases in which the gene rearrangement status was unknown. IHC and subsequent tissue FISH for CCND1, NSD2, and MAF molecules were performed using FFPE sections. All cases were successfully evaluated. This cohort was used to validate the usefulness of IHC for detecting MM‐associated gene rearrangements involving CCND1, NSD2, and MAF loci.
2.2. Tissue FISH analysis using paraffin sections
When a sufficient number of neoplastic plasma cells (MM cells > 30%), as determined with serial H&E and IHC sections, were present in the bone marrow FFPE tissues, standard tissue FISH analysis for gene splits was performed as we previously described.16 In brief, bone marrow sections were deparaffinized, heat‐treated, and digested in pepsin buffer. After denaturation, sections were incubated with the respective FISH probes overnight in a humidified chamber. After post‐hybridization washing, sections were stained with diaminophenilindole and mounted. The FISH probes we used were as follows: t(11q13) Break (Kreatech, Leica Biosystems) for the CCND1 gene, RP11‐709N10 (labeled in green) and RP11‐585J22 (red) for the NSD2 gene, and RP11‐755G18 (green) and RP11‐976M22 (red) for the MAF gene. The frequencies of gene abnormalities were determined by counting 100 non‐overlapping tumor cells. Cut‐off points for FISH analysis were determined by counting 100 non‐overlapping cell nuclei of 13 non‐neoplastic marrow tissues. The samples were considered to be positive for rearrangements in CCND1, NSD2, and MAF genes when more than 10% (mean±3SD, rounded up) of examined nuclei showed split signals.
2.3. Sequential FICTION‐whole‐slide imaging and data processing
When a small number of plasma cells (MM cells < 30%) were present in FFPE specimens, it was necessary to identify MM cell nuclei for accurate FISH evaluation. For this purpose, we used a sequential FICTION‐whole‐slide imaging (WSI) technique (Figure S1), which we recently developed and have described elsewhere.17 This technique enables us to retrieve WSI data of H&E, immunofluorescence (IF), and FISH, using a single FFPE tissue section. Briefly, after deparaffinization, an MM tissue section was stained with H&E. WSI data of the H&E section were obtained using an automated fluorescence image analyzer (IN Cell Analyzer 6000, GE Healthcare), which was equipped with a ×60 objective lens. The H&E‐stained section was then subjected to IF for CD138; the section was heat‐treated in 10 mmol/L citric acid buffer (pH 6.0) to inactivate the H&E dyes and for antigen retrieval. Next, the section was reacted with anti‐CD138 antibody, incubated with Cy3‐labeled second antibody, and finally stained with diaminophenilindole. The IF‐WSI data of the MM section were obtained using the automated fluorescence image analyzer. The IF‐stained section was then subjected to FISH analysis. The tissue section was heat treated and protease digested, and FISH was performed as described above. FISH‐WSI data were obtained again using the automated image analyzer.
Small image tiles obtained with the automated image analyzer were stitched together into a large contiguous montage using image analysis software on a high‐performance computer. The WSI data of H&E, IF for CD138, and FISH for gene splits were analyzed for the presence or absence of gene alterations in CD138‐positive cells using the image editing software, e‐CellAna (e‐Path, Fujisawa, Japan). When 2 FISH signals (a green and a red) were separated in nuclei from a CD138‐positive cell, we considered that the cell possessed had undergone a gene rearrangement. Representative data of FICTION‐WSI are shown in Figure 1.
Figure 1.
