

Abstract
The tumor microenvironment offers favorable conditions for tumor progression, and activated fibroblasts, known as cancer‐associated fibroblasts, play a pivotal role. TP53‐deficient cancer cells are known to induce strong fibroblast activation. We aimed to elucidate the oncogenic role of exosomes derived from TP53‐deficient colon cancer cells in fibroblast proliferation and tumor growth. Cancer cell‐derived exosomes (CDEs) were isolated from the conditioned media of cancer cells using a sequential ultracentrifugation method. The effects of exosomes on tumor growth were evaluated using human cell lines (TP53‐WT colon cancer, HCT116; TP53‐mutant colon cancer, HT29; and fibroblasts, CCD‐18Co and WI‐38) and an immune‐deficient nude mouse xenograft model. HCT116 (HCT116sh p53) cells deficient in TP53 accelerated cocultured fibroblast proliferation compared to TP53‐WT HCT116 (HCT116sh control) cells in vitro. Exosomes from HCT116sh p53 cells suppressed TP53 expression of fibroblasts and promoted their proliferation. Xenografts of HCT116sh p53 cells grew significantly faster than those of HCT116sh control cells in the presence of co‐injected fibroblasts, but this difference was diminished by CDE inhibition. Microarray analysis identified upregulation of several microRNAs (miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p) in TP53‐deficient CDEs, which were functionally proven to suppress TP53 expression in fibroblasts. Exosomes derived from TP53‐mutant HT29 cells also suppressed TP53 expression in fibroblasts and accelerated their growth. The proliferative effect of HT29 on cocultured fibroblasts was diminished by inhibition of these miRNAs in fibroblasts. Our results suggest that CDEs play a pivotal role in tumor progression by fibroblast modification. Cancer cell‐derived exosomes might, therefore, represent a novel therapeutic target in colon cancer.
Keywords: colon cancer, exosomal miRNA, TP53, tumor microenvironment, tumor progression
Abbreviations
- CAF
cancer‐associated fibroblast
- CDE
cancer cell‐derived exosome
- miR
microRNA
- miRNA
microRNA
- qRT‐PCR
quantitative RT‐PCR
- TGF‐β
transforming growth factor‐β
- VEGF
vascular endothelial growth factor
- WST
water‐soluble tetrazolium
1. INTRODUCTION
Activated fibroblasts surrounding cancer cells, known as CAFs, are the main component of cancer stroma and play a critical role in tumor progression.1, 2 Cancer‐associated fibroblasts secrete diverse growth factors, chemokines, and cytokines including TGF‐β or VEGF and accelerate the proliferation of cancer cells directly or indirectly.3, 4, 5 Alteration of TP53 gene expression has been observed in cancer stroma,6, 7, 8 and TP53‐suppressed fibroblasts can promote tumor growth by accelerating the secretion of cytokines or growth factors, in addition to accelerating fibroblast proliferation.9 Cancer‐associated fibroblasts can be considered a promising target for antitumor therapy due to their roles in cancer progression. However, the mechanism by which cells transition from normal tissue fibroblasts to CAFs remains unclear.
The TP53 gene is a crucial tumor suppressor, and its mutational inactivation is observed at a high frequency in all human cancers.10, 11 TP53 is a transcription factor that regulates the expression of genes associated with cell cycle arrest, apoptosis, and senescence. In addition, recent studies suggest that TP53 gene expression acts in a non‐cell‐autonomous fashion and can affect the cellular microenvironment. The functional loss of TP53 in cancer cells activates JAK2‐STAT3 signaling and promotes modification of the tumor stroma and subsequent tumor growth.12 In addition, we previously reported that functional deficiency of TP53 in cancer cells can promote fibroblast‐mediated angiogenesis and tumor growth.13 Significant changes in the secretion levels of a large number of proteins14, 15 and the production of reactive oxygen species12, 13 have been reported as mechanisms by which a cancer cell can affect its surroundings through the alteration of TP53 expression. TP53 expression might also affect surrounding stromal cells by modifying the secretion of miRNAs sequestered in exosomes. Although the exact molecular mechanisms of miRNA recruitment into exosomes are not well understood,16 it is known that changes in TP53 expression in a cancer cell can alter the miRNA profile in CDEs.17 Such changes in miRNA levels have been reported to mediate macrophage repolarization.17 However, to the best of our knowledge, their impact on fibroblast modification has not been reported.
We posited that diverse factors can influence fibroblast modification in the tumor microenvironment; among these, CDEs play a role in fibroblast modification and fibroblast‐mediated tumor growth related to TP53 deficiency in cancer cells. Our goal in this study was to elucidate the role of exosomes derived from TP53‐deficient cancer cells in fibroblast‐mediated tumor growth.
2. MATERIALS AND METHODS
2.1. Cell culture
The human colon cancer cell line HCT116 showing WT TP53 expression, HT29 TP53 mutant cells, nontransformed human colon fibroblasts CCD‐18Co, and WI‐38 human lung fibroblasts were obtained from ATCC. All cell lines used more than 2 years after purchase were authenticated to verify their identity and lack of contamination (National Institute of Biomedical Innovation, Osaka, Japan). All the experiments were carried out using fibroblast cell lines within 15 passages. Cancer cells were cultured in DMEM (D5796; Sigma‐Aldrich) supplemented with 10% FBS, and fibroblasts were grown in Eagle's Minimum Essential Medium (30‐2003; ATCC) with 10% FBS.
2.2. Exosome isolation
Prior to collection of the culture medium, the cancer cells were washed with PBS, and the medium was switched to fresh serum‐free DMEM. After incubation for 48 hours, the conditioned medium was collected, and sequential centrifugation was carried out. The medium was first centrifuged at 300 g for 10 minutes and then at 2000 g for 10 minutes to precipitate the cells. The supernatant was centrifuged at 10 000 g for 30 minutes and then ultracentrifuged at 100 000 g for 70 minutes to pellet extracellular vesicles, which were then washed by suspension in PBS. These vesicles were then ultracentrifuged at 100 000 g for 70 minutes. The final pellet was resuspended in 100 μL PBS. The protein yield was measured using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific).
