

Abstract
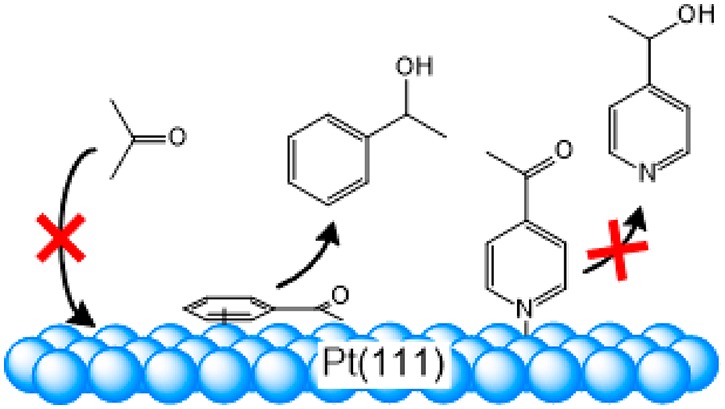
This paper studies the electrochemical hydrogenation of the carbonyl functional group of acetophenone and 4-acetylpyridine at platinum single-crystal electrodes. Comparison with results obtained for the hydrogenation of acetone featuring an isolated carbonyl functional group reveals the influence of the phenyl ring and the pyridine ring, respectively. Lack of acetone adsorption at Pt(111) and Pt(100) due to a weak interaction between surface and carbonyl functional group renders these surfaces inactive for the hydrogenation of acetone. Adsorption through a strong interaction with the phenyl ring of acetophenone activates the Pt(111) and Pt(100) surfaces for hydrogenation of the acetyl substituent. In agreement with previous results for acetone reduction, the Pt(100) surface is specifically active for the hydrogenolysis reaction, breaking the C–O bond, whereas the other surfaces only hydrogenate the carbonyl functionality. In contrast to the phenyl ring, the pyridine ring has a very different effect: due to the dominant interaction of the N atom of the pyridine ring with the platinum electrode, a vertical adsorption mode is realized. The resulting large physical distance between the carbonyl functional group and the electrode surface inhibits the hydrogenation at all platinum surfaces. This also holds for the Pt(110) electrode, which is otherwise active for the electrochemical hydrogenation of the isolated carbonyl functional group of aliphatic ketones. Our results show how the combination of molecular structure of the reactant and surface structure of the catalyst determine the selective electroreduction of functionalized ketones.
Introduction
In recent years there has been increased interest in the electrochemical hydrogenation of organic molecules that feature two or more functional groups. Studies on the electrochemical reduction of acetophenone,1,2 benzaldehyde,2−4 5-hydroxymethyllfurfural,5−7 and ethyl pyruvate8,9 have been performed in the context of the electrorefining of biomass-derived compounds. To design efficient electrochemical processes for these conversions, it is important to utilize our knowledge on the electrochemistry of simple organic molecules10−16 and on adsorption structures13,17−22 to build a conceptual understanding on how (inert) functional groups can influence the electrochemistry of complex organic molecules.
In a previous paper we investigated the electrochemical hydrogenation of acetone at platinum single-crystal electrodes.23 This study showed that acetone reduction only proceeds at step sites, while the crystallographic orientation of the steps determines whether the reduction only yields 2-propanol or in addition also propane. Computational studies indicate that the reduction of the carbonyl functional group is favorable at the Pt(111) and Pt(100) electrode. Nonetheless, no reduction proceeds in practice, since the adsorption of acetone is unfavorable at these surfaces.23 This raises the question whether the hydrogenation of the carbonyl functional group is possible when an (inert) functional group in the substrate molecules interacts strongly with the Pt(111) and Pt(100) surface, thus enabling activation of the carbonyl functional group.
Since benzene binds to metal surfaces through an interaction between its π-electron system and the electrode surface,13,17−20 it can be expected that acetophenone adsorbs to platinum electrodes through the phenyl ring. Pyridine, on the other hand, can adsorb to metal electrodes in two ways: horizontally through an interaction via its π-electron system (similar to benzene)24 or vertically through the N atom.18,21,22 At platinum electrodes, pyridine generally adsorbs in a vertical mode.18,21,22 No changes to this adsorption mode would be expected when an acetyl substituent as in 4-acetylpyridine is introduced.
Here, we compare the hydrogenation of the carbonyl functional group of acetone, acetophenone, and 4-acetylpyridine to establish the impact of an inert functional group (phenyl ring and pyridine ring). We show that adsorption through the phenyl ring in a horizontal mode facilitates the hydrogenation of the carbonyl functional group, while adsorption through the N atom of the pyridine ring in a vertical mode inhibits the hydrogenation reaction. Our results provide a new conceptual understanding about how one functional group in the substrate molecule can alter the reactivity of another functional group through an influence on the adsorption mode. In addition we show that combination of the molecular structure of the reactant and the surface structure of the catalyst determines the selectivity of the electroreduction of functionalized ketones.
Experimental Section
Chemicals and Materials
The blank electrolyte was prepared from Milli-Q water and either H2SO4 or HClO4 (both suprapure, Merck). The ketones used were acetophenone (Laboratory Reagent grade, Fisher Scientific), 4-acetylpyridine (Alfa Aesar 98%), and acetone (Sigma-Aldrich, HPLC garde). All solutions were freed from oxygen by purging with argon (6.0, Linde). All potentials were measured versus a reversible hydrogen electrode (RHE) in contact with the blank electrolyte. A platinum wire was used as a counter electrode. Cyclic voltammograms (CV) were recorded on an Iviumstat potentiostat (Ivium Technologies).