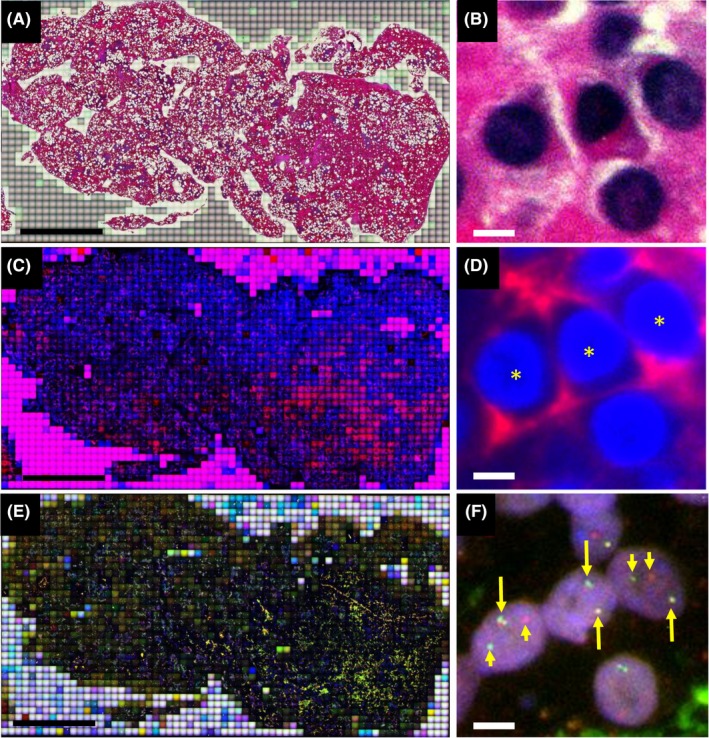
A multiple myeloma case positive for CCND1‐IGH gene rearrangement. Whole‐slide imaging (WSI) data of H&E staining (A and B), immunofluorescence (IF) staining for CD138 (C and D), and fluorescence in situ hybridization (FISH) for the CCND1 gene split (E and F) were sequentially retrieved using a single paraffin section of the tumor. More than 2500 image tiles were stitched together to create a large image (A, C, and E). Three hematopoietic cells, which have mildly eccentric nuclei, are positive for CD138 (indicated by asterisks in D). In these 3 CD138‐positive cells, split FISH signals are present in those on the right and left, but not in the one in the center. Split signals are indicated by short arrows and unsplit signals by long arrows. Length of the scale bars is 3 mm in (A), (C), (E) and 3 μm in (B), (D), (F)
2.4. Double‐immunohistochemistry for CD138 and either CCND1, NSD2, or MAF
To precisely score the IHC expression of CCND1, NSD2, and MAF in tumor cells, all MM cases were analyzed with a double‐IHC technique (Figure 2). Nuclear signals of CCND1, NSD2, and MAF were visualized first with brown chromogen using a Bond‐Max autostainer (Leica Microsystems) and a Bond Polymer Refine Detection kit (Leica Biosystems). Subsequently, using the same tissue sections, CD138 signals were detected with red chromogen using a Bond Polymer AP Red Detection kit (Leica Biosystems). Antibodies used were as follows, anti‐CCND1 (clone SP4, Thermo Fisher Scientific), anti‐NSD2 (clone 29D1, Abcam), anti‐MAF (clone EPR16484, Abcam), and anti‐CD138 (clone B‐A38, Cell Marque). The percentage of positive cells on IHC was determined by calculating the proportion of positively stained nuclear cells divided by CD138‐positive cells (at least 100 cells were examined).
Figure 2.
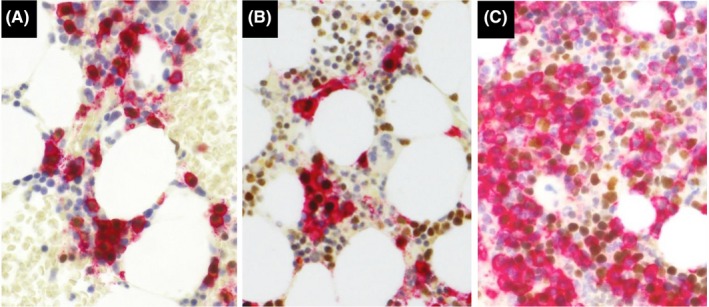
Double immunohistochemistry for CD138 and either CCND1, NSD2, or MAF. CD138 expression is labeled in red and nuclear signals of CCND1 (A), NSD2 (B), and MAF (C) are labeled in brown. Note that nuclear signals are present in CD138‐positive as well as CD138‐negative cells
2.5. Statistical analysis
Statistical evaluation of data from 2 groups was carried out using Fisher's exact test. Cut‐off points for IHC were determined with receiver operating characteristics (ROC) curves. A value of P < .05 in each test was regarded as statistically significant. All analyses were two‐tailed and carried out using statistical packages JMP version 14.2.0 (SAS Institute) and DANS version 10.7 (Sugimoto Data Analysis Service).
