

Abstract
Gastrointestinal cancer is one of the most common types of cancer with high mortality rates. Mutations in several genes are reportedly involved in the progression of gastrointestinal cancer, including tumor protein 53 (TP53), APC regulator of WNT signaling pathway (APC), KRAS proto-oncogene GTPase (KRAS) and erb-b2 receptor tyrosine kinase 2 (ERBB2). Most notably, there are numerous mutations in DNA repair genes, including mismatch repair (MMR) and homologous recombination (HR) genes. The focus of the present study was to investigate the effects of MMR and HR gene mutations on genomic instability in gastrointestinal cancer. Using targeted capture and massively parallel genomic sequencing, 137 gastrointestinal cancer patients were analyzed for somatic single-nucleotide variants (SNVs) and insertion-deletion (indel) mutations in the exon regions of 183 cancer driver genes, including 4 MMR genes [MutL homolog MLH1, MLH2, MLH6 and PMS1 homolog 2, mismatch repair system component (PMS2)] and 15 HR genes [BRCA1 DNA repair associated (BRCA1), BRCA2 DNA repair associated (BRCA2), ATM serine/threonine kinase (ATM), phosphatase and tensin homolog, BLM RecQ like helicase, FA complementation group A, FA complementation group C, FA complementation group D2, FA complementation group E, FA complementation group F, FA complementation group G, nibrin, partner and localizer of BRCA2 and Werner syndrome RecQ like helicase]. A number of frequently mutated genes, including but not limited to, mechanistic target of rapamycin kinase, neurofibromin 1, APC and, in particular, DNA repair genes, including PMS2, ATM and BRCA2, were identified. Frequency analysis was performed based on the SNVs and indels in the 183 genes to indirectly indicate the relative status of genomic instability in each patient. Correlation analysis suggested that MMR and HR gene mutations directly affected the count of SNVs and indels. Overall, 56 of the gastrointestinal cancer patients (40%) were found to have an inactivation mutation (stopgain/frameshift/splicing) in one or more of the four MMR genes, whereas 112 patients (82%) harbored at least one HR gene inactivation mutation. In addition, patients with MMR or HR inactivation variants had more SNVs and indels compared with patients with no such mutations. No other clinical characteristics (including sex and age) appeared to have a statistically significant impact. Further analysis indicated that different MMR or HR genes exerted distinct effects on genomic instability. The results obtained in the current study may lay a foundation for investigations into the tumorigenic process and for the development of novel therapeutic strategies for the treatment of gastrointestinal cancer.
Keywords: gastrointestinal cancer, mismatch repair, homologous recombination repair, inactivation mutation, genomic instability
Introduction
Gastrointestinal (GI) cancer, including colorectal cancer (CRC) and gastric cancer (GC), are common malignant tumors with poor prognosis and high mortality rates among both males and females in China (1). Following lung and liver cancer, CRC and GC result in the greatest numbers of mortalities (2). Despite the use of colonoscopy and gastroscopy in cancer screening, patients with GI cancer tend to be diagnosed at an advanced stage (3). The mortality of gastrointestinal cancer has declined in several Western countries since the introduction of cancer screening programs. Additionally, the removal of adenomas, early detection of cancerous lesions and the availability of more effective therapies for early stage disease have contributed to a decrease in the mortality rate (4). Cancer patients diagnosed at an early stage generally receive prompt treatment and have an improved prognosis compared with those diagnosed at later stages (5,6). Several National Comprehensive Cancer Network member institutions advise the use of molecular screening methods, including immunohistochemical analysis, for mismatch repair (MMR) protein expression or microsatellite instability (MSI) analysis on all newly diagnosed patients with CRCs (7–10). These recommendations reflect the importance of MMR genes and the MSI index in cancer management.
DNA repair can repair damaged DNA and maintain genomic stability. Multiple DNA repair genes, including the MMR and homologous recombination (HR) genes, are involved in the development of GI cancer (11,12). Defective MMR (dMMR) has been revealed to lead to MSI, resulting in high numbers of somatic mutations in intestinal carcinogenesis (13,14).
Clinically, MSI evaluation is recommended in patients with stage II CRC in order to inform treatment decision-making regarding chemotherapy administration (15). Germline mutations in MMR genes have been implicated in Lynch syndrome, which is a highly penetrant, autosomal-dominant inherited cancer predisposition syndrome characterized by the early onset of cancers in the colon as well as at extra-colonic sites such as the endometrium, ovaries, stomach, small intestine, pancreas, urinary tract and brain (16–18). Furthermore, somatic mutations in MMR genes have been reported to result in dMMR (19–22). A number of studies have reported patients with CRC patients harbored germline mutations in HR genes, including ATM serine/threonine kinase (ATM), BRCA1 DNA repair associated (BRCA1), BRCA2 DNA repair associated (BRCA2) and partner and localizer of BRCA2 (PALB2) (23–26). A previous study (27) reported somatic pathogenic mutations among all tumor lineages and revealed that the HR deficiency frequency was ~13% in all solid tumor types (n=53,619) and ~6.3% in CRC. Another report indicated that 41–50% of ovarian carcinomas are estimated to exhibit HR deficiency. However, the frequency of HR deficiency varies according to the method utilized for its evaluation (germline mutations, somatic mutations or HRD score) and histological subtype (28).