The morphology of exosomes was observed using transmission electron microscopy (H‐7600; Hitachi High‐Technologies) after preparation as described here. Approximately 5 μL of sample was placed on Parafilm (Bemis Company, Neenah, WI, USA). Then, a carbon‐coated 400 mesh copper grid was positioned on top of the drop for 10 seconds and washed by a droplet of distilled water. The grid was contrasted by adding a drop of 2% uranyl acetate on Parafilm and incubating the grid on top of the drop for 10 seconds. Excess liquid was removed by gently using an absorbent paper. After drying, the samples were used for observation.
Other materials and methods are described in Appendix S1 and Tables S1,S2. All animal protocols of this study were approved by the Animal Care and Use Committee of Osaka University Graduate School of Medicine (No. 25‐032‐005).
3. RESULTS
3.1. Inactivation of TP53 in cancer cells promoted phenotypic change of fibroblasts and tumor growth
The successful inhibition of TP53 mRNA and protein expression was confirmed by qRT‐PCR and western blotting (Figure 1A). Western blotting revealed that P21, which functions downstream of TP53, was significantly suppressed in HCT116sh p53 cells, indicating that TP53 function was inhibited. The cell viability of CCD‐18Co cells cocultured with HCT116sh p53 cells significantly increased compared to those cocultured with HCT116sh control or CCD‐18Co cells alone (Figure 1B). The expression levels of TGF‐β1 and VEGFA were significantly increased, and TP53 expression was significantly suppressed in CCD‐18Co cells cocultured with HCT116sh p53 cells (Figure 1C,D).
Figure 1.
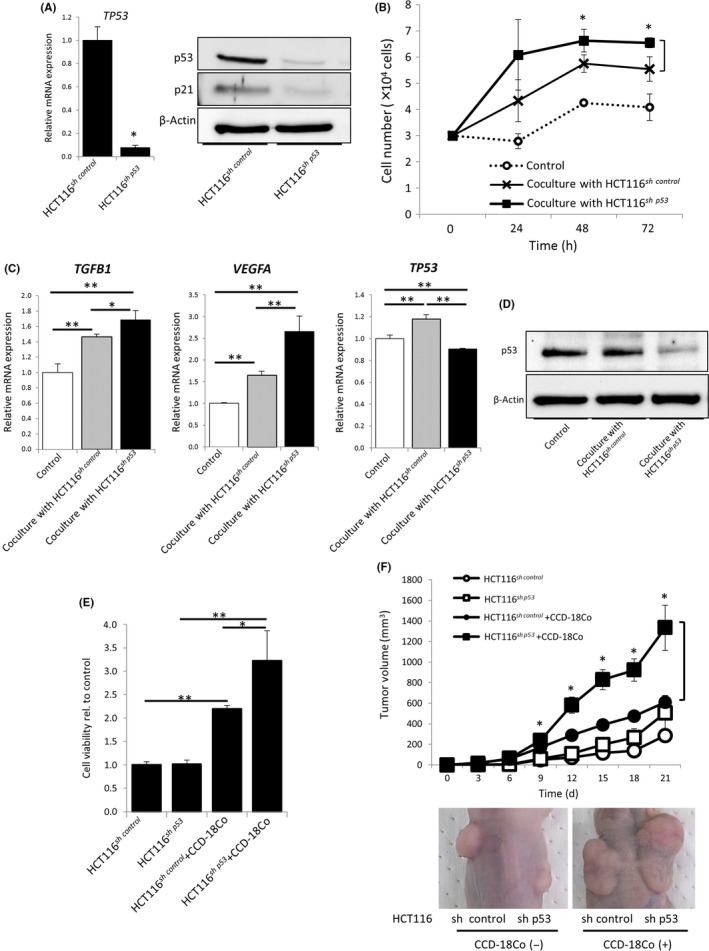
Inactivation of TP53 in cancer cells contributed to fibroblast‐mediated tumor growth. A, Relative expression levels of TP53 mRNA in HCT116sh control or HCT116sh p53 cells (left); *P < 0.01. Western blotting for TP53 and P21 (right). B, Cell proliferation of CCD‐18Co cells cocultured with or without HCT116sh control or HCT116sh p53 cells for 24, 48, and 72 h; *P < 0.05. C, Relative expression levels of TGFB1 (left), VEGFA (center), and TP53 mRNA (right) in CCD‐18Co cells cocultured with or without HCT116sh control or HCT116sh p53 cells; **P < 0.01 and *P < 0.05. D, Western blotting for TP53 in CCD‐18Co cells cocultured with or without HCT116sh control or HCT116sh p53 cells. E, WST assays of HCT116sh control or HCT116sh p53 cells cocultured with or without CCD‐18Co cells for 72 h; *P < 0.05 and **P < 0.01. F, Tumor volume of HCT116sh control or HCT116sh p53 cells injected s.c. into BALB/c nude mice with or without CCD‐18Co cells; *P < 0.05 when compared with HCT116sh control cells injected with CCD‐18Co cells. Representative images are shown in the lower panel
Then we investigated the importance of TP53 in cancer cells for fibroblast‐mediated tumor growth. No difference was observed between HCT116sh control and HCT116sh p53 in the in vitro cell proliferation assay and tumor volume measurements in the xenograft experiments. However, the proliferation of HCT116sh p53 cells cocultured with CCD‐18Co cells was significantly faster than that of HCT116sh control cells. Tumor volumes of coimplanted HCT116sh p53 and CCD‐18Co cells were also significantly bigger than those of HCT116sh control and CCD‐18Co cells (Figure 1E,F).
3.2. Exosomes derived from TP53‐inactivated cancer cells promoted fibroblast‐mediated tumor growth
We next examined the role of CDEs in fibroblast modification. We confirmed that there was no significant difference in the protein levels of isolated pellets from the supernatant of HCT116sh control and HCT116sh p53 cells and that the isolated particles expressed the representative markers for exosomes: Hsp70, Alix, and tetraspanins (CD9, CD63, and CD81) (Figure 2A). Transmission electron microscopy showed the morphology of exosomes (Figure 2B). The distribution of the particle size and particle number (concentration) of the isolated particles from the culture supernatants of HCT116sh control and HCT116sh p53 cells was characterized using a NanoSight instrument (Malvern Panalytical). The diameter of the isolated particles from both cell lines revealed exosomes of typical size (approximately 100 nm), and no significant difference was seen in either particle size or particle number (concentration) from HCT116sh control vs HCT116sh p53 cells (Figure S1).