3. RESULTS
3.1. Cohort 1
In the cohort 1 included were 70 MM cases harboring CCND1 (n = 20), NSD2 (n = 20), MAF (n = 10), or no (n = 20) gene rearrangements, as detected by tissue FISH using either a conventional FISH or a FICTION‐WSI technique. Using these MM cases as tissue FISH‐confirmed controls, we performed double‐IHC for CD138 and either CCND1, NSD2, or MAF. After calculating percentages of positive cells, respective IHC cut‐off points were determined using ROC curves to obtain the highest sensitivity and specificity for estimating the presence of respective gene rearrangements (Table 1). The distribution of IHC‐positive cases is shown in Table S1.
Table 1.
Cohort 1. Receiver operating characteristics analysis for cut‐off points for immunohistochemistry
| Molecule | Cut‐off (%) | AUC (95% CI) | P |
|---|---|---|---|
| CCND1 | 5 | 1.00 (1.00‐1.00) | 8.43E‐13 |
| NSD2 | 10 | .97 (.93‐1.00) | 2.81E‐10 |
| MAF | 10 | .93 (.81‐1.00) | 5.58E‐07 |
For CCND1‐IHC, the cut‐off point was set to 5% by ROC analysis. The area under the curve (AUC) was 1.00 (Table 1), and the sensitivity, specificity, and accuracy for detecting CCND1 gene rearrangement using CCND1‐IHC were calculated to be 1.00, 1.00, and 1.00 (Table 2). For NSD2‐IHC, the cut‐off point was similarly set to 10% and the AUC was .97 (Table 1). The sensitivity, specificity, and accuracy for detecting NSD2 gene rearrangement using NSD2‐IHC were calculated to be .95, .96, and .96 (Table 2). For MAF‐IHC, the cut‐off point was set to 10% by ROC analysis and the AUC was .933 (Table 1). The sensitivity, specificity, and accuracy for detecting MAF gene rearrangement using MAF‐IHC were calculated to be .90, .98, and .97 (Table 2). The results of cohort 1 are summarized in Figure S2.
Table 2.
Cohort 1. Immunohistochemistry for tissue FISH‐confirmed positive and negative controls
| Molecule | Sensitivity (95% CI) | Specificity (95% CI) | Accuracy (95% CI) |
|---|---|---|---|
| CCND1 | 1.00 (.83‐1.00) | 1.00 (.93‐1.00) | 1.00 (.95‐1.00) |
| NSD2 | .95 (.75‐1.00) | .96 (.86‐1.00) | .96 (.88‐.99) |
| MAF | .90 (.56‐1.00) | .98 (.91‐1.00) | .97 (.90‐1.00) |
FISH, fluorescence in situ hybridization.
3.2. Cohort 2
Cohort 2 included 120 MM cases in which the gene status was unknown, and all cases were subjected to double‐IHC for CD138 and either of CCND1, NSD2, or MAF. When the IHC cut‐off points determined in Cohort 1 were employed, CCND1, NSD2, and MAF were positive in 28, 13, and 4 cases, respectively, and the remaining 75 MM cases were negative for all 3 molecules. All MM cases (n = 120) were then subjected to tissue FISH analysis using FFPE tumor sections. In 28 CCND1‐IHC‐positive cases, 26 (93%) cases were positive for CCND1 on FISH, and the remaining 2 cases were negative for all 3 gene rearrangements. All CCND1‐IHC‐negative cases (n = 92) were negative for CCND1 on FISH (P < .0001) with an IHC sensitivity, specificity, and accuracy that was over 98%. Out of the NSD2‐IHC‐positive cases (n = 13), 12 were positive and one was negative on FISH, and out of the NSD2‐IHC‐negative cases (n = 107), 106 were negative and one was positive on FISH (P < .0001), with an IHC sensitivity, specificity, and accuracy of over 92%. Similarly, all MAF‐IHC‐positive cases (n = 4) were positive for MAF on FISH analysis, and all MAF‐IHC‐negative cases (n = 116) were negative for MAF on FISH (P < .0001), and all had an IHC sensitivity, specificity, and accuracy of 100%. The results of Cohort 2 are summarized in Tables 3, S1, and Figure S3. When MM cases included in cohorts 1 and 2 were combined (190 cases in total), an IHC sensitivity, specificity, and accuracy for CCND1, NSD2, and MAF were calculated to be 100%, 99%, and 99%; 94%, 98%, and 97%; and 93%, 99%, and 99%, respectively (Tables S1 and S2).