Single-Crystal Electrodes
Bead-type single-crystal electrodes (Icryst) were used to collect CVs and to conduct electrochemical mass spectroscopy measurements. Experiments involving long-term electrolysis were conducted with disk-shaped single-crystal electrodes of 1 cm in diameter. Both types of single-crystal electrodes were prepared by the Clavilier method:25 The crystals were flame annealed and cooled down over Milli-Q water in an atmosphere of 20% hydrogen and 80% argon. Afterward, the electrode was dipped into water saturated with the hydrogen–argon mixture and transferred with a droplet attached to the surface to the electrochemical cell. Glassware used in this study was kept in a cleaning solution of acidic permanganate for at least 8 h. Prior to use, the glassware was rinsed with acidic peroxide solution and boiled several times in Milli-Q water.
Online Electrochemical Mass Spectrometry (OLEMS)
The details of our OLEMS setup have been reported elsewhere:26 A Teflon frit inserted into an KelF-tip is brought in close contact with the surface of the electrode, which resides in a hanging meniscus configuration. The Teflon frit serves as an interface between the vacuum of the mass spectrometer and the electrolyte. Volatile products formed in the course of the electrochemical reaction evaporate through the tip into the vacuum of the mass spectrometer. A PrismaPlus mass spectrometer (Pfeiffer Vacuum) operating in the multiple-ion detection mode was used in this study.
Surface-Enhanced Raman Spectroscopy (SERS)
Electrodes used for SERS studies were prepared by a protocol established by Zou and Weaver:27 First, a polycrystalline gold electrode is roughened by 20 cycles of potential steps between 1.2 (5 s) and −0.6 V (20 s) vs Ag/AgCl in a solution of 0.5 M KCl. Galvanostatic platinum deposition is conducted with a current density of 400 μA/cm2 (20 s) from a solution of 100 μM H2PtCl6 in 0.35 M Na2HPO4. SERS spectra at this electrode were collected at a LabRam HR800 (Horiba Jobin Yvon) confocal microscope, which featured a HeNe laser (632.81 nm). A detailed description of the setup and the methodology is given by Lai et al.28 Each spectrum shown is the average of 16 spectra taken. In order to avoid beam damage the laser beam was attenuated by a factor of 10 resulting in a measured beam energy at the location of the Raman cell of 0.28 W.
High-Pressure Liquid Chromatography (HPLC) Measurements
Samples for HPLC measurements were generated through long-term electrolysis. Electrolysis was conducted in an H-cell of which the working electrode compartment was equipped with an reference electrode. The compartments of the working electrode and counter electrode were separated with a Nafion membrane. The electrolyte volume of the working electrode compartment was 20 mL. From this volume a sample of 100 μL was taken for HPLC analysis.
For compound separation after long-term electrolysis of acetophenone an Aminex HPX 87-H (Bio-Rad) column filled with Sugar SH1011 (Shodex) in combination with an eluent of 0.5 mM H2SO4 was used. The temperature of the column was kept at 85 °C, and compounds were detected by measuring the refractive index.
For compound separation after long-term electrolysis of acetylpyridine an Phenomenex (Kintex) column was used. In this case the eluent consisted to 30% of acetonitrile and 70% of a solution of 10 mM Na2HPO4 in water with the pH adjusted to 8.2. The temperature of the column was kept at 60 °C, and compounds were detected by measuring the absorption of UV light with a wavelength of 230 nm. Prior to HPLC analysis the samples were neutralized by adding appropriate amounts of Na2HPO4.
Results
Figure 1 compares
the cyclic voltammograms (CV) obtained at Pt(111) in an electrolyte
of 0.1 M H2SO4 with those obtained in the presence
of 0.01 M acetone (Figure 1A), acetophenone (Figure 1B), or 4-acetylpyridine (Figure 1C). In the CV obtained in the blank electrolyte
a sharp spike at approximately 0.5 V appears. This spike is due to
the transition of an unordered sulfate overlayer on the Pt(111) electrode
to a 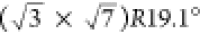 superstructure.29,30 The sulfate spike can only be observed when large (111)-terraces
are present that are not covered by any other adsorbate than sulfate.
In the presence of acetone neither the sulfate spike nor hydrogen
adsorption is suppressed, indicating that acetone does not adsorb
to the Pt(111) surfaces. Indeed, we have shown in a previous publication
that it is thermodynamically unfavorable for the carbonyl functional
group of acetone to interact with surface sites that feature highly
coordinated platinum atoms.23
superstructure.29,30 The sulfate spike can only be observed when large (111)-terraces
are present that are not covered by any other adsorbate than sulfate.
In the presence of acetone neither the sulfate spike nor hydrogen
adsorption is suppressed, indicating that acetone does not adsorb
to the Pt(111) surfaces. Indeed, we have shown in a previous publication
that it is thermodynamically unfavorable for the carbonyl functional
group of acetone to interact with surface sites that feature highly
coordinated platinum atoms.23
Figure 1.

Comparison of the CV obtained at Pt(111) in the blank electrolyte of 0.1 M H2SO4 (black curve) and in the presence of 0.01 M ketone (red curve). (A) CV in the absence (black) and presence of acetone (red). (B) CV in the absence (black) and presence of acetophenone (red) or benzene (0.01 M, blue). (C) CV in the absence (black) and presence of 4-acetylpyridine (red) or pyridne (0.01 M, blue). (Insets) Enlarged potential region just positive of hydrogen evolution. Currents are normalized to the geometric electrode area, which should correspond to the real surface area for an atomically smooth single-crystal electrode.
