

Abstract
Recent immunotherapy advances have convincingly demonstrated complete tumour removal with long-term survival. These impressive clinical responses have rekindled enthusiasm towards immunotherapy and tumour antigen vaccination providing ‘cures’ for melanoma and other cancers. However, many patients still do not benefit; sometimes harmed by severe autoimmune toxicity. Checkpoint inhibitors (anti-CTLA4; anti-PD-1) and interleukin-2 (IL-2) are ‘pure immune drivers’ of pre-existing immune responses and can induce either desirable effector-stimulatory or undesirable inhibitory-regulatory responses. Why some patients respond well, while others do not, is presently unknown, but might be related to the cellular populations being ‘driven’ at the time of dosing, dictating the resulting immune response. Vaccination is in-vivo immunotherapy requiring an active host response. Vaccination for cancer treatment has been skeptically viewed, arising partially from difficulty demonstrating clear, consistent clinical responses. However, this article puts forward accumulating evidence that ‘vaccination’ immunomodulation constitutes the fundamental, central, intrinsic property associated with antigen exposure not only from exogenous antigen (allogeneic or autologous) administration, but also from endogenous release of tumour antigen (autologous) from in-vivo tumour-cell damage and lysis. Many ‘standard’ cancer therapies (chemotherapy, radiotherapy etc.) create waves of tumour-cell damage, lysis and antigen release, thus constituting ‘in-vivo vaccination’ events. In essence, whenever tumour cells are killed, antigen release can provide in-vivo repeated vaccination events. Effective anti-tumour immune responses require antigen release/supply; immune recognition, and immune responsiveness. With better appreciation of endogenous vaccination and immunomodulation, more refined approaches can be engineered with prospect of higher success rates from cancer therapy, including complete responses and better survival rates.
Keywords: cancer, immunotherapy, in vivo vaccination, melanoma, survival, vaccine
Introduction
The consideration of vaccines being applied therapeutically to treat cancer dates back over 100 years or more and constitutes the first form of immunotherapy.1–26 Coley’s toxins could produce complete responses, and induced fever from a nonspecific innate immune response which most likely augmented an existing adaptive immune response to the tumour. The mechanism remains unclear but may have provided ‘danger signals’ through costimulatory pathways to remove regulatory adaptive T-cell suppression. Immune stimulation for cancer therapy has a long and chequered history predating knowledge of an ‘immune system’ and possibly alchemy as a primitive medical science, since many early herbal remedies often had assumed ‘tonic’ elements of inflammatory stimulation or suppression by which they modulated diseases, which is only now being more scientifically realized. This ‘folklore’ history has not helped the modern understanding or acceptance of the immune system’s involvement in cancer therapy. Indeed, as recently as less than a decade ago, many medical oncologists and cancer therapists (now unbelievably) did not seriously consider that the immune system was involved ‘at all’ in cancer or cancer treatment, and that vaccines or vaccination had no role. Historically, vaccination was ascribed a ‘passive’, protective role against diseases, usually infective in origin. However, vaccination is almost always a truly ‘active’ process where the immune system is specifically responding to antigenic signals that stimulate B-cell (plasma) or T-cell responsiveness, and often both. As a consequence, either antibody is produced, or T cells are activated, for example, by production of cytokines or cytotoxicity. The nonspecific or innate immune system is not exempt from this process either, occupying a pivotal role in immune stimulation during vaccination events.
When the term ‘immunotherapy’ is considered, it often has the connotation of a more active exogenous form of treatment where the immune system is overtly stimulated and an effect is observable, compared with ‘vaccination’. However, the two are almost indecipherable in real, practical terms, because the immune system is activated in both, and both influence a wide range of diseases or disorders, not exclusively of infective origin.
Only now is vaccination technology and consideration of ‘vaccines’ for clinical treatment of cancer being seriously reconsidered in light of the recent immunotherapy successes and understanding. Indeed, vaccines may be essential for improving checkpoint-immunotherapy clinical response rates. This article revives understanding about vaccines and vaccination for cancer therapy in this new light, placing it within our contemporary understanding of recent immunotherapies and how they appear to be working for the benefit of the cancer patient.
Melanoma, for which most information exists, has been used as a model for immunotherapies, including therapeutic vaccines, and gradually, many principles of melanoma therapy are being demonstrated for other cancer types. In this review, the recent advances in melanoma, where immunotherapies including cancer vaccine therapies, have been explored to demonstrate technical approaches that have advanced cancer immunotherapy more generally. This presents the case for in vivo vaccination as the basis for most, if not all, cancer (immuno) therapies. Recent knowledge suggests that many of these approaches can be extrapolated and applied to other cancers, including lung, bladder, renal, Merkel cell, colon, haematological, head and neck squamous cell carcinoma (SCC), gastric and breast cancers, revealing common underlying immunotherapy mechanisms between different cancer types, and therefore suggesting that in vivo vaccination is a generalized phenomenon which needs unlocking for great benefit.
Cancer vaccine therapy
Cancer vaccines of various types have been devised and utilized for over 100 years with variable success, but notably with numerous reports of complete clinical responses (CR) with regression of all measurable disease in patients with advanced cancers of different types through immunomodulation.1–38 Such CRs are highly significant events because they often underpin long-term durable survival periods of decades or more, effectively amounting to clinical cancer ‘cure’.23–26 The problem has been the generally low and unpredictable rate of CR induction, or even partial response (PR) or stable disease (SD) outcomes. However, the very fact that these types of clinical useful responses occur is of great importance because they demonstrate convincing evidence that cancer vaccines offer an effective therapeutic approach which requires more intensive investigation and development.23,25 The lack of consistency of results across many cancer vaccine platforms has unfortunately biased the view of many influential commentators against the ‘vaccine’ approach, perhaps understandably. Moreover, largely because of the lack of understanding of the mechanisms of action of cancer vaccines, and the paucity of information on the immune-system functions in general over the years, the pharmaceutical world has tended to favour simpler commercial-scale molecular synthetic options for production and marketing with the security of more financial return, such as cytotoxic chemotherapies and more recently, monoclonal antibodies. However, it should be recognized that monoclonal antibodies were recently considered relatively commercially unfavourable, too, until these mechanisms of action and production were better understood and when enough clinical trials demonstrated efficacy.
Many of the vaccine studies have been performed in the adjuvant setting following surgical removal, for example, higher-risk resected stage II and III patients rendered no evidence of disease.33–38 That assumption undoubtedly arose from the tenet that ‘vaccination’ would be more effective for treating residual low-volume microscopic metastases by ‘preventing’ tumour recurrence, in a similar manner to the way vaccines have been utilized in preventing infection. Interestingly, the highest clinical complete response rates from vaccination have been obtained in surgically unresectable stage IV melanoma patients, indicating that the strength of the active antigenic signal might be of central importance.23,25
Clinical efficacy has been another issue with immunotherapies, including therapeutic vaccines. The ‘clinical effectiveness’ of a treatment is where a clinical response occurs to prolong survival. That is, induction of a CR, PR or SD associated with that treatment, especially where it is the only therapy administered, or where prolonged survival results. The strength of the association of a drug with a useful clinical response makes further use of that agent more compelling for an observer. However, for the specific patients concerned who are successfully treated with resulting CRs, the treatment has been 100% effective. This aspect has made evaluation and comparison of many agents problematic, as most therapies are only ‘clinically effective’ in a small or moderate cohort of patients (even many newer therapies), and selection of responders at the outset is often difficult. For vaccine immunotherapy (and indeed, all immunotherapy until recently), the relative clinical efficacy of inducing CR, PR, SD or prolonged survival has been low, and this has tempered acceptance of immunotherapy in general terms.
Another issue increasingly being recognized is the fact that immune responses are variable in their speed of onset, intensity and duration. Some clinical responses to vaccine therapy for advanced malignancy can be rapid, occurring within a few weeks, with immediate noticeable regression of cancer deposits, although this speed of onset is not commonplace for most vaccine/immunotherapy regimens, it has been observed. Usually, vaccine responses are slower and can take many months to develop. This has been a real issue arising chiefly from our contemporary understanding of chemotherapy and radiotherapy clinical responses which are usually considered to be more immediate, gauged in terms of 2–3 months to observe an effect. With vaccines, and with multiple other forms of immunotherapy, it is being increasingly appreciated that responses take longer, sometimes up to 6–12 months, where partial responses and stable disease can sometimes gradually convert to a complete clinical response. Pseudo progression can also occur where the tumour enlarges due to inflammatory swelling. Therefore, the classical World Health Organization and RECIST criteria perhaps need to be considered and applied differently, or modified, which has led to the newer immunological response criteria,39–41 which is gradually gaining acceptance, including by the US Food and Drug Administration (FDA). Future refinements of the currently available methods of measuring successful responses to immunotherapy might include better volumetric assessments, such as utilization of three-dimensional computed tomography scanning, positron emission tomographic volume scanning, heat mapping or magnetic resonance spectroscopic analyses. Part of the remaining issue with all of these methods has been establishing techniques that are time effective in more accurately assessing changes in multiple metastases, because most current evaluation tools for tumour size assessment only provide approximations for the response to therapies but are rapid, practically. It should be noted, however, that CRs are essentially detectable by most methods, although detection of small tumour volume (<4 mm diameter) remains an issue by any method except pathological sampling.
Tumour antigens and mutational load or burden
Tumours are genetically and antigenically heterogeneous.42,43 A wide range of internal and externally expressed antigens are present within tumour cells, including peptides and glycolipids. Most of the focus has been on peptide antigens and major histocompatibility complex (MHC) antigen presentation; however, glycolipids and CD1 presentation of these has been relatively ignored.44 Moreover, refined selected peptide antigens appear less capable of inducing effective immune responses against tumour than mixed multiple antigens which can provide a stronger signal to the immune system.22,25,32 There is evidence that intense selection pressure is applied from the tumour microenvironment in early clinical untreated non-small-cell lung cancers (NSCLCs) by the immune system which produces multiple routes to immune evasion, leading to different levels of mutational burden, clinical behaviour and disease outcomes.45 Indeed, neoantigen depletion appears to occur in many tumours that have become ‘unresponsive’ to therapies of different types, including inhibitory-checkpoint blockade. This is likely a mechanism for evasion of antitumour immune responses and resistance to therapies. Moreover, the immune microenvironment is highly variable between tumours, patients, and between metastases, and even within the same tumour deposit within patients with almost 30% of tumours showing diverse levels of immune infiltration. This implies that different metastases within the same individual and even parts of the same tumour mass will demonstrate differential susceptibilities to many therapies, including checkpoint blockade, due to their relative expression of tumour antigens, and thus proportionate T-cell stimulatory capacity, leading either to immune evasion (low mutation/antigen expression) or an effective response (higher mutation/antigen expression).45
Higher tumour mutational burden across a number of tumour types showed an association with improved survival. Patients treated with immune-checkpoint inhibitors showed an improved survival among those with a higher mutational burden, although the degree of mutational load required to confer the improved survival varied with tumour type.46 This infers that the greater the mutational load, the greater the antigenic signal and therefore the stronger the in vivo (vaccine) immune response to the tumour. When high-mutational-burden tumour cells are killed, multiple tumour-associated antigens are released and further (potentially stronger) T-cell-mediated (in vivo vaccine) immune responses can proceed.
In patients with NSCLC treated with first-line nivolumab plus ipilimumab, higher tumour mutational burden was associated with significantly longer progression-free survival than for chemotherapy-treated patients, regardless of the degree of programmed cell-death ligand 1 (PD-L1) expression,47 suggesting tumour mutational burden as a potential independent biomarker for response to checkpoint agents. Transfer of ex vivo-cultured autologous tumour-infiltrating lymphocyte (TIL) has been associated with higher CR rates of around 20%, and Prickett and coworkers have also demonstrated that these autologous TILs recognize a range of distinct mutated gene products so that the TILs represent an enriched population of T cells capable of recognizing autologous tumour antigens but are inhibited, so that responsiveness is reduced; however, in the right circumstances (autologous transfer), can elicit a powerful response inducing a CR.48
Balance between T-stimulatory and T-regulatory responses
Most tumours reaching clinical diagnosis appear to exist because of relative immunological tolerance, although the degree and character of the inflammatory infiltration has been observed to vary considerably between different tumours and even among different metastases in the same patient. We do know from experience with the interleukin 2 (IL-2) and checkpoint therapies that induction of ‘overdrive’ of the immune response (as evidenced by autoimmunity which is also induced and generally correlates with an effective antitumour clinical response, e.g. vitiligo) can overcome the tolerant or regulatory state to tip the balance of immune responsiveness towards producing successful clinical responses to a pre-existing in vivo immune response. Higher regulatory T-cell (T-reg) levels and suppressor macrophage levels have been associated with more advanced cancers and poorer overall clinical outcomes.49 However, recent evidence suggests that the Treg population is nonhomogeneous with different functions that may influence antitumour immune responses and clinical outcomes.