Table 3.
Cohort 2, myeloma cases (n = 120)
| Molecule | Sensitivity (95% CI) | Specificity (95% CI) | Accuracy (95% CI) |
|---|---|---|---|
| CCND1 | 1.00 (.87‐1.00) | .98 (.93‐1.00) | .98 (.94‐1.00) |
| NSD2 | .92 (.64‐1.00) | .99 (.95‐1.00) | .98 (.94‐1.00) |
| MAF | 1.00 (.40‐1.00) | 1.00 (.97‐1.00) | 1.00 (.97‐1.00) |
3.3. Cases showing a discordance between FISH and IHC
In cohorts 1 and 2, there were 9 cases in which there were differences between FISH and IHC results (Table 4). One case positive for CCND1 on FISH was positive for CCND1 and NSD2 on IHC. Three cases positive on FISH for NSD2 were positive for NSD2 and MAF (n = 1) and negative for the 3 molecules on IHC (n = 2). Two cases positive for MAF using FISH were positive for NSD2 and MAF (n = 1) and negative for the 3 molecules using IHC (n = 1). Three cases negative for gene rearrangements on FISH were positive for CCND1 (n = 2) and NSD2 (n = 1) using IHC.
Table 4.
Discordant cases between FISH and immunohistochemistry
| Case | Age/Sex | FISH | Immunohistochemistry | ||||
|---|---|---|---|---|---|---|---|
| CCND1 | NSD2 | MAF | CCND1a | NSD2b | MAFb | ||
| 1 | 49/M | Pos | Neg | Neg | (+), 90%c | (+), 40% | (−), <1% |
| 2 | 62/F | Neg | Pos | Neg | (−), 4% | (+), 95% | (+), 30% |
| 3 | 59/F | Neg | Pos | Neg | (−), <1% | (−), 2% | (−), 1% |
| 4 | 83/M | Neg | Pos | Neg | (−), 1% | (−), 5% | (−), <1% |
| 5 | 72/F | Neg | Neg | Pos | (−), <1% | (+), 50% | (+), 95% |
| 6 | 71/F | Neg | Neg | Pos | (−), <1% | (−), 2% | (−), <1% |
| 7 | 70/F | Neg | Neg | Neg | (+), 80% | (−), 1% | (−), <1% |
| 8 | 72/F | Neg | Neg | Neg | (+), 20% | (−), 4% | (−), <1% |
| 9 | 84/F | Neg | Neg | Neg | (−), <1% | (+), 60% | (−), <1% |
neg, negative; pos, positive.
Cut‐off > 5%.
Cut‐off > 10%.
Positive tumor cells.
4. DISCUSSION
In the cohort 1 analysis, we performed IHC for CCND1, NSD2, and MAF in controls and cases proven to be positive and negative by means of FISH. CCND1, NSD2, and MAF overexpression were highly associated with the respective gene rearrangements. Using ROC analysis, we determined the cut‐off points for each IHC, which allowed for estimation of these gene rearrangements with sensitivities ≥ 90% and specificities ≥ 96%. Subsequently, using cohort 2 (120 MM cases in which the gene status was unknown), we performed IHC for CCND1, NSD2, and MAF, and tissue FISH for CCND1, NSD2, and MAF genes. The IHC sensitivity and specificity for the 3 gene rearrangements were calculated to be ≥92% and ≥98%, respectively. Our findings suggested that CCND1, NSD2, and MAF gene rearrangements can be successfully detected using the IHC assay. It should be noted that double‐IHC for CD138 and either of CCND1, NSD2, or MAF should be performed for accurate evaluation of IHC signals in MM cells as these molecules are frequently found in normal hematopoietic cells other than plasma cells and non‐hematopoietic cells localized in the marrow tissue. For example, CCND1 expression is present in endothelial cells18 and in some proliferating cells.19 NSD2 and MAF expression is frequently detected in normal macrophages and T cells, respectively.20, 21 Without a double‐IHC, an accurate evaluation of NSD2‐IHC and MAF‐IHC signals appears to be very difficult, especially when the number of MM cells is small.