Previous studies demonstrated the importance of the MMR and HR genes on GI cancer (29–31). However, the characteristics of mutations in these genes remain unclear, and so is the range of effects they may exert on genomic instability. In the present study, targeted capture and massively parallel next generation sequencing (NGS) technologies were used to study the mutations of 183 cancer-promoting genes in 137 patients with GI cancer, with a particular focus on the mutational status of MMR and HR genes.
Materials and methods
Patients and collection of clinical samples
A total of 137 patients with stage III/IV CRC and GC were included in the current study, which was approved by Ethics Committee of Second Hospital of Anhui Medical University (Hefei, China), between May 2017 and May 2018. Written informed consent was obtained from each patient prior to sample collection. The samples were obtained from 92 males and 45 females (median age, 60 years; age range, 27–84 years). Tissues were fixed with 10% formalin at room temperature for 8 h. A total of 92 formalin-fixed, paraffin-embedded (FFPE) tumor sections collected following surgery were retrieved. In addition, 45 blood samples for circulating cell-free DNA (cfDNA) extraction were collected prior to surgery from patients receiving no prior chemotherapy or radiotherapy. A total of 13 genomic DNA fragments were extracted from patient-matched peripheral blood (when available) and were used as matched normal controls. All FFPE tumor samples were confirmed to have >20% tumor cells upon H&E staining (data not shown). The clinical and pathological characteristics of the patients were obtained from hospital records. Basic patient information is summarized in Table I.
Table I.
Distribution of SNV and indel mutations in patients with gastrointestinal cancer.
| Characteristic | Number of specimens | Number of SNV and indel mutationsa (mean ± SEM) | P-value |
|---|---|---|---|
| Sex | 0.8010 | ||
| Female | 45 | 210.7±24.04 | |
| Male | 92 | 210.0±15.62 | |
| Age | 0.2044 | ||
| ≥55 | 86 | 224.5±17.88 | |
| <55 | 51 | 186.2±17.77 | |
| Tumor type | 0.9105 | ||
| Colorectal | 75 | 213.8±17.54 | |
| Gastric | 62 | 205.8±19.77 | |
| MMR gene mutation status | 4.476×10−08 | ||
| Inactivation (+)b | 56 | 280.2±20.11 | |
| Inactivation (−)c | 81 | 161.8±15.10 | |
| HR gene mutation status | 2.581×10−05 | ||
| Inactivation (+)d | 112 | 232.6±15.06 | |
| Inactivation (−)e | 25 | 110.0±10.23 |
Number of SNV and indel Mutations included all the mutations site of SNV and indel (stopgain/frameshift/splicing/nonsynonymous/synonymous/non-frameshift) in 183 panel genes
Inactivation mutation (+) included individuals with at least one type of stopgain/frameshift/splicing mutation in MMR genes
Inactivation mutation (−) included individuals with none of stopgain/frameshift/splicing mutation in MMR genes
Inactivation mutation (+) included individuals with at least one type of stopgain/frameshift/splicing mutation in HR genes
Inactivation mutation (−) included individuals with none of stopgain/frameshift/splicing mutation in HR genes. SNV, single-nucleotide variant; indel, insertion-deletion; SEM, standard error of the mean; MMR, mismatch repair; HR, homologous recombination. Statistical significance was tested using the Kruskal-Wallis test.
DNA extraction
FFPE tumor sections (40 µm) were treated with 100% xylene (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at room temperature, followed by 100% ethanol (32). Deparaffinized samples were then suspended in proteinase K-containing buffer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Following extraction using phenol-chloroform (V:V=1:1; Sigma-Aldrich; Merck KGaA), DNA samples were treated with ethanol for precipitation and resuspended in deionized water. For cfDNA extraction from blood samples, Cell-Free DNA BCT tubes (Streck, Omaha, NE, USA) were used for the collection of 10 ml blood samples. Validation of the adopted protocols for this study has been performed previously (33). In brief, 1.2 ml of plasma was collected from each patient using two stages of centrifugation at 4°C at 1,600 × g for 10 min, prior to cfDNA extraction. cfDNA extraction was performed with the QIAamp Circulating Nucleic Acid Kit (Qiagen, Inc., Valencia, CA, USA), according to the methodology described by the manufacturer. The processing of white blood cell (WBC) DNA as a control was performed as follows: Using 2 ml of total peripheral blood, DNA was extracted using the Flexigene DNA kit (Qiagen, Inc.). Quantification of isolated DNA samples was performed using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Inc.), and by fluorimetry, using Qubit dsDNA high-sensitivity and/or broad-range assay kits (Thermo Fisher Scientific, Inc.).
Library construction and sequencing
The FD-180 panel targeted for exon regions of 183 tumor driver genes (not shown) was used for the generation of sequencing libraries with the Illumina platforms (Kapa Biosystems; Roche Diagnostics, Basel, Switzerland) and the SeqCap EZ Choice Library (Roche NimbleGen, Inc., Madison, WI, USA), with DNA fragments and cfDNA employed in the library construction following the manufacturer's protocol. DNA sequencing was completed with the Illumina NextSeq 500 system at a depth of 10,000X (cfDNA) and 3,000X (FFPE). All the operations are carried out in accordance with the product manual.