In contrast to acetone, the presence of carbonyl compounds featuring an aromatic ring suppresses the sulfate spike completely. Furthermore, hydrogen adsorption is shifted to lower potentials in the presence of the aromatic compounds. Both the suppression of the hydrogen adsorption and the sulfate spike indicate that acetophenone and 4-acetylpyridene adsorb strongly to the Pt(111) surface. The absence of acetone adsorption at Pt(111) suggests that the adsorption of the aromatic carbonyl compounds does not proceed through an interaction between the surface and the carbonyl functional group. This is supported by the CVs in Figure 1 obtained in the presence of 10 mM benzene (blue curve in Figure 1B) and pyridine (blue curve in Figure 1C). These compounds affect the hydrogen adsorption and the sulfate in a similar way as the respective carbonyl compound. This suggests that the adsorption of the carbonyl compounds is dominated by an interaction between the Pt(111) surface and the phenyl/pyridine ring. In the discussion of Figures S1 and S2 in the Supporting Information, we arrive at the same conclusion for the adsorption at Pt(100) and Pt(110) surfaces. It is difficult to determine from the CVs in Figure 1 whether a reduction proceeds at the Pt(111) electrode. In the potential region near the onset of hydrogen evolution there is no additional shoulder in the CV that would indicate the reduction of the carbonyl compound (cf. insets in Figure 1). The hydrogenation reaction might take place in parallel to hydrogen evolution. Small differences in the current around 0 V between the CVs obtained in the presence and absence of the carbonyl compound might result from experimental inaccuracies when bringing the electrode in the hanging meniscus configuration. Figures S1 and S2 show that the situation is similar for the Pt(100) and the Pt(110) electrodes.
In the presence of acetophenone, a reversible process around 0.03 V is observed in the CV of Pt(111). The linear scan rate dependence of the peak current (as shown in Figure S3 of the Supporting Information) indicates that this pertains to a surface process that is not related to the reduction of acetophenone. The same behavior is observed in the presence of benzene and has been ascribed previously to the adsorption of hydrogen underneath the benzene adlayer.20
In order to check whether acetophenone is hydrogenated at Pt(111) and at other basal planes, we conducted OLEMS measurements and HPLC analysis after long-term electrolysis. Figure 2 shows the OLEMS results obtained for the reduction of acetophenone at the three basal planes (111), (110), and (100) of platinum. Upon cycling into the potential region of hydrogen evolution, a signal evolves for mass 91 at Pt(100), while no such signal appears in the ionic current for the same mass at Pt(111) and Pt(110). The ionic current for mass 91 does not follow the faradaic current in Figure 2C. This is due to a complex diffusion patterns of products in the electrolyte from the electrode surface to the inlet of the mass spectrometer resulting in a smeared out and delayed signal for ethylbenzene. Although it is not possible to quantify our OLEMS data, the appearance of a signal for mass 91 gives qualitative proof for the reduction of acetophenone to the corresponding hydrocarbon (ethylbenzene) at the Pt(100) electrode. Neither the Pt(111) nor the Pt(110) electrode is able to deoxygenate the carbonyl functional group of acetophenone.
Figure 2.

CVs obtained with a sweep rate of 0.5 mV/s at Pt(111) (A), Pt(110) (B), and Pt(100) (C) in an electrolyte of 0.3 M H2SO4 containing 0.03 M acetophenone. (D–F) Ionic current for mass 91. Arrows indicate the direction of the potential scan. Currents are normalized on the geometric electrode area, which should correspond to the real surface area for an atomically smooth single-crystal electrode.
The electrochemical hydrogenation of acetophenone could also yield ethylcyclohexane and cyclohexylmethylketone. Their formation should come along with a signal in the ionic current for masses 112 and 126, respectively. However, no signals were observed for either mass, which shows that ethylbenzene is the only volatile product formed in the course of acetophenone reduction.
Other possible hydrogenation products of acetophenone, such as 1-phenylethanol and 1-cyclohexylethanol, cannot be detected by OLEMS, as alcohols do not pass readily from the aqueous phase into the vacuum of the mass spectrometer. Therefore, we electrolyzed an acetophenone-containing electrolyte for 2 h at each platinum basal plane and conducted an HPLC analysis of the electrolyte afterward. Figure 3A shows the HPLC results of samples obtained for each of the basal planes, and Figure 3B shows standards of 0.5 mM acetophenone, 1-phenylethanol and 1-cyclohexylethanol. After long-term electrolysis all samples contain a compound that elutes after 70.5 min, which indicates the presence of 1-phenylethanol. The compound eluting between 134 and 149 min is acetophenone. Despite the broad signal due to acetophenone, the presence of 1-cyclohexylethanol with a retention time of 132 min should have been detected if it had formed in the course of the electrochemical reduction of acetophenone. The current–time transients corresponding to the data presented in Figure 3 and for electrolysis conducted at 0.01 and 0.05 V are shown in Figures S4–S6 in the SI. Figure S7 in the SI also shows that with decreasing potential the current efficiency for 1-phenylethanol production drops from 35% at 0.05 V to 15% at 0.01 V as hydrogen evolution accelerates faster with potential than ketone reduction.
Figure 3.

(A) HPLC analysis of a sample taken from an electrolyte of 0.1 M H2SO4 containing 0.03 M acetophenone that has been electrolyzed at 0.03 V for 2 h at a platinum electrode (surface structure as indicated). (B) HPLC analysis of 0.5 mM acetophenone (black), 0.5 mM 1-pheylethanol (red), and cyclohexylmethylketone (blue) in water.
