

Abstract
Diabetic neuropathy is one of the most serious complications of diabetes, and its increase shows no sign of stopping. Furthermore, current clinical treatments do not yet approach the best effectiveness. Thus, the development of better strategies for treating diabetic neuropathy is an urgent matter. In this review, we first discuss the advantages and disadvantages of some major mouse models of diabetic neuropathy and then address the targets for mechanism-based treatment that have been studied. We also introduce our studies on each part. Using stem cells as a source of neurotrophic factors to target extrinsic factors of diabetic neuropathy, we found that they present a promising treatment.
Keywords: brain derived neurotrophic factor, diabetes, extrinsic factors, nerve growth factor, nerve regeneration, neurotrophic factors, non-obese type 2 diabetes, phosphatase and tensin homolog, stem cell, streptozotocin
Introduction
About 8.3% of the world’s population is currently estimated to have diabetes mellitus; and by 2050, one third of all American will develop this metabolic disease if its present rate of increase cannot be controlled (Bril, 2014). More seriously, diabetes results in many complications; and diabetic neuropathy is one of the most common of them, affecting 30–50% of diabetic patients (Vuong et al., 2018). The link between diabetes and neuropathy has been noticed for over 100 years, and during this time many types of diabetic neuropathy have been classified. Among them, diabetic sensorimotor polyneuropathy has the highest incidence found in both type 1 and type 2 diabetic patients (Tracy and Dyck, 2008).
Although the advance of functional imaging technology has opened the new horizon for experimentation in human beings, studies on pain based on behavioral animal models are still necessary. Mice and rats are two species that have been widely used for studies on mechanisms, pathogenesis, and treatment of diabetic neuropathy. Both type 1 and type 2 diabetes result in this disorder. In general, chemical induction, nutrition induction, and genetic modification are the three main strategies for establishing diabetic neuropathy models in rodents (Shaikh and Somani, 2010; Islam, 2013). The greatest challenge in early studies on diabetic neuropathy was the availability of sensitive methods to detect neuropathic changes in animal models. The development of modern techniques has played a critical role not only in affording us a better understanding about the pathogenesis of diabetic neuropathy at morphological, biochemical, and electrophysiological levels but also in finding novel treatments. The Diabetic Neuropathy Study Group of the European Association for the Study of Diabetes (EASD) recommended the assessment of three parameters for the identification of the neuropathy phenotype in rodents, i.e., behavior, nerve conduction velocity, and nerve structure with there being a significant difference in two out of these three parameters compared with those for control mice of the same age (Biessels et al., 2014).
An understanding of the mechanisms and pathogenesis of diabetic neuropathy is very important for preventing its progression and finding better treatments. Hyperglycemia is commonly supposed in both type 1 and type 2 diabetes to be the main cause of diabetic neuropathy (Harati, 2007) via four associated pathways found in both the human and mouse consisting of polyol pathway, the advanced glycation end-product (AGE) pathway, the protein kinase C pathway, and the hexosamine pathway (Leinninger et al., 2004). In the polyol pathway, after being activated in a hyperglycemia, aldose reductase converts glucose to sorbitol and then lead to multiple glycolysis reactions that subsequently result in the shortage of cytoplasmic nicotinamide adenine dinucleotide phosphate (NADPH). A reduction in the cytosolic level of NADPH causes a decrease in the most important cellular antioxidant, glutathione (Du et al., 2009). Furthermore, a decreased amount of nicotinamide adenine dinucleotide (NAD+) inhibits the activity of glyceraldehyde-3-phosphate dehydrogenases (GAPDHs), which play a role in keeping the normal flux of glucose through the glycolysis pathway. Inhibition of GAPDHs also causes the accumulation of GAPDH metabolites that then activates the hexosamine pathway (Leinninger et al., 2004). The polyol pathway finally results in the loss of normal energy production and protective systems (Leinninger et al., 2004). AGEs are the products of glycation generated in the polyol pathway; and together with their receptors (RAGEs), they lead to the formation of reactive oxygen species and activation of NF-κB, which is an apoptotic transcription factor (Brownlee, 2000). The protein kinase C pathway is activated by diacylglycerol as a response to a high-glucose environment and has been reported to be tightly linked to many diabetic complications (Koya and King, 1998). As for the hexosamine pathway, its products, such as acylglycosylated proteins, cause an increase in the levels of proteins associated with diabetic complications, especially in the case of type 2 diabetes (Leinninger et al., 2004). In addition to hyperglycemia, other factors such as dyslipidemia (Vincent et al., 2009) and changes in insulin signaling (Murakawa et al., 2002; Kim and Feldman, 2012) have been reported as other contributors to the progression of diabetic neuropathy.
In this review, we first discuss the advantages and disadvantages of some major mouse models of diabetic neuropathy that have been developed and studied extensively. Then in the second part, we address the targets for mechanism-based treatment of diabetic neuropathy that have been studied at both preclinical and clinical levels. We also introduce some results from our previous and present studies in this area.
We have performed a literature search through Pubmed and Scopus with the following keywords: “mouse models of diabetic neuropathy”, “diabetic neuropathy”, “clinical treatment of diabetic neuropathy”, “nerve regeneration”, “intrinsic brakes of nerve regeneration”, and “extrinsic factor of nerve regeneration”. Using these studies, we reviewed mouse models and targets for mechanism-based treatment of diabetic neuropathy.
Experimental Mouse Models of Diabetic Neuropathy
Rodents are commonly used in studies on diabetes and its complications because of their advantages in terms of cost, breeding time, housing and handling, and ethical considerations. There are three main approaches to establish mouse models of diabetic neuropathy: nutritional induction, genetic modification, and chemical induction. Each approach has advantages and disadvantages as well as limitations. In particular, Harati (2007) in a comprehensive review proposed that the major hurdle in studying diabetic neuropathy is the lack of an adequate animal model showing relevant acute and chronic events leading to diabetic neuropathy.
Nutrition-induced diabetic neuropathy mouse model
By mimicking the metabolic syndrome in humans, nutritional induction has been used to establish type 2 diabetic neuropathic pain. In general, these experimental animals are fed a high-fat diet to develop diabetes after a long period associated with obesity. When fed a high-fat diet consisting of 24% fat (from soybean oil and lard), 24% protein and 41% carbohydrate for 12 weeks, C57BL/6 develop symptoms of prediabetes and present signs of neuropathy including decreased sensory nerve conduction velocity, reduced density of intraepidermal nerve fibers (IENF), and thermal hypoalgesia (Coppey et al., 2012). Especially, Sullivan et al. (2007) showed that the hyperglycemia and neuropathy were more robust when C57BL/6 db/db mice were fed a high-fat diet with 17% kcal from fat. Compared to other approaches to establish diabetic neuropathy mouse models, diet/nutrition induction requires a long time for model establishment (Gao and Zheng, 2014). Other factors including variations in neuropathy phenotyping measurements, differences in sex and age, duration of high-fat diet feeding, and the source and percentage of fat content in food were also reported to have an effect on the degree of neuropathy in these models. The Jackson Laboratory reported that male mice are more suitable for diet/nutrition induction of diabetes. In addition, differential sensitivity to pain has been noticed between male and female mice (Stavniichuk et al., 2010). Besides, the type of fat content also has an effect on the severity of diabetes. Compared with unsaturated fat (fish oil), food consisting of saturated and obesogenic fat are more effective than other fat diets in causing weight gain in mouse models (Ikemoto et al., 1996; Wang et al., 2002). These high-fat diet-fed diabetes models show advantages in studies on pre-diabetic or obesity related neuropathy but they seem not to be suitable for studying chronic diabetic neuropathy (Obrosova et al., 2007b).