Figure 2.
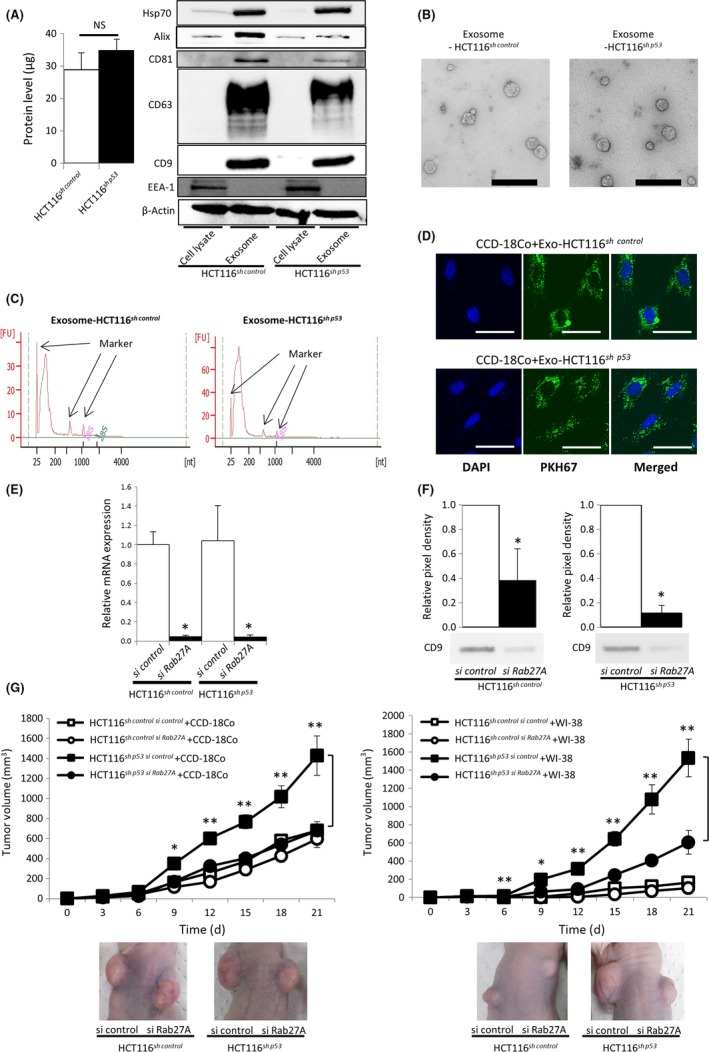
Exosomes derived from TP53‐inactivated cancer cells contributed to fibroblast‐mediated tumor growth. A, Left panel: protein levels of isolated pellets from the supernatant of HCT116sh control and HCT116sh p53 cells. Each sample was prepared from the same number of cells (4 × 107 cells); NS, not significant. Right panel: western blotting for exosome‐positive markers Hsp70, Alix, and tetraspanins (CD9, CD63, and CD81) and the exosome‐negative marker EEA‐1, using cell lysates or isolated pellets from HCT116sh control or HCT116sh p53 cells. Protein levels were normalized to 10 μg per lane. B, Representative transmission electron microscopy images of particles isolated from HCT116sh control or HCT116sh p53 cell culture supernatant. Scale bar = 400 nm. C, RNA content of HCT116sh control‐ or HCT116sh p53‐derived exosomes. RNA was extracted from HCT116sh control‐ or HCT116sh p53‐derived exosomes and analyzed by electrophoresis. D, Localization of exosomes examined by fluorescent microscopy. PKH67‐labeled exosomes from HCT116sh control or HCT116sh p53 cells were incubated with CCD‐18Co cells. Scale bar = 50 μm. E, Relative expression levels of RAB27A mRNA 72 h after transfection of siRNA; *P < 0.01 vs siRNA control. F, Western blotting for CD9 of isolated pellets from the culture supernatant of HCT116sh control or HCT116sh p53 cells transfected with RAB27A siRNA. Culture supernatants were collected 120 h after transfection. Each pellet was normalized not for protein level but for seeded cell number (5 × 107 cells per lane). Bar graphs show the densitometric analysis; *P < 0.05 vs control siRNA. G, Tumor volume of HCT116sh control or HCT116sh p53 cells transfected with control or RAB27A siRNA injected s.c. into BALB/c nude mice along with CCD‐18Co cells (left) or WI‐38 cells (right); *P < 0.05 and **P < 0.01 vs HCT116sh p53 si Rab27A cells injected with CCD‐18Co or WI‐38 cells. Representative images are shown in the lower panels
Figure 2C shows the profile of total RNA extracted from the isolated particles as evaluated by electrophoresis using an Agilent 2100 Bioanalyzer (Agilent Technologies). The RNA did not show clear bands of 18S and 28S rRNA, and only a distinguishable band under 200 bp in size was detected. This result indicates that the RNA was free of impurities from intracellular RNA and that small RNAs such as miRNAs were selectively enriched in the exosomes. To evaluate the potential for uptake and internalization of exosomes by fibroblasts, we labeled exosomes with PKH67, a fluorescent dye. The localization of exosomes from both of HCT116sh control and HCT116sh p53 cells was examined by fluorescent microscopy (Figure 2D). No significant difference was shown in the uptake of PKH67‐labeled exosomes (Figure S2).
To ascertain the role of CDEs in tumor growth, we analyzed the effect of exosome inhibition using siRNA targeting RAB27A (Figures 2E and S3), a regulator of exosome secretion and member of the RAS oncogene family.18 We evaluated the protein levels of the exosome‐associated protein CD9 in isolated pellets obtained from culture media supernatants of HCT116sh control and HCT116sh p53 cells with or without siRAB27A, 5 days after transfection. Expression of CD9 was significantly decreased in the presence of siRAB27A in both HCT116sh control and HCT116sh p53 cells (Figure 2F).
We then evaluated the effect of RAB27A inhibition in cancer cells on fibroblast‐mediated tumor growth. HCT116sh control or HCT116sh p53 cells in which RAB27A was or was not suppressed were s.c. injected into nude mice with fibroblasts. Without inhibition of RAB27A, tumors derived from HCT116sh p53 cells grew faster than tumors derived from HCT116sh control cells in the presence of CCD‐18Co or WI‐38 cells. In sharp contrast, growth acceleration was diminished with RAB27A suppression (Figure 2G), suggesting that exosomes derived from TP53‐deficient cancer cells accelerated tumor growth.
3.3. Exosomes derived from TP53‐inactivated cancer cells suppressed TP53 expression in fibroblasts
To determine the effect of CDEs, we stimulated the fibroblasts with HCT116sh control‐ or HCT116sh p53‐derived exosomes. Contrary to the phenotypic changes observed in the coculture experiments summarized in Figure 1C, the expression levels of TGF‐β1 and VEGFA in CCD‐18Co cells were not increased by HCT116sh p53‐derived exosomes as compared with HCT116sh control‐derived exosomes. However, TP53 expression in CCD‐18Co cells was significantly decreased by HCT116sh p53‐exosomes, which is consistent with the results of the coculture experiments (Figure 3A,B). In addition, CCD‐18Co cells incubated with HCT116sh p53‐derived exosomes showed significantly increased proliferation compared to CCD‐18Co alone or CCD‐18Co incubated with HCT116sh control‐derived exosomes (Figure 3C). Therefore, we hypothesized that TP53‐deficient cancer cells inhibited TP53 expression in fibroblasts and accelerated fibroblast proliferation by altering their exosomal contents. However, the expression of TGF‐β1 and VEGFA might be increased by factors other than exosomes. Thus, we focused on the decreased expression of TP53, as a target gene of exosomes derived from p53‐deficient cancer cells. To investigate the impact of TP53 inhibition in fibroblasts on tumor growth, we targeted TP53 expression in CCD‐18Co cells using siRNA (Figure 3D). The proliferation of CCD‐18Co cells was significantly increased following TP53 suppression (Figure 3E). In addition, coculture with TP53‐suppressed CCD‐18Co cells enhanced the proliferation of TP53‐WT HCT116 cells (Figure 3F) and TP53‐mutant HT29 cells (Figure S4). These results suggest that TP53‐inactivated cancer cells inhibit TP53 expression in fibroblasts through exosome secretion, which leads to the increase in fibroblast proliferation.
Figure 3.
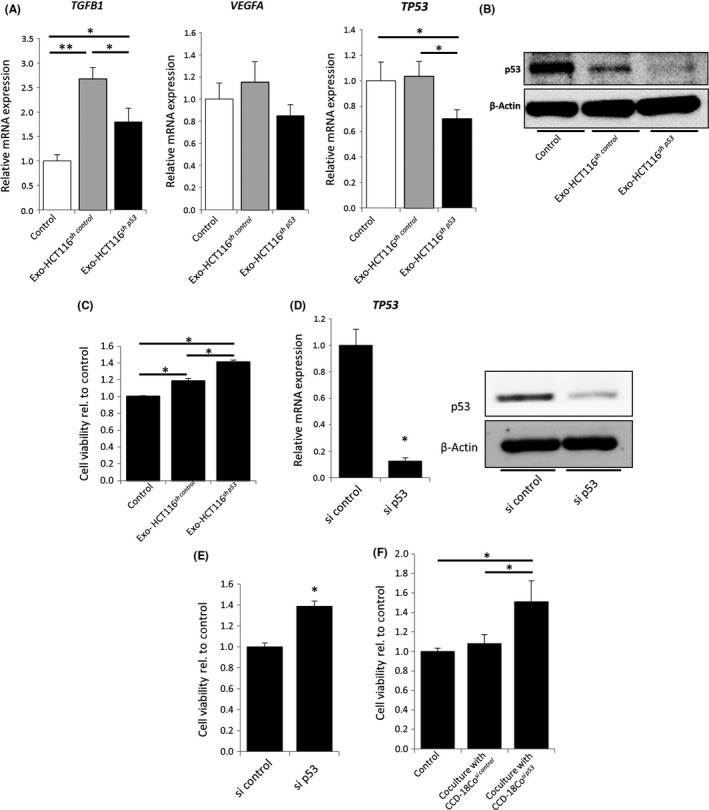
Exosomes derived from TP53‐inactivated cancer cells suppressed TP53 expression in fibroblasts. A, Relative expression levels of TGFB1 (left), VEGFA (center), and TP53 mRNA (right) in CCD‐18Co cells 12 h after incubation with control PBS or 100 μg/mL exosomes derived from either HCT116sh control or HCT116sh p53 cells; *P < 0.05 and **P < 0.01. B, Western blotting for TP53 in CCD‐18Co cells. C, WST assays of CCD‐18Co cells cultured for 48 h. Each sample was stimulated every 12 h by control PBS or 40 μg/mL exosomes derived from either HCT116sh control or HCT116sh p53 cells; *P < 0.01. D, Relative expression levels of TP53 mRNA in CCD‐18Cosi control and CCD‐18Cosi p53 cells (left); *P < 0.01. Western blotting for TP53 (right). E, WST assays of CCD‐18Cosi control and CCD‐18Cosi p53 cells cultured for 72 h in serum‐free Eagle's minimum essential medium; *P < 0.01 vs CCD‐18Cosi control. F, WST assays of HCT116 cells (3 × 104) cocultured for 48 h with or without CCD‐18Cosi control or CCD‐18Cosi p53 cells (5 × 104 cells); *P < 0.05
3.4. Alteration of the miRNA profile in cancer cell‐derived exosomes affected TP53 inhibition in fibroblasts
To clarify the mechanism for the CDE‐induced alteration of TP53 expression in fibroblasts, we examined the miRNA profile of the exosome contents derived from HCT116sh control and HCT116sh p53 cells. The expression profile of human mature miRNAs was obtained using microarray analysis (Human miRNA V21) (Figure 4A). A total of 1547 miRNAs were detected, with 17 and 315 miRNAs exclusively detected in exosomes derived from HCT116sh control and HCT116sh p53 cells, respectively. Therefore, 1215 miRNAs were identified in both HCT116sh control‐ and HCT116sh p53‐derived exosomes (Figure 4B). Furthermore, microarray analysis revealed that HCT116sh p53‐derived exosomes showed a different profile for the 1215 shared miRNAs compared with those obtained from HCT116sh control cells (Figure 4C). Some of the miRNAs were predominantly expressed in HCT116sh p53‐derived exosomes compared to HCT116sh control‐derived exosomes. We used the miRDB database (http://mirdb.org/)19, 20 to obtain a list of the miRNAs that target the TP53 gene. Fifty‐one miRNAs were identified as potentially suppressing TP53 gene expression (Table S3). Ten of these 51 miRNAs were not detected in either the HCT116sh control‐ or HCT116sh p53‐derived exosomes, and 4 of 51 were exclusively detected in HCT116sh p53‐derived exosomes (Figure S5). We then compared the expression of the 37 miRNAs remaining after excluding the 14 miRNAs present in both HCT116sh control‐ and HCT116sh p53‐derived exosomes (Figure 4D). Thirty‐one of 37 miRNAs showed higher expression in HCT116sh p53‐derived exosomes compared with HCT116sh control‐derived exosomes. In contrast, only 6 miRNAs showed a higher expression in HCT116sh control‐derived exosomes.