In cohorts 1 and 2, there were 9 cases that showed discordance between FISH and IHC findings (Table 4). These cases were divided into 3 groups: group 1) positive results on IHC involving molecules other than those expected by FISH (cases 1, 2, and 5); group 2) negative results on IHC involving molecules expected by FISH (cases 3, 4, and 6); and group 3) aberrant expression on IHC of molecules not expected by FISH (cases 7‐9). The significance of these discordant cases may differ depending on how the current findings are used in a clinical setting. When IHC results are to be used as a screening test before subsequent FISH assays, an increase in sensitivity in needed in group 2, for example, by lowering the cut‐off points with a risk of lowing specificity or by employing more than one antibody for IHC. However, it has not been firmly established which is more important for stratification of MM patients, gene rearrangements, or protein overexpression. The clinical significance of protein overexpression should be a subject of future investigations.
Compared with the conventional FISH assays, the detection of abnormal expression by IHC takes much less time, thereby leading to a prompt determination of the best treatment regimen in newly diagnosed MM cases. For example, when NSD2 is positive on IHC as a result of the t(4;14) karyotype abnormality, a regimen that includes PIs rather than IMiDs would be preferred as the initial therapy.6 Patients positive for MAF on IHC and who carry the t(14;16) abnormality are considered to be a high‐risk subpopulation of MM, therefore, they should be treated with a regimen with high‐dose intensity, such as a triple regimen that includes both PI and IMiDs.22 In patients found positive on IHC for CCND1 due to the t(11;14) rearrangement, a combination of Bcl2 inhibitor (venetoclax) with PI may be preferred according to recent reports.23, 24
Compared with using fresh tissue, FFPE tissue specimens have many advantages: they can be routinely prepared, are easily stored, and are superior in morphology. However, there are some issues to be considered when FFPE samples are applied to FISH assays. First, FISH assays using FFPE sections are technically more complicated than those using fresh cells. Second, in some aspirates of cohort 1 (approximately 3%), the quality of the DNA was not sufficiently high. DNA degradation is more prominent in bone marrow biopsy specimens, in which bony tissue decalcification is required. This may be partly resolved by using an EDTA solution, which is a less powerful decalcifier but superior in nucleic acid preservation compared the conventional formic acid.25 Third, the thickness of FFPE sections (4 μm in our study) is usually less than half of the tumor nuclei, resulting in a loss of some FISH signals localized in the nuclei (Figure S4). To partly overcome this issue, we used break‐apart type FISH probes. The number of FISH signals with a break‐apart probe assay is less than that required with a dual‐color fusion probe assay, leading to a more accurate FISH signal evaluation.
Another issue in using FFPE sections for FISH is that it is usually difficult to perform using purified plasma cells or to carry out FISH combined with IF to detect cytoplasmic light chains. These modified FISH techniques are strongly recommended in the Revised International Staging System.26 To improve the rate of abnormality detection, we focused on CD138‐positive cells and evaluated their FISH signals, employing a novel technique that we recently described and designated “sequential FICTION‐WSI.” Using a single FFPE section, we sequentially performed H&E staining, CD138‐IF, and FISH, and each set of WSI data were superimposed. This novel imaging technique enabled us to analyze gene abnormalities much more precisely than by conventional analysis in which H&E, IF, and FISH are separately analyzed using serial sections. The previously described FICTION technique27 is useful for allowing simultaneous visualization of FISH and IF signals on a single FFPE section. However, one of the critical limitations of the FICTION method is that pre‐treatment should be the same for IF and FISH. However with FISH, intense protein digestion of FFPE sections is usually required, and this intense pre‐treatment significantly abolishes the protein antigenicity required for IF staining. We overcame this limitation by performing each stain independently and by sequentially retrieving H&E, IF, and FISH signals of the whole tumor tissue.