Genetically modified diabetic neuropathy mouse model
Transgenic rodent models of type 1 and type 2 diabetic neuropathy have been developed. Non-obese diabetic (NOD) and B6Ins2Akita mice are two of the most investigated models of type 1 diabetes. NOD mice develop autoimmune T cell-mediated insulin-dependent diabetes mellitus spontaneously due to a heritable polygenic immunodeficiency (Leiter, 2001). A previous article from the Jackson Laboratory reported that the progression of diabetes in NOD mice is susceptible to variability and is affected by many factors such as housing conditions, diet, and sex (Leiter, 2001). In contrast to induction by diet/nutrition or streptozotocin (STZ), female mice are frequently used due to their earlier development of type 1 diabetes symptoms (Leiter, 2001). Previous studies on this diabetic mice showed that hyperalgesia occurred early at 8 weeks (Gabra and Sirois, 2005), and hypoalgesia at 12 weeks of age (Obrosova et al., 2005). Carrying a point mutation in the Ins2 insulin gene, C57BL/6 mice, designated as B6Ins2Akita mice, spontaneously develop type 1 diabetes at 7 weeks of age (Yoshioka et al., 1997). Multiple studies on these mice showed that diabetic sensorimotor neuropathy progresses gradually (Choeiri et al., 2005; Sullivan et al., 2007) but they robustly and rapidly develop sympathetic autonomic neuropathy (Schmidt et al., 2009). Both NOD and B6Ins2Akita mice have been shown to develop disease consistent with the pathogenesis of human peripheral diabetic neuropathy (Schmidt et al., 2003, 2009; Choeiri et al., 2005).
Leptin is a hormone secreted from adipocytes after food intake, and it controls appetite via hypothalamic signaling (O’Brien et al., 2014). Thus, mutation of leptin (ob/ob mice) and its receptor (db/db mice) are targets widely investigated to establish type 2 diabetes animal models. According to the literature, there have not been many studies on ob/ob mice. Drel et al. (2006) reported that thermal hypoalgesia was clearly revealed in ob/ob mice. In addition, compared with age-matched control mice by 11 weeks of age, these mice showed a remarkable decrease in density of IENF and in motor and sensory nerve conduction velocities (Drel et al., 2006), thus suggesting that ob/ob mice would be a good choice for a model of hypoalgesia. Established by Sima and Robertson (1978), the C57BKS db/db mouse is known as one of the first mouse models of diabetic peripheral neuropathy. Subsequent studies on this genetic type 2 diabetes model showed that the signs of diabetes and diabetic neuropathy typically occur early. In particular, these animals exhibit hyperalgesia and allodynia between 8–12 weeks and hypoalgesia after 12 weeks of age and present impaired motor and sensory nerve conduction velocity as well as abnormal nerve morphology (Ii et al., 2005; Cheng et al., 2009; Kan et al., 2012). However, by following up on the literature describing studies on this db/db mouse model, we recognized variation in neuropathy characterization of this model. In particular, Wright et al. (2007) showed that whereas tactile allodynia is present in them, significant thermal hypoalgesia or a reduction in IENF density is not found. In contrast, later works reported the presence of both thermal and mechanical hypoalgesia and allodynia in this model (Sullivan et al., 2007; Wang et al., 2011; Dauch et al., 2012). The type of mouse strain was also noted to affect the development of neuropathy. Hyperglycemia is more stable and neuropathy is more severe in C57BKS db/db than in C57BL/6 db/db (Sullivan et al., 2007; Dauch et al., 2012).
These above-mentioned limitations suggest that in studies using genetic type 1 and type 2 diabetic mouse models, a high number of mice and a great amount of time are needed to have a desired cohort having similar diabetic neuropathy phenotypes.
Chemical-induced diabetic neuropathy mouse models
STZ-induced diabetes is the most common model used for the studies on diabetic neuropathy (Pittenger and Vinik, 2003; Colleoni and Sacerdote, 2010; Islam and Wilson, 2012). STZ, known as a powerful alkylating agent, selectively kills pancreatic β cells by interfering with glucose transport and glucokinase function and by inducing DNA strand breaks (Rees and Alcolado, 2005). Many approaches using STZ for establishing diabetes mice have been developed. A single high dose injection of STZ results in extreme destruction in type 1 diabetes; whereas an intermediate one causes partial damage to pancreatic β cells that resembles the characteristic damage seen in type 2 diabetes (Islam and Wilson, 2012). Some studies have described symptoms of neuropathy in diabetes mouse models induced by STZ (Vareniuk et al., 2008; Murakami et al., 2013). Compared with other approaches to prepare diabetic mouse models, this chemical-induced model is not expensive; and optimization of the procedure is easy. In addition, this model is appropriate to use for testing the effects of new drugs, therapies or transplantation on the lowering of blood glucose (King, 2012). The main limitation related to this model is the difference in many features and mechanisms of diabetes induced by STZ from the real diabetes seen in human beings. Furthermore, the mice in these models experience severe distress and impaired general conditions, thus resulting in difficulty in gathering data or measuring pain scores (Colleoni and Sacerdote, 2010).
In particular, with a slight modification of the previous study of Nakamura et al. (2006), we have recently and successfully established a mouse model of non-obese type 2 diabetes that is frequently found in Asia (Vaag and Lund, 2007). Especially, nicotinamide (NA) was used to partially protect pancreatic β cells from the effect of STZ. Compared with type 1 diabetic mice induced by a high dose of STZ, type 2 diabetic mice induced by NA-STZ survive longer and maintain a normal body weight like that of control mice. However, they show significantly higher serum glucose levels by random glucose measuring and glucose tolerance testing. Furthermore, the number of pancreatic β cells and plasma insulin level in these type 2 diabetic mice are remarkably decreased (Vuong et al., 2018). More importantly, our NA-STZ diabetic model mice display the symptoms that meet the criteria for diabetic neuropathy recommended by EASD (Neurodiab), including behavior, physiology, and structure. In particular, as compared with age-matched non-diabetes mice, type 2 diabetic mouse model show: 1) responses to mechanical stimuli by von Frey filaments at a lower threshold of 0.16 g, indicative of mechanical hyperalgesia (Figure 1C & D); 2) a significantly higher cutaneous stimulus threshold, suggesting that the threshold for eliciting action potentials in the periphery is increased in diabetic mice (Table 1); and 3) a significant reduction in the number of IENF (Figure 1A & B) (Vuong et al., 2018). In addition, because sciatic nerve crush has been shown to be an appropriate and widely used model for studies on neuroregenerative therapeutic modalities (Magill et al., 2007), we combined our recently and successfully established sciatic nerve transection-regeneration model (Unezaki et al., 2009) to the non-obese type 2 diabetic mouse model and examined the nerve regeneration in this combined model. The data showed that type 2 diabetic mice display delayed functional recovery and nerve regeneration, suggesting that our model is suitable for the study of the pathogenesis and treatment of early diabetic neuropathy (Vuong et al., 2018).
Figure 1.
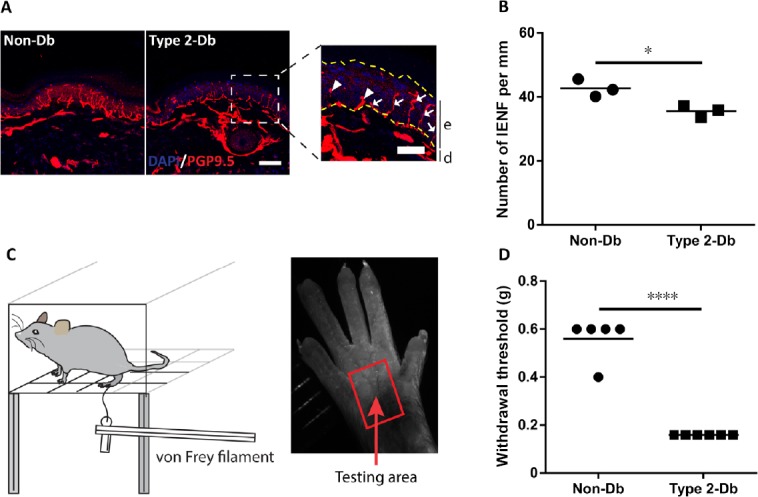
Characterization of type 2 diabetic neuropathy mouse model.
(A) Representative images of intraepidermal nerve fibers (IENF) in the plantar skin of type 2 diabetic mice (type 2 Db) and aged-match normal mice (Non-Db). Immunostaining with anti-PGP9.5 antibody and counterstaining with 4′,6-diamidino-2-phenylindole (DAPI). Scale bar: 50 µm. The enlarged figure presents the epidermis layer (e) (between yellow dashed lines) and dermis layer (d), the counted IENF (arrows), and uncounted IENF (arrowheads). Scale bar: 25 µm. (B) Quantification of the number of IENF per mm. (C) Illustration of behavioral test by von Frey filaments. Von Frey filaments, ranging from 0.02–4 g (cut-off value), are applied to the testing area until they buckle. (D) Withdrawal thresholds to mechanical stimuli. The data are shown as the mean ± SEM. *P < 0.05, ****P < 0.0001 by two tailed t-test. Figure 1A, B, and D are reprinted with permission from Vuong et al. (2018).