Figure 4.
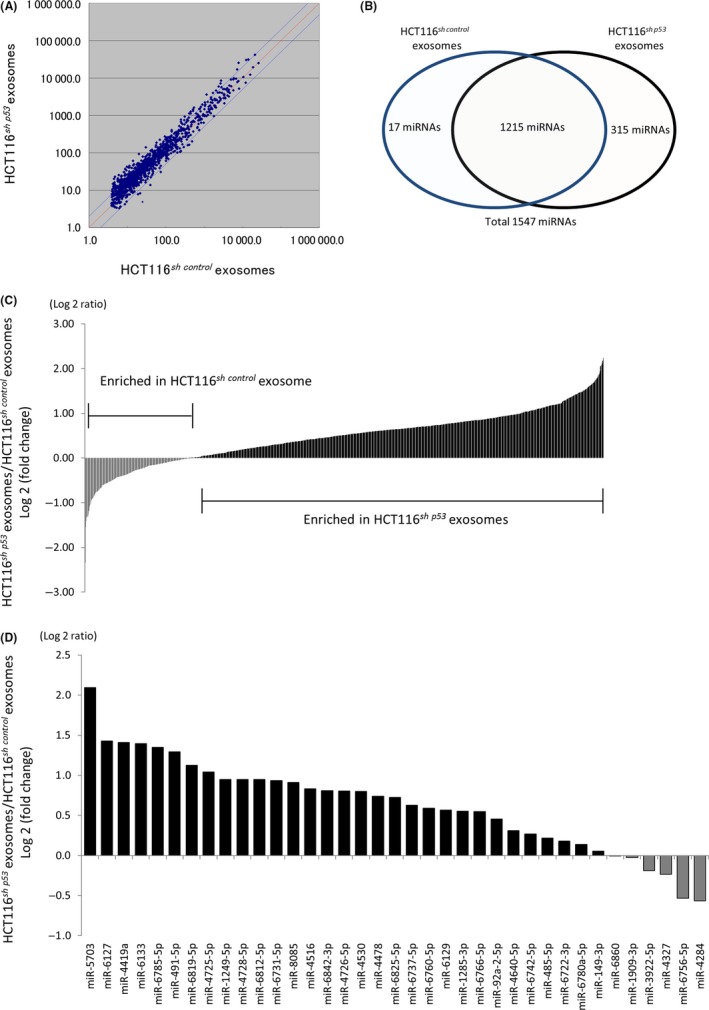
Alterations in the profile of exosomal microRNAs (miRNAs) derived from cancer cells were associated with reduced TP53 expression in fibroblasts. A, miRNA expression profiles of HCT116sh control‐ and HCT116sh p53‐derived exosomes. B, Numbers of miRNAs that were exclusively or commonly detected in HCT116sh control‐ or HCT116sh p53‐derived exosomes. C, Fold change in expression of 1215 miRNAs in HCT116sh p53‐derived exosomes relative to HCT116sh control‐derived exosomes. D, Fold change in expression of detected miRNAs that might suppress TP53 gene activity; expression levels in HCT116sh p53‐derived exosomes relative to those in HCT116sh control‐derived exosomes
3.5. MicroRNA‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p suppressed TP53 expression in fibroblasts
Next, we analyzed the role of the miRNAs in regulating TP53 expression in fibroblasts in vitro. We narrowed down the miRNAs from the previously identified TP53‐targeting miRNAs based on their expression levels, expression changes between HCT116sh p53‐derived and HCT116sh control‐derived exosomes, and the probability of TP53 targeting. Among 51 miRNAs, we identified 3 miRNAs with signal intensity in HCT116sh p53‐derived exosomes of greater than 100, an expression ratio (expression in HCT116sh p53‐derived exosomes/expression in HCT116sh control‐derived exosomes) greater than 1.5, and a target score of 65 or higher as calculated by the miRDB database based on the sequence of the miRNAs. These miRNAs included miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p (Figure 5A).
Figure 5.