Only some 60% of advanced melanomas respond to checkpoint-blockade agents, suggesting that even with efficient immune-driver therapies, the nonresponsiveness cannot be overcome to induce effective clinical responses by those methods. This immunological ‘nonresponsive state’ also encumbers vaccine therapies to cancer, but exactly whether the patient group unresponsive to checkpoint therapies is identical to those not responsive to vaccine therapies is not yet known. Therapies designed to reduce T-reg effects in the tumour microenvironment are currently being explored; however, in about 20% of patients, the balance towards effective antitumour therapy has successfully been achieved by vaccines alone or checkpoint agents alone, to achieve long-term survival.
Higher levels of macrophage density within many types of solid tumours (tumour-associated macrophages; TAMs) with an immunosuppressive phenotype have been correlated with poorer outcome. The mechanisms reported are enhanced angiogenesis, immunosuppression, and inflammation, to facilitate cancer growth and a relatively immunologically tolerant state. However, it is not clear-cut, as macrophages can act in either a stimulatory or inhibitory capacity on other immune cells, and thereby on the overall immune response to influence outcome. TAMs are differentiated from maturation of myeloid-derived suppressor cells (MDSCs), which in turn are derived from circulating monocytes. TAMs then undergo further differentiation into functional subtypes. For example, granulocyte–macrophage colony-stimulating factor (GM-CSF) or cytosine-phosphate-guanine (CpG) can induce toll-like receptor (TLR) expression by TAMs to reduce cancer growth and metastasis; while IL-10 can upregulate PD-L1 on TAM and monocytes causing programmed cell-death 1 (PD-1)-induced immunosuppression.50
MDSCs are immature myeloid cells capable of stimulation of Tregs and strong inhibition of T and natural killer (NK) cells to reduce antitumour responses, and appear to have a role in immunosuppression in many cancers. MDSC levels in patients have been used as a biomarker for resistance to immune-checkpoint-inhibition therapies.51
Therapies aimed at reducing TAMs and MDSCs in the tumour microenvironment are currently being explored to enhance checkpoint and other therapies.
In vivo and in situ vaccination
If the hypothesis is correct that tumour-cell death or injury leads to tumour antigen release, and further, that antigen release and immune stimulation is necessary for an effective antitumour immune response, then it follows that many forms of local (in situ) injury or damage to, or lysis of, the cancer cell ‘in situ’ or ‘in vivo’ (in the patient) can result in multivalent antigen stimulation of the immune system. However, ‘immune symbiosis’ or ‘immune homeostasis’ is what probably prevents elimination of cancers in vivo unless enough immune perturbation occurs and sufficient immunomodulatory stimulation can then reject the cancer (Figure 1). Clearly, cytotoxic therapies such as chemotherapy and radiation are directly aimed at damaging tumour cells. Complete surgical resection of metastases is reported with over 20% of patients surviving 4 years or more, some with durable longer-term survival periods.42,52 It is currently unclear whether this 20% of survivors is similar to the 20% longer-term survival mentioned above for immunotherapy and vaccine therapies. Surgery itself is associated with local and systemic inflammatory responses and in some cases, tumour-cell lysis. Therefore, many forms of cytotoxicity or cellular injury through the spectrum of available and utilized cancer therapies, including traditional chemotherapy, radiation therapy and surgery, or any of the newer therapies, such as inhibitors, receptor blockers or immunotherapies, can exert this effect of antigen release and immune stimulation.24 This means that many of the available cancer therapies are in fact capable of exerting a vaccine effect, intentionally or otherwise. The mechanisms of this vaccination effect through repeated waves of release/exposure of tumour antigen in vivo is not widely appreciated, although it has been observed that tumour antigens are released when cells are lysed or when irradiated, intact, whole tumour cells have been used as vaccines. Further, T-cell antigens specific for the respective tumour have been extracted (eluted) from TIL and these antigens have been used either directly or synthesized from their sequences in peptide- or glycolipid-based vaccines, and combination with the recent immunotherapeutic monoclonal antibody agents is being explored in trials. The processes of antigen spreading, immunoediting, tumour antigen identification and sequencing, the presence of mutations within the tumour and the extent of these (mutational burden), antigenic immune selection with neoantigen emergence, tumour–antigen heterogeneity and multiclonality, the role of pluripotential stem cells, host tumour microenvironment, tumour extracellular matrix and vascular properties, proteomic and metabolic properties and pathways in tumours, mitochondrial function, and tumour microenvironmental (and systemic) regulatory immunomodulation through lymphocytic, dendritic cellular and macrophage pathways are being actively explored. All of these facets are pertinent to the discussions and understanding of tumour immunology and therapeutic vaccine design, and although germane to the argument of in vivo vaccination, some are beyond the scope of this review and are discussed elsewhere.
Figure 1.
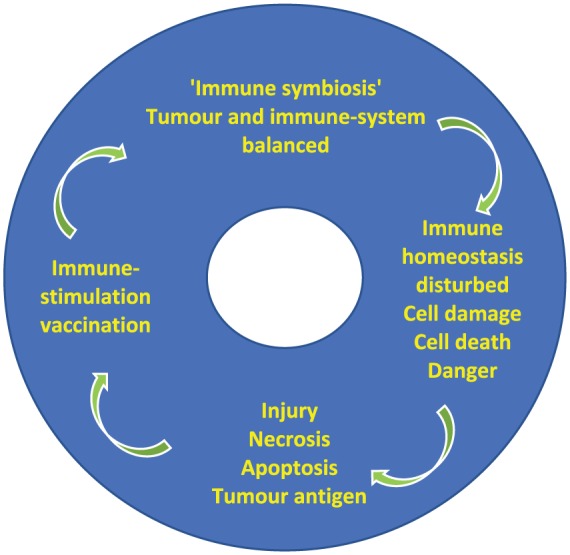
Cancer immunomodulation immunotherapy vaccination cycle.
In vivo natural tumour oncolysis
During the growth of cancers, small, early, cancer-deposit expansion of cancer cells initially keeps pace with the vascularity, so that nutrition is adequately provided to the cancer cells in the growing mass of tumour tissue. However, it is apparent that many cancer cells are not entirely immortal and a proportion of the population of cells within the cancer die through the natural mechanisms of apoptosis and necrosis. This process becomes accentuated when the growth of cancer cells outstrips the vascular supply to the mass of tumour tissue, leading to death of cells that do not receive adequate nutrition. ‘Central necrosis’ of the tumour is a commonly observed phenomenon which is most associated, although not always, with larger tumour masses. It has been assumed that the reason necrosis occurs centrally rather than elsewhere is the progressive loss of vascularity in the deepest and most remote parts of the tumour mass away from the outer vascular shell, leading to effective devascularization of deeper parts. This assumption arises largely because the main vascular supply derives from outside the tumour mass, often with the peritumoural blood supply being observably relatively rich at the tumour surface, with communicating vessels into and out of the tumour, typically becoming progressively less prolific deeper into the tumour mass. However, it is also well recognized, that for some tumours, the cut in vivo tumour surface can bleed freely, demonstrative of high internal vascularity, so there are other mechanisms that produce the necrosis, such as tumour necrosis factor alpha (TNF-α) and other cytokines. Importantly though, the natural cell death typically results in a combination of apoptosis and necrosis, and these processes serve to release tumour antigens and cause in vivo vaccination. Repeated cycles of therapy can produce waves of repeating in vivo vaccination.
Oncolytic viruses
Another wave of intense interest has developed relatively recently concerning viruses, and modifications of them, for treatment of cancer. Virus therapy for cancer, also termed virotherapy, falls into two basic groups: (a) viruses that infect cancer cells and induce expression of viral antigens on the surface of cancers cells, making them more visible to the immune system through improved antigen recognition, resulting in better stimulation of the immune response, thereby eliciting a more effective anticancer response, together with enhanced stimulation of the production of interferons and other cytokines by adjacent immunological and stromal cells (innate immunity activation); and (b) oncolytic viruses, which are termed ‘oncolytic’ because they are capable of lysing tumour cells (more) selectively over normal cells, again with direct in situ stimulation of the immune response and cytokine cascades. The term ‘oncolytic’ has been used less precisely than desirable for viral therapies that induce immunogenic apoptosis rather than true ‘oncolysis’ (bursting) of cancer cells via the necrotic death pathways. Indeed, there is becoming much imprecision in the terminology that is used. This ‘looseness of terminology’ has, and continues to, hamper the proper definitions in the immunological therapeutic virotherapy field. Immunogenic apoptotic-cell death probably should properly be termed ‘oncoptosis’ rather than oncolysis, because no bursting lysis occurs; rather, the cells involute and shrivel. The use of oncological virotherapy has a long history, much greater than many people may think, dating back to the early 1930s, with the first larger-scale research trials being performed and reported over the 1960s and into the 1980s.33–38 Many types of animal and human viruses have been tested, including adenovirus, vaccinia, Newcastle disease virus, reovirus, measles, herpes simplex and zoster, parvo, and ECHO-7, principally as a consequence of their ability to infect and cause selective lysis of tumour cells over normal cells, and thus act as a vaccine. The action of oncolytic viruses can be harnessed either through systemic infection of the patient and in situ oncolysis in vivo, or through ex vivo means first lysing the cultured tumour cells in vitro external to the patient, and then administering the lysate in in vivo doses. The principal effect of oncolytic viruses is typically natural oncolysis of abnormal cell membranes, usually on malignant cells, but oncolytic actions on normal cells can occur as well, for example the vesicular eruptions of normal tissues seen with the cutaneous manifestations of measles or pox-virus infections, as in herpes simplex (coldsores or shingles) or small pox.
Most routes of administration have been used, intravenous, intramuscular, inhaled, oral, subcutaneous and intratumoural, but perhaps the most clinically effective route has been intradermal, probably due to the abundance of dendritic and other antigen-presenting cells in the skin with relatively rapid drainage via lymphatic channels to regional lymph nodes. The most recent addition to the entourage of oncolytic viral agent therapies is T-Vec (talimogene laherparepvec; Imlygic®, Amgen, Thousand Oaks, California, USA) which is a genetically engineered herpes virus with two genes removed and a gene for GM-CSF being added.53 Injection of T-Vec into tumour deposits has been associated with necrosis/apoptosis of the tumour cells with observable clinical responses in the injected tumour deposits, with reported bystander responses in adjacent lesions, and also some systemic responses indicative of a systemic immune response.44,54,55 T-vec has been combined recently with anti-PD-1 therapy with some improvement in the response rates, but the full results are currently not available yet,56–59 and combined with ipilimumab, showing an objective response rate (ORR) of 39% versus 18% with ipilimumab alone.58 Interestingly, there was a doubling of the visceral response rate in the combined versus single-agent ipilimumab arm, a phenomenon (systemic; abscopal) also discussed later, and CRs with combined radiotherapy.58–60 A Coxsackie-virus-based vaccine therapy has been reported and is being commercialized with similar application, injected into cutaneous and subcutaneous melanoma deposits with observable responses in the injected and adjacent non-injected masses.56,61,62
Vaccination from cytotoxic chemotherapy and chemical oncolysis
Most current cytotoxic chemotherapy kills a percentage of cells in the tumour which releases tumour antigens. Cytotoxic chemotherapy primarily interferes with cell division through interrupting nucleic acid replication and processing. The interaction of cytotoxic agents with the deoxyribonucleic acid (DNA), ribonucleic acid (RNA), proteins or cell cytoskeletal functions (mitotic spindle) is generally well understood, but the effects leading to cell death and consequently on cancer growth is perhaps less clearly determined. Further, the effects of cytotoxic chemotherapy on the cell membrane and antigens is currently quite poorly understood. Classical cytotoxic chemotherapy induced cell death is chiefly via an apoptotic pathway by acceleration of senescent cell death pathways (apoptosis). This itself can have immunogenic consequences arising from macrophage and dendritic-cell engulfment and presentation of tumour antigens to the immune system in vivo. However, the resulting onco-apoptosis can follow the usual line of normal senescent cell death with low or little stimulation of host immune mechanisms (even immune-response regulation or suppression), the so-called ‘nonimmunogenic’ apoptotic pathways. This constitutes a type of involutional cell death. The distinction between induction of nonimmunogenic versus an immunogenic route of apoptotic-cell handling is not clearly understood at this time, but the presence of danger signals appears to be important in determining whether the immune system responds and the strength of that response to apoptotic end products of cell death. Consequently, inflammation and immune responses can arise from vaccination by apoptotic cancer-cell bodies as a result of cytotoxic chemotherapy cell death. The critical problem, as far as cancer destruction is concerned, is that apoptosis does not usually act as sufficiently strong a stimulus for inflammation, nor engender an active immune response, in most cases.
However, it is less well appreciated that cytotoxic chemotherapy can damage the cancer-cell membranes and that this can give rise to a different type of immune response.63 When the cancer cell membrane is damaged, and this can occur through a variety of mechanisms related to the membrane structure or function, the cell can no longer maintain its integrity to the degree that the cell lyses or bursts. This process is chemical oncolysis, and necrotic cell death occurs (rather than apoptosis). Lysis of cancer cells produces release of membrane fragments and antigenic molecules associated with the cancer-cell membrane. Not only are a variety of cancer antigens released, but danger signals are typically present as well. Tumours can become erythematous, hot and inflamed, sometimes associated with fever.