In conclusion, CCND1, NSD2, and MAF gene rearrangements can be estimated by IHC with excellent sensitivity and specificity. Compared with the current FISH technique performed in a clinical setting, IHC is much more cost and time efficient, and can be readily applied to routinely prepared FFPE materials. Using the IHC technique, we are now trying to detect other gene translocations, such as MAFB, CCND2, CCND3, and MYC, as well as focal chromosomal abnormalities, such as CKS1B for a 1q gain/amplification.1, 3, 28, 29 In some present MM cases, however, there was some discordance in the results obtained with FISH and those with IHC. More work is warranted to clarify which results are more valuable in the management of MM patients, gene rearrangements detected by FISH or protein overexpression detected by IHC.
DISCLOSURE
The authors declare that they have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors thank Mr. Takeo Sakakibara and Mr. Yuma Sakamoto for their excellent technical assistance. This study was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (15K08351 to T. Murase and H. Inagaki, 16K09855 to M. Ri, and 16K07179 to S. Iida). This work was also partly supported by a grant from the National Cancer Center Research and Development Fund (26‐A‐4), and from the Practical Research for Innovative Cancer Control from Japan Agency for Medical Research and Development, AMED (15ck0106077h0002). Takayuki Murase, Masaki Ri, Shinsuke Iida, and Hiroshi Inagaki designed the research. Takayuki Murase, Masaki Ri, Tomoko Narita, Keiichiro Fujii, Ayako Masaki, Shinsuke Iida, Hiroshi Inagaki performed the experiments. Takayuki Murase, Masaki Ri, Tomoko Narita, Shinsuke Iida, and Hiroshi Inagaki analyzed and interpreted data. All authors wrote and approved the manuscript.
Murase T, Ri M, Narita T, et al. Immunohistochemistry for identification of CCND1, NSD2, and MAF gene rearrangements in plasma cell myeloma. Cancer Sci. 2019;110:2600‐2606. 10.1111/cas.14109
Murase and Ri contributed equally to this work.
REFERENCES
- 1. Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest. 2012;122(10):3456‐3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335‐348. [DOI] [PubMed] [Google Scholar]
- 3. Fonseca R, Bergsagel PL, Drach J, et al. Molecular classification of multiple myeloma: spotlight review. IMWG molecular classification of MM; 2009. [DOI] [PMC free article] [PubMed]
- 4. Palumbo A, Avet‐Loiseau H, Oliva S, et al. Revised International Staging System for multiple myeloma: a report from international Myeloma working group. J Clin Oncol. 2015;33(26):2863‐2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leiba M, Duek A, Amariglio N, et al. Translocation t(11;14) in newly diagnosed patients with multiple myeloma: is it always favorable? Genes Chromosom Cancer. 2016;55(9):710‐718. [DOI] [PubMed] [Google Scholar]
- 6. Avet‐Loiseau H, Leleu X, Roussel M, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol. 2010;28(30):4630‐4634. [DOI] [PubMed] [Google Scholar]
- 7. Gertz MA, Lacy MQ, Dispenzieri A, et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and ‐17p13 in myeloma patients treated with high‐dose therapy. Blood. 2005;106(8):2837‐2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chesi M, Bergsagel PL. Molecular pathogenesis of multiple myeloma: basic and clinical updates. Int J Hematol. 2013;97(3):313‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avet‐Loiseau H, Malard F, Campion L, et al. Translocation t(14;16) and multiple myeloma: is it really an independent prognostic factor? Blood. 2011;117(6):2009‐2011. [DOI] [PubMed] [Google Scholar]
- 10. Chang H, Qi Q, Xu W, Patterson B. c‐Maf nuclear oncoprotein is frequently expressed in multiple myeloma. Leukemia. 2007;21(7):1572‐1574. [DOI] [PubMed] [Google Scholar]
- 11. Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569‐4575. [DOI] [PubMed] [Google Scholar]