Table 1.
Measurement of sensory nerve conduction
| Item | Aged-match normal mice (n = 12) | Type 2 diabetic mice (n = 13) |
|---|---|---|
| Latency (ms) | 7.51±0.72 | 7.90±0.64 |
| Stimulus threshold (mA) | 0.27±0.04 | 0.51±0.09* |
Data was shown the mean ± SEM. *P < 0.05, vs. aged-match normal mice (two-tailed unpaired Student’s t-test). Reprinted with permission from Vuong et al. (2018).
Targets for Treatment of Diabetic Neuropathy
Intrinsic targets
Growth suppressor
Tumor suppressor molecules are known as the brake to the development of cancer by inhibiting cell proliferation and differentiation of dividing cells (Krishnan et al., 2016). The division of Schwann cells and regeneration were reported to involve some common signaling components including certain tumor suppressor genes (Krishnan et al., 2016). Thus, it is a completely logical idea that inhibiting the expression of these tumor suppressor molecules would release the brake on nerve regeneration.
As one of the key members of the phosphoinositide 3-kinase-protein kinase B (Akt) pathway, which is known to play a role in enhancing axon growth, phosphatase and tensin homolog (PTEN) is an interesting target for studies focused on the promotion of nerve regeneration. Many previous studies showed that inhibition of PTEN enhances axon growth in both the central nervous system (Park et al., 2008; Liu et al., 2010) and peripheral nervous system (Christie et al., 2010; Zhou et al., 2012). In particular, we found in our recent work that the expression of PTEN is elevated in the dorsal root ganglions (DRGs) of diabetic mice and that the introduction of a PTEN inhibitor to the distal stump of sciatic nerve promotes nerve regeneration in both diabetic mice and normal mice (Vuong et al., 2018). Previous studies reported that Akt network plays an important role in both mice and humans (Brosius et al., 2010; Manning and Toker, 2017), suggesting that these promising results can be translated to human treatment; however, further studies on this target are needed. In addition to PTEN, other tumor suppressor molecules such as Rb1, p53, and BRCA1 were reported to be involved in inhibiting nerve regeneration (Krishnan et al., 2016, 2018).
Calcium
As an essential component for membrane sealing after axonal transection, calcium plays a critical role in axon regeneration (Xie and Barrett, 1991) and in growth cone formation by rearranging cytoskeletal proteins (Chu and Tator, 2001; Spira et al., 2003; Chierzi et al., 2005). After an injury, calcium influx into the axons is increased until the membrane reseals (McCallum et al., 2006). Other roles of calcium were also documented. Calcium activates kinases such as protein kinase A and extracellular signal-related protein kinases to induce cytoskeletal reorganization and axonal protein synthesis (Doron-Mandel et al., 2015). Furthermore, calcium influx activates proteolytic machinery including calpain, which has been shown to be involved in cleavage of the sub-membrane protein spectrin, releasing membrane tension, rearranging microtubules and neurofilaments, and facilitating lipid vesicle access to the plasma membrane (Gitler and Spira, 1998; Spira et al., 2003). There are two comprehensive reviews in the literature on abnormal calcium signaling affecting axonal degeneration in peripheral diabetic neuropathy (Voitenko, 2004; Fernyhough and Calcutt, 2010). Some studies targeting Ca(V)3.2 T-type calcium channels in preclinical models showed a potentially positive effect of reversing diabetic neuropathic pain (Messinger et al., 2009), however, there has currently been no translation of these results to clinical trials.
Cyclic adenosine monophosphate
The idea of increasing the cyclic adenosine monophosphate (cAMP) level to promote axonal regeneration was raised from the knowledge that cAMP is involved in many key intracellular signaling pathways. Many different mechanisms were reported to be involved in the effects of cAMP on nerve regeneration including increasing translocation of growth factor receptors from the cytoplasm to the cell membrane (Meyer-Franke et al., 1998), indirectly mediating the neuritogenic effects of neurotrophic factors (Gao et al., 2003) and hydrolyzing arginine into urea and ornithine to protect axons from inhibition of myelin formation during regeneration (Lange et al., 2004). Particularly, previous studies on diabetic neuropathy also showed the involvement of cAMP in the progression of diabetic neuropathy (Sanchez and Sharma, 2009; Bai et al., 2014). Notably, a phosphodiesterase-5 inhibitor is one of promising cyclic nucleotide drugs for the treatment of diabetic neuropathy, as revealed by both preclinical and clinical studies (Jain et al., 2001; Patil et al., 2004; Hackett, 2006; Wang et al., 2011, 2017). In clinical trials, the inhibition of phosphodiesterase-5 was proved to have no severe side effects in patients and to attenuate symptoms of diabetic peripheral neuropathy (Hackett, 2006). The increasing number of studies focusing on cAMP suggests that cAMP is a promising target for accelerating nerve regeneration.
Other intrinsic targets
In addition to the major intrinsic factors discussed above, others have also been reported. Given that diabetic neuropathic pain may be the result of changes in both peripheral and central nervous systems (Chen and Pan, 2002), some central factors have been considered as contributors to diabetic neuropathy such as the strengthened level of glutamate released from primary afferents in the spinal cord (Zheng et al., 2010) and the down-regulation of gamma-aminobutyric acid type B receptors (Bai et al., 2014). Many studies have demonstrated that multiple ion channels are altered in both animal models of and human patients with diabetes, including voltage-gated Na+ channels (Nav) (Craner et al., 2002; Misawa et al., 2009), Ca2+ channels (Hall et al., 2001; Jagodic et al., 2007), and voltage-dependent K+ channels (Cao et al., 2010), suggesting the involvement of changes in ion channel expression in diabetic neuropathic pain. Another interesting intrinsic factor that has been noted for a long time (Spuler et al., 1988; Liniger et al., 1989) and revisited recently (Mallik et al., 2017) is ganglioside, which is a membrane-associated molecule playing a role in regeneration.
Extrinsic targets
Glial cell activation
Glia are mainly responsible for maintaining homeostasis, forming myelin, and protecting neurons in both the central and peripheral nervous systems (Mika et al., 2013). All of the glial cells in the spinal cord are affected by diabetes (Schreiber et al., 2015) resulting in detrimental changes in sensory neurons and increased regulation of Nav1.3 sodium channels in DRGs (Cheng et al., 2014). After having been activated, astrocytes and oligodendrocytes near sites of injury produce myelin proteins such as myelin-associated glycoprotein and oligodendrocyte myelin glycoprotein that function as axon growth inhibitors by acting through the intracellular Rho GTPase signaling cascade (Yiu and He, 2006). There are two alternative approaches for targeting this extrinsic factor for the control of diabetic neuropathy, i.e., inhibition of glial activation and inhibition of products formed by the activation of glia cells. Some agents such as minocycline and propentofylline have been used in patients with neuropathic pain, but definitive evidence for their efficacy was not recorded (Ellis and Bennett, 2013). Intrinsic differences between rodents and human microglia is supposedly the main reason for this difference in these results, thus providing a valuable note for future studies on this target (Landry et al., 2012).
Glycation products, oxidative, and nitrosative stress
Glycation is the non-enzymatic reactions of glucose, α-oxoaldehydes, and other saccharide derivatives with proteins, nucleotides, and lipids that subsequently forms early glycation adducts and AGEs (Obrosova, 2009). These glycation products function abnormally to disrupt molecular conformations, alter enzymatic activity, and interfere with receptor functioning; and taken together, they contribute to the pathology of diabetic complications (Dickinson et al., 2002; Ahmed, 2005; Singh et al., 2014). Many drugs have been reported as potential medicines for the inhibition of AGEs and their receptors (RAGEs) (Wada et al., 2001; Hammes et al., 2003; Thornalley, 2005), thus suggesting an alternative approach for improving the status of patients with diabetic neuropathy.
Oxidative stress products are produced from the polyol pathway or by enhanced formation of free radicals due to glucose metabolism itself and/or to deficits in antioxidant defense (Schreiber et al., 2015). Some studies also showed that there is a link between increased formation of AGEs or increased expression of RAGEs and oxidative stress products (Brownlee, 2001; Giacco and Brownlee, 2010). Together with oxidative stress products, reactive nitrogen species are also reportedly contributors to the progression of diabetic neuropathy (Obrosova et al., 2005; Drel et al., 2007b). Especially, reactive nitrogen species were noted to have negative effects on all major cells responsible for peripheral diabetic neuropathy (Obrosova, 2009). Some clinical studies showed that antioxidants are potent for the improvement of diabetic neuropathy (Tang et al., 2007; Ruessmann and German Society of out patient diabetes centres AND (Arbeitsgemeinschaft niedergelassener diabetologisch tätiger Arzte e.V.), 2009; Mijnhout et al., 2010).