Chemical damage to the cancer-cell membrane from cytotoxins can be sufficient to release antigens from the cell surface but without killing the cancer cell, thus liberating antigens that effectively act as an in vivo vaccine to stimulate the immune response.
Classical chemical cell death occurs when damage to the cell membrane, nuclear material or cytosol is sufficiently great to lead to rupture of the cancer cells (oncolysis or necrosis). Cytotoxic chemotherapeutic agents create chemical injury that may damage the nuclear material (e.g. mustard compounds), cytosolic elements (e.g. antibiotic agents like dactinomycin), or the cell membrane structurally or functionally. The intensity of the damage and the capacity of repair mechanisms to correct the injuries determines the ability of the cell to survive or if sufficiently deleterious, causes the death of the cell. For example, if the membrane is damaged such that the sodium/potassium adenosine triphosphate (ATP) pump is affected then the cell swells and bursts, which effectively constitutes lysis and an in vivo vaccine event.
A multitude of mechanisms exist which result in cellular damage to the cancer cell, altering, for example, functional capacity and survival, with the outcomes not always being predictable due to the inherent cellular heterogeneity that exists within most tumours.
Other chemical agents have been reported to act directly on cancer cells to induce cell killing, such as the pink dye Rose Bengal (also known as PV-10; Provectus, Tennesee, USA).64,65 This agent selectively penetrates the cancer-cell membrane, but not the cell membranes of normal cells, to act internally at the lysosomal level, leading to cell death. The cell death appears to be apoptotic, leading to autophagy. PV-10 is injected directly into cutaneous and subcutaneous melanoma masses causing lysis and necrosis of tumour, inducing local inflammatory responses in the injected lesions. Interestingly, ‘bystander’ responses are reported in non-injected lesions and some systemic responses were also noted, indicating generation of a systemic immune response. Although the cellular response to PV-10 is reported to be involutional and apoptotic, clinically, the injected tumour masses undergo overt ulceration more indicative of necrosis. It is therefore likely that the response to direct injection of PV-10 into tumours is a mixed necrotic and apoptotic process. This would explain the observed clinical responses of bystander and systemic responses, which come from induction of local, regional and systemic immune responses, most likely as a result of immunogenic danger signals arising from necrosis. Indeed, this model probably serves as a generalizable model for many agents that locally damage the tumour, causing an element of tumour necrosis, for instance T-Vec, Cavatak® Merck, New Jersey, USA (Coxsackie virus) and radiation therapy.53–66
Vaccination from physical oncolysis
Radiation therapy causes both apoptosis and necrosis in tumour cells exposed to sufficient levels of radiation to damage DNA. The resulting cell-membrane damage and lysis releases tumour antigens from the malignant cells, producing in vivo vaccination events as each cell is damaged or dies. Radiation-induced antitumour immunity has been previously described and can act as an immune adjuvant by exerting systemic immunological effects beyond the local radiation field of delivery.66–71 In another study following radiation exposure in murine and human melanoma, DNA damage-response-marker expression was upregulated days prior to immune-marker expression of death receptors and T-cell costimulatory/co-inhibitory ligand changes indicating a time-dependent effect.69 Complete regression of established murine tumours in most animals using combined local radiation and intratumourally injected IL-2-linked tumour-specific antibody immunocytokine and regression of large tumours was reported with the addition of systemic anti-CTLA-4 T-cell-checkpoint blockade.69 The ‘abscopal effect’ of radiation therapy where tumour regresses outside of the field of radiation has been well recognized both clinically and in animal models, where locally administered radiation can cause systemic immune response against remote tumours not receiving radiation exposure.71 Indeed, MacManus and colleagues showed that 1.1% of 2337 patients with NSCLC receiving localized palliative radiation therapy survived 5 years or more after therapy, indicating that radiation could induce systemic responses with improved survival.72
Radiation and other forms of physical cellular damage induces damage (or danger)-associated molecular patterns (DAMPs), intrinsic danger signals (also known as alarmin) that are capable of eliciting an inflammatory response in a non-infectious situation. These are a diverse variety of predominantly intracellular molecules that are not usually revealed at the external aspect of the cell membrane but can become expressed on the cell surface of damaged cells or are released when cells are lysed when the contents are released into the surrounding tissues and vessels. DAMPs are released in response to trauma or injury to tissues, for example, and when cancer cells are injured from a variety of mechanisms, the DAMP molecules are released into the tissues and are capable of inducing an inflammatory response.
DAMPs are tissue dependent and include intracellular proteins, for example, heat-shock proteins (HSPs), or the chromatin-associated lysosomal protein high-mobility group box 1 (HMGB1), and also include extracellular-matrix-associated stromal proteins, such as hyaluronan fragments. DAMPs also include nonprotein molecules like DNA, adenosine, ATP, uric acid, and heparan sulfate. Calcium regulatory proteins such as S-100 are also able to act as DAMPs in some situations, and even calcium itself, depending on the concentration and location. DAMPs appear to be concentration and location dependent in terms of how great their stimulatory capacity is on inciting danger signals in the immune system in a particular situation.
DAMPs perform a similar function to pathogen-associated molecular patterns (PAMPs) which are a variety of molecules associated with (extrinsic) pathogens usually recognized by the innate immune system through TLRs and other pattern-recognition receptors in plants and animals. Although there are many PAMPs, examples include glycans, bacterial lipopolysaccharides, glycoconjugates, endotoxins, flagellin, lipoteichoic acid, peptidoglycan, viral nucleic acids [e.g. double-stranded ribonucleic acid (dsRNA)] and CpG. Many microbes contain natural danger signals capable of inciting an immune response upon exposure to the animal and human immune system, as an evolutionary adaptation. Such responses have been selected for over millions of years.73 Cancer cells, at least within established tumours, are not only of ‘self’ origin, but are usually lacking the necessary danger signals for eliciting an effective immune response, which forms part of the observed ‘nonresponsiveness’, or natural ‘tolerance’ of tumours in the host. When danger signals are present together with tumour antigens then an immune response can proceed, and this may explain some of the ‘spontaneous regressions’ of cancer observed following an infective or traumatic episode. Many of the current cancer immunotherapy trials are now utilizing adjuvants or danger signals with vaccination, or checkpoint-agent therapy on this basis. Examples are heat-killed vaccines and adjuvants, such as Coley vaccines, site-specific immunomodulators, and systemic immune modulators. Imodulon (IMM-101, Immodulon Therapeutics, Uxbridge, UK) contains heat-killed Mycobacterium obuense and is capable of modulation of the innate and adaptive immune systems in response to cancer, through interaction with a number of receptors (PAMPs) and γδ T cells, granulocytes, and antigen-presenting cells, to downregulate a type 2 (Th2) response bias, in favour of a type 1 (Th1) response.74–79 Radioprotective effects can also occur in tissues through TLRs; either arising from DAMPs or through secondary infection (PAMPs) or when TLR agonists are administered with radiotherapy.80 In essence, radiation therapy can act to cause tumour cell death with in vivo vaccination events through release of antigen in waves with each successive treatment cycle stimulating the immune system; or produce radioprotective responses in target tumour cells, the balance of which can determine the clinical response.
Topical and systemic sensitizing agents
Topical sensitizing agents have been used for many decades as mechanisms for adjuvant boosting for the immune system and have been shown to induce regression of cancer due to stimulation of a pre-existing in vivo immune response. These agents are usually administered percutaneously by painting them onto the skin overlying the tumour deposit such that local absorption of the agent influences the local tumour microenvironment; however, bystander responses have been observed where nontreated metastases also can regress, indicative of enhancement or development of a systemic immune response.81–83
Such immunomodulating agents can act as effective therapies, even after standard therapies have failed, although the response is unpredictable. The agent 2, 4-dinitrochlorobenzene (DNCB) was one of the early agents demonstrating effective clinical responses. Using intralesional DNCB for treating in-transit metastases, CRs occurred in about 60% of patients.81 DNCB has also been used topically.82–84
Diphencyprone (DPCP) is another topically administered agent that has been used more recently to induce immune responses in patients with surgically nonoperable deposits, especially in difficult areas, and has shown remarkable results in some patients with high rates of complete (46%) and partial (38%) regressions being recorded.85–87 DPCP immunotherapy has been used to treat cutaneous warts and alopecia areata, and more recently, has been used as a single agent to successfully treat radiotherapy-resistant extensive, multiple scalp-melanoma metastatic confluent nodules not amenable to surgery.85,86 The fact that complete responses have been reported in locally advanced melanoma, with bystander effects in nontreated lesions and regression of visceral metastases, are highly significant observations.87 Treatment was well tolerated with daily topical applications, was non-invasive and very cost effective.
Both DNCB and DPCP have been utilized combined with other therapies, notably radiation therapy and chemotherapy, and appear to augment these in some situations.88
Dinitrophenyl (DNP) is another agent, used as a hapten, to bind to tumour-cell surface ex vivo (in vitro culture) and are then injected back into the patient to form a melanoma vaccine.89–93 Multiple intradermal injections of DNP-modified autologous tumour cells mixed with Bacillus Calmette–Guérin (BCG) acting as an immunological adjuvant/stimulant were used to induce and enhance local inflammation in the metastatic melanoma deposits to incite a systemic immune response against the patient’s tumour.90 Delayed-type hypersensitivity (DTH) developed in almost all patients against autologous DNP-modified melanoma cells and in about 50% of patients exposed to autologous, unmodified tumour cells. Development of DTH was associated with significantly longer survival. The 5-year overall survival (OS) rate in 214 stage III patients was 44%, compared with a survival of 20–25% after stage IV surgical metastasectomy.92 DNP autologous vaccine treatment following low-dose cyclophosphamide was also used for 97 (83 evaluable) unresectable stage IV melanoma patients with 11 (13%) clinical responses, 2 (2.5%) CRs, 4 (4.5%) PRs and 5 (6%) mixed responses. Survival was prolonged for the responders, and those with DTH to unmodified autologous tumour cells. Regression of tumour required at least 4 months, with some cases not achieving maximum regression until up to 1 year after commencing treatment. Long survivals beyond 2 years and 5 years occurred in some patients developing CR and PR.90–92
Interestingly, antibody responses to DNP have been shown to directly correlate to better outcome. One primary mechanism of action of these agents appears to be haptenation of cell surface and cytoplasmic proteins, which can induce CD8(+) T-lymphocyte-mediated allergic-contact-hypersensitivity responses.
Haptens are small molecules acting as incomplete antigens, often without their own intrinsic activity, but when bound to another, often larger protein, can act with this carrier to generate stronger immune responses. The response strength is determined by several host factors that can determine immunological responses, such as nutrition, tumour burden, the pre-existing immune status and immunosuppression. Haptens can also produce autoimmunity, which is of considerable interest, since the clinical efficacy of checkpoint-inhibitory agents are directly associated with the generation of autoimmune side effects. Haptens can augment or create an effective or partially effective antitumour immune response when bound to a carrier protein that is a suitable tumour antigen93 Another immune stimulant, imiquimod, stimulates TLR 7/8 and has demonstrable antitumour activity, especially against skin cancers [basal cell carcinoma (BCC), SCC and melanomas] which is addressed later94 (Figure 2).
Figure 2.

Mechanism of in vivo cancer vaccination from multiple divergent therapies.
DAMP, danger-associated molecular patterns; PAMP, pathogen-associated molecular patterns; IL, interleukin; TIL, tumour-infiltrating lymphocyte.
Systemic and local intratumoural therapies
Bacillus Calmette–Guérin
Another agent with impressive long-term survival in a notable proportion of treated cases is intradermal and intravesical BCG therapy. In the late 1920s, it was noted that bladder cancer rates were lower in patients dying of tuberculosis, which prompted the initial trials of BCG as a therapy for superficial bladder cancer.95
Intralesional Mycobacterium bovis BCG has been used for local immunotherapy for melanoma nodules, with local complete responses occurring in the directly injected lesions of about 90% of injected lesions and bystander responses in 17% of non-injected, with 25% of patients rendered disease free and alive at 1–6 years postinjection.96,97
In a study of a multivalent melanoma vaccine used systemically, delivered intradermally, together with BCG versus BCG alone as the control group, in resected stage III and stage IV melanoma, the BCG arm performed better than historical controls from a previous study with no therapy, indicating that the therapy BCG had significant systemic efficacy of its own in slowing or preventing recurrence.32,97,98 BCG vaccination therapy has been previously reported as exerting long-term survival effects in melanoma patients.97–100 The role of BCG in bladder cancer treatment has become well established.101,102 Although the precise mechanism of action remains unclear, proposed to be through induction of TRAIL (TNF-related apoptosis-inducing ligand), IL-2, IL-8, IL-18, IL-12, interferon (IFN)-γ, and TNF, NK cells, macrophages, γδ-T cells, augmentation of specific antigen-primed T cells and direct actions of BCG itself.95,100–102 BCG acts as a powerful TLR 2/4 agonist causing immune stimulation, but can also downregulate the immune response. In a placebo-controlled, randomized phase III trial of 254 resected stage II/III colon cancer patients vaccinated with BCG with autologous tumour cells and BCG in the adjuvant setting for all patients, recurrence rates were statistically reduced (44%; p = 0.023) and there was longer recurrence-free survival (42%; p = 0.032) with a trend towards improved OS in the vaccine arm that led to regulatory approval in The Netherlands. However, the benefit resided for the stage II patients rather than the resected stage III patients, where no statistical benefit was demonstrable.103
Interleukin-2
IL-2 has been used for over 20 years, chiefly at high dose for systemic treatment of advanced renal cell carcinoma (RCC),104 metastatic melanoma and a range of other advanced cancers.104–106 It was the first immunotherapy approved by the FDA for advanced RCC and soon after, for advanced melanoma therapy, and has stood the test of time with a small (about 4%), but significant, rate of CRs and PRs. Most notably, when a CR or prolonged PR occurs (and interestingly, like checkpoint agents, some PRs develop into CRs over time), then almost universally, the response leads to long-term survival and in the case of CRs, effective 5- or 10-year (or longer) ‘cure’.104–106 There is some evidence that lower doses of IL-2 (low and intermediate dosing) are also effective in augmenting antitumour immune responses with lower incidence of toxic side effects.104
IL-2 has also been used for intralesional injection into superficial melanoma deposits with good efficacy showing an ORR of 82%, a CR rate of 51% and PR rate of 31%, and an intralesional CR rate of 76%, with a significant in-transit free and OS in partial responders.107–109 Similar results were reported in a systematic analysis of over 2000 patients.109 IL-2 has been combined with a range of other therapies including imiquimod, with a reported 100% intralesional CR rate110 and anti-PD-1 therapy.111 The highest CR rate of 5.0% was associated with IL-2 combined with vaccine therapy in RCC.104 In a separate study, topical daily application of 5% imiquimod cream (Aldara, Bausch, Quebec, Canada) was undertaken for 4 weeks to superficial melanoma lesions, followed by IL-2 injected either intralesionally, subcutaneously or a combination of both, according to three regimens. In six of the eight patients treated where Peripheral Blood Mononuclear Cells (PBMC) could be obtained, the Th1/Th2 balance appeared to be tipped in favour of Th1 cells evidenced by an increased CD4+CD25+ population of PBMC shown to be activated T cells and not Tregs (using CTLA-4, GITR, γ-IFN, and Foxp3). In these studies, the baseline starting point of mean percentage of CD4+CD25+ cells in cancer patients was observed to be lower than for normal controls.112,113 Although IL-2 acts as a pure immune driver augmenting a pre-existing immune response against the active tumour within the patient, when T cells kill tumour cells, antigen release occurs to create another wave of in vivo vaccination.
Electrochemotherapy
A range of cancers, including breast cancer, melanoma, SCC, BCC, sarcoma and lung adenocarcinoma, have been treated with short, high-voltage electric pulse electroporation to aid permeabilization into the tumours of chemotherapeutic agents administered topically or systemically.114 The reported results show induction of CRs and PRs, averaging a CR rate of 52% (range 11–80%) and PR rate of 25% (range 11–49%), with no reported systemic side effects.115,116 Most of the tumours treated are more easily accessible local cutaneous and superficial metastases and in-transit deposits treated with intravenous bleomycin and electroporation, but deeper liver tumours, for example, hepatocellular and colorectal carcinoma, have been treated also using electrochemotherapy. However, there is increasing interest in combining these approaches with systemic-checkpoint immunotherapies.117 Although the cellular responses are not particularly clear, cuffing of chronic inflammatory infiltrates consisting of lymphocytes and plasma cells was observed at the outer edge of the fibrous tissue of the treated colorectal liver metastases, similar to that seen with BRAF inhibitors (below).118 As electroporation kills tumour cells, antigen is released, with each treatment resulting in in vivo vaccination.
OK-432
OK-432 is a vaccine comprising group A Streptococcus pyogenes of human origin that was approved for clinical use in Japan in 1975 for treatment of gastric, primary lung, head and neck, and thyroid cancers, especially when resistant to other chemotherapies. In a recent meta-analysis of 14 trials for patients with stage III or stage IV gastric cancer after curative resection, OK-432 was used in an adjuvant setting against standard chemotherapy controls. A 12% reduction in the risk of recurrence was found with OK-432 treatment with an overall hazard ratio (HR) of 0.88 [95% confidence interval (CI) 0.77–1.00, p = 0.050]. In previous studies, the infiltration of Langerhan’s cells into the tumour after endoscopic OK-432 vaccine injection into stage III gastric cancers prior to resection, was associated with a better clinical response.119 These observations would indicate that injection of OK-432 possibly provides PAMPs to activate innate immunity and thereby stimulate an adaptive T-cell-mediated response, akin to Coley’s and oncolytic viral approaches, modifying a pre-existing in vivo antitumour immune response in the patient.
Indoleamine 2,3-dioxygenase inhibitors
Indoleamine inhibitors block the key enzyme indoleamine 2,3-dioxygenase (IDO) which catabolizes tryptophan to kynurenine which causes immunosuppressive effects. IDO upregulation in multiple cancer types has been associated with poorer survival. IDO inhibitors have been shown in preclinical models and clinical trials to directly and indirectly block IDO. Therefore, blocking immune regulation via the IDO pathway is currently being explored alone and combined with other agents, principally chemotherapy and immunotherapy in a variety of cancers. A proposed mechanism of action of IDO inhibitors is increased production of IL-2 by TIL CD8 T cells to reactivate suppressed TIL.120 IDO inhibitors are capable of reprogramming immunologically ‘cold’ tumours into ‘hot’ tumours, and appear to be well tolerated, but early results are mixed.121–123
Small-molecule inhibitors
BRAF therapies, often combined with MEK inhibitors, have been used in the approximately 40–50% of melanoma patients who have BRAF-mutation-positive tumours. CR rates are variably reported between 0.9 and 6.25%.26 The median OS was 26.1 months for combined dabrafenib plus trametinib therapy, while in the group treated with the combination of vemurafenib and cobimetinib, the median OS was 22.3 months, while vemurafenib alone was 17.8 months (HR = 0.68). While 3-year OS was 45% and 31%, respectively, for patients treated with vemurafenib and cobimetinib, or vemurafenib alone.
Vemurafenib-alone therapy versus dacarbazine was trialled in 675 patients in the BRIM-3 study and showed a median OS of 13.3 months versus 10 months for V600E-mutant advanced melanoma, respectively.124,125 In the final results, 84 dacarbazine-group patients crossed over to vemurafenib, and the OS rates for vemurafenib versus dacarbazine at 4 years were 17% versus 16%, and at 1, 2, 3 years were 56% versus 46%, 30% versus 24%, 21% versus 19%, respectively, by Kaplan–Meier estimates.126
The action of BRAF inhibitory agents is to block the BRAF pathway, reducing cancer-cell proliferation and inducing cell death. Spectacular reductions in active metabolizing melanoma metastases have been demonstrated, although gradual development of resistance to the agents is commonplace over time with a median duration of effect of about 11 months before resistance to the therapy develops.125
The induction of cancer-cell death releases tumour antigens that act also as repeated vaccine events, promoting an immune response. Recent reports indicate that clinical efficacy is associated with infiltration of lymphocytes around metastatic deposits, and removal of lymphocytes close to the tumour results in loss of effectiveness, indicating that these agents are operating through immune mechanisms as well.127–129
Interestingly, breaks in BRAF treatment with later reinstitution, and combination with other immunotherapies, have been associated with durable CRs in patients with BRAF-mutated metastatic melanoma after initial failure of sequential immunotherapies (high-dose IL-2 followed by ipilimumab with or without concurrent radiation therapy).129 The presence of non-Treg, CD4-positive effector-phenotype T cells in these patients was associated with the durable responses. Synergism between conventional or targeted cytotoxic therapy and immunotherapy in cancer treatment is suggested.129 Rechallenge after a break in failed BRAF inhibitor therapy has been demonstrated in several studies to induce clinically useful responses, including PRs and CRs.130–132
The above local and systemic therapies are capable of killing tumour cells, causing release of tumour antigens in vivo, thus creating local vaccination events, with systemic manifestations. Most therapies rely on repeated cycles of treatment for their clinical effects, thus repeated waves of tumour cell death and repeated vaccine events. The immunotherapies are next addressed with similar consideration due to their ability to induce T-cell-mediated tumour-cell killing and in vivo antigen release.
Recent immunotherapies
Checkpoint inhibitors
The literature on the recently approved checkpoint-immunotherapy agents has become extensive and complex, creating many controversies, so this discussion will be abbreviated. The main difficulties with comparisons between treatments rests in the difference in doses, regimens, combinations, sequences, crossovers, pretreatments, biomarkers and staging subgroups used in the various studies which hamper clear interpretation. Many results are also being reported as meeting abstracts rather than peer-reviewed published papers with supplementary information, as the field is advancing so rapidly, so that the detailed data are not always available for full evaluation and comparison. Some of the data is also held commercial in-confidence for a period of time. Indeed, previous evidence shows that many conference abstracts do not reach publication for a variety of reasons.133–137
The inhibitory checkpoint monoclonal antibodies, anti-CTLA-4 and anti-PD-1, operate by taking the brake off of the immune system and by reducing apoptotic lymphocyte death rates, respectively, and have been demonstrated to improve immune responses against the tumour in the patient, resulting in about 10% CR rates and improved disease-free and OS durations.26,138–142 These agents improve survival in about 20–30% of treated patients as monotherapies, and about 30–50% when combined. The main message is that the predominant action of these agents is driving forward pre-existing endogenous immune responses occurring in situ, locally, at the tumour site(s), regionally and systemically within the patients. Although these agents do not supply antigen directly or act as ‘vaccines’ of themselves, their action in causing cancer-cell killing produces tumour antigen release and therefore creates repeated vaccination events. In essence, by inciting immune mechanisms, immunotherapies act in a positive-feedback loop where the immune response is augmenting itself, with immunotherapies causing tumour-cell death, causing subsequent antigen release, inducing stronger immune reactivity, thus producing more tumour-cell death and more immune reactivity in a feed-forward mechanism. Immune responses are not always activatory, however, but can be inhibitory and induce immunological tolerance. The containment of these immune responses remains a significant issue too due to promulgation of autoimmunity, which sometimes has toxicity-related treatment-limiting effects, including a low incidence of treatment-related deaths, as can be seen below.
Anti-CTLA-4 therapy: ipilimumab
Hodi and colleagues reported a median OS of 10 months for ipilimumab plus gp100-treated advanced-melanoma patients, compared with 6.4 months for gp100 alone (HR for death, 0.68; p < 0.001) and 10.1 months for ipilimumab alone. No difference in median OS was detected between the ipilimumab groups with or without gp100.
Serious immune-related adverse events (grade 3 or 4) were reported in 10–15% of patients with ipilimumab treatment, but 3% treated with gp100 alone, with 14 deaths related to the study drugs (2.2%), most being autoimmunity related.139 The OS at 3 years for ipilimumab alone was 34% in the Wolchok and coworkers140 study; the ORR was 10.8% (4/37) in the ipilimumab-alone group (p < 0.001), with a CR rate of 0 in the ipilimumab-alone group.141
Anti-PD-1 therapies
Nivolumab
In the CheckMate 066 study, the nivolumab-alone response rate was higher than chemotherapy in first-line BRAF-wt advanced-melanoma treatment, 40% versus 13.9%108 and 2-year OS was higher at 57.7% versus control 26.7%; and 3-year OS rates were 51.2% and 21.6%, respectively. The CR and PR rates were 19.0% (40 of 210) and 23.8% (50 of 210) for the nivolumab group compared with 1.4% (3 of 208) and 13.0% (27 of 208) in the dacarbazine group, respectively.138,142,143 As an indicator of long-term outcome, the phase I study (CheckMate 003) tested various doses of nivolumab (0.3–10 mg/kg) in 107 patients showing a 5-year OS in 34% of patients, with an apparent plateau at about 2 years, and a median OS in all treated patients of 17.3 months, and 20.3 months (for a 3 mg/kg dose of nivolumab).138
Pembrolizumab
Pembrolizumab was evaluated in the KEYNOTE 001 study in various doses for 655 advanced-melanoma patients with a median OS of 23.5 (2 mg/kg dose), 22.9 (10 mg/kg 2 weekly), and 25.9 months (10 mg/kg 3 weekly). The median OS was 20 months across all doses in patients previously treated with ipilimumab and 28 months in ipilimumab-naïve patients with identical 3-year OS for both groups at 41%. However, in completely treatment-naïve patients, the median OS was 32 months with a 3-year OS of 45%.138 The objective response rate ranged from 8 to 53% being directly related to the degree of programmed death ligand 1 (PD-L1) expression in the pretreatment tumour biopsies144 and baseline tumour size (BTS) was related to OS so that a BTS of <10.2 cm (additive longest dimensions of all target lesions monitored) had a better OS than >10.2 cm (HR 0.38; p < 0.001) and ORR of 44% versus 23%, respectively.145 Durable CRs have been reported in about 16% of patients (105 of 655) in the Keynote-001 study, with about 10% of those patients relapsing after discontinuation of the agent.146
In a pooled analysis of 1012 patients from KEYNOTE-001 and KEYNOTE-002, three dose schedules of pembrolizumab (2 mg/kg every 3 weeks, 10 mg/kg every 2, and 10 mg/kg every 3 weeks) were compared, similar toxicity showing treatment-related adverse events (AE) in 75–83% of patients; most being graded 1 and 2, but grade 3 and 4 AEs were noted in 13.5% of patients [hypothyroidism (7.4%), pneumonitis (2.6%), and hyperthyroidism (2.4%)], and colitis, hypophysitis, nephritis, hepatitis and death in less than 2% of patients.138,147,148 Similar findings were noted in ‘real-world’ settings external to clinical trials.149
Combined CTLA-4 and PD-1 therapy
In the CheckMate 067 trial, untreated advanced-melanoma patients were randomized 1:1:1 to receive nivolumab (1 mg/kg) plus ipilimumab (3 mg/kg) followed by nivolumab (3 mg/ kg); nivolumab (3 mg/kg) plus placebo; or ipilimumab (3 mg/kg) plus placebo, until progression or unacceptable toxicity or withdrawal for other reasons. Stratification was for PD-L1 status, BRAF mutation status, and metastasis stage.
At 36-months minimum follow up, the median OS was not yet reached in the nivolumab-plus-ipilimumab group and was 37.6 months in the nivolumab group and 19.9 months in the ipilimumab group. The OS at 3 years was 58% (53% at 4 years) for nivolumab-plus-ipilimumab therapy and 52% (46% at 4 years) for nivolumab alone, and 34% (30% at 4-years) for ipilimumab alone. The 2-year OS was 64%, 59% and 45%, respectively. In subgroup analysis, the survivals at 4 years were about 5–13% higher for BRAF-mutant versus BRAF-wild-type melanomas; with a similar differential for PD-L1-positive versus -negative melanomas. AEs related to treatment occurred in 96% of the combination group, 86% of the nivolumab-alone group, while grade 3 or 4 treatment-related AEs were reported in 59% of the nivolumab-plus-ipilimumab group, 21% of the nivolumab group and 28% of the ipilimumab group. At 4-year analysis, four treatment-related deaths were reported; two in the combined group and one each in the single-agent groups, from cardiomyopathy, liver necrosis, neutropenia and colon perforation, respectively.146–150
In a double-blind study of 142 metastatic melanoma treatment-naïve patients randomized 2:1 between ipilimumab (3 mg/kg) plus nivolumab 1 mg/kg or placebo; followed by nivolumab or placebo, the ORR was 61.1% (44/72) in the combined nivolumab and ipilimumab group versus 10.8% (4/37) in the ipilimumab-alone group (p < 0.001), with a CR rate of 16 (22.2%) for the combination group and none in the ipilimumab group. The combination group showed 54.3% grade 3–4 drug-related AEs compared with 23.9% in the ipilimumab-monotherapy group; results supported by other studies.141,151,152
Checkpoints combined with other agents
Following the response rate of 84% using topical DPCP for cutaneous melanoma metastases in a 50-patient case series with nodal or visceral metastasis regression in four of the patients,87 DPCP was combined with the PD-1 inhibitor nivolumab treatment which showed internal metastasis regression.153,154
In a small phase Ib clinical trial of 21 stage III/IV patients, 6 (43%) of the 14 patients with visceral metastatic melanoma had complete regression reported following combined intralesional T-Vec and anti-PD-1 therapy.60 The ability of combined intralesional T-Vec and anti-PD-1 therapies to induce complete regression of distant metastases was also demonstrated in two patients with lung metastases in a 10-patient stage III/IV study.57 These are further examples of a local therapy combined with a systemic therapy to induce ‘off-target’ systemic responses. The overall response in injected lesions was reported as 90% with 6 (60%) of the 10 patients achieving lesional CRs.
Other immune-checkpoint protein-modulating agents
A number of clinical trials are currently investigating drugs that target other checkpoint-control proteins such as OX40, B7-H3, and LAG3.155
OX40
OX40 (CD134) is a TNF-receptor superfamily member (TNFRSF-4) appearing on CD4+ cells, and to a lesser degree, on CD8+ cells after activation, and transmits a potent costimulatory signal when engaged. OX40 is minimally expressed on circulating T-reg cells in humans, but is upregulated in inflammation, including in cancer. T-receptor binding to antigen transiently upregulates OX40 on activated T cells about 24–72 h after stimulation (early-to-intermediate activation marker), notably on TILs. The ligand for OX40 (OX40L) is expressed on activated antigen-presenting cells including dendritic cells, B cells, macrophages, and endothelial cells, but also by activated T cells. When OX40 receptors on T cells are bound by OX40L, apoptosis is delayed and cytokine production increases to maintain the cells beyond the initial stimulation to prolong immune response. OX40 induces in vivo T-cell survival and memory functions of both Th1- and Th2-mediated reactions.156 Excessive effects of OX40 have been associated with cytokine storm and autoimmunity. Clinical trials of OX40 agonist antibodies such as GSK3174998, which binds specifically to OX40 to activate TIL, are in progress alone or with pembrolizumab for advanced NSCLC, SCC of the head and neck, RCC, melanoma, bladder, soft tissue sarcoma, triple-negative breast cancer, and colorectal cancer [ClinicalTrials.gov identifier: NCT02528357]. OX40-blocking agents are also being explored to reduce inflammation, for example, in autoimmunity and transplant rejection.
LAG-3
LAG-3 (CD223) is a surface molecule identifying an immune checkpoint physically closely associated with CD4, but having only less than 20% amino-acid homology with CD4; and like CD4, binds to MHC-II as the main ligand on antigen-presenting cells, but with higher affinity. LAG-3 is a member of the immunoglobulin superfamily expressed on TILs, activated CD4+ and CD8+ T cells, as well as Tregs, NK cells, B cells and dendritic cells (DCs). LAG-3 acts to inhibit cellular proliferation and activation of T cells similarly to CTLA-4 and PD-1 and reduces cytokine production. Inhibitory antibodies to LAG-3 take the brakes off the anticancer immune response. Over 20 studies (e.g. anti-LAG3 monoclonal relatlimab; BMS-986016) [ClinicalTrials.gov identifier: NCT02966548] have entered phase II/III trials, and combination trials with LAG-3 and CTLA-4 or PD-1 antibodies are in progress for advanced melanoma, breast, colon, lung, brain, lung, gastric, haematological and renal cancer.157
TLR and STING agonists
The innate immune response is important for anticancer immune responses and for initiating and augmenting adaptive immune responses. Therefore, innate and adaptive immunity are often proceeding synchronously to generate effective immune responses against cancer, leading to tumour regression. Unpicking the precise roles of these responses in antitumour immunity is complex and proving immensely challenging. Many tumours do not demonstrate immunological infiltrates or show low-grade activity and have been termed ‘cold’ tumours. However, initiating innate immune responses can offer a route to activating adaptive T-cell immunity to turn ‘cold’ tumours into ‘hot’ tumours with significant T-cell infiltration. In the process of natural exposure to exogenous antigens, the nonspecific innate immune response provides rapid recognition of danger signals from foreign pathogens such as viruses, bacteria, and parasites. It is this process, detection of foreign antigens through innate immune responses that appears to be the basis of ‘infection-associated’ immune responses to cancer, as discussed above for BCG-, virus- and bacterial-based therapies. Innate immune cellular reactions include macrophages, fixed-tissue histiocytes, NK cells and dendritic cells. Other parts of the innate immune system include basophils, mast cells, eosinophils and neutrophils (granulocytes), all of which can incite immune responses in different settings and have been associated with antitumour immunity. Many of the innate immune cells are responsible for either direct or indirect liberation of cytokines including the ILs. TLRs are proteins comprising at least 10 groups found on the surface of macrophages that can recognize the presence of foreign DNA sequences or small fragments (PAMPs) or aberrant DNA within the cell cytoplasm or external to cells. TLRs can recognize viral proteins and DNA, which appears to be a fundamental evolutionary adaptation for rapid, early detection of danger from viral infection.
TLR9 recognizes the short DNA sequence CpG that is more frequent within bacterial DNA compared with human DNA. With enough CpG, an immune response can be triggered, and incite both an innate immune response through macrophages and an adaptive response by activating T cells.
Levy and colleagues showed that in situ vaccination with a TLR9 ligand (CpG oligodeoxynucleotide) could induce OX40 expression on intratumoural CD4 T cells in spontaneous murine breast cancers, and that administration of an agonist anti-OX40 antibody induced regression of distant metastases in that model.158,159 Furthermore, the in situ vaccination with CpG and anti-OX40 not only caused tumour regression, but offered a protective effect for mice genetically prone to spontaneous breast cancers, and also, those cancer-prone mice showed increased survival.158,159 These studies also demonstrated tumour specificity for these effects, showing protection against the same tumour, but not different tumours. Interestingly, TLR7/8 could replace TLR9, but anti-PD-1 could not replace anti-OX40 to reproduce the effect in those studies. An equivalent CpG agent, SD-101, has entered preliminary clinical trials as monotherapy or in combination with other therapies [ClinicalTrials.gov identifiers: NCT02927964, NCT02266147, NCT01745354, NCT02254772, and NCT02521870]. Anti-OX40 antibody is also under investigation in phase I clinical trials [ClinicalTrials.gov identifiers: NCT02559024, NCT01644968, NCT02221960, NCT02318394, NCT02274155, NCT01862900, NCT01303705 and NCT02205333]. Previously, CpG oligodeoxynucleotide (PF-3512676), in two dose sizes, was tested alone or with dacarbazine for 184 advanced-melanoma patients; however, no CRs were seen and ORRs were low in all groups, indicating that the type of CpG and choice of combination appears critical.160 Other TLR9 agonists in clinical trials are mostly in combination with immune checkpoint drugs: IMO-2125, MGN1703 (lefitolimod) and DV281. Activators of TLR7/8, including NKTR-262 and MEDI9197, have also entered trial phases.
TLRs recognize distinct PAMPs and DAMPs. For example, TLR2 recognizes lipoproteins and peptidoglycans, TLR3 viral dsRNA/viral RNA analogue polyinosinic–polycytidylic acid; TLR4 recognizes lipopolysaccharides, TLR5 bacterial flagellin, TLR7/8 single-stranded RNA; and TLR9 detects CpG-containing oligodeoxynucleotides. TLR2/6, TLR2 and TLR4 recognize endogenous matrix and HSPs. TLRs approved for cancer therapy are the TLR2/4 agonist BCG, TLR4 agonist monophosphoryl lipid A and TLR7 agonist imiquimod. The TLR5 agonist flagellin-derived CBLB502 (entolimod) has entered phase II study in patients with advanced solid tumours.161 As mentioned above, imiquimod stimulates TLR7/8, and when driven by IL-2, can induce CRs (and PRs; SDs) through both local and systemic immune response activation, indicating that innate immune response activation can, in certain circumstances, drive antigen-mediated adaptive systemic vaccine responses.113
Stimulator of interferon genes [STING; also transmembrane protein 173 (TMEM173)], is a protein that is part of the innate immune system specifically capable of responding to intracellular pathogen infection (PAMPs). On exposure to, for example, intracellular viruses, mycobacteria and intracellular parasites, STING induces production of type-I IFNs (IFN-α and IFN-β) that have a cell protective effect for the infected and locally surrounding cells to limit and reduce further infection. DCs are activated by STING which is a mechanism for activation of T-cell adaptive immunity. Agonists based on the STING structure are currently in clinical trials (e.g. ADU-S100/MIW815 and MK-1454) where the agents are being injected intratumourally to stimulate T-cell activation and tumour-cell destruction (DAMP and antigen release). The systemic side effects of STING agonists can be significant with pyrexia, systemic inflammatory responses and autoimmunity.155,162
Tim-3
Another immune checkpoint is T-cell immunoglobulin and mucin domain-3 (Tim-3), which acts as a negative regulatory molecule to induce immunological tolerance and T-cell exhaustion. Tim-3 is expressed on T-effector and regulatory cells, dendritic cells, B cells, macrophages, NK cells and mast cells, and also on a wide range of tumour cells. Tim-3 can reduce antitumour immunity through blocking γ-IFN and IL-2 production, as well as Th1-cell depletion. Tim-3 expression has been related to survival in colorectal, lung, renal, prostate, and cervical cancer, and increased Tim-3 expression has been related to anti-PD-1 resistance to therapy. Anti-Tim-3 antibodies are being clinically trialled in combination with anti-PD-1 antibodies and other agents.163
Numerous other checkpoint molecules and agents are further reviewed in greater detail elsewhere,155 together with how they might influence acquired resistance to the antitumour immune response and immunotherapies.164
Cellular therapies
These include treatments under the categories lymphokine-activated killer cell, TIL, adoptive cell therapy, T-cell receptor TIL therapy, autologous circulating T cells targeting either unspecified tumour-associated antigen or a tumour-specific antigen, chimeric antigen-receptor T-cell (CAR T) therapies, other experimental T-cell therapies based on pluripotent stem cells, CRISPR or γδ-T cells, or cell therapies based on DCs, NK or NK T cells, macrophages or other cell types. Some of these therapies have been associated with a variety of lymphodepletive and other preconditioning regimens such as marrow ablation, using cytarabine and intensive whole-body radiation. The fact that lymphocytes can be cultured or engineered and reinfused to produce homing and specific tumour killing demonstrates the ability of the immune system to treat cancer, and reinforces the in vivo actions of anticancer vaccines to produce effective lymphocyte clones. Combined therapies, including cellular therapies as a component are being trialled clinically. The field is broad and emerging, even though some aspects have been decades in development, and many of the therapies are currently heavily individualized and costly. Toxicity, including death, has been a problem in some studies leading to holds placed on some trials, but the technique looks promising with future refinement and generalization potentially making it more cost effective on a larger scale, especially if allogeneic methods are developed. Cellular therapies have been reviewed in a number of articles and are not detailed further here.165–175
Concluding remarks: vaccination as the basis of immunotherapy
What is so remarkable is that many vastly different treatment modalities for cancer can all induce a definite, but often small, rate of complete clinical regression responses. This fact is highly indicative that a common underlying mechanism is likely responsible and is operating to cause this effect and observation. Moreover, it is rapidly emerging that for almost all cancer therapies, the immune system is either directly or indirectly implicated. For example, small-molecule BRAF inhibitory therapies appear dependent on an immune infiltrate into the tumour occurring and are severely reduced or abrogated when the immune system is depleted. Clearly, the checkpoint inhibitors and IL-2, IFNs, T-Vec and vaccines, all being immune stimulants, function through the immune system for their activity. It is rapidly emerging that radiation therapy, chemotherapy, electrochemotherapy, oncolytics, cell-pathway inhibitors and receptor-blocking agents have an immunomodulatory role too, and that due to the ability of these agents to damage or lyse and kill cancer cells, those apparently cytotoxic therapies can provide release of tumour antigens which constitute active waves of in situ, in vivo vaccine events; in essence, they are performing as vaccines to cause their effects (Figure 3).
Figure 3.

All cancer therapies can lead to in vivo vaccination events.
T-Vec, talimogene laherparepvec; PV-10, Rose Bengal pink dye; IL, interleukin; PD-1, programmed cell-death 1; CTLA, cytotoxic T-lymphocyte antibody; BCG, Bacillus Calmette–Guérin; DNCB, 2, 4-dinitrochlorobenzene; DNP, dinitrophenyl; DCPC, diphencyprone.
The tumour microenvironment (TME) comprises tumour cells, cancer-associated fibroblasts, endothelial cells, myelomonocytic cells, MDSCs, TILs, extracellular matrix, and vessels (arterial, venous and lymphatic), nerves, and a variety of peritumoural stromal vascular and leukocytic cells (some normal and some tumour influenced). This level of complexity has made understanding of the interaction between all of these elements in successful and unsuccessful regression of cancer enigmatic.
The phenomenon of peritumoural cuffing of lymphocytic infiltrates has been observed in a number of therapeutic approaches and has been related to successful outcomes. Indeed, the relationship between lymphocytic infiltration and survival has been historically noted for a long time in breast176, 177 and many other malignancies. A possible future direction would be more detailed studies of the TME across many different modalities of therapy (and tumours) in order to investigate for possible common features or differences that might more clearly indicate similar underlying mechanisms associated with response or failure. For example, the state of the TME before therapy, such as the activation status of the T cells prior to any therapy, has emerged as pivotal. In general terms, tumours that are ‘hot’ and have higher levels of inflammation with activated T cells and low regulatory-cell densities appear to be better positioned to respond to immunotherapies, while tumours that are ‘cold’ and are in a suppressed state with a low infiltration of T cells respond less well to immunotherapies. One of the key approaches is therefore to modulate ‘cold’ tumours into ‘hot’ ones to prepare the TME for more successful immunotherapeutic intervention.
As we understand more about the immunological mechanisms associated with the therapies above capable of causing tumour regression, that is, antigen release, neoantigen unmasking/exposure, the control of antigen recognition, immune responsiveness, immunosuppressive T-reg responses to inhibit TIL activation, suppressor macrophages, augmentation of initially weak antitumour responses into effective tumour killing, how innate immunity interacts with adaptive responses and how these relate to natural vaccination responses that continually occur to a vast range of exogenous and endogenous antigens throughout life, the prospect of in vivo vaccination and immunomodulation will become clearer. To generate an effective antitumour immune response, antigen must be released/supplied, immune recognition must occur and immune responsiveness proceed.
In essence, whenever tumour cells are killed, antigen release can provide repeated in vivo vaccination events. Most of the treatments used for cancer outlined above require repetitive cycles of therapy to achieve cancer regression when it occurs. This aspect is now emerging as possibly the most important mechanism by which perhaps the entire range of standard and newer cancer therapies are exerting their anticancer effects, by causing waves of antigen release and repetitive in vivo vaccination events.
Acknowledgments
The author would like to thank the Australian Melanoma Research Foundation for part support of the melanoma vaccine studies together with the SA Government Blue Sky Beat Cancer Research Grant Awards, Cancer Council SA, South Australian Health & Medical Research Institute (SAHMRI), the Marjorie Hooper & Florence Cancer Research Fellowship Awards of the Royal Australasian College of Surgeons Research Foundation, National Health and Medical Research Foundation, Florey Research Award of the University of Adelaide and many private donors, all of which have contributed over many years to the vaccine immunomodulation immunotherapy knowledge base.
Footnotes
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement: The authors declare that there is no conflict of interest.
ORCID iD: Brendon J Coventry  https://orcid.org/0000-0002-3596-7735
https://orcid.org/0000-0002-3596-7735
References
- 1. Coley W. The treatment of malignant tumors by repeated inoculations of erysipelas: with report of ten original cases. Am J Med Sci 1893; 105: 487–511. [PubMed] [Google Scholar]
- 2. Coley WB. Treatment of inoperable malignant tumors with toxins of erysipelas and the bacillus prodigiosus. Trans Am Surg Assn 1894; 12: 183–212. [Google Scholar]
- 3. Coley WB. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the streptococcus erysipelas and the bacillus prodigiosus). Proc R Soc Med 1910; 3: 1–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coley WB. Disappearance of a recurrent carcinoma after injections of mixed toxins. Ann Surg 1912; 55: 897–898. [Google Scholar]
- 5. Coley WB IX. Contribution to the study of sarcoma of the femur: periosteal round-celled sarcoma of the femur, involving two-thirds of the shaft, with very extensive multiple metastases-apparent cure by the mixed toxins of erysipelas and bacillus prodigiosus. Well 10(1/2) years, when a malignant tumor (sarcoma and epithelioma) developed in the thigh at the site of an old X-ray dermatitis. Ann Surg 1913; 58: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coley WB. End results in Hodgkin’s disease and lymphosarcoma treated by the mixed toxins of erysipelas and bacillus prodigiosus, alone or combined with radiation. Ann Surg 1928; 88: 641–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anonymous. Erysipelas and prodigiosus toxins (Coley). JAMA 1934; 103: 1067–1069. [Google Scholar]
- 8. Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res 1991; 3–11. [PubMed] [Google Scholar]
- 9. Nauts HC, Swift WE, Coley BL. The treatment of malignant tumors by bacterial toxins as developed by the late William B Coley, MD, reviewed in the light of modern research. Cancer Res 1946; 6: 205–216. [PubMed] [Google Scholar]
- 10. Nauts HC, Fowler GA, Bogatko FH. A review of the influence of bacterial infection and of bacterial products (Coley’s toxins) on malignant tumors in man; a critical analysis of 30 inoperable cases treated by Coley’s mixed toxins, in which diagnosis was confirmed by microscopic examination selected for special study. Acta Med Scand Suppl 1953; 276: 1–103. [PubMed] [Google Scholar]
- 11. Pelner L, Fowler GA, Nauts HC. Effects of concurrent infections and their toxins on the course of leukemia. Acta Med Scand Suppl 1958; 338: 1–47. [PubMed] [Google Scholar]
- 12. Nauts HC. Bacterial vaccine therapy of cancer. Dev Biol Stand 1977; 38: 487–494. [PubMed] [Google Scholar]
- 13. Nauts HC. Bacterial pyrogens: beneficial effects on cancer patients. Prog Clin Biol Res 1982; 107: 687–696. [PubMed] [Google Scholar]
- 14. Nauts HC. Bacteria and cancer–antagonisms and benefits. Cancer Surv 1989; 8: 713–723. [PubMed] [Google Scholar]
- 15. Nauts HC, McLaren JR. Coley toxins–the first century. Adv Exp Med Biol 1990; 267: 483–500. [DOI] [PubMed] [Google Scholar]
- 16. Tang ZY, Zhou HY, Zhao G, et al. Preliminary result of mixed bacterial vaccine as adjuvant treatment of hepatocellular carcinoma. Med Oncol Tumor Pharmacother 1991; 8: 23–28. [DOI] [PubMed] [Google Scholar]
- 17. Hoption Cann SA, Van Netten JP, Van Netten C, et al. Spontaneous regression: a hidden treasure buried in time. Med Hypotheses 2002; 58: 115–119. [DOI] [PubMed] [Google Scholar]
- 18. Hoption Cann SA, Van Netten JP, Van Netten C. Dr William Coley and tumour regression: a place in history or in the future postgrad. Med J 2003; 79: 672–680. [PMC free article] [PubMed] [Google Scholar]
- 19. Morton DL, Eilber FR, Storm FK, et al. New advances in surgical oncology. West J Med 1983; 139: 342–350. [PMC free article] [PubMed] [Google Scholar]
- 20. North RJ, Awwad M. T cell suppression as an obstacle to immunologically mediated tumor regression: elimination of suppression results in regression. Prog Clin Biol Res 1987; 244: 345–358. [PubMed] [Google Scholar]
- 21. Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA 1993; 90: 3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hersey P, Menzies SW, Coventry B, et al. Phase I/II study of immunotherapy with T-cell peptide epitopes in patients with stage IV melanoma. Cancer Immunol Immunother 2005; 54: 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coventry BJ, Hersey P, Halligan AM, et al. Immuno-chemotherapy using repeated vaccine treatment can produce successful clinical responses in advanced metastatic melanoma. J Cancer Ther 2010; 1: 205–213. [Google Scholar]
- 24. Coventry BJ, Ashdown ML. Complete clinical responses to cancer therapy caused by multiple divergent approaches: a repeating theme lost in translation. Cancer Manag Res 2012; 4: 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Coventry BJ, Lilly C, Hersey P, et al. Prolonged repeated vaccine immuno-chemotherapy induces long-term clinical responses and survival for advanced metastatic melanoma. J Immunother Cancer 2014; 2: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coventry BJ, Baum D, Lilly CA. Long-term survival in advanced melanoma patients using repeated therapies: successive immunomodulation improving the odds? Cancer Manag Res 2015; 7: 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cassel WA, Murray DR, Torbin AH, et al. Viral oncolysate in the management of malignant melanoma. I. Preparation of the oncolysate and measurement of immunological responses. Cancer 1977; 40: 672–679. [DOI] [PubMed] [Google Scholar]
- 28. Murray DR, Cassel WA, Torbin AH, et al. Viral oncolysate in the management of malignant melanoma. II. Clinical studies. Cancer 1977; 40: 680–686. [DOI] [PubMed] [Google Scholar]
- 29. Cassel WA, Murray DR. Treatment of stage II malignant melanoma patients with a Newcastle disease virus oncolysate. Nat Immun Cell Growth Regul 1988; 7: 351–352. [PubMed] [Google Scholar]
- 30. Cassel WA, Murray DR. A ten-year follow-up on stage II malignant melanoma patients treated post-surgically with Newcastle disease virus oncolysate. Med Oncol Tumor Pharmocother 1992; 9: 169–171. [DOI] [PubMed] [Google Scholar]
- 31. Morton DL, Barth A. Vaccine therapy for malignant melanoma. CA Cancer J Clin 1996; 45: 225–244. [DOI] [PubMed] [Google Scholar]
- 32. Faries MB, Mozzillo N, Kashani-Sabet M, et al. Long-term survival after complete surgical resection and adjuvant immunotherapy for distant melanoma metastases. Ann Surg Oncol 2017; 24: 3991–4000. [DOI] [PubMed] [Google Scholar]
- 33. Batliwalla FM, Bateman BA, Serrano D, et al. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med 1998; 4: 783–794. [PMC free article] [PubMed] [Google Scholar]
- 34. Berd D, Sato T, Cohn H, et al. Treatment of metastatic melanoma with autologous, hapten-modified melanoma vaccine: regression of pulmonary metastases. Int J Cancer 2001; 94: 531–539. [DOI] [PubMed] [Google Scholar]
- 35. Hersey P, Coates AS, McCarthy WH, et al. Adjuvant immunotherapy of patients with high risk melanoma using vaccinia viral lysates of melanoma. Results of a randomized trial. J Clin Oncol 2002; 20: 4181–4190. [DOI] [PubMed] [Google Scholar]
- 36. Mitchell MS, Kan-Mitchell J, Morrow PR, et al. Phase I trial of large multivalent immunogen derived from melanoma lysates in patients with disseminated melanoma. Clin Cancer Res 2004; 10: 76–83. [DOI] [PubMed] [Google Scholar]
- 37. Wallack MK, Sivanandham M, Balch CM, et al. Surgical adjuvant active specific immunotherapy for patients with stage III melanoma: the final analysis of data from a phase III, randomized, double-blind, multicenter vaccinia melanoma oncolysate trial. J Am Coll Surg 1998; 187: 69–77; discussion 77–79. [DOI] [PubMed] [Google Scholar]
- 38. Suriano R, Rajoria S, George AL, et al. Follow-up analysis of a randomized phase III immunotherapeutic clinical trial on melanoma. Mol Clin Oncol 2013; 1: 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009; 15: 7412–7420. [DOI] [PubMed] [Google Scholar]
- 40. Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017; 18: e143–e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sullivan RJ, Atkins MB, Kirkwood JM, et al. An update on the society for immunotherapy of cancer consensus statement on tumor immunotherapy for the treatment of cutaneous melanoma: version 2.0. J Immunother Cancer 2018; 6: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Caswell DR, Swanton C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 2017; 15: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol 2019; 30: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coventry B, Heinzel S. CD1a in human cancers: a new role for an old molecule. Trends Immunol 2004; 25: 242–248. [DOI] [PubMed] [Google Scholar]