Our HPLC data show that all three basal planes of platinum can reduce the carbonyl functional group of acetophenone to the corresponding alcohol. The OLEMS data show that the Pt(100) electrode has the unique ability among the platinum basal planes to catalyze in addition to the hydrogenolysis. Neither HPLC nor OLEMS gives any indication for the formation of cyclohexylmethylketone, 1-cyclohexylethanol, or ethylcyclohexane which would form if the phenyl ring was hydrogenated in addition to the carbonyl functional group. This is in line with literature results that show that platinum single crystals hydrogenate benzene only at potentials lower than 0 V.13,17 Hence, selective reduction of the carbonyl functional group in acetophenone is possible at platinum electrodes, and the selective formation of 1-phenylethanol is possible by the appropriate choice of the surface structure of the electrocatalyst.
As for acetophenone, a solution of 0.1 M acid containing 0.03 M 4-acetylpyridine was electrolyzed for 2 h at a potential of 0.03 V at each of the three platinum basal planes. Because of their basic character, pyridine derivatives and piperidine derivatives, which are potential hydrogenation products of acetylpyridine, interact very strongly with water and do not pass from the aqueous phase into the gas phase. Therefore, OLEMS is not a suitable tool to detect hydrogenation products of 4-acetylpyridine, and we only conducted HPLC analysis after long-term electrolysis. The HPLC results for each sample taken after electrolysis of a 4-acetylpyridne containing electrolyte with the indicated single-crystal electrode are shown in Figure 4A, and Figure 4B shows the HPLC data for the 10 mM standards of 4-acetylpyridine eluting after 7.25 min (black curve), of 1-(pyridine-4-yl)ethan-1-ol eluting after 5.87 min (red curve), and of 4-ethylpyridne eluting after 13.36 min (blue curve). Aside from a broad peak around 7 min due to the elusion of 4-acetylpyridine, no signal is observed due to a compound eluting after 5.87 min (alcohol), 13.36 min (hydrocarbon), or any other compound. The results of Figure 4 show that 4-acetylpyridne cannot be reduced at any of the three platinum basal planes under conditions that show the hydrogenation of acetophenone at each of the three platinum basal planes.
Figure 4.

(A) HPLC analysis of a sample taken from an electrolyte of 0.1 M H2SO4 containing 0.03 M 4-acetylpyridine that has been electrolyzed at 0.03 V for 2 h with a platinum electrode (surface structure as indicated). Corresponding current time transients are shown in Figure S8 in the SI. (B) HPLC analysis of 0.01 M 4-acetylpyridne (black), 0.01 M 1-(pyridine-4-yl)ethan-1-ol (red), and 4-ethylpyridne (blue) in water.
Surface-enhanced Raman spectroscopy (SERS) was employed to obtain information on the adsorption of acetophenone and 4-acetylpyridine on a polycrystalline platinum electrode. Figure 5 shows the SERS spectra of acetophenone and 4-acetylpyridine adsorbed on a platinum electrode at 0.4 V. The full spectra (from 200 to 3500 cm–1) of acetophenone and 4-acetylpyridine are given in Figures S9 and S10, respectively. We assign the intense band at 1022 cm–1 in the red curve of Figure 5 to the in-plane ring-breathing mode of 4-acetylpyridine that occurs at 993 cm–1 in the spectrum of the free compound.31 Upon adsorption the ring-breathing mode of pyridine derivatives blue shifts due to the surface bonding through the N atom.32 The significant blue shift of 29 cm–1 of the ring-breathing mode indicates therefore that 4-acetylpyridine binds through the N atom to the platinum surface in a vertical adsorption mode. This is in agreement with the general tendency of pyridine to bind vertically to platinum surfaces.18,21,22 However, considering the molecular geometry of 4-acetylpyridine, an interaction between the surface and the carbonyl functional group cannot be realized when the molecule adsorbs in a vertical adsorption mode. This is also reflected by the band position of the symmetrical C=O stretch vibration occurring at 1691 cm–1 in the red curve of Figure 5. The band is red shifted by only 2 cm–1 with respect to its location in the spectrum of the pure substance.31 This suggests that the strength of the C=O bond is not reduced significantly as would be expected if the carbonyl functional group interacted strongly with the platinum surface.
Figure 5.
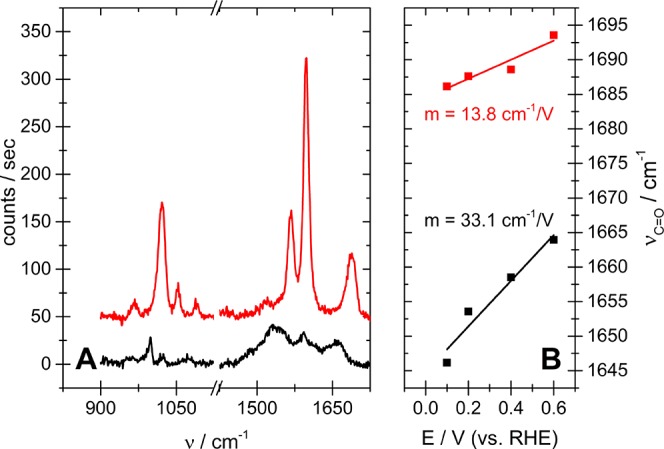
(A) SERS spectra obtained at a platinum-covered gold electrode in 0.1 M H2SO4 containing 10 mM 4-acetylpyridin (red curve) or 0.01 M acetophenone (black curve). (B) Band position of the C=O stretch vibration as a function of the applied potential. Slope of the linear regression (m) gives the Stark tuning experienced by the carbonyl functional group.