Variant calling and analysis
Raw data were processed into clean FASTQ output with Flexbar (https://sourceforge.net/projects/flexbar/) through trimming of adapter sequences and the removal of low-quality reads (average quality score <15). Raw reads were checked for data quality using FASTQ (www.bioinformatics.babraham.ac.uk). Plots of quality (Q) scores across all bases in reads indicated the majority of positions had Q≥20. Q scores are logarithmically related to the base calling error probabilitie s(P), Q=−10 log10P. A lower base call accuracy of 99% (Q20) will have an incorrect base call probability of 1 in 100, meaning that every 100 bp sequencing read will likely contain an error. Raw reads were then trimmed for adapter contamination with Trimmomatic (version 0.32; http://usadellab.org/cms/). Leading and trailing low-quality bases (Q<3) were removed. Reads were also scanned with a 4-base-wide sliding window and the following bases were cut when the average Q per base dropped to <15. Finally, only reads >50 bases were kept for subsequent analysis. Either patient-matched peripheral blood (when available) or in-batch pooled FFPE normal controls were used for mutation calling. Paired clean reads, following Trimmomatic treatment, were aligned against the reference genome hg19 (hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips) using Burrows-Wheelers Aligner (version 0.7.13) (34). Calibration was then performed on the remaining reads, which were realigned via the Genome Analysis Toolkit (version 4.0.3.0) (35). Genome Analysis Toolkit was used to perform base quality score recalibration (BQSR). BQSR is a process by which machine learning is applied to a model to score errors empirically and adjust the quality scores accordingly.
Analysis of the realigned BAM files and detection of somatic single-nucleotide variants (SNVs) and insertion/deletion (indel) mutations was performed using MuTect (version 1.7.0) (36). Normal germline variants were filtered out using the Single Nucleotide Polymorphism Database (37) or the Exome Aggregation Consortium database (http://exac.broadinstitute.org/). Default parameter settings were used for all programs. The elimination of erroneous base calls and generation of final mutations was performed by variation frequency (>0.5%).
Variation analysis
The SNV and indel mutations (including stopgain, frameshift, splicing, synonymous, non-synonymous and non-frameshift types) of the 183-gene panel were analyzed in the 137 patients with GI cancer. MMR and HR genes were then investigated for SNV and indel mutations, respectively.
MMR and HR genes inactivation analysis
A total of four MMR (MLH1, MSH2, MSH6 and PMS2), and 15 HR [(BRCA1, BRCA2, ATM, phosphatase and tensin homolog (PTEN), BLM RecQ like helicase (BLM), BRCA1 interacting protein C-terminal helicase 1 (BRIP1), FA complementation group A (FANCA), FA complementation group C (FANCC), FA complementation group D2 (FANCD2), FA complementation group E (FANCE), FA complementation group F (FANCF), FA complementation group G (FANCG), nibrin (NBN), PALB2 and Werner syndrome RecQ like helicase (WRN)] genes were selected and analyzed according to previous studies (18–24). In the present study, inactivation mutations included stopgain, frameshift and splicing somatic mutations. Any MMR or HR gene with an inactivation mutation was defined as MMR inactivation-positive (MMR+) or HR inactivation-positive (HR+).
Statistical analysis
The results are presented as mean ± standard deviation. Statistical significance was tested using the Kruskal-Wallis test. P<0.05 indicated a statistically significant difference. A number of SNV and indel mutation sites in all MMR or HR genes to represent the MMR or HR mutational status were used. The number of SNV and indel mutation sites of the 183-gene panel in each patient was used to predict the degree of genomic instability. To investigate the effects of the mutational status of MMR and HR genes, the Pearson's correlation test between MMR and HR mutational status and genomic instability was used. All statistical analyses were performed using GraphPad Prism (version 5.0; GraphPad, San Diego, CA, USA) and SPSS software (version 17.0; SPSS Inc., Chicago, IL, USA).
Results
Molecular mutation characteristics in 137 patients with GI cancer
To investigate the molecular characteristics of GI cancer, we analyzed the SNV and indel mutations of 183 genes in 137 patients using NGS technology. As shown in Fig. 1, the number of mutations among the 137 patients was 24–613 (median, 115). Several MMR gene inactivation mutations were identified. Certain HR genes had a high frequency of inactivation mutations including, BRCA2 (32.9%; n=45), ATM (29.2%; n=40), BRCA1 (17.5%; n=24) and FANCD2 (15.3%; n=21). These results indicated the different effects of MMR and HR genes on genomic instability.
Figure 1.
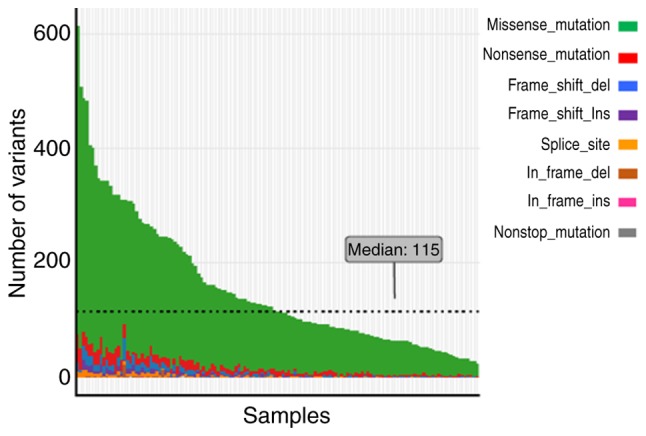
Gene mutations in 183 genes among 137 patients with gastrointestinal cancer. The eight colors represent the different types of mutations as presented on the right hand side of the figure. Ordinate and abscissa represent the total number of mutations and samples respectively. Ins, insertion; del, deletion.