- 12. Narita T, Inagaki A, Kobayashi T, et al. t(14;16)‐positive multiple myeloma shows negativity for CD56 expression and unfavorable outcome even in the era of novel drugs. Blood Cancer J. 2015;5:e285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang H, Stewart AK, Qi XY, Li ZH, Yi QL, Trudel S. Immunohistochemistry accurately predicts FGFR3 aberrant expression and t(4;14) in multiple myeloma. Blood. 2005;106(1):353‐355. [DOI] [PubMed] [Google Scholar]
- 14. McKenna RW, Kyle RA, Kuehl WM, Harris HL, Coupland RW, Fned F. Plasma cell neoplasms In: Swerdlow SH, Campo E, Harris NL, eds. World Health Organization & International Agency for Research on Cancer WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2017:241‐258. [Google Scholar]
- 15. van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T‐cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED‐2 Concerted Action BMH4‐CT98‐3936. Leukemia. 2003;17(12):2257‐2317. [DOI] [PubMed] [Google Scholar]
- 16. Xia H, Nakayama T, Sakuma H, et al. Analysis of API2‐MALT1 fusion, trisomies, and immunoglobulin VH genes in pulmonary mucosa‐associated lymphoid tissue lymphoma. Hum Pathol. 2011;42(9):1297‐1304. [DOI] [PubMed] [Google Scholar]
- 17. Fujii K, Ishibashi KI, Kato J, et al. Cellular‐level characterization of B cells infiltrating pulmonary MALT lymphoma tissues. Virchows Arch. 2016;469(5):575‐580. [DOI] [PubMed] [Google Scholar]
- 18. Abdulla Z, Turley H, Gatter K, Pezzella F. Immunohistological recognition of cyclin D1 expression by non‐lymphoid cells among lymphoid neoplastic cells. APMIS. 2014;122(3):183‐191. [DOI] [PubMed] [Google Scholar]
- 19. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: cyclin D1: normal and abnormal functions. Endocrinology. 2004;145(12):5439‐5447. [DOI] [PubMed] [Google Scholar]
- 20. Rani A, Afzali B, Kelly A, et al. IL‐2 regulates expression of C‐MAF in human CD4 T cells. J Immunol. 2011;187(7):3721‐3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang P, Guo L, Duan ZJ, et al. Histone methyltransferase NSD2/MMSET mediates constitutive NF‐κB signaling for cancer cell proliferation, survival, and tumor growth via a feed‐forward loop. Mol Cell Biol. 2012;32(15):3121‐3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mikhael JR, Dingli D, Roy V, et al. Management of newly diagnosed symptomatic multiple myeloma: updated Mayo Stratification of Myeloma and Risk‐Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc. 2013;88(4):360‐376. [DOI] [PubMed] [Google Scholar]
- 23. Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar S, Vij R, Kaufman JL, et al. Venetoclax monotherapy for relapsed/refractory multiple myeloma: safety and efficacy results from a phase I study. Blood. 2016;128:488.27235136 [Google Scholar]
- 25. Neat MJ, Moonim MT, Dunn RG, Geoghegan H, Foot NJ. Fluorescence in situ hybridisation analysis of bone marrow trephine biopsy specimens; an additional tool in the diagnostic armoury. J Clin Pathol. 2013;66(1):54‐57. [DOI] [PubMed] [Google Scholar]
- 26. Kastritis E, Terpos E, Roussou M, et al. Evaluation of the Revised International Staging System in an independent cohort of unselected patients with multiple myeloma. Haematologica. 2017;102(3):593‐599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Korac P, Jones M, Dominis M, et al. Application of the FICTION technique for the simultaneous detection of immunophenotype and chromosomal abnormalities in routinely fixed, paraffin wax embedded bone marrow trephines. J Clin Pathol. 2005;58(12):1336‐1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Avet‐Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109(8):3489‐3495. [DOI] [PubMed] [Google Scholar]
- 29. Chang H, Qi X, Trieu Y, et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression‐free survival post‐autologous stem cell transplantation. Br J Haematol. 2006;135(4):486‐491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials