

Abstract
Aims
Most patients with acute heart failure (AHF) are treated with supplemental oxygen during hospitalization. In this study, we investigated the effect of oxygen titrated to high vs. low pulse oximetry targets in patients hospitalized with AHF.
Methods and results
In a pilot, open‐label randomized controlled trial (RCT), 50 patients who were admitted with AHF were randomized to either high (≥96%) or low (90–92%) SpO2 targets. Oxygen was manually titrated to the assigned target ranges for 72 h. The primary endpoint was the change in N‐terminal pro‐brain‐type natriuretic peptide (NT‐proBNP) from randomization to 72 h, and secondary endpoints included patient‐reported dyspnoea by visual analogue scale (VAS), patient global assessment (PGA), peak expiratory flow (PEF) within 72 h, and clinical outcomes up to 30 days following hospital discharge. The median age was 73.5 years, and 42% were women. The change in NT‐proBNP was −6963 (−13 345, −1253) pg/mL in the high SpO2 group and −2093 (−5692, −353) pg/mL in the low SpO2 group (P = 0.46), and the 72 h to baseline NT‐proBNP ratio was similar between groups (0.7 vs. 0.6, P = 0.51). There were no differences between arms in change in dyspnoea VAS (P = 0.86), PGA (P = 0.91), PEF (P = 0.52), in‐hospital mortality (4.0% vs. 8.0%, P = 0.50), or 30 day heart failure readmission rates (20.8% vs. 8.7%, P = 0.22).
Conclusions
In this study, no differences were observed in the primary or secondary outcomes for patients randomized to high vs. low SpO2 targets. Further RCTs with larger sample sizes are warranted to determine the efficacy and safety of oxygen therapy in patients with AHF.
Keywords: Supplemental oxygen, Heart failure, Randomized controlled trial
Introduction
Supplemental oxygen (O2) therapy is a routine treatment in the management of many patients with dyspnoea, including those with acute heart failure (AHF).1 Regardless of the arterial O2 saturation levels, O2 is often administered in these patients on the basis of the clinicians' or patients' belief that it will ameliorate dyspnoea, or that improving oxygenation of the myocardial tissue will improve cardiac function.2, 3 However, given the lack of high‐quality evidence, there is an ongoing debate regarding the role that O2 plays in the treatment of patients with AHF.
While there is consensus among clinicians regarding the treatment of hypoxaemia (low O2 saturation levels or SpO2) in the acute setting, it is unclear whether O2 should be administered in AHF patients who have normal O2 saturation. Several physiologic studies have suggested deleterious effects of hyperoxia (i.e. high O2) on cardiac function.4, 5, 6, 7 These effects are thought to be due to high O2 stimulating the overproduction of reactive O2 species, and hyperoxia‐induced vasoconstriction that can lead to decreased coronary blood flow, and eventually to cardiac dysfunction.3 Previous studies have shown that the patients' perception of dyspnoea is not directly correlated with SpO2.2
Several major randomized controlled trials (RCTs) have shown O2 therapy to have no clinical benefits in patients without hypoxaemia presenting with acute myocardial infarction,8, 9 and others suggested possible harms.10 Recent heart failure (HF) guidelines have taken a cautious, yet variable, approach regarding recommendations on the use of supplemental O2 therapy in normoxaemic patients with AHF.11, 12, 13
We designed the High vs. Low SpO2 oxygen therapy in patients with acute Heart Failure (HiLo‐HF) pilot trial to investigate the feasibility of conducting an RCT as well as to explore the effects of supplemental O2 therapy in patients who were hospitalized with AHF.
Methods
HiLo‐HF trial was a single‐centre, pilot, open‐label RCT designed to test the feasibility, efficacy, and safety of targeting a high (high SpO2) vs. low (low SpO2) O2 saturation range. The study was approved by the Health Research Ethics Board of the University of Alberta, and written informed consent was obtained from all subjects prior to study participation. The Canadian VIGOUR Centre (thecvc.ca) managed the trial. The trial was registered at ClinicalTrials.gov (NCT03110042).
Participants
Patients who presented to the emergency department (ED) at University of Alberta Hospital with AHF were screened for this study. The inclusion and exclusion criteria of the HiLo‐HF pilot RCT were as follows.
Inclusion criteria
Patients who met the inclusion criteria were >40 years of age presenting to the ED with objective AHF (BNP > 400 pg/mL and/or chest X‐ray with pulmonary congestion) and with a planned admission for the treatment of HF as the primary diagnosis. Patients were eligible for randomization within 16 h of presenting to the ED.
Exclusion criteria
Patients on home O2, known prior hypercapnic failure (PaCO2 > 50 mmHg), asthma, primary pulmonary hypertension, requiring urgent positive pressure ventilation or intubation, or on >10 L/min O2 were excluded.
Patients who did not meet the inclusion criteria for the HiLo‐HF trial were potentially eligible for the HiLo‐HF registry (eligibility criteria for HiLo‐HF registry provided in Table S1). The pilot RCT included 50 patients (25 patients in each arm) as a demonstration of feasibility (Figure 1).
Figure 1.
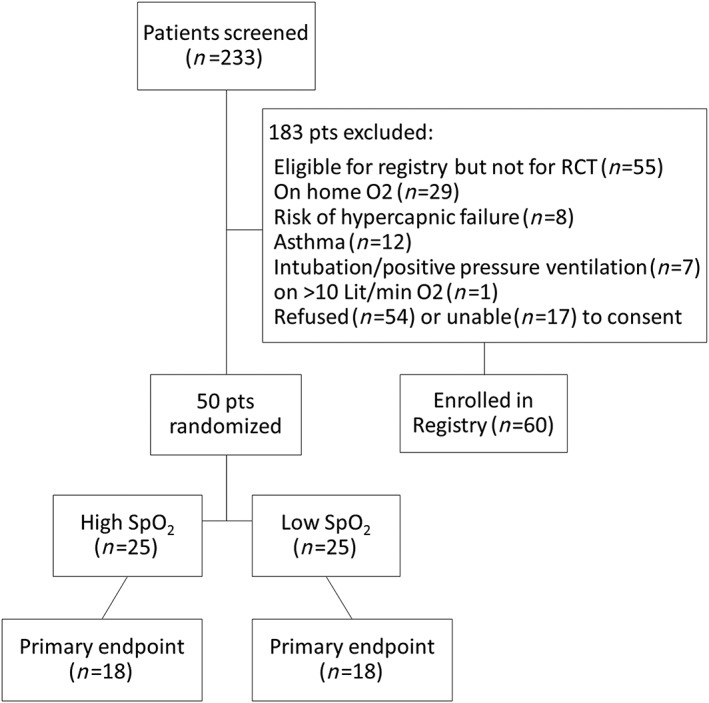
Patient flow diagram. Note: O2, oxygen; pts, patients; SpO2, peripheral oxygen saturation level.
Intervention
Patients were randomized in the ED to either high SpO2 or low SpO2 groups after providing informed written consent (Figure 2). All patients had nasal cannula placed as the usual standard of care, and patients were titrated to the pre‐specified target ranges according to the detailed instructions provided in the Supporting Information.
High SpO2 group: In the high SpO2 arm, patients were manually titrated by a trained research co‐ordinator to a target SpO2 range of ≥96%.
Low SpO2 group: In the low SpO2 arm, patients were manually titrated by a trained research co‐ordinator to the target SpO2 range of 90–92%.
Figure 2.
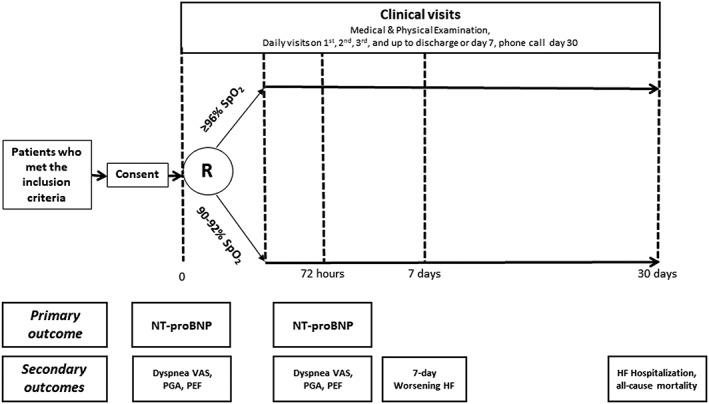
Study groups and primary/secondary endpoints. Note: HF, heart failure; NT‐proBNP, N‐terminal pro‐brain‐type natriuretic peptide; PEF, peak expiratory flow; PGA, patient global assessment; R, randomization; SpO2, peripheral oxygen saturation level; VAS, visual analogue scale.
Consented patients were randomly allocated to study groups via the automated web‐based system within REDCap.14 The allocation was concealed. Time at randomization was considered as study time zero (T0). All patients received usual standard of care with the exception of their O2 management. After 72 h, patients were switched over to usual care for O2 therapy at the discretion of the treating physician. We selected the 72 h time frame because in previous studies most patients with AHF were no longer on O2 by 72 h.2
Follow‐up
Patients were assessed on a daily basis while in hospital and on day of discharge to assess for in‐hospital safety events [clinically assessed worsening HF (WHF), or other clinical events]. Patients were followed up by telephone and health records review for a period of 30 days after hospital discharge.
Endpoints
The primary endpoint of this study was the change in N‐terminal pro‐brain‐type natriuretic peptide (NT‐proBNP) from baseline to 72 h (expressed as an absolute change and as a ratio of the baseline value).15 Secondary endpoints included (i) change in dyspnoea on visual analogue scale (VAS) from baseline to 72 h [area under the curve (AUC), mm/h]16, 17; (ii) change in global symptoms using patient global assessment (PGA) measure to 72 h (AUC, mm/h)18; (iii) change in peak expiratory flow (PEF) at 72 h (L/min)2; (iv) WHF at 7 days; (v) diuretic response as defined by weight loss up to 72 h per 40 mg of furosemide or equivalent19; and (vi) clinical event at 30 days following hospital discharge (all‐cause mortality and HF readmission).
Worsening HF was defined as signs and/or symptoms of HF that require intensification of intravenous therapy for HF, or new institution of mechanical ventilator support (continuous positive airway pressure (CPAP), non‐invasive ventilation (NIV), or intubation) or circulatory support (mechanical circulatory assist devices).16, 17
Directly measured patient‐reported outcomes (e.g. dyspnoea VAS and PGA) were collected at set time points as per Figure 2. For dyspnoea VAS, patients were asked to evaluate their breathing by marking a 10 cm vertical line, with the top labelled ‘best you have ever had’ and the bottom labelled ‘worst you have ever had’. We scored the patients' markings on a scale of 0 to 100 by measuring the distance in millimetres from the bottom of the line. A similar approach was used for PGA to evaluate patients' general well‐being. Given the open‐label design, a research co‐ordinator who was blinded to the patient's group allocation was assigned to perform or record the subjective endpoints evaluations (i.e. dyspnoea VAS and PGA). Samples for NT‐proBNP were collected via standardized laboratory procedures, processed, and frozen for batch analysis at the end of the trial. The Roche NT‐proBNP assays were performed by the University of Alberta Clinical Trials laboratory on the Elecsys 2010 (Roche Diagnostics, Manheim, Germany; reporting range 5 to 35 000 pg/mL).
Statistical analysis
All analyses were performed on the basis of intention‐to‐treat principle. Categorical variables were summarized as frequency and percentages and compared between groups using the Pearson χ 2 test or Fisher's exact test, as appropriate. Continuous variables were summarized as median with inter‐quartile range (IQR) and compared using the Mann–Whitney test. No data imputation has been performed when data are missing in one or more data points. Analysis of covariance (ANCOVA) was applied for the primary endpoint analysis. Given the non‐normal distribution, the values were log transformed prior to ANCOVA. Summary results were reported in the original scale, while the significance test result was that of the changes in log scale applying Wald statistic.
The AUC representing the change in VAS, PGA, and PEF from baseline to 72 h was computed according to the trapezoidal rule for each patient18 and was compared between the study arms using ANCOVA. Similarly, ANCOVA was applied to compare the relative changes of dyspnoea VAS AUC, PGA, and PEF from baseline to 72 h. The 30 day clinical events were estimated using the Kaplan–Meier method and were compared between the intervention arms using the log‐rank test. Patients who remained alive and without hospital readmission were censored at their last available study date. All statistical analyses were conducted using SAS statistical software (version 9.4; SAS Institute, Cary, North Carolina).
Results
Two‐hundred thirty‐three patients who presented to ED with AHF were screened for eligibility between 24 November 2016 and 27 February 2018, and 50 patients were enrolled into the HiLo‐HF pilot trial (25 per arm). Patients excluded are presented in Figure 1.
The patients enrolled in the trial had a median age of 73.5 years, 42% were women, 70% had prior history of HF, 56% had coronary artery disease (CAD), 18% had chronic obstructive pulmonary disease (COPD), and 62% were current/past smokers (Table 1). There were no clinically important differences in demographic or clinical features between arms.
Table 1.
Baseline centeracteristics of the patients in HiLo‐HF trial and registry
| HiLo registry (n = 60) | HiLo pilot RCT | ||||
|---|---|---|---|---|---|
| All (n = 50) | High SpO2 target (n = 25) | Low SpO2 target (n = 25) | P‐value | ||
| Age, year | 77 (65.5, 86) | 73.5 (67, 84) | 73.0 (70, 77) | 74 (59, 86) | 0.75 |
| Women, n (%) | 21 (35) | 21 (42) | 11 (44) | 10 (40) | 0.77 |
| Race | |||||
| Aboriginal | 1 (1.7) | 1 (2) | 0 (0) | 1 (4) | 0.14 |
| Caucasian | 49 (81.7) | 39 (78) | 20 (80) | 19 (76) | 0.73 |
| Other | 10 (16.6) | 10 (20) | 5 (20) | 5 (20) | 0.27 |
| Medical history | |||||
| Heart failure | 34 (56.7) | 35 (70) | 15 (60) | 20 (80) | 0.12 |
| Ischaemic | 21 (35) | 22 (44) | 9 (36) | 13 (52) | 0.25 |
| Non‐ischaemic | 13 (21.7) | 13 (26) | 6 (24) | 7 (28) | |
| AF/flutter | 35 (58.3) | 26 (52) | 12 (48) | 14 (56) | 0.57 |
| Cardiac devicesa | 11 (18.3) | 10 (20) | 5 (20) | 5 (20) | 1.00 |
| CAD | 26 (43.3) | 28 (56) | 14 (56) | 14 (56) | 1.00 |
| MI | 11 (42.3) | 14 (50) | 8 (57.1) | 6 (42.9) | 0.45 |
| PCI | 11 (42.3) | 10 (35.7) | 4 (28.6) | 6 (42.9) | 0.43 |
| CABG | 11 (42.3) | 9 (32.1) | 5 (35.7) | 4 (28.6) | 0.68 |
| Prior stroke | 15 (25) | 10 (20.4) | 6 (25) | 4 (16) | 0.43 |
| Diabetes | 21 (35) | 22 (44) | 8 (32) | 14 (56) | 0.08 |
| HTN | 45(75) | 35 (70) | 16 (64) | 19 (76) | 0.35 |
| COPD | 19 (31.7) | 9 (18) | 6 (24) | 3 (12) | 0.27 |
| Asthma | 2 (3.3) | 0 (0) | 0 (0) | 0 (0) | n/a |
| Smoking | 36 (60) | 31 (62) | 18 (72) | 13 (52) | 0.14 |
| Current | 4 (11.1) | 9 (29) | 6 (33.3) | 3 (23.1) | 0.53 |
| Pack/year | 20 (7.3, 38.8) | 27.5 (10, 40) | 33.8 (12.5, 45) | 25.0 (6.3, 31) | 0.14 |
| Cancer within past 5 years | 11 (18.3) | 4 (8) | 2 (8) | 2 (8) | 1.00 |
| Charlson Index | 4 (3.5, 6) | 4 (3, 6) | 4 (3, 5) | 5 (4, 6) | 0.10 |
| Baseline LVEF, n (%) | 0.33 | ||||
| ≤20% | 8 (13.3) | 4 (8) | 1 (4) | 3 (12) | |
| 21–40% | 12 (20) | 20 (40) | 9 (36) | 11 (44) | |
| 41–45% | 7 (11.7) | 3 (6) | 3 (12) | 0 (0) | |
| 46–50% | 3 (5) | 5 (10) | 3 (12) | 2 (8) | |
| ≥51% | 24 (40) | 16 (32) | 9 (36) | 7 (28) | |
| Missing | 6 (10) | 2 (4) | 0 (0) | 2 (8) | |
| Mode of ED arrival, n (%) | 0.59 | ||||
| Direct admission from clinic | 1 (1.7) | 1 (2) | 0 (0) | 1 (4) | |
| Self‐presentation | 30 (50) | 27 (54) | 14 (56) | 13 (52) | |
| EMS | 29 (48.3) | 22 (44) | 11 (44) | 11 (44) | |
| O2 in EMS, n (%) | 16 (55.2) | 12 (54.5) | 7 (63.6) | 5 (45.5) | 0.39 |
| O2 in EMS, L/min | 2.0 (2, 4) | 5.5 (3, 7) | 5.5 (4, 8) | 4.0 (2, 6) | 0.61 |
| Pre‐randomization SpO2, % | 95 (93, 97) | 94.5 (93, 97) | 94 (92, 96) | 96 (93, 98) | 0.12 |
| Pre‐randomization O2, n (%) | 26 (43.3) | 21 (42) | 11 (44) | 10 (40) | 0.77 |
| Pre‐randomization O2, L/min | 2 (2, 3.2) | 2 (2, 3) | 2 (2, 3) | 2.2 (2, 4) | 0.37 |
| Time | |||||
| From triage to admission order, h | 11.2 (8, 13.8) | 7.2 (5, 11.7) | 7.5 (4.7, 11.7) | 7.0 (5.1, 10.6) | 0.67 |
| From triage to enrolment, h | 19.2 (7.2, 21.5) | 11.4 (7.3, 13.5) | 11.4 (6.2, 13.5) | 11.3 (8.1, 14.2) | 0.47 |
| From triage to first NT‐proBNP test, h | — | 13.2 (8.0, 15.3) | 13.1 (7.5, 15.4) | 13.2 (8.2, 15.3) | 0.44 |
| From triage to disposition from ED, h | 16.2 (11.3, 21.8) | 12.8 (9, 15.7) | 12.7 (6.6, 15.3) | 14.6 (9.4, 16.8) | 0.24 |
| From triage to discharge from hospital, days | 6 (2.8, 12.4) | 6.3 (3.7, 11) | 4.7 (2.7, 6.7) | 9.5 (4.9, 19.9) | 0.01 |
Note: AF, atrial fibrillation; CABG, coronary artery bypass grafting; CAD, coronary artery disease; COPD, chronic obstructive pulmonary disease; ED, emergency department; EMS, emergency medical services; HiLo, high‐dose oxygen/low‐dose oxygen; HTN, hypertension; MI, myocardial infarction; NT‐proBNP, N‐terminal pro‐brain‐type natriuretic peptide; O2, oxygen; PCI, percutaneous coronary intervention; SpO2, peripheral oxygen saturation level.
Cardiac devices including pacemaker, implantable cardioverter defibrillator, and cardiac resynchronization therapy. Unless described otherwise, median (25th percentile, 75th percentile) are reported.
Twenty‐two (44%) patients presented via ambulance, and the median rate of administered O2 in the ambulance was 5.5 L/min among the 12 who received O2 before the ED.
Pre‐randomization SpO2 was 94% (IQR 92, 96) and 96% (IQR 93, 98), respectively, in the high and low SpO2 groups with 11 (44%) and 10 (40%) patients receiving O2 (median 2 L/min O2; IQR 2, 3).
The median time from triage to disposition from ED was 12.8 (IQR 9.0, 15.7) h, which was not different between study arms (P = 0.24).
Adherence to study protocol
At individual assessment time points, 83–94% of patients in the high SpO2 group and 5–30% of patients in the low SpO2 group were at the assigned SpO2 ranges. However, when we accounted for supplemental O2 volumes, only 14.5%, 18.7%, 6.9%, and 10.2% had non‐adherence to the study protocol (defined as SpO2 levels out of the target range ±1% with inappropriate O2 volumes administered) at 6, 24, 48, and 72 h after randomization, respectively. The rate of non‐adherence was not significantly different between study groups (Table 2 and Figure S1).
Table 2.
Adherence to study protocol in HiLo‐HF pilot randomized controlled trial
| All (n = 50) | High SpO2 target (n = 25) | Low SpO2 target (n = 25) | |
|---|---|---|---|
| % on determined SpO2 range ±1%, n (%) | |||
| 6 h | 27 (56.2) | 20 (83.3) | 7 (29.2) |
| 24 h | 25 (53.2) | 20 (87) | 5 (20.8) |
| 48 h | 25 (58.1) | 18 (85.7) | 7 (31.8) |
| 72 h | 18 (46.1) | 17 (94.4) | 1 (4.8) |
| % on O2, n (%) | |||
| 6 h | 27 (56.2) | 18 (75) | 9 (37.5) |
| 24 h | 18 (38.3) | 12 (52.2) | 6 (25) |
| 48 h | 17 (39.5) | 13 (61.9) | 4 (18.2) |
| 72 h | 16 (41) | 12 (66.6) | 4 (19) |
| O2 volume in pts treated with O2, L/min | |||
| 6 h | 2 (2, 3) | 2.5 (2, 4) | 2 (2, 3) |
| 24 h | 2.5 (2, 4) | 3 (2, 4) | 2 (1.3, 2.6) |
| 48 h | 2 (2, 3.5) | 2.5 (2, 4.7) | 2 (1.2, 3.1) |
| 72 h | 2.2 (1.1, 3.5) | 2.5 (1.6, 6.1) | 1.7 (1, 3.2) |
| Number of pts with SpO2 out of target range ±1% with inappropriate O2 volume, n (%) | |||
| 6 h | 7/48 (14.5) | 3/24 (12.5) | 4/24 (16.6) |
| 24 h | 9/48 (18.7) | 3/24 (12.5) | 6/24 (25) |
| 48 h | 3/43 (6.9) | 2/21 (9.5) | 1/22 (4.5) |
| 72 h | 4/39 (10.2) | 1/18 (5.5) | 3/21 (14.2) |
Note: HiLo, high‐dose oxygen/low‐dose oxygen; O2, oxygen; pts, patients; SpO2, peripheral oxygen saturation level.
Primary endpoint
Follow‐up (i.e. 72 h) NT‐proBNP tests were missed in 14 patients: seven patients were discharged before 72 h, one patient left against medical advice, one patient refused further blood tests, one was withdrawn from the study for safety reasons, and four were missed because of staff error. Hence, the analysis for primary endpoint was limited to the remaining 36 patients with available baseline and follow‐up NT‐proBNP results.
Baseline and 72 h NT‐proBNP levels were not statistically significantly different between groups with high and low SpO2 targets [Table 3 and Figure 3(A)]. Although numerically higher in the high SpO2 arm, NT‐proBNP change was not significantly different between study arms. Ratio change of NT‐proBNP to 72 h was also similar between trial arms (P = 0.51). Moreover, there was no difference between groups in terms of change in NT‐proBNP after adjustment for age, sex, past history of diabetes mellitus (DM), chronic kidney disease (CKD), COPD, cerebrovascular accident (CVA), and prior HF (P = 0.74).
Table 3.
Primary and secondary endpoints
| HiLo‐HF registry | HiLo‐HF pilot RCT | ||||
|---|---|---|---|---|---|
| All (n = 36) | High SpO2 target (n = 18) | Low SpO2 target (n = 18) | P‐value | ||
| NT‐proBNP | |||||
| Baseline, pg/mL | — | 14 140.1 (5570.6, 27 806.6) | 15 987.9 (6025.6, 29 785.5) | 10 262.5 (4355.3, 27 223.0) | 0.45a |
| 72 h, pg/mL | — | 7108.9 (4310.5, 17 007.0) | 6479.7 (4529.5, 12 304.9) | 10 156.8 (4001.8, 17 133.8) | 0.72a |
| ΔNT‐proBNP, pg/mL | — | −3971.4 (−11 194.5, −1049.5) | −6963.5 (−13 345.1, −1 253.3) | −2093.1 (−5692.1, −353.5) | 0.46a |
| 72 h to baseline ratio | — | 0.7 (0.5, 0.8) | 0.7 (0.3, 0.8) | 0.6 (0.5, 0.9) | 0.51b |
| Secondary endpoints | |||||
|---|---|---|---|---|---|
| HiLo‐HF registry (n = 43) | All (n = 39) | High SpO2 target (n = 18) | Low SpO2 target (n = 21) | P‐value | |
| VAS | |||||
| ΔVAS, mm | 15 (5, 35) | 10 (5, 25) | 10 (5, 25) | 10 (7, 20) | 0.86b |
| VAS AUC, mm·h | 5295 (4537, 5983) | 5160 (4380, 5932) | 5160 (4050, 6150) | 5167 (4552, 5703) | 0.73 |
| PGA | |||||
| ΔPGA, mm | 20 (10, 30) | 10 (0, 20) | 5 (0, 20) | 10 (0, 15) | 0.91b |
| PGA AUC, mm·h | 4320 (3360, 5280) | 4620 (3840, 5760) | 4860 (4290, 5775) | 4320 (3795, 5670) | 0.63 |
| PEF | |||||
| ΔPEF, L/min | 45 (0–80) | 47.5 (20, 75) | 52.5 (20, 90) | 42.5 (25, 67.5) | 0.52b |
| PEF AUC, L/min·h | 19 920 (12 795, 24 840) | 16 740 (14 610, 22 110) | 15 660 (12 540, 19 500) | 18 960 (16 080, 24 840) | 0.19 |
| Diuretic response | |||||
| 72 h/baseline weight ratio | 0.96 (0.94, 0.98) | 0.96 (0.94, 0.98) | 0.97 (0.94, 0.98) | 0.96 (0.93, 0.97) | 0.55 |
Note: AUC, area under the curve; HiLo, high‐dose oxygen/low‐dose oxygen; NT‐proBNP, N‐terminal pro‐brain‐type natriuretic peptide; PEF, peak expiratory flow; PGA, patient global assessment; RCT, randomized controlled trial; VAS, visual analogue scale.
P‐value based on comparison of high and low SpO2 groups on logarithmic scale.
Test of difference at 72 h adjusting for baseline applying the analysis of covariance model. Unless described otherwise, median (25th percentile, 75th percentile) are reported.
Figure 3.
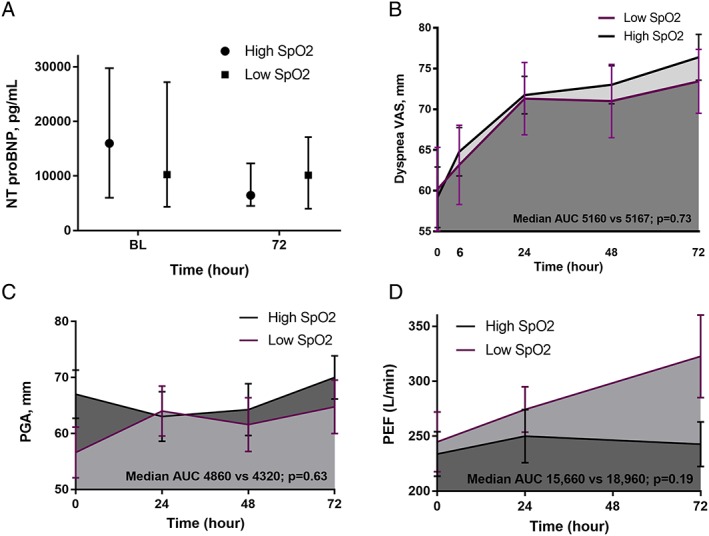
Change in NT‐proBNP levels (A), dyspnoea VAS (B), patient global assessment (C), and peak expiratory flow (D) from baseline to 72 h in groups with high and low SpO2 targets. Note: AUC, area under the curve; BL, baseline; NT‐proBNP, N‐terminal pro‐brain‐type natriuretic peptide; PEF, peak expiratory flow; PGA, patient global assessment; SpO2, peripheral oxygen saturation level; VAS, visual analogue scale.
Secondary endpoints
Dyspnoea scores
The dyspnoea VAS was not different between study arms in different study time points from 6 to 72 h after randomization (all P > 0.05) (Table 3). The change in dyspnoea from baseline to 72 h was not different between study groups, and VAS AUC was similar between groups with high and low SpO2 set points [Figure 3(B)].
Patient global assessment
Similarly, the patient symptoms according to PGA were not different between study arms in different study time points from 6 to 72 h after randomization (all P > 0.05). The change in PGA from baseline to 72 h was not different between study groups, and PGA AUC was similar between two arms of the trial [Table 3 and Figure 3(C)].
Peak expiratory flow
The PEF was not significantly different between study groups in different time points [Figure 3(D)]. In ANCOVA, adjusting for baseline values, the change in PEF from baseline to 72 h (P = 0.52) and PEF AUC (P = 0.19) was not different between two study arms.
Diuretic response
Data for both the baseline and 72 h weight were only available for 39 patients. Follow‐up/baseline weight ratio was similar between study groups [median (IQR) 0.97 (0.94, 0.98) vs. 0.96 (0.93, 0.97) in the high vs. low SpO2 arm, respectively; P = 0.55].
Worsening heart failure
Worsening HF occurred in one patient (4%) from the low SpO2 group, and there was no difference between study arms in terms of WHF (P = 1.0).
Clinical outcomes
For 30 day clinical events, no missing patient values occurred following health records surveillance, so the analysis included all 50 patients. One patient in the high SpO2 arm and two patients in the low SpO2 arm died in hospital (4.0% vs. 8.0%, P = 0.50). Among those who survived to be discharged, five patients in the high SpO2 arm and two patients in the low SpO2 arm were re‐hospitalized within 30 days after hospital discharge (20.8% vs. 8.7%, P = 0.22). The Kaplan–Meier curve showed no difference between study groups in death/re‐hospitalization at 30 days following hospital discharge (P‐value for log‐rank test = 0.36) ( Figure S3).
Length of stay
The median length of hospital stay (LOS) was 6.3 (IQR 3.7, 11.0) days in the pilot RCT, and it was significantly longer in the low SpO2 group than in the high SpO2 group (9.5 vs. 4.7 days, P = 0.011) ( Figure S2). However, after adjustment for age, sex, residence type (home vs. long‐term care facility), prior history of HF, CAD, DM, hypertension, CKD, cerebrovascular disease (CVA), atrial fibrillation, and the use of cardiac devices, the difference in the LOS was not significant between groups (P = 0.070).
Safety
One patient in the high SpO2 group was withdrawn after randomization because of high partial pressures of CO2 and potential risk of hypercapnic failure. Epistaxis related to the use of nasal cannula was reported in one patient in the high SpO2 arm, but no significant adverse event was reported in any of the two groups.
HiLo‐HF registry
Patients in the registry (n = 60) had a median age of 77 years, and 35% were women. Thirty‐four (56.7%), 26 (43.3%), and 19 (31.7%) patients had past medical history of HF, CAD, and COPD, respectively. The median time from triage to enrolment was 19.2 (IQR 7.2, 21.5) h, which was longer than that among the trial patients (median 11.4; IQR 7.3, 13.5; P < 0.001). Baseline symptoms were similar between registry and trial populations, and there was no difference in terms of VAS AUC, PGA AUC, PEF AUC, and diuretic response (all P‐values > 0.05).
Pooled cohort
Given that the trial was neutral for primary and secondary endpoints, as the next step, we pooled both trial arms and the HiLo‐HF registry to form a cohort of 110 patients who presented to ED with AHF.
In the first 24 h after randomization, SpO2 levels were inversely correlated with patients' perception of symptom measured by either dyspnoea VAS or PGA (r = −0.36, P = 0.014), but there was no correlation after that. At 24 h, patients with SpO2 < 94% had a higher (i.e. better) dyspnoea VAS (84 vs. 67, P = 0.003) and PGA (82 vs. 57, P < 0.001) than had those with SpO2 levels ≥ 94%.
Baseline BNP (n = 110) or NT‐proBNP (n = 50) levels had no correlations with SpO2 levels, dyspnoea VAS, PGA, or PEF at baseline. There was no correlation between NT‐proBNP levels and SpO2 levels, dyspnoea VAS, PGA, or PEF at follow‐up (i.e. 72 h) ( Table S6).
There was no correlation between ΔSpO2 and ΔNT‐proBNP from baseline to 72 h in the pilot cohort ( Figure S4). There was no correlation between O2 administered from baseline to 72 h and the change in NT‐proBNP levels (ΔNT‐proBNP) or the ratio change of NT‐proBNP (Δ/baseline NT‐proBNP) at the same study period ( Figure S5).
Discussion
The HiLo‐HF pilot trial is the first RCT to explore the effects of supplemental O2 therapy in patients with AHF. In this trial, titrating O2 therapy to high or low SpO2 targets did not result in changes in biomarkers, symptoms, or clinical outcomes. Regardless of group allocation, NT‐proBNP levels, patient‐reported symptoms (e.g. VAS and PGA), and pulmonary function (i.e. PEF) improved over time. In addition, while the pilot demonstrated success in recruitment, the protocol resulted in missing information for a variety of reasons. Overall, these lessons suggest that while a definitive trial is warranted, the protocol and operation of the trial should be further adjusted for pragmatic implementation.
Three small studies provided the foundation of what we know currently about the possible effects of hyperoxygenation in patients with HF. A study by Mak et al. including patients with stable CAD (n = 12) and those with HF (n = 16) showed that extreme hyperoxia (FiO2 = 1.0, PaO2 ~ 300 mmHg) was associated with impairment of cardiac relaxation and increased left ventricular filling pressures in patients with and without HF.6 Another study showed that high‐flow O2 (~5 L/min, FiO2 ~ 0.40) reduced both cardiac output and heart rate and caused a trend towards increased systemic vascular resistance than did room air (FiO2 = 0.21).7 Finally, the study of Haque et al. showed a decrease in stroke volume and an increase in pulmonary capillary wedge pressure with hyperoxia in patients admitted with AHF, and this effect started at an FiO2 level of 0.24—equivalent to 1 L/min of supplemental O2.5
The SpO2 levels in the high SpO2 arm of this study rose over time with AHF treatment, but these remained steady in the low SpO2 arm from baseline to 72 h. The manual SpO2 titration method did not induce a proper separation in SpO2 levels between the two trial arms. There were some adherence issues, mostly related to the health care professionals' non‐adherence to follow the protocol in down‐titrating O2 for those with SpO2 levels above the assigned range. These issues could be partially addressed by utilizing automated closed‐loop systems for controlling supplemental O2 delivery. These systems provide a potential solution to this problem with near‐constant adjustments and less variability of blood O2 saturations.20 They can regulate the flow of O2 on a second‐by‐second basis through a sophisticated closed‐loop algorithm that receives data input regarding peripheral O2 saturation level from pulse oximetry and reacts to that immediately with increasing or decreasing O2 flow in order to prevent under‐delivery or over‐delivery of O2.
Other studies have attempted to understand the effects associated with supplemental O2 therapy in other clinical settings.21, 22, 23 A recent meta‐analysis, pooling 7998 patients with acute myocardial infarction from eight RCTs, showed no clinical benefits on mortality or infarct size with supplemental O2 therapy as compared with room air.9 Although the only two small RCTs in patients with cardiac arrest showed no mortality difference between groups treated with high (FiO2 = 1.0) vs. conservative levels of O2,24, 25 large cohort studies and meta‐analysis of observational studies suggested decreased survival after resuscitation from cardiac arrest with hyperoxia.26, 27 Studies from the stroke setting demonstrated no benefit of liberal O2 therapy in those patients.28, 29 A total of 11 RCTs including 6366 patients with acute stroke showed a non‐significant increase in mortality at 3, 6, and 12 months with normobaric O2 as compared with room air.30 A study in the critical care setting reported an absolute risk reduction of 8.6% for the primary outcome of intensive care unit mortality with conservative O2 therapy (PaO2 = 70–100 mmHg or SpO2 = 94–98%) as compared with usual care (FiO2 ≥ 0.40, PaO2 = 100–150 mmHg, and SpO2 ≥ 97%).31 A multi‐centre RCT in patients with stable COPD and moderate desaturation at rest or during exercise showed no benefit of long‐term supplemental O2 therapy in terms of time to death or hospitalization.32 A meta‐analysis of 25 RCTs (16 037 patients) compared the outcomes of liberal vs. conservative O2 treatment in acutely ill patients and showed liberal oxygenation to increase mortality by roughly 20% in a dose‐dependent way, without improving other patient‐important outcomes such as disability or LOS.33
These findings have both clinical and health policy implications. Changes in SpO2 levels might be a harbinger of patients' deterioration in patients with AHF and hence hyperoxygenation, with masking those changes, decreases the likelihood of timely detection and intervention.33 On the other hand, given the cost of O2 therapy and the ubiquitous use of O2 in hospitalized or ED patients with AHF,2, 34 a lack of clinical benefit could mean that by departing from this practice, health care systems could save significant amount of funds from being wasted on a potentially futile intervention and directed towards other treatments with proven efficacies.
There are several limitations to this study that are noteworthy. The study is a pilot trial, and hence, it is underpowered to detect small differences between study groups. We used a relatively cautious approach of titrating O2 delivery to a specific saturation. Hence, even patients in the high SpO2 group did not experience extreme hyperoxia. The use of manual titration method and reliance on the treating team to do that were not associated with proper separation of SpO2 levels in this study. A device approach using automated closed‐loop systems has the potential to solve that issue. In this study, we did not restrict the patient population to patients with AHF who were normoxaemic at presentation and have included patients with hypoxaemia as well. This will increase the representativeness of our study population to the actual AHF population. However, there is less controversy about the use of O2 in hypoxaemic patients compared with those with normoxaemia at rest or minimal activity. We lacked data regarding patients' baseline SpO2, given that patients were recruited at ED and a proportion of patients had already received O2 in ambulance or in the ED prior to recruitment. Finally, a change in the timeline for follow‐up NT‐proBNP test from a fixed timeline (72 h) to sampling at 72 h or at discharge if earlier could have prevented a significant proportion of missing data on primary endpoint in this study.
In conclusion, we found no differences in improvements in NT‐proBNP or patient symptoms between high and low SpO2 targets in the first 72 h after admission for AHF. Further RCTs with larger sample size are warranted to determine the comparative efficacy and safety of treatment with supplemental O2 in patients with AHF.
Conflict of Interest
None.
Funding
This study was supported by operating grants from the Heart and Stroke Foundation of Canada (HSF) and Alberta Innovates ‐ Health Solutions (AIHS). None of the funding agencies had any input into design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript. F.A.M. is supported by the University of Alberta/Capital Health Chair in Cardiovascular Outcomes Research. B.H.R. was supported by a salary award as a Tier I Canada Research Chair in Evidence‐based Emergency Medicine from the Canadian Institutes of Health Research and the Government of Canada (Ottawa, ON). N.S. received a graduate studentship from Alberta Innovates ‐ Health Solutions.
Supporting information
Appendix S1. Oxygen dose adjustment in the Spo2 titration arms of study.
Table S1. Eligibility criteria for HiLo‐HF RCT and registry.
Table S2. Guideline recommendations regarding oxygen therapy in hypoxemic and normoxemic patients with heart failure.
Table S3. Baseline medications in the study populations of HiLo‐HF trial and registry.
Table S4. Total Furosemide dose at home, during the first 72 hours in hospital and on discharge.
Figure S1. SpO2 levels and O2 volumes in two study groups in different study timepoints.
Figure S2. Comparison of the distribution of length of hospital stay (in days & log‐scale) between the intervention groups.
Figure S3. Kaplan–Meier curves of clinical events consisted of in‐hospital mortality and 30‐day death/re‐hospitalization.
Table S5. Patient characteristics in the HiLo patients with available NT‐proBNP data for primary endpoint (n = 36).
Table S6. Correlation between natriuretic peptide levels and patient's saturation level and symptoms at baseline and follow‐up.
Figure S4. The relationship between ΔSpO2 and ΔNT‐BNP in HiLo‐HF cohort (n = 50).
Figure S5. The relationship between O2 from baseline to 72 hours and ΔNT‐BNP in HiLo‐HF cohort (n = 50).
Acknowledgements
We would like to thank all the participants, the Emergency Medicine Research Group (EMeRG) in the Department of Emergency Medicine at the University of Alberta for their assistance with patient screening; project leads Nubia Zepeda and Melisa Spaling; Quentin Kushnerik; and Dr. Paul M. Brown for his assistance with statistical analysis.
Sepehrvand, N. , Alemayehu, W. , Rowe, B. H. , McAlister, F. A. , van Diepen, S. , Stickland, M. , and Ezekowitz, J. A. (2019) High vs. low oxygen therapy in patients with acute heart failure: HiLo‐HF pilot trial. ESC Heart Failure, 6: 667–677. 10.1002/ehf2.12448.
References
- 1. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJV, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WHW, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62: e147–e239. [DOI] [PubMed] [Google Scholar]
- 2. Ezekowitz JA, Hernandez AF, O'Connor CM, Starling RC, Proulx G, Weiss MH, Bakal JA, Califf RM, McMurray JJV, Armstrong PW. Assessment of dyspnea in acute decompensated heart failure: insights from ASCEND‐HF (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) on the contributions of peak expiratory flow. J Am Coll Cardiol 2012; 59: 1441–1448. [DOI] [PubMed] [Google Scholar]
- 3. Sepehrvand N, Ezekowitz JA. Oxygen therapy in patients with acute heart failure friend or foe? Jacc‐Heart Failure 2016; 4: 783–790. [DOI] [PubMed] [Google Scholar]
- 4. Farquhar H, Weatherall M, Wijesinghe M, Perrin K, Ranchord A, Simmonds M, Beasley R. Systematic review of studies of the effect of hyperoxia on coronary blood flow. Am Heart J 2009; 158: 371–377. [DOI] [PubMed] [Google Scholar]
- 5. Haque WA, Boehmer J, Clemson BS, Leuenberger UA, Silber DH, Sinoway LI. Hemodynamic effects of supplemental oxygen administration in congestive heart failure. J Am Coll Cardiol 1996; 27: 353–357. [DOI] [PubMed] [Google Scholar]
- 6. Mak S, Azevedo ER, Liu PP, Newton GE. Effect of hyperoxia on left ventricular function and filling pressures in patients with and without congestive heart failure. Chest 2001; 120: 467–473. [DOI] [PubMed] [Google Scholar]
- 7. Park JH, Balmain S, Berry C, Morton JJ, McMurray JJ. Potentially detrimental cardiovascular effects of oxygen in patients with chronic left ventricular systolic dysfunction. Heart 2010; 96: 533–538. [DOI] [PubMed] [Google Scholar]
- 8. Hofmann R, James SK, Jernberg T, Lindahl B, Erlinge D, Witt N, Arefalk G, Frick M, Alfredsson J, Nilsson L, Ravn‐Fischer A, Omerovic E, Kellerth T, Sparv D, Ekelund U, Linder R, Ekström M, Lauermann J, Haaga U, Pernow J, Östlund O, Herlitz J, Svensson L, DETO2X–SWEDEHEART Investigators . Oxygen therapy in suspected acute myocardial infarction. N Engl J Med 2017; 377: 1240–1249. [DOI] [PubMed] [Google Scholar]
- 9. Sepehrvand N, James SK, Stub D, Khoshnood A, Ezekowitz JA, Hofmann R. Effects of supplemental oxygen therapy in patients with suspected acute myocardial infarction: a meta‐analysis of randomised clinical trials. Heart 2018; 104: 1691–1698. [DOI] [PubMed] [Google Scholar]
- 10. Stub D, Smith K, Bernard S, Nehme Z, Stephenson M, Bray JE, Cameron P, Barger B, Ellims AH, Taylor AJ, Meredith IT, Kaye DM, AVOID Investigators . Air versus oxygen in ST‐segment‐elevation myocardial infarction. Circulation 2015; 131: 2143–2150. [DOI] [PubMed] [Google Scholar]
- 11. Ezekowitz JA, O'Meara E, McDonald MA, Abrams H, Chan M, Ducharme A, Giannetti N, Grzeslo A, Hamilton PG, Heckman GA, Howlett JG. 2017 Comprehensive update of the Canadian Cardiovascular Society guidelines for the management of heart failure. Can J Cardiol 2017; 33: 1342–1433. [DOI] [PubMed] [Google Scholar]
- 12. Lindenfeld J, Albert NM, Boehmer JP, Collins SP, Ezekowitz JA, Givertz MM, Katz SD, Klapholz M, Moser DK, Rogers JG, Starling RC. HFSA 2010 comprehensive heart failure practice guideline. J Card Fail 2010; 16: e1–e194. [DOI] [PubMed] [Google Scholar]
- 13. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18: 891–975. [DOI] [PubMed] [Google Scholar]
- 14. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009; 42: 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zannad F, Garcia AA, Anker SD, Armstrong PW, Calvo G, Cleland JGF, Cohn JN, Dickstein K, Domanski MJ, Ekman I, Filippatos GS, Gheorghiade M, Hernandez AF, Jaarsma T, Koglin J, Konstam M, Kupfer S, Maggioni AP, Mebazaa A, Metra M, Nowack C, Pieske B, Piña IL, Pocock SJ, Ponikowski P, Rosano G, Ruilope LM, Ruschitzka F, Severin T, Solomon S, Stein K, Stockbridge NL, Stough WG, Swedberg K, Tavazzi L, Voors AA, Wasserman SM, Woehrle H, Zalewski A, McMurray JJV. Clinical outcome endpoints in heart failure trials: a European Society of Cardiology Heart Failure Association consensus document. Eur J Heart Fail 2013; 15: 1082–1094. [DOI] [PubMed] [Google Scholar]
- 16. Felker GM, Pang PS, Adams KF, Cleland JG, Cotter G, Dickstein K, Filippatos GS, Fonarow GC, Greenberg BH, Hernandez AF, Khan S, Komajda M, Konstam MA, Liu PP, Maggioni AP, Massie BM, McMurray J, Mehra M, Metra M, O'Connell J, O'Connor CM, Pina IL, Ponikowski P, Sabbah HN, Teerlink JR, Udelson JE, Yancy CW, Zannad F, Gheorghiade M, International AHFS Working Group . Clinical trials of pharmacological therapies in acute heart failure syndromes: lessons learned and directions forward. Circ Heart Fail 2010; 3: 314–325. [DOI] [PubMed] [Google Scholar]
- 17. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M. Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet 2013; 381: 29–39. [DOI] [PubMed] [Google Scholar]
- 18. Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, LeWinter M, Deswal A, Rouleau JL, Ofili EO, Anstrom KJ, Hernandez AF, McNulty S, Velazquez EJ, Kfoury AG, Chen HH, Givertz MM, Semigran MJ, Bart BA, Mascette AM, Braunwald E, O'Connor CM, NHLBI Heart Failure Clinical Research Network . Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med 2011; 364: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Voors AA, Davison BA, Teerlink JR, Felker GM, Cotter G, Filippatos G, Greenberg BH, Pang PS, Levin B, Hua TA, Severin T, Ponikowski P, Metra M, for the RELAX‐AHF Investigators . Diuretic response in patients with acute decompensated heart failure: characteristics and clinical outcome—an analysis from RELAX‐AHF. Eur J Heart Fail 2014; 16: 1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lellouche F, L'Her E. Automated oxygen flow titration to maintain constant oxygenation. Respir Care 2012; 57: 1254–1262. [DOI] [PubMed] [Google Scholar]
- 21. Clark AL, Johnson M, Fairhurst C, Torgerson D, Cockayne S, Rodgers S, Griffin S, Allgar V, Jones L, Nabb S, Harvey I. Does home oxygen therapy (HOT) in addition to standard care reduce disease severity and improve symptoms in people with chronic heart failure? A randomised trial of home oxygen therapy for patients with chronic heart failure. Health Technol Assess 2015; 19: 1–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koshy A, Pellicori P, Clark AL. The effect of increasing inspired oxygen on exercise performance in patients with chronic heart failure. Heart 2016; 102: 597–601. [DOI] [PubMed] [Google Scholar]
- 23. Shah P, Pellicori P, Rimmer S, Rigby AS, Clark AL. Effect of increased inspired oxygen on exercise performance in patients with heart failure and normal ejection fraction. Int J Cardiol 2018; 268: 166–169. [DOI] [PubMed] [Google Scholar]
- 24. Young P, Bailey M, Bellomo R, Bernard S, Dicker B, Freebairn R, Henderson S, Mackle D, McArthur C, McGuinness S, Smith T, Swain A, Weatherall M, Beasley R. HyperOxic Therapy OR NormOxic Therapy after out‐of‐hospital cardiac arrest (HOT OR NOT): a randomised controlled feasibility trial. Resuscitation 2014; 85: 1686–1691. [DOI] [PubMed] [Google Scholar]
- 25. Kuisma M, Boyd J, Voipio V, Alaspaa A, Roine RO, Rosenberg P. Comparison of 30 and the 100% inspired oxygen concentrations during early post‐resuscitation period: a randomised controlled pilot study. Resuscitation 2006; 69: 199–206. [DOI] [PubMed] [Google Scholar]
- 26. Damiani E, Adrario E, Girardis M, Romano R, Pelaia P, Singer M, Donati A. Arterial hyperoxia and mortality in critically ill patients: a systematic review and meta‐analysis. Crit Care 2014; 18: 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Helmerhorst HJF, Roos‐Blom M‐J, van Westerloo DJ, Abu‐Hanna A, de Keizer NF, de Jonge E. Associations of arterial carbon dioxide and arterial oxygen concentrations with hospital mortality after resuscitation from cardiac arrest. Crit Care 2015; 19: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ali K, Warusevitane A, Lally F, Sim J, Sills S, Pountain S, Nevatte T, Allen M, Roffe C. The stroke oxygen pilot study: a randomized controlled trial of the effects of routine oxygen supplementation early after acute stroke—effect on key outcomes at six months. PLoS ONE 2014; 8: e59274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pountain SJ, Roffe C. Does routine oxygen supplementation in patients with acute stroke improve outcome? BMJ 2012; 345: e6976. [DOI] [PubMed] [Google Scholar]
- 30. Ding J, Zhou D. The effect of normobaric oxygen in patients with acute stroke: a systematic review and meta‐analysis. Neurol Res 2018; 40: 433–444. [DOI] [PubMed] [Google Scholar]
- 31. Girardis M, Busani S, Damiani E, Donati A, Rinaldi L, Marudi A, Morelli A, Antonelli M, Singer M. Effect of conservative vs conventional oxygen therapy on mortality among patients in an intensive care unit: the Oxygen‐ICU Randomized Clinical Trial. JAMA 2016; 316: 1583–1589. [DOI] [PubMed] [Google Scholar]
- 32. Albert RK, Au DH, Blackford AL, Casaburi R, Cooper JA Jr, Criner GJ, Diaz P, Fuhlbrigge AL, Gay SE, Kanner RE, MacIntyre N, Martinez FJ, Panos RJ, Piantadosi S, Sciurba F, Shade D, Stibolt T, Stoller JK, Wise R, Yusen RD, Tonascia J, Sternberg AL, Bailey W. A randomized trial of long‐term oxygen for COPD with moderate desaturation. N Engl J Med 2016; 375: 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chu DK, Kim LH, Young PJ, Zamiri N, Almenawer SA, Jaeschke R, Szczeklik W, Schünemann HJ, Neary JD, Alhazzani W. Mortality and morbidity in acutely ill adults treated with liberal versus conservative oxygen therapy (IOTA): a systematic review and meta‐analysis. Lancet 2018; 391: 1693–1705. [DOI] [PubMed] [Google Scholar]
- 34. Siemieniuk RAC, Chu DK, Kim LH, Güell‐Rous MR, Alhazzani W, Soccal PM, Karanicolas PJ, Farhoumand PD, Siemieniuk JL, Satia I, Irusen EM. Oxygen therapy for acutely ill medical patients: a clinical practice guideline. BMJ 2018; 363: k4169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Oxygen dose adjustment in the Spo2 titration arms of study.
Table S1. Eligibility criteria for HiLo‐HF RCT and registry.
Table S2. Guideline recommendations regarding oxygen therapy in hypoxemic and normoxemic patients with heart failure.
Table S3. Baseline medications in the study populations of HiLo‐HF trial and registry.
Table S4. Total Furosemide dose at home, during the first 72 hours in hospital and on discharge.
Figure S1. SpO2 levels and O2 volumes in two study groups in different study timepoints.
Figure S2. Comparison of the distribution of length of hospital stay (in days & log‐scale) between the intervention groups.
Figure S3. Kaplan–Meier curves of clinical events consisted of in‐hospital mortality and 30‐day death/re‐hospitalization.
Table S5. Patient characteristics in the HiLo patients with available NT‐proBNP data for primary endpoint (n = 36).
Table S6. Correlation between natriuretic peptide levels and patient's saturation level and symptoms at baseline and follow‐up.
Figure S4. The relationship between ΔSpO2 and ΔNT‐BNP in HiLo‐HF cohort (n = 50).
Figure S5. The relationship between O2 from baseline to 72 hours and ΔNT‐BNP in HiLo‐HF cohort (n = 50).