Neurotrophic suppor
Neurotrophic factors play a crucial role in the regulation of survival and differentiation of neural cells (Afshari et al., 2009). In the progression of diabetic neuropathy, a number of neurotrophic factors are impaired or altered (Leinninger et al., 2004; Obrosova, 2009). Thus, many studies have approached the improvement of diabetic neuropathy by introducing exogenous neurotrophic factors to diabetic mouse models. In a previous study, Christianson et al. (2007) showed that intrathecal administration of recombinant nerve growth factor (NGF) and neurotrophin-3 to diabetic mice promoted the innervation of myelinated fibers in their plantar skin. Other studies have also supported the potential of recombinant growth factors such as NGF (Apfel et al., 1994; Whitworth et al., 1995), glial cell-derived neurotrophic factor (Akkina et al., 2001), and neurotrophin-3 (Sayers et al., 2003) as therapeutics to prevent or relieve diabetic complications. Especially, some clinical studies using neurotrophic factors for the treatment of diabetic neuropathy have been summarized in a critical review by Leinninger et al. (2004).
Another source for neurotrophic factors that has been noted recently is stem cells. Many kinds of mesenchymal stem cells have been reported as potential sources secreting neurotrophic factors that provide beneficial effects of neuroprotection and neuroregeneration (Koh et al., 2008; Nesti et al., 2011; Zhou et al., 2016). In our recent study, we focused on adipose-derived stem cells (ADSCs) because of their advantages compared to other kinds of mesenchymal stem cells. These cells are an abundant source, easily harvested and cultured, and show rapid proliferation (Luna et al., 2014). In addition, previous studies showed that from an equivalent amount of tissue, the number of stem cells isolated from adipose tissue is 500 times greater than that isolated from bone marrow (Hass et al., 2011). Furthermore, there are many reports showing that ADSCs also secrete various neurotrophic factors such as NGF and brain derived neurotrophic factor (Lopatina et al., 2011; Lu et al., 2011; Hofer and Tuan, 2016), thus suggesting their use as a potential approach for both treatment of neurological diseases and repair of damaged peripheral nerves. Some studies collected neurotrophic factors secreted into the culture media under an hypoxic condition (Lopatina et al., 2011; Lu et al., 2011) whereas others used certain supplements to induce ADSCs to differentiate into neural cells (Kingham et al., 2014; Georgiou et al., 2015). Strikingly, previous studies of Lattanzi et al. (2011) and Lu et al. (2011) on human ADSCs and our recent study on mouse ADSCs (mADSCs) found that ADSCs also secrete neurotrophic factors under normal culture conditions. In particular, quantitative real time polymerase chain reaction results on mADSCs at passage 3 showed significant increases in the expression of NGF, brain derived neurotrophic factor, glial cell-derived neurotrophic factor, and ciliary neurotrophic factor compared with those in adipose tissue; and the expression of neurotrophin-4 tended to increase, but this increase was not significant (Figure 2). We then established a culture system for mADSCs in which the cells were cultured in Eagle’s minimal essential medium supplemented with fetal bovine serum from primary to secondary culture passage 2 and subsequently in serum-free Eagle’s minimal essential medium at passage 3. Equal volumes of mADSCs-media collected at passage 3 and Neurobasal A (21103-049, Gibco, Waltham, MA, USA) were mixed (conditioned media) for use in DRG neuron cultures by the protocol described previously (Tu et al., 2016). The results showed that the neurite extension of DRG neurons was promoted significantly when the cells were cultured in this mixture (Figure 3). These results suggest that mADSCs secreted neurotrophic factors into their culture media.
Figure 2.

Increased expression of nerve growth factor (NGF) (A), brain-derived neurotrophic factor (BDNF) (B), and glial cell-derived neurotrophic factor (GDNF), neurotrophin-4 (NT4), and ciliary neurotrophic factor (CNTF) (C) in mouse adipose-derived stem cells (mADSCs) at passage 3 compared with that in adipose tissue (AT).
Glyceraldehyde-3-phosphate dehydrogenase was used as a reference gene. Each gene was assayed triply. *P < 0.05, **P < 0.01, ***P < 0.001, vs. AT (two tailed t-test).
Figure 3.

Promotion of neurite extension by conditioned media containing equal volume of mouse adipose-derived stem cell-media and Neurobasal A (conditioned media).
(A, B) Representatives of dorsal root ganglion neurons cultured in Dulbecco’s modified Eagle medium (DMEM) (A) or in conditioned media (B). Scale bar: 100 µm. (C) Graph shows that the conditioned media promoted neurite extension of dorsal root ganglion neurons. **P < 0.01, vs. DMEM (two tailed t-test).
Discussion
Like other diseases, the greatest hurdle in studies on diabetes is establishing the appropriate animal models that have symptoms, progression, and complications similar to those of this disease in actual human patients. There are three main approaches for preparing animal models of diabetes: chemical induction, nutritional induction, and genetic modification. Each approach has its own advantages and disadvantages. In general, the strain, sex, and age of mice have been noted as factors affecting the success in establishing diabetic mouse models. Until now, there has been no animal model that completely and accurately reflects the real progression and symptoms of diabetic neuropathy seen in human beings. Thus, appropriate model selection must be made with regards to the aim and scope of the study. Careful and detailed experimental procedures are thus also always critical. In addition, although there is a great amount of literature on studies using animal models of diabetic neuropathy, optimization of all procedures for establishing and characterizing diabetic neuropathy models is needed prior to examining the treatment targets. Together with efforts in establishing more suitable models of diabetic neuropathy, scientists around the world also set the criteria for charactering such models. The criteria recommended by The Diabetic Neuropathy Study Group of the EASD (Neurodiab) is widely used (Biessels et al., 2014). Islam (2013) also recommended that sensitivity to anti-diabetic and anti-neuropathic drugs should be added as additional criteria of successful models of diabetes. In this regard, the use of appropriate and sensitive methods to characterize the established diabetic neuropathy models is one of the most important considerations for a successful study. It is generally suggested to use more than one method for each purpose. The combination of a diabetic model with other models was also reported in some articles to serve the specific aim of the studies. In a previous study of ours, we established a combined sciatic nerve transection-regeneration and type 2 diabetic neuropathy mouse model and showed that this combination was suitable for studies on the treatment of pre-diabetic and early stages of diabetic neuropathy in humans, known as stocking-glove neuropathy (Vuong et al., 2018).
Following up the literature regarding research on diabetic neuropathy, we have noted that some treatment strategies gave great results at the clinical level (Hackett, 2006; Tang et al., 2007; Mijnhout et al., 2010). However, there is a controversy regarding some strategies. For instance, there was an inconsistency in results between some groups on the effect of oxidative stress on neuronal morphology. Especially, the works of Russell et al. (2002) showed that hyperglycemia and oxidative stress result in neuronal death by apoptosis, whereas Schmidt (2001) found no evidence of neuronal apoptosis. The duration of diabetes, the time points for analyses, and the preferred subset of neuronal targets of hyperglycemia were assumed as reasons for these contradictory results (Leinninger et al., 2004). In addition, nitrosative stress was put into focus as a target for diabetic treatment, however some studies reported that the severity of this stress was less in diabetic rats than in mice with their diabetic condition induced by STZ (Drel et al., 2007a; Obrosova et al., 2007a).
Both prevention of the progression of neuropathic symptoms and nerve impairment and degeneration, as well as the promotion of regeneration of degenerated nerve fibers should be considered equally in treating diabetic neuropathy (Yasuda et al., 2003). In addition, external factors strongly affect the intrinsic growth capacity of an injured neuron (Ferguson and Son, 2011). Furthermore, even though stem cell therapy has identified itself as a promising therapy for regenerative medicine, it may not be a standard treatment option for all stages of diabetic neuropathy. Because the progression of diabetic neuropathy is so complex, with its wide spectrum and different phases, such therapy might have different effects on different axonal structures or functions (Zhou et al., 2016). Thus, a combined strategy seems to be a better approach for the treatment of diabetic neuropathy. In this fashion, neurotrophic factors are likely one of the essential components for any combinatorial therapeutic strategy of axon regeneration (Afshari et al., 2009). In an ongoing study, we found that mADSCs showed an increased expression of neurotrophic factors and secreted them into the culture media under normal culture conditions. These results were supported by the effect of the conditioned media on promoting the neurite extension of DRG neurons (Figures 2 and 3). However, as with any other approaches, further studies using carefully designed experiments are required for a better understanding of the actions of neurotrophins and their receptors on both neurons and glia (Ferguson and Son, 2011).
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: None.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Yu J; T-Editor: Jia Y
References
- 1.Afshari FT, Kappagantula S, Fawcett JW. Extrinsic and intrinsic factors controlling axonal regeneration after spinal cord injury. Expert Rev Mol Med. 2009;11:e37. doi: 10.1017/S1462399409001288. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed N. Advanced glycation endproducts--role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67:3–21. doi: 10.1016/j.diabres.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Akkina SK, Patterson CL, Wright DE. GDNF rescues nonpeptidergic unmyelinated primary afferents in streptozotocin-treated diabetic mice. Exp Neurol. 2001;167:173–182. doi: 10.1006/exnr.2000.7547. [DOI] [PubMed] [Google Scholar]
- 4.Apfel SC, Arezzo JC, Brownlee M, Federoff H, Kessler JA. Nerve growth factor administration protects against experimental diabetic sensory neuropathy. Brain Res. 1994;634:7–12. doi: 10.1016/0006-8993(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 5.Bai HP, Liu P, Wu YM, Guo WY, Guo YX, Wang XL. Activation of spinal GABAB receptors normalizes N-methyl-D-aspartate receptor in diabetic neuropathy. J Neurol Sci. 2014;341:68–72. doi: 10.1016/j.jns.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Biessels GJ, Bril V, Calcutt NA, Cameron NE, Cotter MA, Dobrowsky R, Feldman EL, Fernyhough P, Jakobsen J, Malik RA, Mizisin AP, Oates PJ, Obrosova IG, Pop-Busui R, Russell JW, Sima AA, Stevens MJ, Schmidt RE, Tesfaye S, Veves A, et al. Phenotyping animal models of diabetic neuropathy: a consensus statement of the diabetic neuropathy study group of the EASD (Neurodiab) J Peripher Nerv Syst. 2014;19:77–87. doi: 10.1111/jns5.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bril V. Neuromuscular complications of diabetes mellitus. Continuum (Minneap Minn) 2014;20:531–544. doi: 10.1212/01.CON.0000450964.30710.a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brosius FC, Khoury CC, Buller CL, Chen S. Abnormalities in signaling pathways in diabetic nephropathy. Expert Rev Endocrinol Metab. 2010;5:51–64. doi: 10.1586/eem.09.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brownlee M. Negative consequences of glycation. Metabolism. 2000;49:9–13. doi: 10.1016/s0026-0495(00)80078-5. [DOI] [PubMed] [Google Scholar]
- 10.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 11.Cao XH, Byun HS, Chen SR, Cai YQ, Pan HL. Reduction in voltage-gated K+ channel activity in primary sensory neurons in painful diabetic neuropathy: role of brain-derived neurotrophic factor. J Neurochem. 2010;114:1460–1475. doi: 10.1111/j.1471-4159.2010.06863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen SR, Pan HL. Hypersensitivity of spinothalamic tract neurons associated with diabetic neuropathic pain in rats. J Neurophysiol. 2002;87:2726–2733. doi: 10.1152/jn.2002.87.6.2726. [DOI] [PubMed] [Google Scholar]
- 13.Cheng HT, Dauch JR, Hayes JM, Hong Y, Feldman EL. Nerve growth factor mediates mechanical allodynia in a mouse model of type 2 diabetes. J Neuropathol Exp Neurol. 2009;68:1229–1243. doi: 10.1097/NEN.0b013e3181bef710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng KI, Wang HC, Chuang YT, Chou CW, Tu HP, Yu YC, Chang LL, Lai CS. Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P-p38 mitogen-activated protein kinase activation in streptozotocin-induced diabetic rats. Eur J Pain. 2014;18:162–173. doi: 10.1002/j.1532-2149.2013.00356.x. [DOI] [PubMed] [Google Scholar]
- 15.Chierzi S, Ratto GM, Verma P, Fawcett JW. The ability of axons to regenerate their growth cones depends on axonal type and age, and is regulated by calcium, cAMP and ERK. Eur J Neurosci. 2005;21:2051–2062. doi: 10.1111/j.1460-9568.2005.04066.x. [DOI] [PubMed] [Google Scholar]
- 16.Choeiri C, Hewitt K, Durkin J, Simard CJ, Renaud JM, Messier C. Longitudinal evaluation of memory performance and peripheral neuropathy in the Ins2C96Y Akita mice. Behav Brain Res. 2005;157:31–38. doi: 10.1016/j.bbr.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Christianson JA, Ryals JM, Johnson MS, Dobrowsky RT, Wright DE. Neurotrophic modulation of myelinated cutaneous innervation and mechanical sensory loss in diabetic mice. Neuroscience. 2007;145:303–313. doi: 10.1016/j.neuroscience.2006.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci. 2010;30:9306–9315. doi: 10.1523/JNEUROSCI.6271-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu GK, Tator CH. Calcium influx is necessary for optimal regrowth of transected neurites of rat sympathetic ganglion neurons in vitro. Neuroscience. 2001;102:945–957. doi: 10.1016/s0306-4522(00)00514-5. [DOI] [PubMed] [Google Scholar]
- 20.Colleoni M, Sacerdote P. Murine models of human neuropathic pain. Biochim Biophys Acta. 2010;1802:924–933. doi: 10.1016/j.bbadis.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Coppey L, Lu B, Gerard C, Yorek MA. Effect of inhibition of angiotensin-converting enzyme and/or neutral endopeptidase on neuropathy in high-fat-fed C57Bl/6J mice. J Obes. 2012;2012:326806. doi: 10.1155/2012/326806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol. 2002;52:786–792. doi: 10.1002/ana.10364. [DOI] [PubMed] [Google Scholar]
- 23.Dauch JR, Yanik BM, Hsieh W, Oh SS, Cheng HT. Neuron-astrocyte signaling network in spinal cord dorsal horn mediates painful neuropathy of type 2 diabetes. Glia. 2012;60:1301–1315. doi: 10.1002/glia.22349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickinson PJ, Carrington AL, Frost GS, Boulton AJ. Neurovascular disease, antioxidants and glycation in diabetes. Diabetes Metab Res Rev. 2002;18:260–272. doi: 10.1002/dmrr.305. [DOI] [PubMed] [Google Scholar]
- 25.Doron-Mandel E, Fainzilber M, Terenzio M. Growth control mechanisms in neuronal regeneration. FEBS Lett. 2015;589:1669–1677. doi: 10.1016/j.febslet.2015.04.046. [DOI] [PubMed] [Google Scholar]
- 26.Drel VR, Mashtalir N, Ilnytska O, Shin J, Li F, Lyzogubov VV, Obrosova IG. The leptin-deficient (ob/ob) mouse: a new animal model of peripheral neuropathy of type 2 diabetes and obesity. Diabetes. 2006;55:3335–3343. doi: 10.2337/db06-0885. [DOI] [PubMed] [Google Scholar]
- 27.Drel VR, Pacher P, Vareniuk I, Pavlov IA, Ilnytska O, Lyzogubov VV, Bell SR, Groves JT, Obrosova IG. Evaluation of the peroxynitrite decomposition catalyst Fe(III) tetra-mesitylporphyrin octasulfonate on peripheral neuropathy in a mouse model of type 1 diabetes. Int J Mol Med. 2007a;20:783–792. [PMC free article] [PubMed] [Google Scholar]
- 28.Drel VR, Pacher P, Vareniuk I, Pavlov I, Ilnytska O, Lyzogubov VV, Tibrewala J, Groves JT, Obrosova IG. A peroxynitrite decomposition catalyst counteracts sensory neuropathy in streptozotocin-diabetic mice. Eur J Pharmacol. 2007b;569:48–58. doi: 10.1016/j.ejphar.2007.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Du ZX, Zhang HY, Meng X, Guan Y, Wang HQ. Role of oxidative stress and intracellular glutathione in the sensitivity to apoptosis induced by proteasome inhibitor in thyroid cancer cells. BMC Cancer. 2009;9:56. doi: 10.1186/1471-2407-9-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellis A, Bennett DL. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111:26–37. doi: 10.1093/bja/aet128. [DOI] [PubMed] [Google Scholar]
- 31.Ferguson TA, Son YJ. Extrinsic and intrinsic determinants of nerve regeneration. J Tissue Eng. 2011;2:2041731411418392. doi: 10.1177/2041731411418392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernyhough P, Calcutt NA. Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium. 2010;47:130–139. doi: 10.1016/j.ceca.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabra BH, Sirois P. Hyperalgesia in non-obese diabetic (NOD) mice: a role for the inducible bradykinin B1 receptor. Eur J Pharmacol. 2005;514:61–67. doi: 10.1016/j.ejphar.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 34.Gao F, Zheng ZM. Animal models of diabetic neuropathic pain. Exp Clin Endocrinol Diabetes. 2014;122:100–106. doi: 10.1055/s-0033-1363234. [DOI] [PubMed] [Google Scholar]
- 35.Gao Y, Nikulina E, Mellado W, Filbin MT. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci. 2003;23:11770–11777. doi: 10.1523/JNEUROSCI.23-37-11770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Georgiou M, Golding JP, Loughlin AJ, Kingham PJ, Phillips JB. Engineered neural tissue with aligned, differentiated adipose-derived stem cells promotes peripheral nerve regeneration across a critical sized defect in rat sciatic nerve. Biomaterials. 2015;37:242–251. doi: 10.1016/j.biomaterials.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 37.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gitler D, Spira ME. Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron. 1998;20:1123–1135. doi: 10.1016/s0896-6273(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 39.Hackett G. PDE5 inhibitors in diabetic peripheral neuropathy. Int J Clin Pract. 2006;60:1123–1126. doi: 10.1111/j.1742-1241.2006.01087.x. [DOI] [PubMed] [Google Scholar]
- 40.Hall KE, Liu J, Sima AA, Wiley JW. Impaired inhibitory G-protein function contributes to increased calcium currents in rats with diabetic neuropathy. J Neurophysiol. 2001;86:760–770. doi: 10.1152/jn.2001.86.2.760. [DOI] [PubMed] [Google Scholar]
- 41.Hammes HP, Du X, Edelstein D, Taguchi T, Matsumura T, Ju Q, Lin J, Bierhaus A, Nawroth P, Hannak D, Neumaier M, Bergfeld R, Giardino I, Brownlee M. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med. 2003;9:294–299. doi: 10.1038/nm834. [DOI] [PubMed] [Google Scholar]
- 42.Harati Y. Diabetic neuropathies: unanswered questions. Neurol Clin. 2007;25:303–317. doi: 10.1016/j.ncl.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Hass R, Kasper C, Bohm S, Jacobs R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal. 2011;9:12. doi: 10.1186/1478-811X-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hofer HR, Tuan RS. Secreted trophic factors of mesenchymal stem cells support neurovascular and musculoskeletal therapies. Stem Cell Res Ther. 2016;7:131. doi: 10.1186/s13287-016-0394-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ii M, Nishimura H, Kusano KF, Qin G, Yoon YS, Wecker A, Asahara T, Losordo DW. Neuronal nitric oxide synthase mediates statin-induced restoration of vasa nervorum and reversal of diabetic neuropathy. Circulation. 2005;112:93–102. doi: 10.1161/CIRCULATIONAHA.104.511964. [DOI] [PubMed] [Google Scholar]
- 46.Ikemoto S, Takahashi M, Tsunoda N, Maruyama K, Itakura H, Ezaki O. High-fat diet-induced hyperglycemia and obesity in mice: differential effects of dietary oils. Metabolism. 1996;45:1539–1546. doi: 10.1016/s0026-0495(96)90185-7. [DOI] [PubMed] [Google Scholar]
- 47.Islam MS. Animal models of diabetic neuropathy: progress since 1960s. J Diabetes Res. 2013;2013:149452. doi: 10.1155/2013/149452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Islam MS, Wilson RD. Experimentally induced rodent models of type 2 diabetes. Methods Mol Biol. 2012;933:161–174. doi: 10.1007/978-1-62703-068-7_10. [DOI] [PubMed] [Google Scholar]
- 49.Jack MM, Ryals JM, Wright DE. Protection from diabetes-induced peripheral sensory neuropathy--a role for elevated glyoxalase I? Exp Neurol. 2012;234:62–69. doi: 10.1016/j.expneurol.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic-Todorovic V, Todorovic SM. Cell-specific alterations of T-type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci. 2007;27:3305–3316. doi: 10.1523/JNEUROSCI.4866-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jain NK, Patil CS, Singh A, Kulkarni SK. Sildenafil-induced peripheral analgesia and activation of the nitric oxide-cyclic GMP pathway. Brain Res. 2001;909:170–178. doi: 10.1016/s0006-8993(01)02673-7. [DOI] [PubMed] [Google Scholar]
- 52.Kan M, Guo G, Singh B, Singh V, Zochodne DW. Glucagon-like peptide 1, insulin, sensory neurons, and diabetic neuropathy. J Neuropathol Exp Neurol. 2012;71:494–510. doi: 10.1097/NEN.0b013e3182580673. [DOI] [PubMed] [Google Scholar]
- 53.Kim B, Feldman EL. Insulin resistance in the nervous system. Trends Endocrinol Metab. 2012;23:133–141. doi: 10.1016/j.tem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.King AJ. The use of animal models in diabetes research. Br J Pharmacol. 2012;166:877–894. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kingham PJ, Kolar MK, Novikova LN, Novikov LN, Wiberg M. Stimulating the neurotrophic and angiogenic properties of human adipose-derived stem cells enhances nerve repair. Stem Cells Dev. 2014;23:741–754. doi: 10.1089/scd.2013.0396. [DOI] [PubMed] [Google Scholar]
- 56.Koh SH, Kim KS, Choi MR, Jung KH, Park KS, Chai YG, Roh W, Hwang SJ, Ko HJ, Huh YM, Kim HT, Kim SH. Implantation of human umbilical cord-derived mesenchymal stem cells as a neuroprotective therapy for ischemic stroke in rats. Brain Res. 2008;1229:233–248. doi: 10.1016/j.brainres.2008.06.087. [DOI] [PubMed] [Google Scholar]
- 57.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes. 1998;47:859–866. doi: 10.2337/diabetes.47.6.859. [DOI] [PubMed] [Google Scholar]
- 58.Krishnan A, Duraikannu A, Zochodne DW. Releasing ‘brakes’ to nerve regeneration: intrinsic molecular targets. Eur J Neurosci. 2016;43:297–308. doi: 10.1111/ejn.13018. [DOI] [PubMed] [Google Scholar]
- 59.Krishnan A, Purdy K, Chandrasekhar A, Martinez J, Cheng C, Zochodne DW. A BRCA1-dependent DNA damage response in the regenerating adult peripheral nerve milieu. Mol Neurobiol. 2018;55:4051–4067. doi: 10.1007/s12035-017-0574-7. [DOI] [PubMed] [Google Scholar]
- 60.Landry RP, Jacobs VL, Romero-Sandoval EA, DeLeo JA. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp Neurol. 2012;234:340–350. doi: 10.1016/j.expneurol.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Lange PS, Langley B, Lu P, Ratan RR. Novel roles for arginase in cell survival, regeneration, and translation in the central nervous system. J Nutr. 2004;134:2812S–2817S. doi: 10.1093/jn/134.10.2812S. discussion 2818S-2819S. [DOI] [PubMed] [Google Scholar]
- 62.Lattanzi W, Geloso MC, Saulnier N, Giannetti S, Puglisi MA, Corvino V, Gasbarrini A, Michetti F. Neurotrophic features of human adipose tissue-derived stromal cells: in vitro and in vivo studies. J Biomed Biotechnol. 2011;2011:468705. doi: 10.1155/2011/468705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leinninger GM, Vincent AM, Feldman EL. The role of growth factors in diabetic peripheral neuropathy. J Peripher Nerv Syst. 2004;9:26–53. doi: 10.1111/j.1085-9489.2004.09105.x. [DOI] [PubMed] [Google Scholar]
- 64.Leiter EH. The NOD mouse: a model for insulin-dependent diabetes mellitus. Curr Protoc Immunol Chapter. 2001;15:Unit 15.9. doi: 10.1002/0471142735.im1509s24. [DOI] [PubMed] [Google Scholar]
- 65.Liniger C, Pernet A, Moody JF, Assal JP. Effect of gangliosides on diabetic peripheral neuropathy. Diabetes Res Clin Pract. 1989;7:251–258. doi: 10.1016/0168-8227(89)90012-0. [DOI] [PubMed] [Google Scholar]
- 66.Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lopatina T, Kalinina N, Karagyaur M, Stambolsky D, Rubina K, Revischin A, Pavlova G, Parfyonova Y, Tkachuk V. Adipose-derived stem cells stimulate regeneration of peripheral nerves: BDNF secreted by these cells promotes nerve healing and axon growth de novo. PLoS One. 2011;6:e17899. doi: 10.1371/journal.pone.0017899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu S, Lu C, Han Q, Li J, Du Z, Liao L, Zhao RC. Adipose-derived mesenchymal stem cells protect PC12 cells from glutamate excitotoxicity-induced apoptosis by upregulation of XIAP through PI3-K/Akt activation. Toxicology. 2011;279:189–195. doi: 10.1016/j.tox.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 69.Luna AC, Madeira ME, Conceicao TO, Moreira JA, Laiso RA, Maria DA. Characterization of adipose-derived stem cells of anatomical region from mice. BMC Res Notes. 2014;7:552. doi: 10.1186/1756-0500-7-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Magill CK, Tong A, Kawamura D, Hayashi A, Hunter DA, Parsadanian A, Mackinnon SE, Myckatyn TM. Reinnervation of the tibialis anterior following sciatic nerve crush injury: a confocal microscopic study in transgenic mice. Exp Neurol. 2007;207:64–74. doi: 10.1016/j.expneurol.2007.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mallik S, Kallis C, Lunn MPT, Smith AG. Gangliosides for the treatment of diabetic peripheral neuropathy. Cochrane Database Syst Rev. 2017;3:CD011028. [Google Scholar]
- 72.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McCallum JB, Kwok WM, Sapunar D, Fuchs A, Hogan QH. Painful peripheral nerve injury decreases calcium current in axotomized sensory neurons. Anesthesiology. 2006;105:160–168. doi: 10.1097/00000542-200607000-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Messinger RB, Naik AK, Jagodic MM, Nelson MT, Lee WY, Choe WJ, Orestes P, Latham JR, Todorovic SM, Jevtovic-Todorovic V. In vivo silencing of the Ca(V)3.2 T-type calcium channels in sensory neurons alleviates hyperalgesia in rats with streptozocin-induced diabetic neuropathy. Pain. 2009;145:184–195. doi: 10.1016/j.pain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mijnhout GS, Alkhalaf A, Kleefstra N, Bilo HJ. Alpha lipoic acid: a new treatment for neuropathic pain in patients with diabetes? Neth J Med. 2010;68:158–162. [PubMed] [Google Scholar]
- 77.Mika J, Zychowska M, Popiolek-Barczyk K, Rojewska E, Przewlocka B. Importance of glial activation in neuropathic pain. Eur J Pharmacol. 2013;716:106–119. doi: 10.1016/j.ejphar.2013.01.072. [DOI] [PubMed] [Google Scholar]
- 78.Misawa S, Sakurai K, Shibuya K, Isose S, Kanai K, Ogino J, Ishikawa K, Kuwabara S. Neuropathic pain is associated with increased nodal persistent Na(+) currents in human diabetic neuropathy. J Peripher Nerv Syst. 2009;14:279–284. doi: 10.1111/j.1529-8027.2009.00239.x. [DOI] [PubMed] [Google Scholar]
- 79.Murakami T, Iwanaga T, Ogawa Y, Fujita Y, Sato E, Yoshitomi H, Sunada Y, Nakamura A. Development of sensory neuropathy in streptozotocin-induced diabetic mice. Brain Behav. 2013;3:35–41. doi: 10.1002/brb3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murakawa Y, Zhang W, Pierson CR, Brismar T, Ostenson CG, Efendic S, Sima AA. Impaired glucose tolerance and insulinopenia in the GK-rat causes peripheral neuropathy. Diabetes Metab Res Rev. 2002;18:473–483. doi: 10.1002/dmrr.326. [DOI] [PubMed] [Google Scholar]
- 81.Nakamura T, Terajima T, Ogata T, Ueno K, Hashimoto N, Ono K, Yano S. Establishment and pathophysiological characterization of type 2 diabetic mouse model produced by streptozotocin and nicotinamide. Biol Pharm Bull. 2006;29:1167–1174. doi: 10.1248/bpb.29.1167. [DOI] [PubMed] [Google Scholar]
- 82.Nesti C, Pardini C, Barachini S, D’Alessandro D, Siciliano G, Murri L, Petrini M, Vaglini F. Human dental pulp stem cells protect mouse dopaminergic neurons against MPP+ or rotenone. Brain Res. 2011;1367:94–102. doi: 10.1016/j.brainres.2010.09.042. [DOI] [PubMed] [Google Scholar]
- 83.O’Brien PD, Sakowski SA, Feldman EL. Mouse models of diabetic neuropathy. ILAR journal / National Research Council, Institute of Laboratory Animal Resources. 2014;54:259–272. doi: 10.1093/ilar/ilt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Obrosova IG. Diabetes and the peripheral nerve. Biochim Biophys Acta. 2009;1792:931–940. doi: 10.1016/j.bbadis.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 85.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabo C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 86.Obrosova IG, Drel VR, Oltman CL, Mashtalir N, Tibrewala J, Groves JT, Yorek MA. Role of nitrosative stress in early neuropathy and vascular dysfunction in streptozotocin-diabetic rats. Am J Physiol Endocrinol Metab. 2007a;293:E1645–1655. doi: 10.1152/ajpendo.00479.2007. [DOI] [PubMed] [Google Scholar]
- 87.Obrosova IG, Ilnytska O, Lyzogubov VV, Pavlov IA, Mashtalir N, Nadler JL, Drel VR. High-fat diet induced neuropathy of pre-diabetes and obesity: effects of “healthy” diet and aldose reductase inhibition. Diabetes. 2007b;56:2598–2608. doi: 10.2337/db06-1176. [DOI] [PubMed] [Google Scholar]
- 88.Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patil CS, Singh VP, Singh S, Kulkarni SK. Modulatory effect of the PDE-5 inhibitor sildenafil in diabetic neuropathy. Pharmacology. 2004;72:190–195. doi: 10.1159/000080104. [DOI] [PubMed] [Google Scholar]
- 90.Pittenger G, Vinik A. Nerve growth factor and diabetic neuropathy. Exp Diabesity Res. 2003;4:271–285. doi: 10.1155/EDR.2003.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabet Med. 2005;22:359–370. doi: 10.1111/j.1464-5491.2005.01499.x. [DOI] [PubMed] [Google Scholar]
- 92.Ruessmann HJ. German Society of out patient diabetes centres AND (Arbeitsgemeinschaft niedergelassener diabetologisch tätiger Arzte e.V.) (2009) Switching from pathogenetic treatment with alpha-lipoic acid to gabapentin and other analgesics in painful diabetic neuropathy: a real-world study in outpatients. J Diabetes Complications. 23:174–177. doi: 10.1016/j.jdiacomp.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 93.Russell JW, Golovoy D, Vincent AM, Mahendru P, Olzmann JA, Mentzer A, Feldman EL. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002;16:1738–1748. doi: 10.1096/fj.01-1027com. [DOI] [PubMed] [Google Scholar]
- 94.Sanchez AP, Sharma K. Transcription factors in the pathogenesis of diabetic nephropathy. Expert Rev Mol Med. 2009;11:e13. doi: 10.1017/S1462399409001057. [DOI] [PubMed] [Google Scholar]
- 95.Sayers NM, Beswick LJ, Middlemas A, Calcutt NA, Mizisin AP, Tomlinson DR, Fernyhough P. Neurotrophin-3 prevents the proximal accumulation of neurofilament proteins in sensory neurons of streptozocin-induced diabetic rats. Diabetes. 2003;52:2372–2380. doi: 10.2337/diabetes.52.9.2372. [DOI] [PubMed] [Google Scholar]
- 96.Schmidt RE. Neuronal preservation in the sympathetic ganglia of rats with chronic streptozotocin-induced diabetes. Brain Res. 2001;921:256–259. doi: 10.1016/s0006-8993(01)03155-9. [DOI] [PubMed] [Google Scholar]
- 97.Schmidt RE, Green KG, Snipes LL, Feng D. Neuritic dystrophy and neuronopathy in Akita (Ins2(Akita)) diabetic mouse sympathetic ganglia. Exp Neurol. 2009;216:207–218. doi: 10.1016/j.expneurol.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmidt RE, Dorsey DA, Beaudet LN, Frederick KE, Parvin CA, Plurad SB, Levisetti MG. Non-obese diabetic mice rapidly develop dramatic sympathetic neuritic dystrophy: a new experimental model of diabetic autonomic neuropathy. Am J Pathol. 2003;163:2077–2091. doi: 10.1016/S0002-9440(10)63565-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schreiber AK, Nones CF, Reis RC, Chichorro JG, Cunha JM. Diabetic neuropathic pain: physiopathology and treatment. World J Diabetes. 2015;6:432–444. doi: 10.4239/wjd.v6.i3.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shaikh AS, Somani RS. Animal models and biomarkers of neuropathy in diabetic rodents. Indian J Pharmacol. 2010;42:129–134. doi: 10.4103/0253-7613.66833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sima AA, Robertson DM. Peripheral neuropathy in mutant diabetic mouse [C57BL/Ks (db/db)] Acta Neuropathol. 1978;41:85–89. doi: 10.1007/BF00689757. [DOI] [PubMed] [Google Scholar]
- 102.Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. 2014;18:1–14. doi: 10.4196/kjpp.2014.18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Spira ME, Oren R, Dormann A, Gitler D. Critical calpain-dependent ultrastructural alterations underlie the transformation of an axonal segment into a growth cone after axotomy of cultured Aplysia neurons. J Comp Neurol. 2003;457:293–312. doi: 10.1002/cne.10569. [DOI] [PubMed] [Google Scholar]
- 104.Spuler M, Dimpfel W, Tullner HU. Ganglioside therapy in experimental diabetic neuropathy. Arzneimittelforschung. 1988;38:881–884. [PubMed] [Google Scholar]
- 105.Stavniichuk R, Drel VR, Shevalye H, Vareniuk I, Stevens MJ, Nadler JL, Obrosova IG. Role of 12/15-lipoxygenase in nitrosative stress and peripheral prediabetic and diabetic neuropathies. Free Radic Biol Med. 2010;49:1036–1045. doi: 10.1016/j.freeradbiomed.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, Brosius F, 3rd, Feldman EL. Mouse models of diabetic neuropathy. Neurobiol Dis. 2007;28:276–285. doi: 10.1016/j.nbd.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang J, Wingerchuk DM, Crum BA, Rubin DI, Demaerschalk BM. Alpha-lipoic acid may improve symptomatic diabetic polyneuropathy. Neurologist. 2007;13:164–167. doi: 10.1097/01.nrl.0000263703.78318.2b. [DOI] [PubMed] [Google Scholar]
- 108.Thornalley PJ. The potential role of thiamine (vitamin B1) in diabetic complications. Curr Diabetes Rev. 2005;1:287–298. doi: 10.2174/157339905774574383. [DOI] [PubMed] [Google Scholar]
- 109.Tracy JA, Dyck PJ. The spectrum of diabetic neuropathies. Phys Med Rehabil Clin N Am. 2008;19:1–26, v. doi: 10.1016/j.pmr.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tu NH, Katano T, Matsumura S, Pham VM, Muratani T, Minami T, Ito S. Role of c-Jun N-terminal kinase in late nerve regeneration monitored by in vivo imaging of thy1-yellow fluorescent protein transgenic mice. Eur J Neurosci. 2016;43:548–560. doi: 10.1111/ejn.13139. [DOI] [PubMed] [Google Scholar]
- 111.Unezaki S, Yoshii S, Mabuchi T, Saito A, Ito S. Effects of neurotrophic factors on nerve regeneration monitored by in vivo imaging in thy1-YFP transgenic mice. J Neurosci Methods. 2009;178:308–315. doi: 10.1016/j.jneumeth.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 112.Vaag A, Lund SS. Non-obese patients with type 2 diabetes and prediabetic subjects: distinct phenotypes requiring special diabetes treatment and (or) prevention? Appl Physiol Nutr Metab. 2007;32:912–920. doi: 10.1139/H07-100. [DOI] [PubMed] [Google Scholar]
- 113.Vareniuk I, Pavlov IA, Obrosova IG. Inducible nitric oxide synthase gene deficiency counteracts multiple manifestations of peripheral neuropathy in a streptozotocin-induced mouse model of diabetes. Diabetologia. 2008;51:2126–2133. doi: 10.1007/s00125-008-1136-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst. 2009;14:257–267. doi: 10.1111/j.1529-8027.2009.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Voitenko N. Calcium signaling in diabetic neuropathy. Neurophysiology. 2004;36:310–314. [Google Scholar]
- 116.Vuong PM, Nguyen HT, Katano T, Matsumura S, Saito A, Yamada A, Furue H, Ito S. Impaired peripheral nerve regeneration in type-2 diabetic mouse model. Eur J Neurosci. 2018;47:126–139. doi: 10.1111/ejn.13771. [DOI] [PubMed] [Google Scholar]
- 117.Wada R, Nishizawa Y, Yagihashi N, Takeuchi M, Ishikawa Y, Yasumura K, Nakano M, Yagihashi S. Effects of OPB-9195, anti-glycation agent, on experimental diabetic neuropathy. Eur J Clin Invest. 2001;31:513–520. doi: 10.1046/j.1365-2362.2001.00826.x. [DOI] [PubMed] [Google Scholar]
- 118.Wang H, Storlien LH, Huang XF. Effects of dietary fat types on body fatness, leptin, and ARC leptin receptor, NPY, and AgRP mRNA expression. Am J Physiol Endocrinol Metab. 2002;282:E1352–1359. doi: 10.1152/ajpendo.00230.2001. [DOI] [PubMed] [Google Scholar]
- 119.Wang L, Chopp M, Zhang ZG. PDE5 inhibitors promote recovery of peripheral neuropathy in diabetic mice. Neural Regen Res. 2017;12:218–219. doi: 10.4103/1673-5374.200804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang L, Chopp M, Szalad A, Liu Z, Bolz M, Alvarez FM, Lu M, Zhang L, Cui Y, Zhang RL, Zhang ZG. Phosphodiesterase-5 is a therapeutic target for peripheral neuropathy in diabetic mice. Neuroscience. 2011;193:399–410. doi: 10.1016/j.neuroscience.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Whitworth IH, Terenghi G, Green CJ, Brown RA, Stevens E, Tomlinson DR. Targeted delivery of nerve growth factor via fibronectin conduits assists nerve regeneration in control and diabetic rats. Eur J Neurosci. 1995;7:2220–2225. doi: 10.1111/j.1460-9568.1995.tb00643.x. [DOI] [PubMed] [Google Scholar]
- 122.Wright DE, Johnson MS, Arnett MG, Smittkamp SE, Ryals JM. Selective changes in nocifensive behavior despite normal cutaneous axon innervation in leptin receptor-null mutant (db/db) mice. J Peripher Nerv Syst. 2007;12:250–261. doi: 10.1111/j.1529-8027.2007.00144.x. [DOI] [PubMed] [Google Scholar]
- 123.Xie XY, Barrett JN. Membrane resealing in cultured rat septal neurons after neurite transection: evidence for enhancement by Ca(2+)-triggered protease activity and cytoskeletal disassembly. J Neurosci. 1991;11:3257–3267. doi: 10.1523/JNEUROSCI.11-10-03257.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 126.Zheng W, Honmou O, Miyata K, Harada K, Suzuki J, Liu H, Houkin K, Hamada H, Kocsis JD. Therapeutic benefits of human mesenchymal stem cells derived from bone marrow after global cerebral ischemia. Brain Res. 2010;1310:8–16. doi: 10.1016/j.brainres.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 127.Zhou JY, Zhang Z, Qian GS. Mesenchymal stem cells to treat diabetic neuropathy: a long and strenuous way from bench to the clinic. Cell Death Discov. 2016;2:16055. doi: 10.1038/cddiscovery.2016.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhou S, Shen D, Wang Y, Gong L, Tang X, Yu B, Gu X, Ding F. microRNA-222 targeting PTEN promotes neurite outgrowth from adult dorsal root ganglion neurons following sciatic nerve transection. PLoS One. 2012;7:e44768. doi: 10.1371/journal.pone.0044768. [DOI] [PMC free article] [PubMed] [Google Scholar]