The ring-breathing mode of acetophenone appears at 999 cm–1 in the black curve of Figure 5, corresponding to a red shift of 1 cm–1 compared to the band position at 1000 cm–1 in the spectrum of the bulk compound (cf. Figure S10B in the Supporting Information). Benzene derivatives experience a red shift of the ring-breathing mode by about 1–14 cm–1 upon adsorption through the aromatic π-electron system.33 The red shift of the ring-breathing mode of acetophenone indicates therefore that the compound binds horizontally to the platinum surface. This adsorption mode agrees with other benzene derivatives, which generally realize flat adsorption geometries on metal electrodes.13,17−22,34 Different from the spectrum of 4-acetylpyridine, for which the ring C–C stretching vibration (at 1567 and 1599 cm–1, respectively) and the symmetrical C=O-stretching vibration are well separated, bands corresponding to these modes merge into a multiplet in the spectrum of acetophenone. However, in the multiplet, a peak at 1664 cm−1 appears, which is close to the band position of the symmetrical C=O stretch vibration at 1678 cm−1 in the spectrum of bulk acetophenone, and which is therefore assigned to the same mode of adsorbed acetophenone. The red shift of 12 cm–1 indicates a slight weakening of the C=O bond. This might be due to the adsorption of acetophenone at those surface sites that do not interact strongly with carbonyl functional groups (such as Pt(111) and Pt(100) terraces23). Adsorption at step and kink sites that interact more strongly with the carbonyl functional group should result in a considerable weakening of the C=O bond and a stronger red shift of the corresponding band. However, these bands would be hidden in the multiplet.
The SERS spectra of Figure 5 support the notion derived from the CVs in Figure 1 as well as those in Figures S1 and S2 of the Supporting Information: adsorption of acetophenone and 4-acetylpyridine is dominated by an interaction between the surface and the phenyl and pyridine ring, respectively. Furthermore, we can derive from the band position of the ring-breathing mode that acetophenone realizes a horizontal adsorption geometry at platinum, while 4-acetylpyridine binds vertically to the same surface. In the discussion of Figures S9 and S10 in the Supporting Information we derive these adsorption geometries also from the relative band intensity of the ring-breathing mode.
Additional information on the adsorption mode can be derived from Figure 5B, which shows how the applied potential affects the band position of the symmetrical carbonyl stretch vibration. The carbonyl functional group of 4-acetylpyridine experiences a relatively weak Stark tuning slope of 13.8 cm–1/V as compared to a Stark tuning slope of 33.1 cm–1/V for acetophenone. The relatively small Stark shift indicates that the electric field caused by the potential difference between the electrode and the electrolyte has already declined considerably at the location of the carbonyl functional group. Electrodes covered with pyridine in the vertical adsorption mode have a small double-layer capacity since the electric field is shielded effectively by the pyridine ring.24,35−38 The pyridine ring of adsorbed 4-acetylpyridine can shield the carbonyl functional group from the electric field only when the ring resides between the electrode and the carbonyl functional group. Such a geometry is only realized in a more or less vertical adsorption mode. Hence, the low Stark tuning slope agrees with a vertical adsorption mode of 4-acetylpyridine. The higher Stark tuning slope experienced by the carbonyl functional group of acetophenone means that it is located closer to the surface where a change in the electrode potential causes a large change of the electric field. This is in line with the flat adsorption mode derived from the band position of the ring-breathing mode, though it is difficult to derive from the Stark effect alone more information on the adsorption mode.
Discussion
We first compare the results obtained for the electrochemical hydrogenation of acetophenone with our previous results for the electrochemical hydrogenation of acetone at platinum single-crystal electrodes.23 Stepped Pt(111) crystals reduce acetone nearly exclusively to 2-propanol, while stepped Pt(100) electrodes reduce acetone in addition to propane. Qualitatively the same selectivity pattern is observed here for the hydrogenation of acetophenone. HPLC and OLEMS data in Figures 2 and 3 show that acetophenone is reduced selectively to the corresponding alcohol at Pt(111) and Pt(110) electrodes, while ethylbenzene is formed (in addition to 1-phenylethanol) in the course of acetophenone reduction at the Pt(100) electrode. The finding that the Pt(100) electrode catalyzes the deoxygenation/hydrogenolysis of acetophenone is another example of the interesting ability of (100) terraces to make and break bonds.39,40 An important difference with acetone reduction is that all three basal planes of platinum show at least some moderate activity for the hydrogenation of the carbonyl functional group of acetophenone, while pristine Pt(111) and Pt(100) electrodes do not display any activity for acetone reduction.23 The inactivity of these surfaces for acetone reduction has been assigned to their inability to bind acetone.23Figure 1 shows that acetophenone binds to the Pt(111) surface, whereas acetone does not. CV and SERS data suggest that adsorption of acetophenone to Pt(111) and Pt(100) is brought about by an interaction between the surface and the phenyl ring. The horizontal adsorption mode of acetophenone forces the carbonyl functional group in close contact with the Pt(111) surface. In this situation, the Pt(111) surface has the ability to reduce the carbonyl functional group.
The important implication of this conclusion is that the electrochemical conversion of one functional group can be enabled by a strong interaction between the electrode surface and another inert functional group in the same substrate molecule. On the other hand, our results obtained for 4-acteylpyridine show that this also requires a certain adsorption geometry. Although the interaction between the electrode surface and the pyridine ring brings about adsorption as well, no activation of the carbonyl functional group can be achieved. In the vertical adsorption mode the acetyl functional group is directed away from the surfaces, which impairs the electron transfer. This is also true for the Pt(110) electrode, which is otherwise active for the hydrogenation of the isolate carbonyl functional group in acetone, that is, adsorption through the N atom of 4-acetylpyridne inhibits the hydrogenation reaction.
Conclusion
In this paper, we investigated the electrochemical hydrogenation of functionalized ketones on platinum single-crystal electrodes. The insight that the a secondary functional group affects the reducibility of the carbonyl functional group through an influence on the adsorption geometry is rather intuitive: the vertical adsorption mode brought about by the adsorption through the N atom of the pyridine ring in 4-acetylpyridine creates a physical distance between the carbonyl functional group and the electrode surface. This renders the Pt(110) electrode, otherwise most active for the reduction of acetone, inactive for the reduction of the carbonyl functional group of 4-acetylpyridine. The horizontal adsorption mode of the phenyl ring in acetophenone activates the otherwise inactive Pt(111) electrode. Here adsorption through the phenyl ring reduces the physical distance between the electrode and the carbonyl functional group. Despite its simplicity, the finding that a secondary, inert functional group (here phenyl and pyridine ring) affects the reactivity of another (here carbonyl functional group) by defining the adsorption mode on the electrode poses a new concept for electrocatalysis.
In addition, we showed that the electrocatalytic hydrogenation of acetophenone displays the same sensitivity toward the crystallographic orientation of the platinum electrode as observed earlier for acetone.23 Selective formation of the alcohol is observed when electrodes with an (111) or an (110) surface structure are employed. Platinum electrodes with an (100) surface structure perform in addition to the hydrogenolysis to the hydrocarbon. The high structural sensitivity of (100) surfaces for hydrogenolysis is in agreement with previous results on acetone electroreduction23 and more generally with the ability of (100) surface sites to break or make bonds between C, N, and O.39,40
Acknowledgments
We gratefully acknowledge financial support from The Netherlands Organization for Scientific Research (NWO) in the framework of the fund New Chemical Innovations (project: 731.015.204 ELECTROGAS) with financial support of Akzo Nobel Chemicals, Shell Global Solutions, Magneto Special Anodes (Evoqua Water Technologies) and Elson Technologies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b05397.
CV data at Pt(100) and Pt(110) in the presence of acetone, acetophenenone, 4-acetylpyridine, benzene, and pyridine; scan rate dependence of CV at Pt(15,15,14) in the presence of acetophenone; full SERS spectra of acetophenone and 4-acetylpyridine (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Villalba M.; del Pozo M.; Calvo E. J. Electrocatalytic hydrogenation of acetophenone and benzophenone using palladium electrodes. Electrochim. Acta 2015, 164, 125–131. 10.1016/j.electacta.2015.02.113. [DOI] [Google Scholar]
- Sanyal U.; Lopez-Ruiz J.; Padmaperuma A. B.; Holladay J.; Gutiérrez O. Y. Electrocatalytic Hydrogenation of Oxygenated Compounds in Aqueous Phase. Org. Process Res. Dev. 2018, 22 (12), 1590–1598. 10.1021/acs.oprd.8b00236. [DOI] [Google Scholar]
- Cantu D. C.; Padmaperuma A. B.; Nguyen M.-T.; Akhade S. A.; Yoon Y.; Wang Y.-G.; Lee M.-S.; Glezakou V.-A.; Rousseau R.; Lilga M. A. A Combined Experimental and Theoretical Study on the Activity and Selectivity of the Electrocatalytic Hydrogenation of Aldehydes. ACS Catal. 2018, 8 (8), 7645–7658. 10.1021/acscatal.8b00858. [DOI] [Google Scholar]
- Song Y.; Sanyal U.; Pangotra D.; Holladay J. D.; Camaioni D. M.; Gutiérrez O. Y.; Lercher J. A. Hydrogenation of benzaldehyde via electrocatalysis and thermal catalysis on carbon-supported metals. J. Catal. 2018, 359, 68–75. 10.1016/j.jcat.2017.12.026. [DOI] [Google Scholar]
- Kwon Y.; de Jong E.; Raoufmoghaddam S.; Koper M. T. M. Electrocatalytic Hydrogenation of 5-Hydroxymethylfurfural in the Absence and Presence of Glucose. ChemSusChem 2013, 6 (9), 1659–1667. 10.1002/cssc.201300443. [DOI] [PubMed] [Google Scholar]
- Kwon Y.; Birdja Y. Y.; Raoufmoghaddam S.; Koper M. T. M. Electrocatalytic Hydrogenation of 5-Hydroxymethylfurfural in Acidic Solution. ChemSusChem 2015, 8 (10), 1745–1751. 10.1002/cssc.201500176. [DOI] [PubMed] [Google Scholar]
- Chadderdon X. H.; Chadderdon D. J.; Matthiesen J. E.; Qiu Y.; Carraher J. M.; Tessonnier J.-P.; Li W. Mechanisms of Furfural Reduction on Metal Electrodes: Distinguishing Pathways for Selective Hydrogenation of Bioderived Oxygenates. J. Am. Chem. Soc. 2017, 139 (40), 14120–14128. 10.1021/jacs.7b06331. [DOI] [PubMed] [Google Scholar]
- Rees N. V.; Taylor R. J.; Jiang Y. X.; Morgan I. R.; Knight D. W.; Attard G. A. In Situ Surface-Enhanced Raman Spectroscopic Studies and Electrochemical Reduction of α-Ketoesters and Self Condensation Products at Platinum Surfaces. J. Phys. Chem. C 2011, 115 (4), 1163–1170. 10.1021/jp106495b. [DOI] [Google Scholar]
- Hazzazi O. A.; Huxter S. E.; Taylor R.; Palmer B.; Gilbert L.; Attard G. A. Electrochemical studies of irreversibly adsorbed ethyl pyruvate on Pt{h k l} and epitaxial Pd/Pt{h k l} adlayers. J. Electroanal. Chem. 2010, 640 (1–2), 8–16. 10.1016/j.jelechem.2009.12.026. [DOI] [Google Scholar]
- Zinola C. F.; Rodríguez J. L.; Arévalo M. C.; Pastor E. A DEMS study of the electroreduction and oxidation of 3-buten-2-one and 2-butanone adsorbates on platinum in sulphuric solutions. J. Electroanal. Chem. 1998, 454 (1–2), 161–172. 10.1016/S0022-0728(98)00265-4. [DOI] [Google Scholar]
- Zinola C. F.; Castro Luna A. M. Electrochemical Behavior and Semiempirical Approach to the Electroadsorption of 3-Buten-2-one and 2-Butanone on Platinum. J. Colloid Interface Sci. 1999, 219 (2), 260–274. 10.1006/jcis.1999.6480. [DOI] [PubMed] [Google Scholar]
- Schmiemann U.; Müller U.; Baltruschat H. The influence of the surface structure on the adsorption of ethene, ethanol and cyclohexene as studied by DEMS. Electrochim. Acta 1995, 40 (1), 99–107. 10.1016/0013-4686(94)00243-T. [DOI] [Google Scholar]
- Baltruschat H.; Ernst S. Molecular Adsorbates at Single-Crystal Platinum-Group Metals and Bimetallic Surfaces. ChemPhysChem 2011, 12 (1), 56–69. 10.1002/cphc.201000644. [DOI] [PubMed] [Google Scholar]
- Lai S. C. S.; Koper M. T. M. Electro-oxidation of ethanol and acetaldehyde on platinum single-crystal electrodes. Faraday Discuss. 2009, 140 (0), 399–416. 10.1039/B803711F. [DOI] [PubMed] [Google Scholar]
- Lai S. C. S.; Koper M. T. M. The Influence of Surface Structure on Selectivity in the Ethanol Electro-oxidation Reaction on Platinum. J. Phys. Chem. Lett. 2010, 1 (7), 1122–1125. 10.1021/jz100272f. [DOI] [Google Scholar]
- Bondue C. J.; Koper M. T. M. A mechanistic investigation on the electrocatalytic reduction of aliphatic ketones at platinum. J. Catal. 2019, 369, 302–311. 10.1016/j.jcat.2018.11.019. [DOI] [Google Scholar]
- Hartung T.; Baltruschat H. Differential electrochemical mass spectrometry using smooth electrodes: adsorption and hydrogen/deuterium exchange reactions of benzene on platinum. Langmuir 1990, 6 (5), 953–957. 10.1021/la00095a012. [DOI] [Google Scholar]
- Soriaga M. P.; Hubbard A. T. Determination of the orientation of adsorbed molecules at solid-liquid interfaces by thin-layer electrochemistry: aromatic compounds at platinum electrodes. J. Am. Chem. Soc. 1982, 104 (10), 2735–2742. 10.1021/ja00374a008. [DOI] [Google Scholar]
- Rodríguez J. L.; Pastor E. A comparative study on the adsorption of benzyl alcohol, toluene and benzene on platinum. Electrochim. Acta 2000, 45 (25–26), 4279–4289. 10.1016/S0013-4686(00)00561-2. [DOI] [Google Scholar]
- Jerkiewicz G.; DeBlois M.; Radovic-Hrapovic Z.; Tessier J.-P.; Perreault F.; Lessard J. Underpotential Deposition of Hydrogen on Benzene-Modified Pt(111) in Aqueous H2SO4. Langmuir 2005, 21 (8), 3511–3520. 10.1021/la048290a. [DOI] [PubMed] [Google Scholar]
- Cai W.-B.; She C.-X.; Ren B.; Yao J.-L.; Tian Z.-W.; Tian Z.-Q. Surface Raman spectroscopic investigation of pyridine adsorption at platinum electrodes—effects of potential and electrolyte. J. Chem. Soc., Faraday Trans. 1998, 94 (20), 3127–3133. 10.1039/a803240h. [DOI] [Google Scholar]
- Bilmes S. A.; Rubim J. C.; Otto A.; Arvia A. J. SERS from pyridine adsorbed on electrodispersed platinum electrodes. Chem. Phys. Lett. 1989, 159 (1), 89–96. 10.1016/S0009-2614(89)87460-3. [DOI] [Google Scholar]