The 20 most frequently mutated genes are shown in Fig. 2A. The mechanistic target of rapamycin kinase was the most commonly mutated gene, detected in 85% of the patients. Other genes, including PMS2 and regulator of WNT signaling pathway (APC), also exhibited a high mutation frequency. Certain HR genes were among the 20 most commonly mutated genes, including ATM, FANCD2, ATR serine/threonine kinase (ATR), BRCA2, BRCA1 and FANCA (Fig. 2A). The mutational status of MMR and HR genes is illustrated in Fig. 2B. As presented, PMS2 mutation was detected in 83% of the patients. ATM, FANCD2 and BRCA2 were also found to have a mutation frequency of ~80% (Fig. 2B), whereas FANCF had a relatively lower (29%) mutation frequency.
Figure 2.


Molecular characteristics of somatic mutations in the 137 patients with gastrointestinal cancer. (A) All SNV and indel mutations (stopgain/frameshift/splicing/nonsynonymous/synonymous/non-frameshift) of the 20 most commonly mutated genes in patients and gene mutation frequency were analyzed. The x-axis represents each of the 137 samples and the y-axis represents the proportion of the gene mutation samples. The highest numbers of SNV and indel mutations in each patient; left, gene mutation frequency in the 137 patients; right, number of patients with gene mutations. (B) The mutational status of mismatch repair and homologous recombination genes. The x-axis represents each of the 137 samples; the y-axis represents the proportion of the gene mutation samples. SNV, single-nucleotide variant; indel, insertion-deletion; del, deletion; ins, insertion; MTOR, mechanistic target of rapamycin kinase; PMS2, PMS1 homolog 2 mismatch repair system component; NF1, neurofibromin 1; APC, APC regulator of WNT signaling pathway; ATM, ATM serine/threonine kinase; FANCD2, FA complementation group D2; ATR, ATR serine/threonine kinase; BRCA2, BRCA2 DNA repair associated; DICER1, dicer 1 ribonuclease III; ROS1, ROS proto-oncogene 1 receptor tyrosine kinase; FLT1, fms related tyrosine kinase 1; TSC2, TSC complex subunit 2; SETBP1, SET binding protein 1; ALK, ALK receptor tyrosine kinase; IGF1R, insulin like growth factor 1 receptor; PLCG2, phospholipase C γ 2; FANCA, FA complementation group A; BRCA1, BRCA1 DNA repair associated; ERBB3, erb-b2 receptor tyrosine kinase 3; JAK1, Janus kinase 1; BRIP1, BRCA1 interacting protein C-terminal helicase 1; MSH6, mutS homolog 6; PALB2, partner and localizer of BRCA2; BLM, BLM RecQ like helicase; WRN, Werner syndrome RecQ like helicase; MSH2, mutS homolog 2; MLH1, mutL homolog 1; FANCE, FA complementation group E; FANCG, FA complementation group G; NBN, nibrin; FANCF, FA complementation group F.
Mutational status of MMR and HR genes positively indicates genomic instability in GI cancer
A Pearson's correlation test between MMR and HR mutational status and genomic instability was performed. The results of correlation analysis indicated a significant association between MMR gene mutational status and genomic instability (R2=0.703; P=2.2×10−16). Patients harboring high numbers of SNV and indel mutations in MMR genes had a higher frequency of mutations in all 183-gene panels (Fig. 3A). A significant correlation was also observed in the HR group (R2=0.901; P=2.2×10−16; Fig. 3A). These results demonstrated that the mutational status of MMR and HR genes is positively correlated with genomic instability.
Figure 3.

Effects of MMR and HR gene deficiency on genomic instability in gastrointestinal cancer. (A) Number of SNV and indel mutation sites in MMR or HR genes positively correlated with mutation sites in the 183-gene panel. (B) Inactivation mutation frequency in MMR and HR genes in the 137 patients with gastrointestinal cancer. (C) Differences in the number of SNV and indel mutations in a panel of 183 genes between MMR or HR gene inactivation mutation positive and negative groups. The numbers in brackets on the x-axis is number of samples with inactivation mutations. MMR, mismatch repair; HR, homologous recombination; SNV, single-nucleotide variant; indel, insertion-deletion; MLH1, mutL homolog 1; MSH2, mutS homolog 2; MSH6, mutS homolog 6; PMS2, PMS1 homolog 2 mismatch repair system component; BRCA1, BRCA1 DNA repair associated; BRCA2, BRCA2 DNA repair associated; ATM, ATM serine/threonine kinase; PTEN, phosphatase and tensin homolog; BLM, BLM RecQ like helicase; BRIP1, BRCA1 interacting protein C-terminal helicase 1; FANCA, FA complementation group A; FANCC, FA complementation group C; FANCD2, FA complementation group D2; FANCE, FA complementation group E; FANCF, FA complementation group F; FANCG, FA complementation group G; NBN, nibrin; PALB2, partner and localizer of BRCA2; WRN, Werner syndrome RecQ like helicase.