- 45. Rosenthal R, Cadieux EL, Salgado R, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019; 567: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019; 51: 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hellmann MD, Callahan MK, Awad MM, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 2019; 35: 329. [DOI] [PubMed] [Google Scholar]
- 48. Prickett TD, Crystal JS, Cohen CJ, et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol Res 2016; 4: 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ward-Hartstonge KA, Kemp RA. Regulatory T-cell heterogeneity and the cancer immune response. Clin Transl Immunology 2017; 6: e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol 2018; 8: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weber R, Fleming V, Hu X, et al. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front Immunol 2018; 9: 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Howard JH, Thompson JF, Mozzillo N, et al. Metastasectomy for distant metastatic melanoma: analysis of data from the first multicenter selective lymphadenectomy trial (MSLT-I). Ann Surg Oncol 2012; 19: 2547–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 2015; 33: 2780–2788. [DOI] [PubMed] [Google Scholar]
- 54. Fountzilas C, Patel S, Mahalingam D. Review: oncolytic virotherapy, updates and future directions. Oncotarget 2017; 8: 102617–102639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Collins JM, Redman JM, Gulley JL. Combining vaccines and immune checkpoint inhibitors to prime, expand, and facilitate effective tumor immunotherapy. Expert Rev Vaccines 2018; 17: 697–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Senior M. Checkpoint inhibitors go viral. Nat Biotechnol 2019; 37: 12–17. [DOI] [PubMed] [Google Scholar]
- 57. Sun L, Funchain P, Song JM, et al. Talimogene laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: a case series. J Immunother Cancer 2018; 6: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chesney J, Puzanov I, Collichio F, et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J Clin Oncol 2018; 36: 1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blake Z, Marks DK, Gartrell RD, et al. Complete intracranial response to talimogene laherparepvec (T-Vec), pembrolizumab and whole brain radiotherapy in a patient with melanoma brain metastases refractory to dual checkpoint-inhibition. J Immunother Cancer 2018; 6: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ribas A, Dummer R, Puzanov I, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017; 170: 1109–1119.e10. Erratum in: Cell 2018; 174: 1031–1032. [DOI] [PubMed] [Google Scholar]
- 61. Au GG, Beagley LG, Haley ES, et al. Oncolysis of malignant human melanoma tumors by coxsackie viruses A13, A15 and A18. Virol J 2011; 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Annels NE, Arif M, Simpson GR, et al. Oncolytic immunotherapy for bladder cancer using coxsackie A21 virus. Mol Ther Oncolytics 2018; 9: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hato SV, Khong A, De Vries IJ, et al. Molecular pathways: the immunogenic effects of platinum-based chemotherapeutics. Clin Cancer Res 2014; 20: 2831–2837. [DOI] [PubMed] [Google Scholar]
- 64. Ross MI. Intralesional therapy with PV-10 (Rose Bengal) for in-transit melanoma. J Surg Oncol 2014; 109: 314–319. [DOI] [PubMed] [Google Scholar]
- 65. Thompson JF, Agarwala SS, Smithers BM, et al. Phase 2 study of intralesional PV-10 in refractory metastatic melanoma. Ann Surg Oncol 2015; 22: 2135–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Finkelstein SE, Timmerman R, McBride WH, et al. The confluence of stereotactic ablative radiotherapy and tumor immunology. Clin Dev Immunol 2011; 2011: 439752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schaue D, McBride WH. T lymphocytes and normal tissue responses to radiation. Front Oncol 2012; 2: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McBride WH, Ganapathy E, Lee MH, et al. A perspective on the impact of radiation therapy on the immune rheostat. Br J Radiol 2017; 90: 20170272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Werner LR, Kler JS, Gressett MM, et al. Transcriptional-mediated effects of radiation on the expression of immune susceptibility markers in melanoma. Radiother Oncol 2017; 124: 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Morris ZS, Guy EI, Francis DM, et al. In situ tumor vaccination by combining local radiation and tumor-specific antibody or immunocytokine treatments. Cancer Res 2016; 76: 3929–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buchwald ZS, Wynne J, Nasti TH, et al. Radiation, immune checkpoint blockade and the abscopal effect: a critical review on timing, dose and fractionation. Front Oncol 2018; 8: 612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mac Manus MP, Matthews JP, Wada M, et al. Unexpected long-term survival after low-dose palliative radiotherapy for non-small cell lung cancer. Cancer 2006; 106: 1110–1116. [DOI] [PubMed] [Google Scholar]
- 73. Coventry BJ, Ashdown M, Henneberg M, et al. The Immune System and Responses to Cancer: Coordinated Evolution. F1000Research 2015, 4: 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fowler DW, Copier J, Wilson N, et al. Mycobacteria activate γδ T-cell anti-tumour responses via cytokines from type 1 myeloid dendritic cells: a mechanism of action for cancer immunotherapy. Cancer Immunol Immunother 2012; 61: 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stebbing J, Dalgleish A, Gifford-Moore A, et al. An intra-patient placebo-controlled phase I trial to evaluate the safety and tolerability of intradermal IMM-101 in melanoma. Ann Oncol 2012; 23: 1314–1319. [DOI] [PubMed] [Google Scholar]
- 76. Bazzi S, Modjtahedi H, Mudan S, et al. Analysis of the immunomodulatory properties of two heat-killed mycobacterial preparations in a human whole blood model. Immunobiology 2015; 220: 1293–1304. [DOI] [PubMed] [Google Scholar]
- 77. Dalgleish AG, Mudan S, Fusi A. Enhanced effect of checkpoint inhibitors when given after or together with IMM-101: significant responses in four advanced melanoma patients with no additional major toxicity. J Transl Med 2018; 16: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Forbes NS, Coffin RS, Deng L, et al. White paper on microbial anti-cancer therapy and prevention. J Immunother Cancer 2018; 6: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bazett M, Costa AM, Bosiljcic M, et al. Harnessing innate lung anti-cancer effector functions with a novel bacterial-derived immunotherapy. Oncoimmunology 2017; 7: e1398875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu Z, Lei X, Li X, et al. Toll-like receptors and radiation protection. Eur Rev Med Pharmacol Sci 2018; 22: 31–39. [DOI] [PubMed] [Google Scholar]
- 81. Goodnight JEJ, Morton DL. The role of immunotherapy in the management of patients with malignant melanoma. World J Surg 1979; 3: 309–320. [DOI] [PubMed] [Google Scholar]
- 82. Wack C, Kirst A, Becker JC, et al. Chemoimmunotherapy for melanoma with dacarbazine and 2,4-dinitrochlorobenzene elicits a specific T cell dependent immune response. Cancer Immunol Immunother 2002; 51: 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. von Nida J, Quirk C. Successful treatment of in-transit melanoma metastases using topical 2-4 dinitrochlorobenzene. Australas J Dermatol 2003; 44: 277–280. [DOI] [PubMed] [Google Scholar]
- 84. Strobbe LJ, Hart AA, Rümke P, et al. Topical dinitrochlorobenzene combined with systemic dacarbazine in the treatment of recurrent melanoma. Melanoma Res 1997; 7: 507–512. [DOI] [PubMed] [Google Scholar]
- 85. Damian DL, Thompson JF. Treatment of extensive cutaneous melanoma metastases with topical diphencyprone. J Am Acad Dermatol 2007; 56: 869–871. [DOI] [PubMed] [Google Scholar]
- 86. Damian DL, Shannon KF, Saw RP, et al. Topical diphencyprone immunotherapy for cutaneous metastatic melanoma. Australas J Dermatol 2009; 50: 266–271. [DOI] [PubMed] [Google Scholar]
- 87. Damian DL, Saw RPM, Thompson JF. Topical immunotherapy with diphencyprone for in transit and cutaneously metastatic melanoma. J Surg Oncol 2014; 109: 308–313. [DOI] [PubMed] [Google Scholar]
- 88. Trowbridge RM, Mitkov MV, Pittelkow MR, et al. Immunomodulation of malignant melanoma by contact sensitizing agents. Expert Rev Clin Immunol 2014; 10: 63–76. [DOI] [PubMed] [Google Scholar]
- 89. Berd D, Kairys J, Dunton C, et al. Autologous, hapten-modified vaccine as a treatment for human cancers. Semin Oncol 1998; 25: 646–653. [PubMed] [Google Scholar]
- 90. Berd D, Sato T, Cohn H, et al. Treatment of metastatic melanoma with autologous, hapten-modified melanoma vaccine: regression of pulmonary metastases. Int J Cancer 2001; 94: 531–539. [DOI] [PubMed] [Google Scholar]
- 91. Berd D. Autologous, hapten-modified vaccine as a treatment for human cancers. Vaccine 2001; 19: 2565–2570. [DOI] [PubMed] [Google Scholar]
- 92. Berd D. M-Vax: an autologous, hapten-modified vaccine for human cancer. Expert Rev Vaccines 2004; 3: 521–527. [DOI] [PubMed] [Google Scholar]
- 93. Erkes DA, Selvan SR. Hapten-induced contact hypersensitivity, autoimmune reactions, and tumor regression: plausibility of mediating antitumor immunity. J Immunol Res 2014; 2014: 175265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Steinman A, Funk JO, Schuler G, et al. Topical imiquimod treatment of a cutaneous melanoma metastasis. J Am Acad Dermatol 2000; 43: 555–556. [PubMed] [Google Scholar]
- 95. Fuge O, Vasdev N, Allchorne P, et al. Immunotherapy for bladder cancer. Res Rep Urol 2015; 7: 65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Morton D, Eilber FR, Malmgren RA, et al. Immunological factors which influence response to immunotherapy in malignant melanoma. Surgery 1970; 68:158–163; discussion 63–64. [PubMed] [Google Scholar]
- 97. Morton DL, Eilber FR, Holmes EC, et al. BCG immunotherapy of malignant melanoma: summary of a seven-year experience. Ann Surg 1974; 180: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hsueh EC, Essner R, Foshag LJ, et al. Active immunotherapy by reinduction with a polyvalent allogeneic cell vaccine correlates with improved survival in recurrent metastatic melanoma. Ann Surg Oncol 2002; 9: 486–492. [DOI] [PubMed] [Google Scholar]
- 99. Yang J, Jones MS, Ramos RI, et al. Insights into local tumor microenvironment immune factors associated with regression of cutaneous melanoma metastases by Mycobacterium bovis Bacille Calmette-Guérin. Front Oncol 2017; 7: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kidner TB, Morton DL, Lee DJ, et al. Combined intralesional Bacille Calmette-Guérin (BCG) and topical imiquimod for in-transit melanoma. J Immunother 2012; 35: 716–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer - a current perspective. Nat Rev Urol 2014; 11: 153–162. [DOI] [PubMed] [Google Scholar]
- 102. Monteiro LL, Witjes JA, Agarwal PK, et al. ICUD-SIU International Consultation on Bladder Cancer 2017: management of non-muscle invasive bladder cancer. World J Urol 2019; 37: 51–60. [DOI] [PubMed] [Google Scholar]
- 103. Vermorken JB, Claessen AM, Van Tinteren H, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. Lancet 1999; 353: 345–350. [DOI] [PubMed] [Google Scholar]
- 104. Bright R, Coventry BJ, Eardley-Harris N, et al. Clinical response rates from interleukin-2 therapy for metastatic melanoma over 30 years’ experience: a meta-analysis of 3312 patients. J Immunother 2017; 40: 21–30. [DOI] [PubMed] [Google Scholar]
- 105. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014; 192: 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jiang T, Zhou C, Ren S. Role of IL-2 in cancer immunotherapy. Oncoimmunology 2016; 5: e1163462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Boyd KU, Wehrli BM, Temple CL. Intra-lesional interleukin-2 for the treatment of in-transit melanoma. J Surg Oncol 2011; 104: 711–717. [DOI] [PubMed] [Google Scholar]
- 108. Temple-Oberle CF, Byers BA, Hurdle V, et al. Intra-lesional interleukin-2 therapy for in transit melanoma. J Surg Oncol 2014; 109: 327–331. [DOI] [PubMed] [Google Scholar]
- 109. Byers BA, Temple-Oberle CF, Hurdle V, et al. Treatment of in-transit melanoma with intra-lesional interleukin-2: a systematic review. J Surg Oncol 2014; 110: 770–775. [DOI] [PubMed] [Google Scholar]
- 110. Shi VY, Tran K, Patel F, et al. 100% Complete response rate in patients with cutaneous metastatic melanoma treated with intralesional interleukin (IL)-2, imiquimod, and topical retinoid combination therapy: results of a case series. J Am Acad Dermatol 2015; 73: 645–654. [DOI] [PubMed] [Google Scholar]