- Bondue C. J.; Calle-Vallejo F.; Figueiredo M. C.; Koper M. T. M. Structural principles to steer the selectivity of the electrocatalytic reduction of aliphatic ketones on platinum. Nature Catalysis 2019, 2 (3), 243–250. 10.1038/s41929-019-0229-3. [DOI] [Google Scholar]
- Yang D. F.; Stolberg L.; Lipkowski J.; Irish D. E. Adsorption of pyridine at the Au(210)—solution interface. J. Electroanal. Chem. 1992, 329 (1), 259–278. 10.1016/0022-0728(92)80221-O. [DOI] [Google Scholar]
- Clavilier J.; Armand D.; Sun S. G.; Petit M. Electrochemical adsorption behaviour of platinum stepped surfaces in sulphuric acid solutions. J. Electroanal. Chem. Interfacial Electrochem. 1986, 205 (1), 267–277. 10.1016/0022-0728(86)90237-8. [DOI] [Google Scholar]
- Wonders A. H.; Housmans T. H. M.; Rosca V.; Koper M. T. M. On-line mass spectrometry system for measurements at single-crystal electrodes in hanging meniscus configuration. J. Appl. Electrochem. 2006, 36 (11), 1215–1221. 10.1007/s10800-006-9173-4. [DOI] [Google Scholar]
- Zou S.; Weaver M. J. Surface-Enhanced Raman Scattering on Uniform Transition-Metal Films: Toward a Versatile Adsorbate Vibrational Strategy for Solid-Nonvacuum Interfaces?. Anal. Chem. 1998, 70 (11), 2387–2395. 10.1021/ac9800154. [DOI] [PubMed] [Google Scholar]
- Lai S. C. S.; Kleyn S. E. F.; Rosca V.; Koper M. T. M. Mechanism of the Dissociation and Electrooxidation of Ethanol and Acetaldehyde on Platinum As Studied by SERS. J. Phys. Chem. C 2008, 112 (48), 19080–19087. 10.1021/jp807350h. [DOI] [Google Scholar]
- Funtikov A. M.; Linke U.; Stimming U.; Vogel R. An in-situ STM study of anion adsorption on Pt(111) from sulfuric acid solutions. Surf. Sci. 1995, 324 (1), L343–L348. 10.1016/0039-6028(94)00774-8. [DOI] [Google Scholar]
- Funtikov A. M.; Stimming U.; Vogel R. Anion adsorption from sulfuric acid solutions on Pt(111) single crystal electrodes. J. Electroanal. Chem. 1997, 428 (1), 147–153. 10.1016/S0022-0728(96)05051-6. [DOI] [Google Scholar]
- Zhai C.; Cui F.; Liu X. A combined experimental and theoretical study on vibrational spectra of 2-acetylpyridine. Spectrochim. Acta, Part A 2015, 134, 90–95. 10.1016/j.saa.2014.06.086. [DOI] [PubMed] [Google Scholar]
- Xu M.-M.; Yan L.-J.; Wei C.; Yang F.-Z.; Yuan Y.-X.; Yao J.-L.; Han S.-Y.; Wang M.; Gu R.-A. Isomer-dependent adsorption of cyanopyridines on platinum probed by surface-enhanced Raman spectroscopy. J. Electroanal. Chem. 2011, 662 (2), 426–431. 10.1016/j.jelechem.2011.09.020. [DOI] [Google Scholar]
- Gao P.; Weaver M. J. Surface-enhanced Raman spectroscopy as a probe of adsorbate-surface bonding: benzene and monosubstituted benzenes adsorbed at gold electrodes. J. Phys. Chem. 1985, 89 (23), 5040–5046. 10.1021/j100269a031. [DOI] [Google Scholar]
- Singh N.; Nguyen M.-T.; Cantu D. C.; Mehdi B. L.; Browning N. D.; Fulton J. L.; Zheng J.; Balasubramanian M.; Gutiérrez O. Y.; Glezakou V.-A.; Rousseau R.; Govind N.; Camaioni D. M.; Campbell C. T.; Lercher J. A. Carbon-supported Pt during aqueous phenol hydrogenation with and without applied electrical potential: X-ray absorption and theoretical studies of structure and adsorbates. J. Catal. 2018, 368, 8–19. 10.1016/j.jcat.2018.09.021. [DOI] [Google Scholar]
- Stolberg L.; Morin S.; Lipkowski J.; Irish D. E. Adsorption of pyridine at the Au(111)-solution interface. J. Electroanal. Chem. Interfacial Electrochem. 1991, 307 (1), 241–262. 10.1016/0022-0728(91)85552-Z. [DOI] [Google Scholar]
- Stolberg L.; Lipkowski J.; Irish D. E. Adsorption of pyridine at the Au (100)-solution interface. J. Electroanal. Chem. Interfacial Electrochem. 1987, 238 (1), 333–353. 10.1016/0022-0728(87)85184-7. [DOI] [Google Scholar]
- Stolberg L.; Lipkowski J.; Irish D. E. Adsorption of pyridine at the Au(110)-solution interface. J. Electroanal. Chem. Interfacial Electrochem. 1990, 296 (1), 171–189. 10.1016/0022-0728(90)87241-B. [DOI] [Google Scholar]
- Stolberg L.; Lipkowski J.; Irish D. E. Adsorption of pyridine at the Au(311)—solution interface. J. Electroanal. Chem. 1992, 322 (1), 357–372. 10.1016/0022-0728(92)80088-L. [DOI] [Google Scholar]
- Li H.; Calle-Vallejo F.; Kolb M. J.; Kwon Y.; Li Y.; Koper M. T. M. Why (1 0 0) Terraces Break and Make Bonds: Oxidation of Dimethyl Ether on Platinum Single-Crystal Electrodes. J. Am. Chem. Soc. 2013, 135 (38), 14329–14338. 10.1021/ja406655q. [DOI] [PubMed] [Google Scholar]
- Li H.; Li Y.; Koper M. T. M.; Calle-Vallejo F. Bond-Making and Breaking between Carbon, Nitrogen, and Oxygen in Electrocatalysis. J. Am. Chem. Soc. 2014, 136 (44), 15694–15701. 10.1021/ja508649p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.