Prevalence and influence of MMR and HR deficiency in GI cancer
The current study aimed to summarize the inactivation mutation ratio of MMR and HR genes in patients with GI cancer. Different inactivation mutation frequencies of MMR and HR genes were detected, as shown in Fig. 3B. BRCA2 exhibited a high deficiency frequency (45/137, 32.85%) in GI cancer (Table II). Other genes, including ATM (29.2%), PTEN (23.36%), BRCA1 (17.52%), MSH6 (20.44%), FANCD2 (15.33%) and MSH2 (13.87%) also exhibited high proportions of inactivation mutations (Fig. 3B, Table II). The current study revealed that 40.9% (56/137) of the patients with GI cancer harbored at least one inactivation mutation site in one or more MMR genes (Table I). A total of 81.8% (112/137) of the patients also exhibited HR gene deficiencies (Table I). Moreover, as shown in Fig. 3C, patients positive for MMR gene inactivation mutations had significantly increased genomic instability compared with negative cases (SNV and indel count, 280.2±20.11 vs. 161.8±15.10, respectively; P<0.0001), as well as HR gene inactivation mutation-positive patients (SNV and indel count, 232.6±15.06 vs. 110.0±10.23, respectively; P<0.0001) (Table I). By contrast, no significant association between genomic instability and other clinical characteristics, including sex, age and tumor type, were identified in GI cancer (Table I). The results obtained suggested the contribution of MMR and HR gene deficiency to genomic instability in GI cancer.
Table II.
Molecular mutation characteristics of MMR and HR genes in 137 patients with gastrointestinal cancer.
| Proportion of samples with MMR and HR genes inactivation mutation | Number of SNV and indel mutations in 183 panel genesa (mean ± SEM) | ||||||
|---|---|---|---|---|---|---|---|
| Gene | Stopgain | Frameshift | Splicing | Proportion of samples with inactivation mutation % (n) | Inactivation mutation (+)b (n)c | Inactivation mutation (−)d (n) | P-value |
| MLH1 | 7 | 4 | 0 | 6.57 (9) | 283.56±53.32 (9) | 205.0±13.43 (128) | 0.0576 |
| MSH2 | 8 | 11 | 0 | 13.87 (19) | 276.6±30.99 (19) | 199.5±14.15 (118) | 0.0053 |
| MSH6 | 12 | 17 | 0 | 20.44 (28) | 315.6±32.77 (28) | 183.1±12.99 (109) | <0.0001 |
| PMS2 | 9 | 5 | 0 | 9.49 (13) | 266.0±31.59 (13) | 204.3±14.00 (124) | 0.0263 |
| BRCA1 | 19 | 5 | 0 | 17.52 (24) | 365.6±42.67 (24) | 177.2±10.82 (113) | <0.0001 |
| BRCA2 | 30 | 22 | 0 | 32.85 (45) | 305.3±25.74 (45) | 163.7±12.32 (92) | <0.0001 |
| ATM | 31 | 16 | 0 | 29.2 (40) | 315.1±31.22 (40) | 166.9±10.60 (97) | <0.0001 |
| PTEN | 3 | 30 | 0 | 23.36 (32) | 208.3±24.39 (32) | 210.8±15.42 (105) | 0.9089 |
| BLM | 17 | 6 | 0 | 16.06 (22) | 281.5±27.90 (22) | 196.6±14.34 (115) | 0.0019 |
| BRIP1 | 13 | 7 | 0 | 13.87 (19) | 321.3±33.13 (19) | 192.3±13.57 (118) | 0.0002 |
| FANCA | 7 | 12 | 0 | 13.14 (18) | 378.4±43.08 (18) | 184.7±12.03 (119) | <0.0001 |
| FANCC | 5 | 8 | 0 | 8.76 (12) | 282.1±61.22 (12) | 203.3±13.02 (125) | 0.3094 |
| FANCD2 | 16 | 7 | 0 | 15.33 (21) | 318.0±46.56 (21) | 190.7±12.21 (116) | 0.0076 |
| FANCE | 0 | 3 | 0 | 2.19 (3) | 529.7±62.86 (3) | 203.0±12.65 (134) | 0.0071 |
| FANCF | 5 | 4 | 0 | 6.57 (9) | 268.4±38.93 (9) | 206.1±13.69 (128) | 0.0443 |
| FANCG | 2 | 15 | 0 | 11.68 (16) | 347.6±49.84 (16) | 192.0±12.45 (121) | 0.0023 |
| NBN | 6 | 6 | 0 | 8.03 (11) | 329.4±44.71 (11) | 199.8±13.33 (126) | 0.0045 |
| PALB2 | 9 | 7 | 0 | 10.95 (15) | 248.8±35.28 (15) | 205.5±14.02 (122) | 0.1789 |
| WRN | 11 | 9 | 0 | 13.14 (18) | 367.1±43.27 (18) | 186.5±12.25 (119) | <0.0001 |
Number of SNV and indel mutations included all the mutations site of SNV and indel (stopgain/frameshift/splicing/nonsynonymous/synonymous/non-frameshift) in 183 panel genes
Inactivation mutation (+) included individuals with at least one type of stopgain/frameshift/splicing mutation in MMR and HR genes
Number of samples with inactivation mutations
Inactivation mutation negative included individuals with no stopgain/frameshift/splicing mutations in MMR and HR genes. SNV, single-nucleotide variant; indel, insertion-deletion; SEM, standard error of the mean; MMR, mismatch repair; HR, homologous recombination; SEM, standard error of the mean. Statistical significance was tested using the Kruskal-Wallis test.