- 111. Zhang X, Shi X, Li J, et al. Combination immunotherapy with interleukin-2 surface-modified tumor cell vaccine and programmed death receptor-1 blockade against renal cell carcinoma. Cancer Sci 2019; 110: 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Green DS, Bodman-Smith MD, Dalgleish AG, et al. Phase I/II study of topical imiquimod and intralesional interleukin-2 in the treatment of accessible metastases in malignant melanoma. Br J Dermatol 2007; 156: 337–345. [DOI] [PubMed] [Google Scholar]
- 113. Green DS, Dalgleish AG, Belonwu N, et al. Topical imiquimod and intralesional interleukin-2 increase activated lymphocytes and restore the Th1/Th2 balance in patients with metastatic melanoma. Br J Dermatol 2008; 159: 606–614. [DOI] [PubMed] [Google Scholar]
- 114. Probst U, Fuhrmann I, Beyer L, et al. Electrochemotherapy as a new modality in interventional oncology: a review. Technol Cancer Res Treat 2018; 17: 1533033818785329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Sersa G, Cemazar M, Snoj M. Electrochemotherapy of solid tumors—preclinical and clinical experience. Conf Proc IEEE Eng Med Biol Soc 2011; 2011: 728–731. [DOI] [PubMed] [Google Scholar]
- 116. Sersa G, Cufer T, Paulin SM, et al. Electrochemotherapy of chest wall breast cancer recurrence. Cancer Treat Rev 2012; 38: 379–386. [DOI] [PubMed] [Google Scholar]
- 117. Campana LG, Edhemovic I, Soden D, et al. Electrochemotherapy - Emerging applications technical advances, new indications, combined approaches, and multi-institutional collaboration. Eur J Surg Oncol 2019; 45: 92–102. [DOI] [PubMed] [Google Scholar]
- 118. Gasljevic G, Edhemovic I, Cemazar M, et al. Histopathological findings in colorectal liver metastases after electrochemotherapy. PLoS One 2017; 12: e0180709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Oba MS, Teramukai S, Ohashi Y, et al. The efficacy of adjuvant immunochemotherapy with OK-432 after curative resection of gastric cancer: an individual patient data meta-analysis of randomized controlled trials. Gastric Cancer 2016; 19: 616–624. [DOI] [PubMed] [Google Scholar]
- 120. Spranger S, Koblish HK, Horton B, et al. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer 2014; 2: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Prendergast GC, Malachowski WJ, Mondal A, et al. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol 2018; 336: 175–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Prendergast GC, Mondal A, Dey S, et al. Inflammatory reprogramming with IDO1 inhibitors: turning immunologically unresponsive ‘cold’ tumors ‘hot’. Trends Cancer 2018; 4: 38–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Yentz S, Smith D. Indoleamine 2,3-dioxygenase (IDO) inhibition as a strategy to augment cancer immunotherapy. BioDrugs 2018; 32: 311–317. [DOI] [PubMed] [Google Scholar]
- 124. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomized open-label study. Lancet Oncol 2014; 15: 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chapman PB, Robert C, Larkin J, et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: final overall survival results of the randomized BRIM-3 study. Ann Oncol 2017; 28: 2581–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012; 18: 1386–1394. [DOI] [PubMed] [Google Scholar]
- 128. Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res 2013; 19: 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wyluda EJ, Cheng J, Schell TD, et al. Durable complete responses off all treatment in patients with metastatic malignant melanoma after sequential immunotherapy followed by a finite course of BRAF inhibitor therapy. Cancer Biol Ther 2015; 16: 662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Mackiewicz-Wysocka M, Krokowicz L, Kocur J, et al. Resistance to vemurafenib can be reversible after treatment interruption: a case report of a metastatic melanoma patient. Medicine (Baltimore) 2014; 93: e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ibrahim T, Routier E, Weill A, et al. Successful re-challenge with anti-BRAF and anti-MEK in a patient with symptomatic melanoma flare. Eur J Cancer 2017; 82: 25–26. [DOI] [PubMed] [Google Scholar]
- 132. Valpione S, Carlino MS, Mangana J, et al. Rechallenge with BRAF-directed treatment in metastatic melanoma: a multi-institutional retrospective study. Eur J Cancer 2018; 91: 116–124. Erratum in: Eur J Cancer 2018 Mar 13. [DOI] [PubMed] [Google Scholar]
- 133. Takeda A, Loveman E, Harris P, et al. Time to full publication of studies of anti-cancer medicines for breast cancer and the potential for publication bias: a short systematic review. Health Technol Assess 2008; 12: iii, ix-x, 1–46. [DOI] [PubMed] [Google Scholar]
- 134. Krzyzanowska M, Pintilie M, Tannock I. Factors associated with failure to publish large randomized trials presented at an oncology meeting. JAMA 2003; 290: 495–501. [DOI] [PubMed] [Google Scholar]
- 135. Harris P, Takeda A, Loveman E, et al. Time to full publication of studies of anticancer drugs for breast cancer, and the potential for publication bias. Int J Technol Assess Health Care 2010; 26: 110–116. [DOI] [PubMed] [Google Scholar]
- 136. Moher D. Reporting research results: a moral obligation for all researchers. Can J Anesth 2007; 54: 331–335. [DOI] [PubMed] [Google Scholar]
- 137. Scherer RW, Meerpohl JJ, Pfeifer N, et al. Full publication of results initially presented in abstracts. Cochrane Database Syst Rev 2018; 11: MR000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mackiewicz J, Mackiewicz A. Programmed cell death 1 checkpoint inhibitors in the treatment of patients with advanced melanoma. Contemp Oncol (Pozn) 2017; 21: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363: 711–723. Erratum in: N Engl J Med 2010; 363: 1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017; 377: 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Postow MA, Chesney J, Pavlick AC, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 2015; 372: 2006–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Ascierto PA, Long GV, Robert C, et al. Survival outcomes in patients with previously untreated BRAF wild-type advanced melanoma treated with nivolumab therapy: three-year follow-up of a randomized phase 3 trial. JAMA Oncol 2018; 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Long GV, Weber JS, Larkin J, et al. Nivolumab for patients with advanced melanoma treated beyond progression: analysis of 2 phase 3 clinical trials. JAMA Oncol 2017; 3: 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Daud AI, Wolchok JD, Robert C, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol 2016; 34: 4102–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Joseph RW, Elassaiss-Schaap J, Kefford R, et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res 2018; 24: 4960–4967. Erratum in: Clin Cancer Res 2018; 24: 6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Robert C, Ribas A, Hamid O, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol 2018; 36: 1668–1674. [DOI] [PubMed] [Google Scholar]
- 147. Hamid O, Puzanov I, Dummer R, et al. Final analysis of a randomised trial comparing pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory advanced melanoma. Eur J Cancer 2017; 86: 37–45. [DOI] [PubMed] [Google Scholar]
- 148. Schachter J, Ribas A, Long GV, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017; 390: 1853–1862. [DOI] [PubMed] [Google Scholar]
- 149. So AC, Board RE. Real-world experience with pembrolizumab toxicities in advanced melanoma patients: a single-center experience in the UK. Melanoma Manag 2018; 5: MMT05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hodi FS, Chiarion-Sileni V, Gonzalez R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 2018; 19: 1480–1492. Erratum in: Lancet Oncol 2018; 19: e668. Lancet Oncol 2018; 19: e581. [DOI] [PubMed] [Google Scholar]
- 151. Sznol M, Ferrucci PF, Hogg D, et al. Pooled analysis safety profile of nivolumab and ipilimumab combination therapy in patients with advanced melanoma. J Clin Oncol 2017; 35: 3815–3822. [DOI] [PubMed] [Google Scholar]
- 152. Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015; 33: 1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Fujimura T, Furudate S, Kakizaki A, et al. Contact immunotherapy enhances the therapeutic effects of nivolumab in treating in-transit melanoma: two cases reports. J Dermatol 2016; 43: 686–689. [DOI] [PubMed] [Google Scholar]
- 154. Gulati N, Carvajal RD, Postow MA, et al. Definite regression of cutaneous melanoma metastases upon addition of topical contact sensitizer diphencyprone to immune checkpoint inhibitor treatment. Exp Dermatol 2016; 25: 553–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Marin-Acevedo JA, Dholaria B, Soyano AE, et al. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol 2018; 11: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Weinberg AD, Morris NP, Kovacsovics-Bankowski M, et al. Science gone translational: the OX40 agonist story. Immunol Rev 2011; 244: 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Long L, Zhang X, Chen F, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer 2018; 9: 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Sagiv-Barfi I, Czerwinski DK, Levy S, et al. Eradication of spontaneous malignancy by local immunotherapy. Sci Transl Med 2018; 10: eaan4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Brody JD, Ai WZ, Czerwinski DK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol 2010; 28: 4324–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Weber JS, Zarour H, Redman B, et al. Randomized phase 2/3 trial of CpG oligodeoxynucleotide PF-3512676 alone or with dacarbazine for patients with unresectable stage III and IV melanoma. Cancer 2009; 115: 3944–3954. [DOI] [PubMed] [Google Scholar]
- 161. Li K, Qu S, Chen X, et al. Promising targets for cancer immunotherapy: TLRs, RLRs, and STING-mediated innate immune pathways. Int J Mol Sci 2017; 18: 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Sokolowska O, Nowis D. STING signaling in cancer cells: important or not? Arch Immunol Ther Exp (Warsz) 2018; 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. He Y, Cao J, Zhao C, et al. TIM-3, a promising target for cancer immunotherapy. Onco Targets Ther 2018; 11: 7005–7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Syn NL, Teng MWL, Mok TSK, et al. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol 2017; 18: e731–e741. [DOI] [PubMed] [Google Scholar]
- 165. Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol 2009; 21: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Yang JC, Rosenberg SA. Adoptive T-cell therapy for cancer. Adv Immunol 2016; 130: 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Rosenberg SA. CCR 20th anniversary commentary: autologous T cells-the ultimate personalized drug for the immunotherapy of human cancer. Clin Cancer Res 2015; 21: 5409–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Feldman SA, Assadipour Y, Kriley I, et al. Adoptive cell therapy–tumor-infiltrating lymphocytes, T-cell receptors, and chimeric antigen receptors. Semin Oncol 2015; 42: 626–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Goff SL, Dudley ME, Citrin DE, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol 2016; 34: 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Andersen R, Donia M, Ellebaek E, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res 2016; 22: 3734–3745. [DOI] [PubMed] [Google Scholar]
- 171. Klemen ND, Feingold PL, Goff SL, et al. Metastasectomy following immunotherapy with adoptive cell transfer for patients with advanced melanoma. Ann Surg Oncol 2017; 24: 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Tang J, Pearce L, O’Donnell-Tormey J, et al. Trends in the global immuno-oncology landscape. Nat Rev Drug Discov 2018; 17: 783–784. Erratum in: Nat Rev Drug Discov 2018 Oct 26. [DOI] [PubMed] [Google Scholar]
- 173. Guedan S, Alemany R. CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front Immunol 2018; 9: 2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Guedan S, Ruella M, June CH. Emerging Cellular Therapies for Cancer. Annu Rev Immunol 2019; 26: 145–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. June CH, O’Connor RS, Kawalekar OU, et al. CAR T cell immunotherapy for human cancer. Science 2018; 359: 1361–1365. [DOI] [PubMed] [Google Scholar]
- 176. Coventry BJ, Weightman M, Skinner JM, et al. Improving evaluation of the distribution and density of immunostained cells in breast cancer using computerized video image analysis. Cancer Manag Res 2011; 3: 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Coventry BJ, Weightman MJ, Bradley J, Skinner JM. Immune profiling in human breast cancer using high-sensitivity detection and analysis techniques. JRSM Open 2015; 6: 2054270415603909. [DOI] [PMC free article] [PubMed] [Google Scholar]