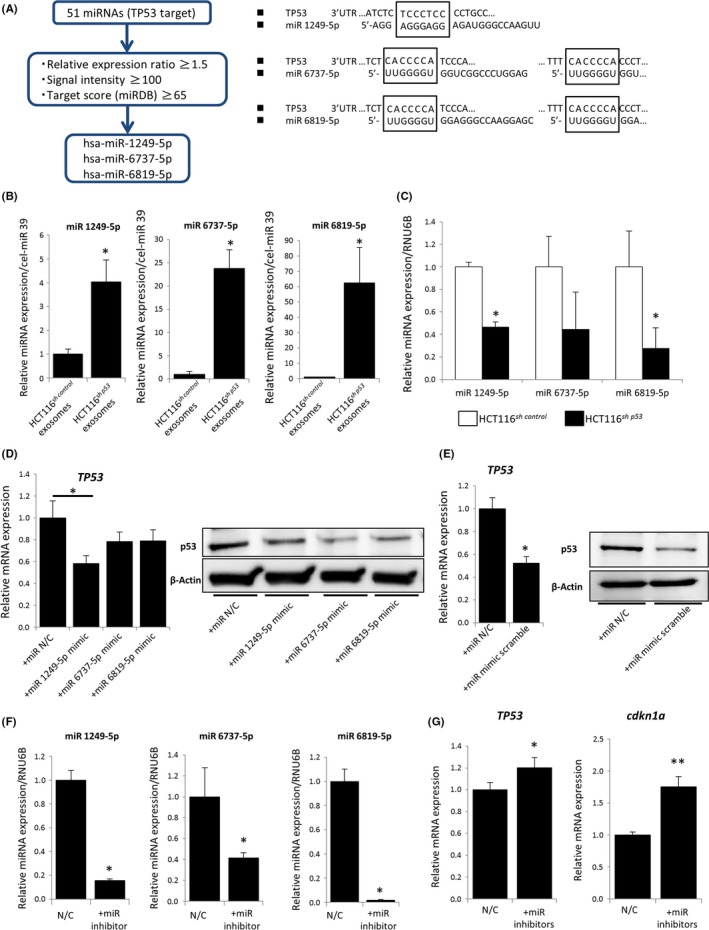
MicroRNA (miR)‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p suppressed TP53 expression in fibroblasts. A, Three miRNAs targeting TP53, which predicted that the 3′‐UTR of TP53 is a direct target of miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p. B, Relative expression levels of miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p in HCT116sh control‐ or HCT116sh p53‐derived exosomes using quantitative RT‐PCR (qRT‐PCR); *P < 0.05 vs HCT116sh control‐derived exosomes. C, Relative expression levels of miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p in HCT116sh control and HCT116sh p53 cells vs RNU6B as an internal control, evaluated by qRT‐PCR; *P < 0.05 vs HCT116sh control. D, Relative expression of TP53 in CCD‐18Co cells with or without miRNA mimics of miR‐1249‐5p, 6737‐5p, or 6819‐5p; *P < 0.05 using qRT‐PCR and western blotting. E, Relative TP53 expression in CCD‐18Co cells with or without scrambled miRNA mimics of the 3 miRNAs, assayed using qRT‐PCR and western blotting; *P < 0.01. F, Relative expression of intracellular miRNAs (miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p) with specific miRNA inhibitors for miR‐1249‐5p, 6737‐5p or 6819‐5p determined by qRT‐PCR using RNU6B as an internal control; *P < 0.05 vs CCD‐18Co cells without inhibitor (negative control [N/C]). G, Relative expression of TP53 and CDKN1A in CCD‐18Co cells with or without miRNA inhibitors, determined using qRT‐PCR; *P < 0.05 and **P < 0.01 vs negative control CCD‐18Co cells (N/C)
We validated the expression profile of the 3 miRNAs in HCT116sh p53 exosomes and HCT116sh control‐derived exosomes by qRT‐PCR analysis using syn‐cel‐miR‐39 as an external control (Figure 5B). Differences in the miRNA profile between the 2 cell lines were observed regardless of the miRNA profile of the overall profile of intracellular miRNAs (Figure 5C). To determine the effect of the 3 miRNAs on TP53 expression in fibroblasts, miRNA mimics were used to overexpress miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p in CCD‐18Co cells. Our qRT‐PCR results indicated that miR‐1249‐5p significantly downregulated TP53 mRNA expression in fibroblasts, whereas miR‐6737‐5p and miR‐6819‐5p overexpression did not lead to a significant downregulation of TP53 mRNA expression (Figure 5D). All 3 miRNAs showed downregulation of TP53 protein in CCD‐18Co cells by western blotting (Figure 5D). Scrambled miRNA mimics (a mixture of the 3 miRNA mimics in the ratio shown in the microarray) showed a marked downregulation of TP53 in CCD‐18Co cells by both qRT‐PCR and western blotting (Figure 5E).
We next introduced specific inhibitors for the 3 miRNAs into CCD‐18Co cells, and the expression of miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p was successfully downregulated by using a specific inhibitor for each miRNA in CCD‐18Co cells (Figure 5F). With the mixture of the 3 miRNA inhibitors, both TP53 and its downstream target CDKN1A were significantly upregulated (Figure 5G).
3.6. Exosomes derived from TP53‐mutant cancer cells modified fibroblasts to promote tumor growth
We utilized HT29 TP53‐mutant cancer cells to further ascertain the role of TP53 in cancer cells in fibroblast‐mediated tumor growth. The proliferation of CCD‐18Co cells was increased when cocultured with HT29 cells (Figure 6A). This result was consistent with that using HCT116sh p53 cells.
Figure 6.
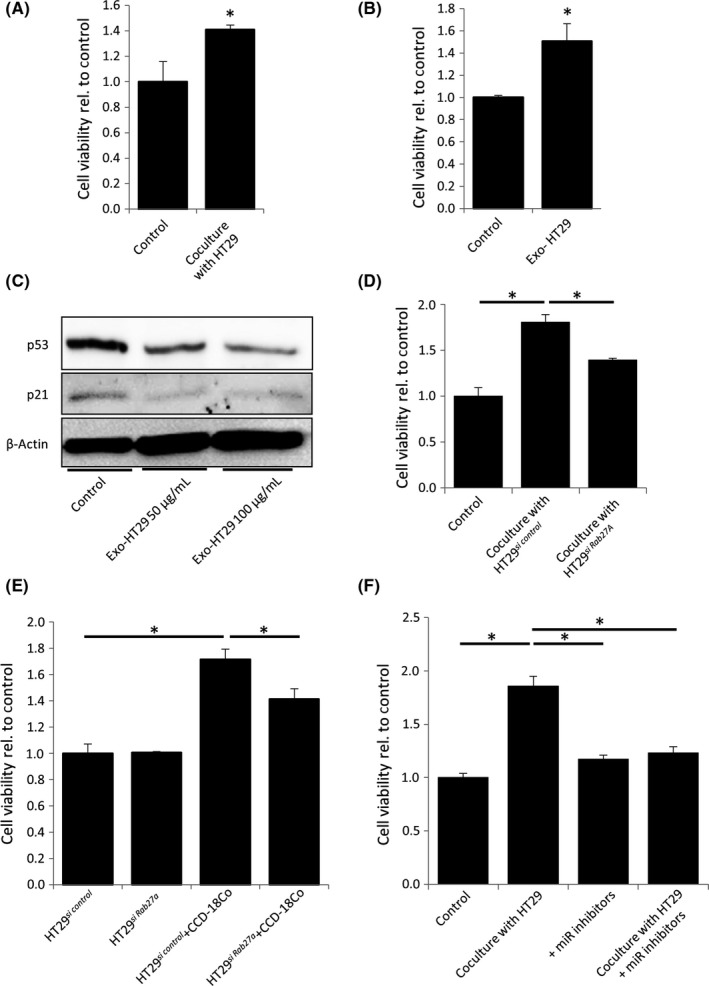
Exosomes derived from TP53‐mutant cancer cells modified fibroblasts and promoted tumor growth. A, WST assays of CCD‐18Co cells cocultured for 48 h with or without HT29 cells; *P < 0.05. B, WST assays of CCD‐18Co cells cultured for 48 h. Each sample was stimulated every 12 h by control PBS or 40 μg/mL exosomes derived from HT29 cells; *P < 0.01. C, Western blotting for TP53 and P21 in CCD‐18Co cells 12 h after stimulation with 50 or 100 μg/mL exosomes derived from HT29 cells. D, WST assays of CCD‐18Co cells cocultured for 48 h with or without HT29si control or HT29si Rab27A cells; *P < 0.01. E, WST assays of HT29si control or HT29si Rab27A cells cultured for 72 h with or without CCD‐18Co cells; *P < 0.01. F, WST assays of CCD‐18Co cells containing 3 microRNA (miR) inhibitors cocultured with or without HT29 cells for 48 h; *P < 0.01
Next, we stimulated the fibroblasts with HT29‐derived exosomes, which also significantly accelerated the proliferation of CCD‐18Co cells (Figure 6B). The TP53 and P21 expression levels of CCD‐18Co cells were significantly suppressed by HT29‐derived exosomes (Figure 6C). When CCD‐18Co cells were cocultured with HT29 cells in which the expression of RAB27A was suppressed, the proliferation of HT29 cells, as well as fibroblasts, was significantly inhibited (Figure 6D,E).
We investigated the role of miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p in mediating the enhancement of CCD‐18Co cell proliferation by HT29 cells. The expression levels of these 3 miRNAs were higher in exosomes derived from HT29 cells than from TP53‐WT HCT116 cells (Figure S6). When we introduced inhibitors for these 3 miRNAs into CCD‐18Co cells, the proliferation of these cells was significantly inhibited in the presence of HT29 cells (Figure 6F). These results suggest that exosomes derived from TP53‐mutant HT29 cancer cells modify fibroblasts in a similar fashion as those from HCT116sh p53 cells.
4. DISCUSSION
The present study shows that TP53 deficiency in cancer cells can promote stroma‐mediated tumor growth and CDEs play a critical role in this process. We also discovered that the TP53 expression in donor cancer cells affects the miRNA profile of CDEs and that exosomes derived from TP53‐deficient cancer cells can inhibit TP53 expression and the associated enhanced proliferation of recipient fibroblasts. In addition, we found that several specific miRNAs in CDEs can suppress TP53 expression in fibroblasts and that specific inhibitors of these miRNAs can restore TP53 expression.
Altered TP53 gene expression has been observed not only in cancer cells but also in stromal cells in cancer tissues.6, 7, 8, 21, 22, 23 TP53 gene expression levels were found to be negatively correlated with those of ACTA2 (α‐smooth muscle actin) in clinical samples of breast cancer stroma and in fibroblast cell lines.23 The present study found TP53 suppression in fibroblasts caused by CDEs but did not elucidate the underlying mechanism of tumor growth mediated by TP53‐suppressed fibroblasts. However, previous reports have shown that TP53‐suppressed fibroblasts behave like CAFs and that these cells have greater growth‐promoting activity toward cancer cells than WT fibroblasts.9, 22, 23
In cellular processes other than cancer, the regulation of fibroblasts by TP53 has also been reported. MicroRNA‐mediated TP53 suppression in cardiac fibroblasts plays a central role in cardiac fibrosis by promoting an increase in fibroblast proliferation as well as fibroblast‐to‐myofibroblast transition.24 These findings provide further evidence that TP53 expression in fibroblasts can be regulated by an miRNA‐mediated mechanism.
Normal tissue fibroblasts have been shown to acquire a CAF‐like phenotype following exposure to soluble factors secreted from cancer cells,25, 26 and exosomes have emerged as one of the most crucial players in tumor progression.27, 28 Exosomes can transport miRNAs from the secreting cells to the recipient cells and can inhibit specific gene expression in the recipient cells.29 In the present study, we show that CDEs produced from cancer cells deficient in TP53 can interact with stromal cells and contain higher levels of miRNAs that can suppress TP53 expression in recipient cells. In addition, transfection of WT TP53 into TP53 mutant cancer cells has been reported to induce reprogramming of the global miRNA profiles in secreted exosomes, which in turn can affect the activation of genes associated with apoptosis, including TP53, in the recipient cells.17 These data support our findings that reduced TP53 expression in donor cancer cells can modify their exosomal miRNA profile and that this, in turn, can affect the expression of a number of genes involved in various molecular pathways, including TP53 expression in surrounding recipient cells. Thus, our study provides further evidence for a novel cascade of interactions between cancer cells with TP53 deficiency and surrounding fibroblasts through inhibition of TP53 expression.
We have identified 3 specific miRNAs in CDEs obtained from TP53‐deficient colon cancer cells, which can downregulate TP53 expression in fibroblasts, miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p. However, microarray analysis and the miRDB database revealed that as many as 41 miRNAs in such CDEs could suppress TP53 gene expression, suggesting that additional miRNAs have the potential to downregulate TP53 expression in fibroblasts in the cancer microenvironment. Details regarding the involvement of additional miRNAs and the clinical applications of exosomal miRNAs are still unclear. The exact regulatory mechanism of exosomal miRNA expression is not yet well understood,16 and alterations in the profile of these miRNAs in CDEs were not associated with changes in the intracellular miRNA profile of cancer cells in this study. TP53 was reported to facilitate the processing of primary miRNAs to precursor miRNAs, and TP53 deficiency of cancer cells showed broad downregulation of mature miRNAs.30 We speculate that TP53 inhibition in cancer cells might alter the packaging of miRNAs into CDEs, in addition to affecting the processing of intracellular miRNAs. The modification of exosomal miRNAs could be a mechanism by which the loss of p53 affects surrounding cells and promotes cancer progression associated with the tumor microenvironment. In addition, we have shown that inhibition of miR‐1249‐5p, miR‐6737‐5p, and miR‐6819‐5p in fibroblasts can restore TP53 expression. Further research is needed to expand our understanding of miRNAs that affect tumor progression and develop a delivery system for inhibitors of such miRNAs that could be used in the clinical setting as a therapeutic tool.