Different MMR or HR genes may exert different effects on genomic instability
Among the 137 patients with GI cancer, 56 were found to be positive for MMR gene inactivation mutations and 117 were found to be positive with HR gene inactivation mutations (36 of the patients were double-positive). Although the results obtaine in the current study demonstrated that patients positive for either MMR or HR gene inactivation mutations had higher genomic instability compared with negative patients, as shown in Fig. 4A, there was no significant difference between patients with MMR gene inactivation mutations and those with HR gene deficiencies in the current study (SNV and indel count, 280.2±20.11 vs. 232.6±15.06, respectively; P=0.019). Additionally, the positive and negative groups according to the inactivation mutation status of 4 MMR and 15 HR genes were compared, and the number of SNV and indel mutations in the 183-gene panel of patients with any MMR or HR inactivation mutations was revealed to be higher compared with those without such mutations, including those positive for inactivation mutations in MSH6, BRCA1, BRCA2, ATM or FANCD2 (Table II). Additionally, patients with MLH1, PTEN, FANCC or PALB2 inactivation mutations did not exhibit significant differences in the number of SNV and indel mutations compared with patients without inactivation mutations in these genes (Table II). No significant effects on genomic instability among the four groups patients with MLH1, MSH2, MSH6 and PMS2 inactivation mutations (Fig. 4B). However, the effect on genomic instability of certain genes, including BRCA1, BRCA2, ATM, FANCA and BRIP1, was markedly more pronounced compared with that of PTEN in patients with HR deficiency (Fig. 4C). These results reflected the different effect on genomic instability among MMR and HR genes.
Figure 4.

Different inactivated MMR and HR genes had different influence on genomic instability. (A) Number of SNV and indel mutations in 183-gene panel in inactivation positive groups for MMR and HR genes. Inactivation positive mutations included individuals with at least one type of stopgain/frameshift/splicing mutation. Inactivation negative mutations included individuals with no stopgain/frameshift/splicing mutations. The numbers in brackets on the x-axis represents the number of samples with inactivation mutation. There was no significant difference between patients with MMR gene inactivation mutations and those with HR gene deficiencies in the current study. (B) Parallel comparison of the effects of MMR genes on genomic instability. MMR, mismatch repair; HR, homologous recombination; SNV, single-nucleotide variant; indel, insertion-deletion; MLH1, mutL homolog 1; MSH2, mutS homolog 2; MSH6, mutS homolog 6; PMS2, PMS1 homolog 2 mismatch repair system component. The numbers in brackets on the x-axis is number of samples with inactivation mutation. (C) The effects of HR genes on genomic instability. BRCA1, BRCA1 DNA repair associated; BRCA2, BRCA2 DNA repair associated; ATM, ATM serine/threonine kinase; PTEN, phosphatase and tensin homolog; BLM, BLM RecQ like helicase; BRIP1, BRCA1 interacting protein C-terminal helicase 1; FANCA, FA complementation group A; FANCC, FA complementation group C; FANCD2, FA complementation group D2; FANCF, FA complementation group F; FANCG, FA complementation group G; NBN, nibrin; PALB2, partner and localizer of BRCA2; WRN, Werner syndrome RecQ like helicase; FANCE, FA complementation group E. The numbers in brackets on the x-axis represents the number of samples with inactivation mutations.
Discussion
GI cancer is a leading cause of morbidity and mortality, and accounts for 25% of new cancer cases and cancer-associated mortalities worldwide (1,38). Despite modern improvements in treatment, the majority of patients with GI cancer have a poor prognosis due to late-stage diagnosis (3). To date, early screening and diagnosis based on the understanding of the molecular mechanisms underlying GI cancer is becoming the best way to benefit patients with GI cancer (39). MMR deficiency and MSI in particular, which are suspected to be associated with the efficacy of immune checkpoint inhibitors (40). Approximately 15% of patients with CRC exhibit high MSI, mostly as a result of dMMR (41). In the present study, a systematic analysis of 183 gene mutations in 137 patients with GI cancer was performed to evaluate novel molecular characteristics in GI cancer. Furthermore, the effects of MMR and HR mutational status on genomic instability were investigated.
Several studies have proposed APC as one of the most prominent tumor promoting genes, regulating the Wnt/β-catenin signaling pathway and participating in the tumorigenesis of CRC (42–44). The results obtained in the current study revealed that the APC gene was mutated in 81% of patients with GI cancer. Other genes, including PMS2, neurofibromin 1 and ATM, also exhibited a high mutation frequency. Notably, seven of the 20 most frequently mutated genes in the 183-gene panel were DNA repair genes (MMR gene, MSH2; HR genes, ATM, FANCD2, ATR, BRCA2, FANCA and BRCA1). This indicated that homologous recombination repair is an important event during the progression of GI cancer, in addition to MMR. With the high mutation frequency of the MMR and HR genes, correlation analysis was performed and a significant association between the mutational status of MMR or HR genes and genomic instability in GI cancers was identified. The higher the number of mutations in MMR or HR genes, the higher the number of mutations in the 183-gene panel.
The DNA MMR system regulates genetic fidelity, the accumulation of genetic errors, MSI and intestinal carcinogenesis (13,14). In the present study, 40.9% (n=56,137) of patients with GI cancer had one or more MMR gene inactivating mutations, whereas HR deficiency occurred in 81.8% (112/137) of the patients. A previous study indicated that 35% of patients with pancreatic cancer (n=109) harbored pathogenic or likely pathogenic mutations in HR genes (45). In addition, no significant association was observed between genomic instability and other clinical characteristics, including sex, age and tumor type in GI cancer. One large-scale analysis suggested that 13% of all tumor types had HR deficiency (n=53,619), including PTEN (5.8%), BRCA2 (2.8%), BRCA1 (2.6%) and ATM (1.2%) (27). However, the data obtained in the current study revealed that MSH6 was the most frequently mutated MMR gene, with an inactivation mutation in 20.44% of the cases. A high deficiency frequency of certain HR genes, including BRCA2, ATM, BRCA1, PTEN and FANCD2, was detected in the current study. These results demonstrated the molecular characteristics of HR gene inactivation mutations, in addition to dMMR. Furthermore, almost no splicing mutations in MMR and HR genes were detected in GI cancer. Further investigation revealed that patients with MLH1, PTEN, FANCC or PALB2 inactivation mutations did not exhibit significant differences in the numbers of SNV and indel mutations compared with patients without inactivation mutations in these genes.
No notably different effects on genomic instability among the four groups with MLH1, MSH2, MSH6 and PMS2 inactivation mutations were observed in the current study. However, the effect on genomic instability of certain genes, including BRCA1, BRCA2, ATM, FANCA and BRIP1, was more pronounced compared with that of PTEN in patients with HR deficiency.
The present study demonstrated the association between genomic instability and the mutational status of MMR and HR genes in GI cancer. The results obtained reveal a novel model based on the mutational status of DNA repair genes for predicting response to therapy. The higher the number of mutations detected in DNA repair genes, the more tumor neoantigens may be induced; however, further research and evaluation of this model is required.
Acknowledgements
The authors would like to thank Professor Jian Xu [First Dimension Biosciences (Suzhou) Co., Ltd., Suzhou, China] for contributing to revising the manuscript.
Funding
No funding was received.
Availability of data and material
The datasets generated and/or analyzed during the present study are not publicly available due to confidential personal information that could compromise research participant privacy.
Authors' contributions
XL, TT and DL designed the study. KH, PJ and WZ clinically diagnosed the patients, obtained the informed consent, harvested tissue samples, examined the archives and identified the cases included in the study, examined the slides and collected pathological information. HY, XW and PM performed the data analysis. PM and HY wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The First Affiliated Hospital of Anhui Medical University Ethics Committee approved the study and all patients provided written informed consent prior to enrollment into the project.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 2.Brody H. Colorectal cancer. Nature. 2015;521(Suppl):S1. doi: 10.1038/519S1a. [DOI] [PubMed] [Google Scholar]
- 3.Ng SC, Wong SH. Colorectal cancer screening in Asia. Br Med Bull. 2013;105:29–42. doi: 10.1093/bmb/lds040. [DOI] [PubMed] [Google Scholar]
- 4.Labianca R, Nordlinger B, Beretta GD, Mosconi S, Mandalà M, Cervantes A, Arnold D, ESMO Guidelines Working Group Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;6(Suppl):vi64–vi72. doi: 10.1093/annonc/mdt354. [DOI] [PubMed] [Google Scholar]
- 5.Lutgens MW, Oldenburg B, Siersema PD, van Bodegraven AA, Dijkstra G, Hommes DW, de Jong DJ, Stokkers PC, van der Woude CJ, Vleggaar FP. Colonoscopic surveillance improves survival after colorectal cancer diagnosis in inflammatory bowel disease. Br J Cancer. 2009;101:1671–1675. doi: 10.1038/sj.bjc.6605359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lansdorp-Vogelaar I, van Ballegooijen M, Zauber AG, Habbema JD, Kuipers EJ. Effect of rising chemotherapy costs on the cost savings of colorectal cancer screening. J Natl Cancer Inst. 2009;101:1412–1422. doi: 10.1093/jnci/djp319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beamer LC, Grant ML, Espenschied CR, Blazer KR, Hampel HL, Weitzel JN, MacDonald DJ. Reflex immunohistochemistry and microsatellite instability testing of colorectal tumors for Lynch syndrome among US cancer programs and follow-up of abnormal results. J Clin Oncol. 2012;30:1058–1063. doi: 10.1200/JCO.2011.38.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burt RW. Who should have genetic testing for the Lynch syndrome? Ann Intern Med. 2011;155:127–128. doi: 10.7326/0003-4819-155-2-201107190-00009. [DOI] [PubMed] [Google Scholar]
- 9.Matloff J, Lucas A, Polydorides AD, Itzkowitz SH. Molecular tumor testing for Lynch syndrome in patients with colorectal cancer. J Natl Compr Canc Netw. 2013;11:1380–1385. doi: 10.6004/jnccn.2013.0161. [DOI] [PubMed] [Google Scholar]
- 10.Ward RL, Hicks S, Hawkins NJ. Population-based molecular screening for Lynch syndrome: Implications for personalized medicine. J Clin Oncol. 2013;31:2554–2562. doi: 10.1200/JCO.2012.46.8454. [DOI] [PubMed] [Google Scholar]
- 11.Broustas CG, Lieberman HB. DNA damage response genes and the development of cancer metastasis. Radiat Res. 2014;181:111–130. doi: 10.1667/RR13515.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spies M, Fishel R. Mismatch repair during homologous and homeologous recombination. Cold Spring Harb Perspect Biol. 2015;7:a022657. doi: 10.1101/cshperspect.a022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010;7:153–162. doi: 10.1038/nrclinonc.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vilar E, Tabernero J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov. 2013;3:502–511. doi: 10.1158/2159-8290.CD-12-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R, Shepherd LE, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin Cancer Res. 2016;22:813–820. doi: 10.1158/1078-0432.CCR-15-1678. [DOI] [PubMed] [Google Scholar]
- 17.Kastrinos F, Stoffel EM. The history, genetics, and strategies for cancer prevention in Lynch syndrome. Clin Gastroenterol Hepatol. 2014;12:715–727.e41-e43. doi: 10.1016/j.cgh.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynch HT, de la Chapelle A. Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet. 1999;36:801–818. [PMC free article] [PubMed] [Google Scholar]
- 19.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, Markowitz SD. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang YC, Lu YP, Tseng RC, Lin RK, Chang JW, Chen JT, Shih CM, Chen CY. Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J Clin Invest. 2003;111:887–895. doi: 10.1172/JCI15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu HS, Wen CK, Tang YA, Lin RK, Li WY, Hsu WH, Wang YC. Promoter hypermethylation is the predominant mechanism in hMLH1 and hMSH2 deregulation and is a poor prognostic factor in nonsmoking lung cancer. Clin Cancer Res. 2005;11:5410–5416. doi: 10.1158/1078-0432.CCR-05-0601. [DOI] [PubMed] [Google Scholar]
- 23.Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, Roa BB, Wenstrup RJ, Hartman AR, Syngal S. Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology. 2015;149:604–613.e20. doi: 10.1053/j.gastro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol. 2017;3:464–471. doi: 10.1001/jamaoncol.2016.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, Taylor M, Easton DF. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- 27.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous recombination deficiency: Exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–1154. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.da Cunha Colombo Bonadio RR, Fogace RN, Miranda VC, Diz MDPE. Homologous recombination deficiency in ovarian cancer: A review of its epidemiology and management. Clinics (Sao Paulo) 2018;73(Suppl 1):e450s. doi: 10.6061/clinics/2018/e450s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao EC, Lipkin SM. Molecular models for the tissue specificity of DNA mismatch repair-deficient carcinogenesis. Nucleic Acids Res. 2006;34:840–852. doi: 10.1093/nar/gkj489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gentles L, Goranov B, Matheson E, Herriott A, Kaufmann A, Hall S, Mukhopadhyay A, Drew Y, Curtin NJ, O'Donnell RL. Exploring the frequency of homologous recombination DNA repair dysfunction in multiple cancer types. Cancers (Basel) 2019;11(pii):E354. doi: 10.3390/cancers11030354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerbinskaite A, Mukhopadhyay A, Plummer ER, Curtin NJ, Edmondson RJ. Defective homologous recombination in human cancers. Cancer Treat Rev. 2012;38:89–100. doi: 10.1016/j.ctrv.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 32.Miranda E, Destro A, Malesci A, Balladore E, Bianchi P, Baryshnikova E, Franchi G, Morenghi E, Laghi L, Gennari L, Roncalli M. Genetic and epigenetic changes in primary metastatic and nonmetastatic colorectal cancer. Br J Cancer. 2006;95:1101–1107. doi: 10.1038/sj.bjc.6603337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malapelle U, Mayo de-Las-Casas C, Rocco D, Garzon M, Pisapia P, Jordana-Ariza N, Russo M, Sgariglia R, De Luca C, Pepe F, et al. Development of a gene panel for next-generation sequencing of clinically relevant mutations in cell-free DNA from cancer patients. Br J Cancer. 2017;116:802–810. doi: 10.1038/bjc.2017.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 39.Vucic EA, Thu KL, Robison K, Rybaczyk LA, Chari R, Alvarez CE, Lam WL. Translating cancer ‘omics’ to improved outcomes. Genome Res. 2012;22:188–195. doi: 10.1101/gr.124354.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Copija A, Waniczek D, Witkos A, Walkiewicz K, Nowakowska-Zajdel E. Clinical significance and prognostic relevance of microsatellite instability in sporadic colorectal cancer patients. Int J Mol Sci. 2017;18(pii):E107. doi: 10.3390/ijms18010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Näthke IS. A role for the adenomatous polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001;3:429–432. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- 43.Leoz ML, Carballal S, Moreira L, Ocana T, Balaguer F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl Clin Genet. 2015;8:95–107. doi: 10.2147/TACG.S51484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26:570–579. doi: 10.1016/j.cellsig.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 45.Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the present study are not publicly available due to confidential personal information that could compromise research participant privacy.