Our research has revealed a novel regulator of fibroblast modification related to cross‐talk among cancer cells in the tumor microenvironment. We propose that specific miRNAs in CDEs could play an essential role in stroma‐mediated tumor growth and that targeting such miRNAs could represent a possible therapeutic strategy.
DISCLOSURE
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by JSPS KAKENHI grant numbers JP17K15944 (to S. Yoshii) and JP15K19326 (to Y. Hayashi) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Yoshii S, Hayashi Y, Iijima H, et al. Exosomal microRNAs derived from colon cancer cells promote tumor progression by suppressing fibroblast TP53 expression. Cancer Sci. 2019;110:2396–2407. 10.1111/cas.14084
Yoshii and Hayashi contributed equally to this work.
REFERENCES
- 1. Junttila MR, de Sauvage FJ. Influence of tumour micro‐environment heterogeneity on therapeutic response. Nature. 2013;501:346‐354. [DOI] [PubMed] [Google Scholar]
- 2. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF‐1/CXCL12 secretion. Cell. 2005;121:335‐348. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki S, Ishii G, Matsuwaki R, et al. Ezrin‐expressing lung adenocarcinoma cells and podoplanin‐positive fibroblasts form a malignant microenvironment. J Cancer Res Clin Oncol. 2015;141:475‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor‐promoting cell type. Cell Cycle. 2006;5:1597‐1601. [DOI] [PubMed] [Google Scholar]
- 6. Wernert N, Locherbach C, Wellmann A, et al. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res. 2001;21:2259‐2264. [PubMed] [Google Scholar]
- 7. Hawsawi NM, Ghebeh H, Hendrayani SF, et al. Breast carcinoma‐associated fibroblasts and their counterparts display neoplastic‐specific changes. Cancer Res. 2008;68:2717‐2725. [DOI] [PubMed] [Google Scholar]
- 8. Eng C, Leone G, Orloff MS, et al. Genomic alterations in tumor stroma. Cancer Res. 2009;69:6759‐6764. [DOI] [PubMed] [Google Scholar]
- 9. Addadi Y, Moskovits N, Granot D, et al. p53 status in stromal fibroblasts modulates tumor growth in an SDF1‐dependent manner. Cancer Res. 2010;70:9650‐9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal‐tumor development. N Engl J Med. 1988;319:525‐532. [DOI] [PubMed] [Google Scholar]
- 11. Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303‐312. [DOI] [PubMed] [Google Scholar]
- 12. Wormann SM, Song L, Ai J, et al. Loss of P53 function activates JAK2‐STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016;151:180‐193. e12. [DOI] [PubMed] [Google Scholar]
- 13. Hayashi Y, Tsujii M, Kodama T, et al. p53 functional deficiency in human colon cancer cells promotes fibroblast‐mediated angiogenesis and tumor growth. Carcinogenesis. 2016;37:972‐984. [DOI] [PubMed] [Google Scholar]
- 14. Khwaja FW, Svoboda P, Reed M, et al. Proteomic identification of the wt‐p53‐regulated tumor cell secretome. Oncogene. 2006;25:7650‐7661. [DOI] [PubMed] [Google Scholar]
- 15. Komarova EA, Diatchenko L, Rokhlin OW, et al. Stress‐induced secretion of growth inhibitors: a novel tumor suppressor function of p53. Oncogene. 1998;17:1089‐1096. [DOI] [PubMed] [Google Scholar]
- 16. Hagiwara K, Katsuda T, Gailhouste L, et al. Commitment of Annexin A2 in recruitment of microRNAs into extracellular vesicles. FEBS Lett. 2015;589:4071‐4078. [DOI] [PubMed] [Google Scholar]
- 17. Trivedi M, Talekar M, Shah P, et al. Modification of tumor cell exosome content by transfection with wt‐p53 and microRNA‐125b expressing plasmid DNA and its effect on macrophage polarization. Oncogenesis. 2016;5:e250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ostrowski M, Carmo NB, Krumeich S, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12:19‐30. sup pp 1‐13. [DOI] [PubMed] [Google Scholar]
- 19. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146‐D152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang X. Improving microRNA target prediction by modeling with unambiguously identified microRNA‐target pairs from CLIP‐ligation studies. Bioinformatics. 2016;32:1316‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo G, Marrero L, Rodriguez P, et al. Trp53 inactivation in the tumor microenvironment promotes tumor progression by expanding the immunosuppressive lymphoid‐like stromal network. Cancer Res. 2013;73:1668‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bar J, Moskovits N, Oren M. Involvement of stromal p53 in tumor‐stroma interactions. Semin Cell Dev Biol. 2010;21:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Otomo R, Otsubo C, Matsushima‐Hibiya Y, et al. TSPAN12 is a critical factor for cancer‐fibroblast cell contact‐mediated cancer invasion. Proc Natl Acad Sci USA. 2014;111:18691‐18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nagpal V, Rai R, Place AT, et al. MiR‐125b is critical for fibroblast‐to‐myofibroblast transition and cardiac fibrosis. Circulation. 2016;133:291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu X, Chen X, Zhou Q, et al. Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer Lett. 2013;335:128‐135. [DOI] [PubMed] [Google Scholar]
- 26. Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169‐184. [DOI] [PubMed] [Google Scholar]
- 27. Becker A, Thakur BK, Weiss JM, et al. Extracellular vesicles in cancer: cell‐to‐cell mediators of metastasis. Cancer Cell. 2016;30:836‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peinado H, Aleckovic M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro‐metastatic phenotype through MET. Nat Med. 2012;18:883‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melo SA, Sugimoto H, O'Connell JT, et al. Cancer exosomes perform cell‐independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014;26:707‐721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki HI, Yamagata K, Sugimoto K, et al. Modulation of microRNA processing by p53. Nature. 2009;460:529‐533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials