

Abstract
Post-translational modification of proteins is a strategy widely used in biological systems. It expands the diversity of the proteome, and allows for the tailoring of both function and localization of proteins within cells as well as the material properties of structural proteins and matrices. Despite their ubiquity in biology, with a few exceptions, the potential of post-translational modifications in biomaterials synthesis has remained largely untapped. As a proof-of-concept to demonstrate the feasibility of creating a genetically encoded biohybrid material through post-translational modification, herein we report the generation of a family of three stimulus-responsive hybrid materials — fatty-acid-modified elastin-like polypeptides — using a one-pot recombinant expression and post-translational lipidation methodology. These hybrid biomaterials contain an amphiphilic domain, comprised of a β-sheet-forming peptide that is post-translationally functionalized with a C14 alkyl chain, fused to a thermally responsive elastin-like polypeptide. They exhibit temperature-triggered, hierarchical self-assembly across multiple length scales with varied structure and material properties that can be controlled at the sequence level.
Developing new biomaterials is a vibrant area of research, with applications in tissue engineering, regenerative medicine, and drug-delivery.1 In particular, protein-and peptide-based materials are attractive candidates for these applications because of their well-defined composition, lack of toxicity, and biodegradability.2–5 However, compared to synthetic polymers, the precision offered by recombinant expression is offset by a limited compositional repertoire that consists of the canonical amino acids.6
One strategy to expand the diversity of protein-based materials is post-translational modification, a large and diverse class of chemical transformations carried out on proteins within cells after their expression, which nature uses to diversify the proteome.7,8 Post-translational modifications play an important role in modifying the function and localization of polypeptides in the cellular environment, as well as the material properties of structural proteins and biological matrices.9,10 Despite their ubiquity in biology, with a few exceptions, such as the recombinant expression of collagen and mussel foot protein, 11,12 the use of post-translational modifications to synthesize hybrid biomaterials is still untapped.
As a proof-of-concept and to demonstrate the feasibility of preparing a genetically encoded biohybrid material, we created a series of three lipid-modified polypeptides (which we refer to as fatty acid-modified elastin-like polypeptides (FAMEs)) using one-pot recombinant expression and post-translational lipidation in E. coli. These FAMEs are thermally responsive hybrid materials whose hierarchical self-assembly can be triggered by a change in temperature and encoded at the sequence level.
The FAME introduced here contains three components (Figure 1a). The first component is a myristoyl group (M), which consists of a linear C14 alkyl chain, chosen because it can be genetically incorporated at the N-terminus of proteins in a single reaction catalyzed by the N-myristoyl transferase (NMT) enzyme.13 The second component is a short, structure-directing (β-sheet forming) peptide sequence of 5 to 10 amino acids (B) that, when conjugated to an alkyl tail such as the myristoyl group, together form a peptide amphiphile (PA). PAs are known to self- assemble into diverse morphologies depending on their peptide and lipid combination14,15,16,17 and as such have been used in a number of biomedical applications, including scaffolds for tissue engineering.17,18 The third component of the FAME is an elastin-like polypeptide (ELP), which can be fused to the C-terminus of the structure-directing peptide at the gene level. ELPs consist of repeat units of the tropoelastin-derived pentapeptides [Val-Pro-Gly-Xaa-Gly], where Xaa can be any amino acid, except proline.19,20 Additionally, we included a short and flexible linker — (Gly-Gly-Ser)n (n = 2–3) — between the PA and the ELP domains to ensure that the N-terminal peptide (B) is accessible to the active site of the NMT enzyme and not sterically hindered by the ELP (Figure 1b).
Figure 1.
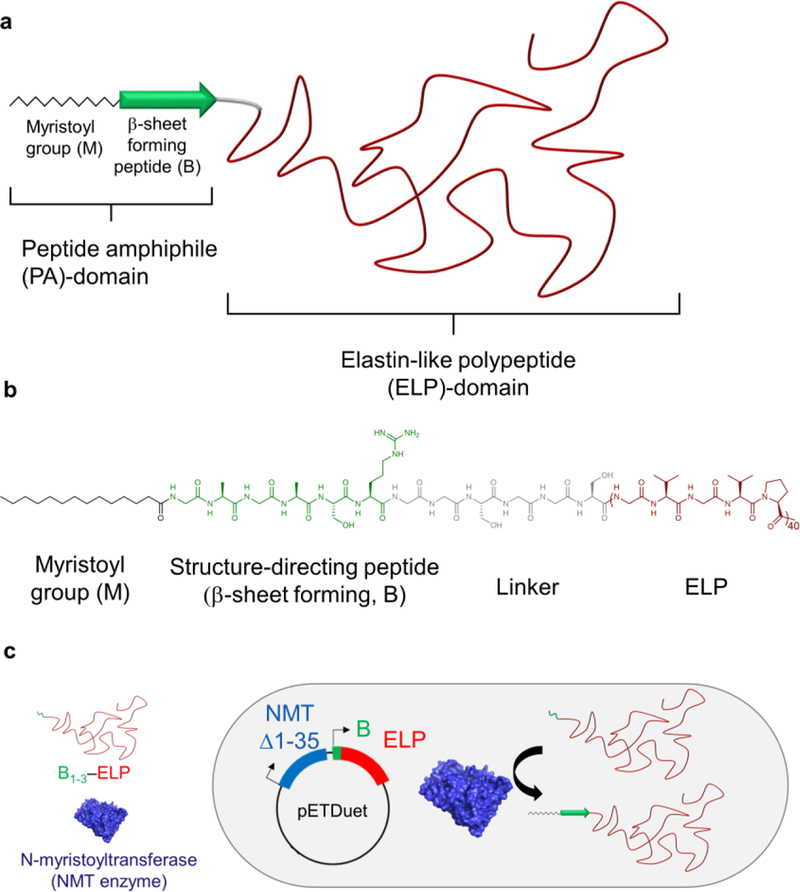
Schematic of the structure and synthesis of FAMEs through post-translational modification of ELPs. a) FAMEs consist of three main components: a myristoyl group (zigzag chain) and a structure-directing peptide sequence (green arrow, B) — which together form a PA domain — and an ELP domain (shown in red). b) Molecular structure of M-B1-ELP shown as an example. In addition to the three main components, a short, flexible linker is also incorporated into the FAMEs, to ensure that myristoylation of the B domain is not sterically hindered by ELP (see text). c) Schematic representation of the one-pot expression and post-translational lipidation by tandem expression of the NMT enzyme (Δ1–35) and B1–3–ELP using pETDuet expression vector in which B is a peptide that is designed de novo to be recognized by the NMT enzyme as a substrate, and myristoylated.
In this design, the ELP moiety serves several purposes. (1) ELPs can be conveniently synthesized in high yield by recombinant expression in E. coli. (2) ELPs are monodisperse, non-toxic,21 and biodegradable22. They have been used as a purification tag for fusion proteins23 and for many biomedical applications, including injectable controlled release depots,23,24 tissue-engineering scaffolds,25,26 and for thermally triggered targeting of tumors.27 (3) They exhibit a lower critical solution temperature (LCST) phase transition, which enables their transition from a soluble state to an insoluble coacervate by: i) increasing the solution temperature above their transition temperature (Tt) or ii) isothermally depressing their transition temperature below the operating temperature by the addition of kosmotropic ions in the Hofmeister series (which lower the Tt of FAMEs by reducing hydration shielding and increasing surface tension of the biopolymer’s hydrophobic regions).28 The Tt can be precisely tuned to within a narrow temperature range at the molecular level by manipulating two orthogonal and genetically encoded variables of the ELP — the composition of Xaa and the chain length.29 Here, we show that this tunable phase transition can be used as a convenient trigger to control the hierarchical self-assembly of FAMEs.
Co-expression of the NMT enzyme with a peptide substrate results in the covalent attachment of a myristoyl group to the N-terminus of the substrate through the formation of an amide bond. This technique has been previously used to generate natively myristoylated eukaryotic proteins in E. coli for structural and functional studies.30 Inspired by this precedent, we use the simultaneous expression of NMT and its substrate (B) in E. coli to recombinantly create a PA. Because the substrate is also fused to an ELP, this process results in the in vivo, one-pot synthesis of FAMEs (Figure 1c). These hybrid materials retain the defining characteristics of both the ELP and PA, namely thermal responsiveness and hierarchical self-assembly, respectively, yielding a unique material with programmable behavior.
The first major challenge to address was the identification of suitable sequences for the peptides (B), which needed to be structure-directing (capable of β-sheet formation) and also serve as a myristoylation substrate. Previous studies have shown that it is possible to modify heterologous proteins in E. coli lysate with myristic acid and analogues containing bioorthogonal handles such as azides. This robust bioconjugation was achieved by co-expression of a protein of interest fused to an 11-amino acid signal peptide from ADP-ribosylation factor (a natively myristoylated yeast protein) and NMT.31–34 However, to date, the ability of NMT to myristoylate other sequences, and in particular structure-directing peptides, has not been investigated.
Analysis of the N-terminal sequence of myristoylated proteins revealed no universal consensus sequence except the presence of an N-terminal Gly residue.35 This lack of consensus, we hypothesized, should enable diverse sequences to serve as suitable myristoylation substrates without the need for site-directed mutagenesis or directed evolution of the NMT enzyme. For our de novo designed sequences, we always positioned a Gly residue at the N-terminus. To ensure that these sequences would also, upon myristoylation, form PAs that would themselves act as structure-directing self-assembly domains, we turned to previous studies, which have established that interactions among the first 4–6 residues after the alkyl tail play a crucial role in controlling the formation of β-sheets.36,37
According to these principles, three sequences (B1–3) were designed (Table 1) and confirmed as reliable potential sites for modification by an online software predictor38,39, which uses a machine-learning algorithm from verified NMT substrates to predict a sequence’s likelihood of myristoylation. Sequence B1 consists of Gly-Ala-Gly-Ala-Ser, whose alternating Gly/Ala residues were inspired by a repeat unit commonly found in spider silk and represent the minimum structural motif necessary for β-sheet formation.40 Sequence B2, consisting of Gly-Ala-Gly-Ala-Gly-Ala-Tyr, was designed to enhance inter-strand interactions by increasing the number of Gly-Ala repeat units to three, as well as introducing an aromatic residue, Tyr.41 Sequence B3, Gly-Leu-Ser-Leu-Ser, combines the bulky hydrophobic amino acid, Leu, with the hydrogen-bonding residue, Ser, which increases the peptide’s overall propensity to form β-sheets.42 Based on two experimentally-derived thermodynamic scales, B3 shows the highest propensity to form P-sheets, followed by B2 and B1 (Supplementary Information, section 2).43,44
Table 1.
A summary of the ELPs, FAMEs, and control PAs used in this study.
|
Identifier |
N-terminal Modification |
Sequence |
||
|---|---|---|---|---|
| NMT Recognition sequences a) |
ELP | |||
| B1-ELP | N/A | GAGASRGGSGGS | ||
| ELPs | B2-ELP | N/A | GAGAGAYRGGSGGSGGS | |
| B3-elp | N/A | GLSLSRGGSGGS | (GVGVP)40GY | |
| M-B1-ELP | Myristoyl | GAGASRGGSGGS | ||
| FAMEs | M-B2-ELP | Myristoyl | GAGAGAYRGGSGGSGGS | |
| M-B3-ELP | Myristoyl | GLSLSRGGSGGS | ||
| M-B1 | Myristoyl | GAGASR | ||
| M-B2 | Myristoyl | GAGAGAYR | N/A | |
| Controls | M-B3 | Myristoyl | GLSLSR | |
| ELP | N/A | N/A | (GVGVP)40GY | |
| M-ELP | Myristoyl | N/A | (GVGVP)40GY | |
Amino acid abbreviations: Gly, G; Ala, A; Ser, S; Arg, R; Tyr, Y; Leu, L, Val, V and Pro, P. De novo designed recognition sequences contain a β-sheet forming domain (bold), trypsin cleavage site (Arg, underlined), and a flexible linker (italics).
In addition, to help with the characterization of these materials and verify recombinant myristoylation by mass spectrometry, we also included an Arg residue at the C-terminus of all three sequences, which enables selective cleavage of the polypeptide by the protease trypsin. In this study, we always used an ELP with the sequence [Gly-Val-Gly-Val-Pro]40, but this design could be easily applied to other ELPs, featuring different lengths and guest residue compositions, to tune the LCST of the resulting FAME. Additionally, this approach should be broadly applicable such that the ELP could be substituted with other protein polymers, such as resilin- like polypeptides (RLP)45 and collagens.
The genes for the NMT enzyme from S. cerevisiae and the polypeptide (B1–3–linker–ELP sequences, hereafter referred to as B1–3–ELPs) were cloned into tandem expression cassettes of the bicistronic pETDuet-1 vector (Figure 1b). This plasmid was then transformed into E. coli BL21(DE3) cells, which were cultured in the presence of myristic acid and then chemically induced to express both genes by the addition of isopropyl-β-D-thiogalactopyranoside. The FAMEs were purified by taking advantage of their temperature-triggered phase transition using inverse transition cycling (ITC), a non-chromatographic method for purification of ELP fusion proteins.46 The details of cloning, expression, purification, and characterization are provided in Supplementary Information.
Results and Discussion
FAMEs display LCST phase behavior
We first sought to investigate whether FAMEs exhibit the desired temperature-triggered LCST phase behaviour that would enable extrinsic control of their hierarchical self-assembly. The LCST phase transition of unmodified ELPs and FAMEs was quantified for each polypeptide by measuring the turbidity at 350 nm of varying polypeptide concentrations while gradually increasing the temperature from 10 °C to 50 °C (Figure 2). In the absence of myristoylation, all the constructs (B1–3–ELP) exhibited a similar LCST Tt, demonstrating that incorporation of the short NMT enzyme recognition sequences (B) and linker at the N-terminus of the ELP did not significantly alter the phase-transition behaviour (Figure 2a,d,g, and Supplementary Figure 20). Additionally, the Tt of all three constructs exhibited an identical inverse dependence on the concentration of the ELP in solution, consistent with previous studies on ELPs and their fusions.47 Finally, the phase transition of non-myristoylated ELPs was completely reversible.
Figure 2.
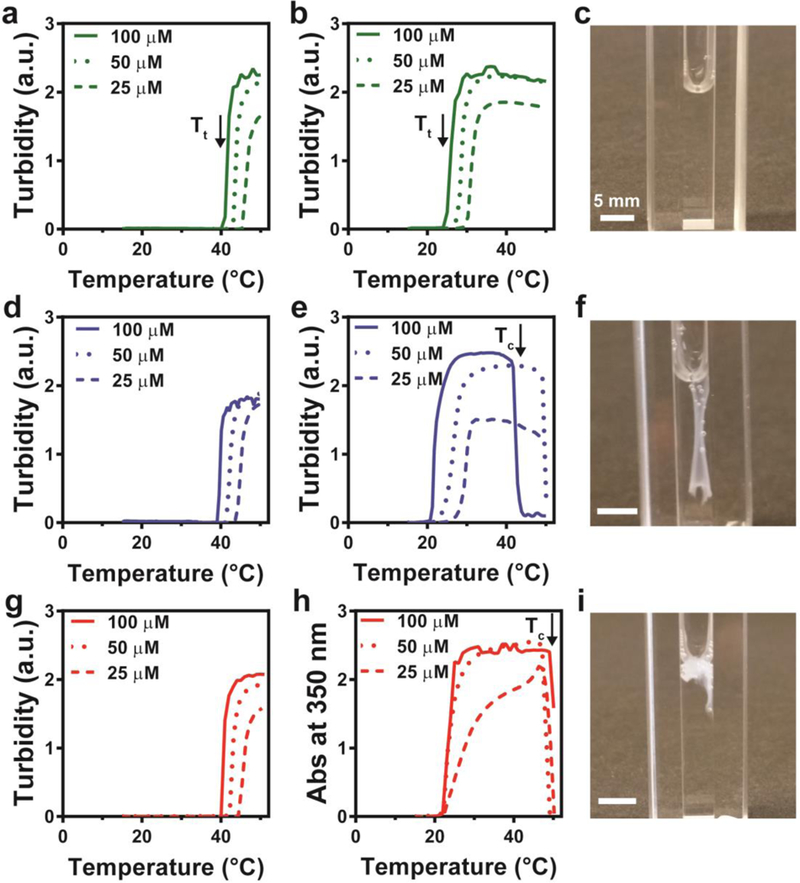
Temperature-triggered macroscale self-assembly of FAMEs. a-i, Temperature-programmed turbidimetry assays and images (c, f, i) obtained after heating above Tc and cooling below Tt for B1-ELP (a), M-B1-ELP (b, c), B2-ELP (d), M-B2-ELP (e, f), B3-ELP (g), and M-B3-ELP (h, i). Each turbidimetry assay was carried out at three concentrations: 100 μM (solid lines), 50 μM (dotted lines), 25 μM (dashed lines). The arrows denote the onset of LCST behavior (Tt) (a,b) and the onset of hysteretic behavior and morphogenesis of M-B2-ELP (e) and M-B3-ELP (h) into macroscopic structures above the critical temperature (Tc). The images in c, f, and i demonstrate that the self-assembled structures formed by M-B2-ELP and M-B3-ELP on heating are stable upon cooling (f,i), whereas that formed by M-B1-ELP resolubilizes (c).
All three FAMEs exhibited a Tt lower than that of their non-myristoylated counterparts (B1–3–ELPs). For example, the Tt of all FAMEs were in the range of 20–25 °C at 100 μM (Figure 2b, e, h), which is 15 °C lower than that of the non-myristoylated parent polypeptides (Figure 2a, d, g). This behavior can be explained by the fact that Tt inversely scales with hydrophobicity,19 and myristoylation increases the chain hydrophobicity and also removes the charged N-terminal amine.
We noted that subtle differences in the NMT recognition sequence (B) significantly altered the concentration dependence and reversibility of the LCST phase behavior. The Tt of M-B1-ELP and M-B2-ELP both exhibited steep concentration dependence (Figure 2b, e), while that of M-B3-ELP did not change when its concentration was reduced from 100 μM to 25 μM (Figure 2h). All FAMEs exhibited reversible LCST phase behaviour up to 30 °C (Supplementary Figure 21), but only M-B1-ELP had reversible phase behaviour across the entire experimental temperature range of 15–50 °C. M-B2-ELP and M-B3-ELP displayed hysteretic phase transition behavior (Supplementary Figure 22) after the temperature had reached a critical point (Tc~45 °C). The onset of this hysteretic behavior was marked by a sudden decrease in the turbidity of the solution (indicated by vertical arrows in Figure 2e, h), which is due to M-B2-ELP and M-B3-ELP self-assembling into macroscopic objects (mm to cm length scale) that drifted out of the light path.
While both M-B1-ELP and the M-ELP control (which lacks a B domain) resolubilized completely upon cooling (Figure 2c and Supplementary Figure 24), we observed that structures formed by M-B2-ELP above Tc (long, flat sheets, Figure 2f, and Supplementary Figure 23) remained stable even when cooling back below Tt. M-B3-ELP formed weaker amorphous aggregates above Tc (Figure 2i, and Supplementary Figure 23); vide infra for nanoscale morphological characterization. These results demonstrate that temperature can be used as an external cue to trigger the hierarchical self-assembly of FAMEs. It is also intriguing that the differences in these self-assembled macroscopic structures were the result of only minor sequence variations in the short B domain of the PA, highlighting the potent ability of PAs to direct hierarchical self-assembly.
Myristoylation induces FAME self-assembly
To understand the self-assembly mechanism at the molecular level, we utilized various spectroscopic techniques to study the effect of myristoylation on the structure and self-assembly of FAMEs below their Tt, beginning with dynamic light scattering (DLS). As shown in Figure 3a, in the absence of myristoylation (colored dashed lines), B1–3–ELP exists as unimers in solution, as seen by the autocorrelation decay at short time scales that is consistent with the behavior of canonical ELPs with comparable length (Figure 3a). In contrast, DLS conclusively showed that all three FAMEs, M-B1–3–ELP, as well as the M-ELP control, self-assembled to form significantly larger aggregates, as shown by the shift in their autocorrelation decay function to longer time scales. There are two important conclusions from the DLS data. First, the incorporation of a single myristoyl group into a FAME is enough to trigger the self-assembly of FAME’s even at temperatures below their Tt. Second, as the FAMEs only differ in the short peptide sequences of their PA domain, the differences in these nanoscale assemblies observed below the Tt stem from subtle differences in the B1–3 sequences.
Figure 3.
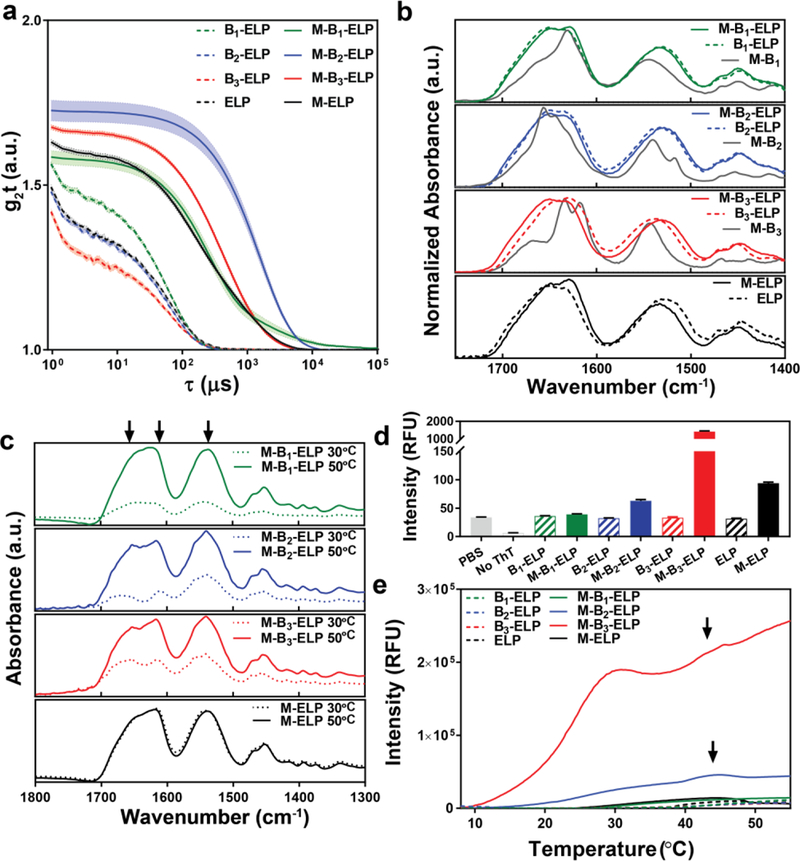
Spectroscopic and dynamic light scattering (DLS) characterization of the effect of myristoylation on the structure and the self-assembly of the FAMEs. a) DLS of FAMEs (M-B1–3–ELPs), unmodified B1–3–ELP, and controls at T < Tt. The shift in the DLS autocorrelation functions of the FAME’s to longer timescales compared to that of controls confirms the nanoscale aggregation of FAMEs below their Tt. Error bars represent the mean ± standard error of the mean (s.e.m., shown as a shaded band around each line) calculated from 12 measurements. b) ATR-IR of lyophilized FAMEs, B1–3–ELPs, control PAs (M-B1–3), ELP, and M-ELP. The secondary structure of controls and FAMEs have significant similarities, and myristoylation does not result in major changes in the secondary structure. Control PAs (grey solid line in each panel) form β-sheets in the lyophilized powder, and the internal structure of each PA is dictated by its B sequence. c) Variable temperature ATR-IR of FAMEs and control M-ELP at T = 30 °C (Tc > T > Tt, dashed lines) and at T = 50 °C (T > Tc, solid lines). The conformation of the ELP domain does not change significantly in each FAME above its Tt; however, the PA-domain has a subtle effect on the conformation of the ELP. Arrows mark the position of characteristic ELP peaks at amide I and II bands. d) Static ThT fluorescence quantifies the propensity to form β-sheets at T = 20 °C (T < Tt). Error bars represent mean ± standard deviations calculated from three measurements. e) Dynamic ThT fluorescence assay, which show that the temperature-triggered phase transition of ELPs triggers the self-assembly of PA-domains, and in turn the FAMEs (arrows mark the final stage in the hierarchical self-assembly of M-B2-ELP and M-B3-ELP).
Characterization of FAME secondary structure
We next investigated the effect of myristoylation on the constructs’ secondary structure using circular dichroism (CD, Supplementary Figure 26) and infrared spectroscopy (FTIR, Figure 3b). Both techniques demonstrated that myristoylation did not result in a global change in the secondary structure of the ELP domain, as FAMEs and their non-myristoylated controls exhibited similar CD and FTIR spectra, which are also consistent with previous reports of other ELPs showing a substantial amount of random coils and a subpopulation of β-turns resulting from its Pro-Gly motif.48 While we could not collect CD spectra for the control PAs (synthetic M-B1–3 without the ELP) due to their limited solubility, their FTIR spectra (Figure 3b, solid grey lines) were noticeably different from those of the ELPs and FAMEs. The complex absorption pattern of amide carbonyls observed for PAs is consistent with the presence of β-sheet structures in the lyophilized powder.49,50
To investigate the effect of temperature on the secondary structure of FAMEs, we then performed variable temperature ATR-IR in solution (100 μM in PBS, Figure 3c). At this concentration and below the Tt, we could not observe any signal for the FAMEs after subtracting the buffer background signal. However, as the temperature was increased to 30 °C (Tc > T > Tt) the signal intensity dramatically increased, which is consistent with the formation of polymer-rich coacervates over the ATR crystal. The increase in the signal intensity with temperature points to continued dehydration of the FAMEs’ ELP domains in the coacervate. Given the size difference between the PA and ELP domains, the recorded IR signature is dominated by characteristic ELP signatures (amide I band containing a low-frequency component around 1615 cm−1 and a high frequency component near 1650 cm−1 and a broad amide II band at 1540 cm−1, positions marked by vertical arrows in Figure 3c). While the spectra for each FAME resembles control M-ELP (Figure 3c, black curves) and ELP (Supplementary Figure 28), closer inspection reveals sharper peaks and the presence of small features, appearing as discernible shoulders around major peaks for M-B2-ELP and M-B3-ELP. These effects are present in both amide I and II bands at 30 and 50 °C, but the peaks in the amide I band are more distinct. This is in contrast to M-B1-ELP and M-ELP, which showed broad peaks with no discernible sharp features at all temperatures. These data also point to the PA-domains influencing the conformation of the ELP domains. This indicates that the self-assembly of FAMEs in solution may be altered by the PA-domains alone, consistent with DLS (Figure 3a). Finally, ATR-IR spectroscopy shows that the conformation of the ELP domain does not appreciably change above the Tc; the peaks observed at 30 °C only grow in intensity at 50 °C.
Because FTIR spectroscopy suggests that the PA-domains have a propensity to adopt a β-sheet secondary structure (Figure 3b), we performed a thioflavin T (ThT) fluorescence assay to investigate the role of the M-B1–3 domains in directing hierarchical self-assembly of the FAMEs. ThT is a benzothioazole salt that is commonly used to visualize and quantify the presence of fibrillar-or amyloid-like aggregates. The fluorescence of free ThT in solution is strongly quenched by water, but upon binding to aggregates rich in β-sheets, the fluorescence is significantly enhanced by up to 1000-fold.51 We began with a single time-point “static” ThT assay at 20 °C, a temperature below the Tt of each FAME (Figure 3d). The static ThT assay is a fluorescent probe of the initial nanoscale self-assembled structures that are directed by the PA component of each FAME. The static ThT assay clearly shows that myristoylation is necessary for strong fluorescence of the ThT dye because the fluorescence of ThT is strongly quenched in the absence of myristoylation. The comparison of ThT uptake between the FAMEs demonstrates that the peptide sequence (B) following the myristoyl group also strongly influences the interaction of the FAME with ThT. Below Tt, ThT did not interact strongly with the structures formed by M-B1-ELP, which features flexible and small amino acids in the recognition sequence. In comparison, M-B2-ELP exhibited a 2-fold increase in ThT uptake and fluorescence, likely due to an increase in β-sheet propensity, and possibly through the added effect of inter-chain interactions from π–π stacking via the Tyr residue. Strikingly, ThT fluorescence increased 43-fold in the presence of the M-B3-ELP construct, which contains alternating hydrophobic Leu and polar Ser residues. Overall, the results of the static ThT assay are consistent with our calculated thermodynamic β-sheet propensity scales (Supplementary Table 2).
To understand the hierarchical self-assembly of the nanoscale structures into larger macroscopic objects that is driven by the LCST behavior of the ELP component, we monitored the evolution of ThT fluorescence as a function of temperature. The differences between the FAMEs were more pronounced with this dynamic ThT assay (Figure 3e). Uptake of ThT did not increase significantly as non-myristoylated ELP was heated, as we observed only a minor increase in the fluorescence after 40 °C, corresponding to its Tt at 100 μM. This is presumably due to non-specific sequestration of ThT into the hydrophobic polypeptide-rich coacervate phase. In contrast, at T = 30 °C (Tc > T > Tt), even M-B1-ELP — the FAME that did not exhibit macroscopic assembly — exhibited a six-fold increase in ThT fluorescence compared to its non- myristoylated control (B1-ELP). This increase in ThT fluorescence above Tt, was significantly greater for the other two FAMEs that form macroscopic objects (Figure 2f, i). At 30 °C (Tc > T > Tt), M-B2-ELP showed a 150-fold and M-B3-ELP showed a 270-fold increase in fluorescence compared to non-myristoylated controls, supporting our original hypothesis that the temperature-triggered phase-transition of ELP domain can be used as a trigger of hierarchical FAME self-assembly. Both M-B2-ELP and M-B3-ELP displayed another transition in the ThT fluorescence close to 45 °C (marked by an arrow in Figure 3e). This second transition (Tc) coincided with the onset of hysteretic behavior and the formation of the macroscopic objects shown in Figure 2. We recognize that the ThT fluorescence signal also depends on factors such as the viscosity of the environment52,53 and the increased viscosity of the ELP coacervates can also increase the ThT fluorescence above Tt. Taken together, these studies suggest that FAMEs self-assemble in three stages.
Structural characterization of FAMEs self-assembly pathway
To complement the spectroscopic characterization, we carried out extensive structural characterization of the self-assembled FAMEs with imaging techniques that allowed us to visualize morphology at various temperature regimes and across different length scales. We started by using cryogenic transmission electron microscopy (cryo-TEM) to image the nanoscale morphology of FAMEs in their near-native hydrated state with nanometer spatial resolution. Control PAs (M-B1–3) at 20 °C were found to form fibers and sheets with large aspect ratios, consistent with the behavior of previously reported PAs (Supplementary Figure 33).16,54 In contrast, the FAMEs exhibited a diverse range of nanoscale morphologies at 20 °C (T < Tt), with overall length of assemblies scaling with β-sheet propensity. M-B1-ELP formed short, worm-like nanostructures 88.1 ± 5.2 nm in length and 11.4 ± 0.5 nm in width (Figure 4a). M-B2-ELP formed fibers with an average diameter of 21 ± 0.73 nm, and lengths ranging from 10–1500 nm (Figure 4b); M-B3-ELP formed an extended network of long fibers >3 μM in length and 22.89 ± 0. 68 nm in width (Figure 4c).
Figure 4.
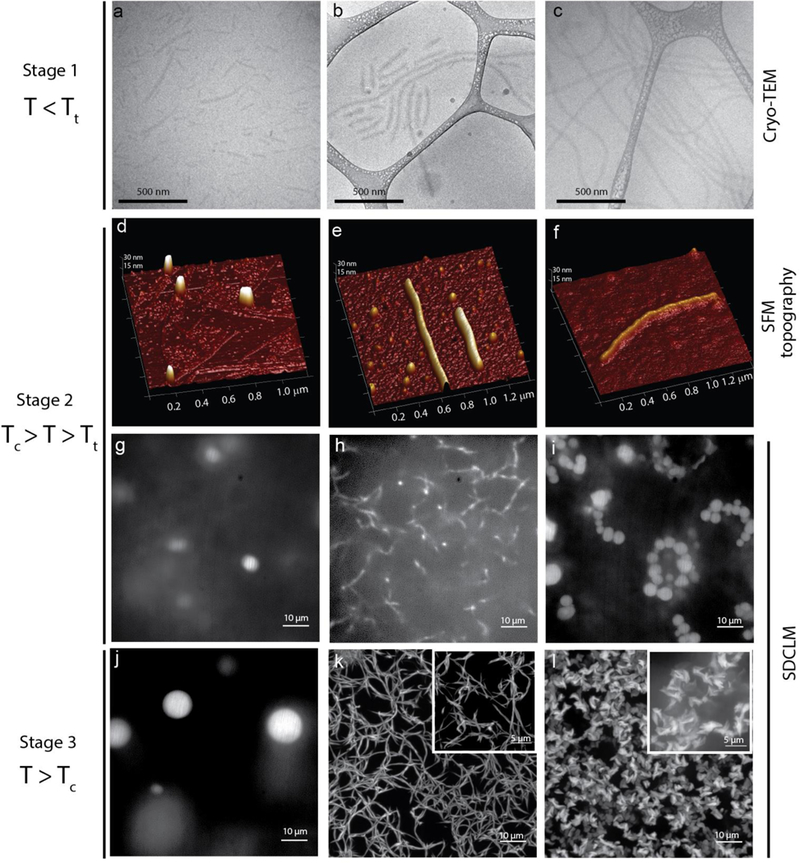
Characterization of the morphology of the FAME aggregates and visualization of their temperature-triggered phase transition and self-assembly across different length scales and temperatures. a-c, Cryo-TEM of the FAMEs dissolved in PBS at 20 °C (T < Tt): M-B1-ELP (a), M-B2-ELP (b), and M-B3-ELP (c), showing cylindrical micelle morphology with an average length increasing with the β-sheet-formation propensity of the PA-domain. d-f, SFM topography of the FAMEs, drop cast from solution at 30 °C (Tc > T > Tt): M-B1-ELP (d), M-B2-ELP (e), and M-B3-ELP (f), showing that FAME nano-aggregates are rod-like polymeric micelles and fibers. g-l, M-B1-ELP transitions into liquid coacervates at 30 °C (g) that remain stable up to 50 °C (j) but reach a larger equilibrium volume. M-B2-ELP transitions into a network of fibers above Tt (h). At higher temperatures (T > Tc), these fibers form a stable interconnected network of fibers (k). Above Tt, M-B3-ELP forms a “beads-on-a-string” morphology that is likely due to the arrested coalescence of the initial liquid coacervates (i). Above Tc, these droplets form fractal-like amorphous aggregates (l). The inset in k and l provide a higher magnification obtained using the super-resolution mode of the microscope.
Based on previous reports and our experience, we know that solvated ELPs are difficult to visualize in cryo-TEM due to poor electron contrast resulting from their high water content.55 Therefore, the observed structures are likely reflective of the dehydrated core of the FAME assemblies, which consists of the PA-domain. The hydrated ELP corona (not seen in cryo-TEM) surrounds the M-B1–3 core and prevents further assembly at lower temperatures (T < Tt). We did not observe major differences in the nanoscale morphology of the aggregates below versus above the Tt using cryo-TEM (structures with similar widths were observed at Tc > T > Tt), although the cross-section of larger scale M-B2-ELP and M-B3-ELP assemblies was visible as a shadow of dehydrated PA-domains on the TEM grid (Supplementary Figure 34).
Next, scanning force microscopy (SFM) was used to visualize the nanoscale morphology of the aggregates at 30 °C (Tc > T > Tt, stage 2). The topography showed rod-like structures for M-B2-ELP and M-B3-ELP that consist of a PA-core and an ELP-corona (Figure 4d–f). Mapping the mechanical response of all three FAMEs by SFM confirmed a structure composed of a stiffer core surrounded by a softer corona (Supplementary Figure 36). On the mica surface, M-B1-ELP (Figure 4d) formed small spherical aggregates and no rods were observed, although it is possible that these smaller structures are caused by surface-induced aggregation. In contrast, M-B2-ELP (Figure 4e) primarily formed elongated fibers (10–20 nm in height, length > 1 μm), although a sub-population of spherical aggregates were also visible. M-B3-ELP (Figure 4f) formed elongated fibers (6–10 nm in height, length > 1 μm).
The temperature-triggered assembly process of FAMEs in the hydrated state was visualized in real time at longer length scale by spinning disk confocal laser microscopy (SDCLM). We genetically encoded a Lys residue at the C-terminus of the M-B1–3–ELP constructs to enable labeling with Alexa Fluor® 488 dye. The turbidity profile of these labeled FAMEs was identical to their parent sequences (Supplementary Figure 25), indicating that the addition of the Lys residue and the fluorophore at a position distant from the PA domain did not perturb self-assembly. While heating fluorescently labeled protein samples from ~4 °C (T < Tt) to 30 °C (Tc > T > Tt), we monitored the second stage of self-assembly by taking consecutive images from a focal plane distant from the cover slip to avoid surface-induced artifacts (Figure 4g–i). The temperature was then raised to 50 °C to monitor the third and final stage of self-assembly (T > Tc. Figure 4j–l).
Before reaching thermal equilibrium, as expected, a homogeneous fluorescent signal was observed across the viewing field, suggesting that at stage 1 (T < Tt) each population of the FAMEs exists as a nanoscale assembly below the diffraction limit, consistent with DLS and cryo-TEM data. However, the PA domain significantly altered the morphogenesis of aggregates at higher temperatures. During stage 2, at 30 °C (Tc > T > Tt), M-B1-ELP, whose PA domain has the lowest propensity to form β-sheets, transitioned into liquid-like droplets similar to canonical ELPs (Supplementary Figure 31).56 These droplets moved quickly in and out of the focal plane, which rendered the accurate measurement of their diameter difficult. However, as expected, these liquid-like smaller coacervates coalesced with each other over time and equilibrated to a larger coacervate ~4–5 μm in diameter (Figure 4g). At 50 °C (T > Tc), these liquid-like coacervates further coalesced to form slightly larger droplets ~8–12 μm in diameter (Figure 4j).
In the case of M-B2-ELP at 30 °C, we did not observe liquid droplets but instead observed the anisotropic growth of small fibers with a width of 0.1–0.2 μm (Figure 4h). These fibrous structures exhibited high mobility, but at 50 °C formed a stable interconnected network of fibers (Figure 4k). To visualize the finer details of the entangled network, we used the microscope’s super-resolution mode, recording multiple images at different focal planes and then merging these z-stack images (Figure 4k inset). This higher resolution image demonstrates the stacking of narrow fibers into a bundle of fibers exhibiting a larger width and some polydispersity. The smallest fiber width observed was ~130 nm, which is close to the resolution limit of the instrument.
Interestingly, M-B3-ELP initially transitioned into liquid-like droplets at 30 °C (Figure 4i), but these droplets had distinct features compared to M-B1-ELP (Figure 4g) and canonical ELP coacervates (Supplementary Figure 31). We observed many polydisperse droplets that form a “beads-on-a-string” morphology (Figure 4i). Upon increasing the temperature to 50 °C, these arrested coacervates formed aggregates with a “fractal-like” morphology that we propose is a consequence of increased desolvation of the ELP chains within the coacervates, leading to network formation through aggregated ELP chains (Figure 4l).
We are mindful that slight differences in the heating profile used in the spectroscopic characterization and SDCLM may impact the final structure of the aggregates. Nevertheless, these results confirm the hierarchical self-assembly process of M-B2-ELP and M-B3-ELP that drives the formation of macroscopic objects through three distinct stages, consistent with the results of the turbidimetry and spectroscopic characterization. These experiments confirm the influence of the PA domain on the interactions between the coacervates and the dynamics of their coalescence.
Similar observations have been made by others studying the maturation of ELP coacervates with different proline periodicity.57 Keely and co-workers observed that changes in the proline distribution near the C-terminus of multi-domain ELPs affected the dynamics of maturation, assembly, and spreading of the coacervates. They attributed these observations to the effect of secondary structure content inside the ELP coacervates.58 In our system, we propose that the nanoscale strcuture of the PA domain drives the morphological differences observed in the later stages of FAME coacervation, since the spectroscopic data suggest that temperature has very little effect on the protein secondary structure in all constructs.
To gain a better understanding of the third stage of the self-assembly process, we fixed the macroscopic objects formed by M-B2-ELP and M-B3-ELP above their Tc and investigated their structure with scanning electron microscopy (SEM). M-B2-ELP showed a network of narrow interwoven fibers (~ 50 nm) that extended in the fiber axis direction to form long bundles with much larger overall diameters (1 μm, Figure 5a). We observed a similar bundle of fibers using cryo-SEM (Supplementary Figure 37), demonstrating that the sample preparation process, cross-linking and dehydration, did not perturb the overall morphology. In contrast, the M-B3-ELP structure was composed of amorphous aggregates, which were made of entangled fibers that were visible upon closer inspection (Figure 5b).
Figure 5.

SEM morphological characterization of the macroscopic aggregates formed by heating M-B2-ELP (a) and M-B3-ELP (b) above their Tc and fixing the samples with glutaraldehyde and dehydration. The insets are at 10x magnification.
As pointed out earlier, unlike canonical ELP coacervates, structures formed by M-B2-ELP and M-B3-ELP above Tc do not dissolve upon re-cooling (Supplementary Figure 22), and in the case of M-B2-ELP they were able to withstand moderate mechanical agitation (inversion and brief mixing by vortex). These observations, coupled with SEM, point to the PA domains undergoing an irreversible transition during the self-assembly process, perhaps forming a network of connected PA domains that is further reinforced by the interaction among the ELP chains. This hypothesis would explain the observed thermal hysteresis, as structures held together by just the ELP chains are expected to dissolve back into solution once the temperature is lowered below the Tt (Supplementary Figure 24). We discuss this phenomenon in further detail in section 22 of the Supplementary Information.
Combining the insights gained from the spectroscopic and structural studies, we propose the following three-stage self-assembly mechanism (Figure 6). At temperatures below their Tt, the nanostructure formed by each FAME is determined by the fine balance between the attractive forces of the PA-domain at the core (hydrophobic interactions, β-sheet propensity, and secondary interactions such as hydrogen bonds) and the repulsive steric force of the hydrated ELP domain at the corona. This notion is consistent with the mechanism proposed for the self-assembly of canonical PAs,16 which is traditionally controlled by fine-tuning similar attractive interactions against ionic repulsive forces that can be turned off using triggers such as pH or ionic strength. At this stage, the ELP chains in the nanostructures of the FAMEs are hydrated and responsible for stabilizing the nanostructures in aqueous solution.
Figure 6.
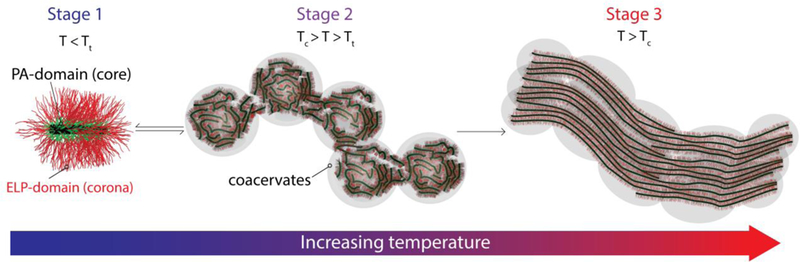
The proposed three-step mechanism of FAME self-assembly. Stage 1 (T < Tt): below Tt, the nanostructure of the aggregates is determined by the fine balance between the attractive forces of the PA-core and the repulsive force of the hydrated ELP-corona. Stage 2 (Tc > T > Tt): the dehydrated ELP domain undergoes a LCST phase transition into a liquid-like coacervate and tends to form spherical droplets. The PA domain modulates the size of, and interactions between, the ELP coacervates. Stage 3 (T > Tc): the repulsion between the ELP coronas —further dehydrated— is lessened, in turn decreasing the core-core distances inside the coacervates and drives macroscale self-assembly. The cores likely dynamically rearrange and non-covalently cross-link to form macroscopic aggregates.
In stage 2 (Tc > T > Tt), the ELP domain in the nanostructure dehydrates and undergoes an LCST phase-transition into a liquid-like coacervate with a preference to form spherical droplets to minimize the surface tension of the polymer-rich colloidal particle suspended in the solvent. This dehydration step has several consequences. (1) Above their Tt, the ELP domains are more desolvated, rendering the corona more hydrophobic and thus increasing the interactions between the FAME nanostructures. (2) The self-assembly of the ELP chains into polymer-rich coacervates concentrates the PA domain in a polymer rich medium, thereby reducing the water content and increasing the strength of the core-core interactions. (3) The nano-aggregation of the cores inside the coacervates consequently controls the kinetics of coacervate maturation and coalescence. If the cores are held together by weak interactions, as in the case of M-B1-ELP, the overall assembly behavior is similar to canonical ELPs. For FAMEs with stronger core interactions, the equilibrium size and coalescence of the coacervates is significantly affected by the PA-domain self-assembly at stage 1 as seen by the anisotropic growth of M-B2-ELP fibers (Figure 4h) and the disrupted coalescence of M-B3-ELP stemming from a large coacervate size and slow coalescence dynamics (Figure 4i).
The third and final stage of self-assembly occurs at temperatures T > Tc, where the repulsion between the ELP coronas is reduced due to further dehydration, which results in a decrease in the core-core distances inside the coacervates. In M-B2-ELP, we propose that the cores are connected (non-covalently cross-linked) in this stage through a thermally-driven dynamic rearrangement leading to the formation of bundled fibers. In contrast, if the cores are held together by very strong forces as for M-B3-ELP, this dynamic rearrangement may not be competitive with non-specific aggregation of the ELP chains, resulting in the formation of ill-defined macroscopic aggregates.
We recognize that our proposed mechanism is only the first step in understanding the self-assembly of FAMEs. Further investigation is necessary to better understand the interplay between the PA and ELP domains, as well as the molecular details of the hierarchical self-assembly process. Those will be carried out using additional spectroscopic techniques including NMR spectroscopy59, stochastic optical reconstruction microscopy60,61, and computational simulations.
Conclusion
We have demonstrated that exploiting post-translational modification is a powerful strategy to create novel responsive, biohybrid materials with emergent behavior. We have demonstrated that hierarchical self-assembly of myristoylated peptide polymers can be directed by the de novo design of myristoylation substrates, which function as peptide amphiphiles when recombinantly fused to an ELP.
We wish to highlight that the synthesis of this hybrid biomaterial would not have been possible in one step through chemical synthesis. This is because PAs are typically made through iterative solid-phase peptide synthesis methods and the production of a macromolecule that combines the structural domains of both PAs and ELPs would require sequential solid-phase synthesis62 followed by protein-ligation strategies63, which is technically challenging, time consuming, and expensive. Consequently, lipid modification of high molecular weight biopolymers has remained largely unexplored in the field of materials science. In contrast, this genetically encoded, one-pot synthesis that is carried out by E. coli is easy to scale-up64. Additionally, it should also be possible to decorate the C-terminal end of FAMEs with bioactive peptide and protein motifs. This would enable the production of recombinant, post-translationally modified bioactive materials with unprecedented control over their hierarchical structure and function.
Finally, FAMEs are a valuable addition to the subset of materials based on PAs and ELPs. Two other notable examples include short lipidated ELPs62 prepared by solid-phase synthesis, which exhibited LCST behavior and assembled into nanofibers with tunable length in combination with phospholipids, and robust dynamic membranes65, prepared by co-assembly of peptide amphiphiles and ELPs with opposite charges in solution. We are intrigued by the possibility of combining FAMEs with these systems and leveraging their hierarchical self-assembly. We hope that this work will serve as the starting point to a more detailed exploration of the structure and function of FAMEs.
Methods
A description of materials and methods are provided in the Supplementary Information.
Statistics
Technical replicates were measured for characterization techniques. The error bars in Figure 3a (shown as shaded bands) represent mean ± standard error of the mean (SEM), n = 12. The error bars in Figure 3d represent mean ± standard deviation, n = 3. The width and length (mean ± SEM, n = 25) of constructs were quantified with ImageJ using a representative image.
Data availability statement
The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information.
Supplementary Material
4. Acknowledgements
This research was funded by the NSF through the Research Triangle MRSEC (DMR-1121107) and by the NIH (R01 GM-061232). Duke University Shared Materials Instrumentation Facility (SMIF) and Analytical Instrumentation facility (AIF) at North Carolina State University are members of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the NSF (ECCS-1542015) as part of the National Nanotechnology Coordinated Infrastructure (NNCI). The authors thank Katherine Franz for access to chromatography instrumentation and peptide synthesizer. We are grateful to Jennifer G. Mark for conducting SDCLM experiments and Michelle Plue for the SEM imaging. We thank Helma Burg for her support in SFM sample preparation and analysis as well as Marc-Jan van Zadel for assistance with the variable temperature ATR-IR experiments. We are thankful to Michael Rubinstein for valuable suggestions regarding the mechanism of self-assembly.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Langer R & Tirrell DA Designing materials for biology and medicine. Nature 428, 487–492 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Maskarinec SA & Tirrell DA Protein engineering approaches to biomaterials design. Curr. Opin. Biotechnol. 16, 422–426 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Chilkoti A, Dreher MR & Meyer DE Design of thermally responsive, recombinant polypeptide carriers for targeted drug delivery. Adv. Drug Deliv. Rev. 54, 1093–1111 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Haider M, Megeed Z & Ghandehari H Genetically engineered polymers: Status and prospects for controlled release. J. Control. Release 95, 1–26 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Chow D, Nunalee ML, Lim DW, Simnick AJ & Chilkoti A Peptide-based biopolymers in biomedicine and biotechnology. Mater. Sci. Eng. R-Rep. 62, 125–155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hochkoeppler A Expanding the landscape of recombinant protein production in Escherichia coli. Biotechnol. Lett. 35, 1971–1981 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Mann M & Jensen ON Proteomic analysis of post-translational modifications. Nat. Biotechnol. 21, 255–61 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Walsh CT, Garneau-Tsodikova S & Gatto GJ Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. 44, 7342–7372 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Wold F In vivo chemical modification of proteins. Annu. Rev. Med. 50, 783–814 (1981). [DOI] [PubMed] [Google Scholar]
- 10.Walsh G & Jefferis R Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 24, 1241–1252 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Pinkas DM, Ding S, Raines RT & Barron AE Tunable, post-translational hydroxylation of collagen domains in Escherichia coli. ACS Chem. Biol. 6, 320–324 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim S et al. In vivo post-translational modifications of recombinant mussel adhesive protein in insect cells. Biotechnol. Prog. 27, 1390–1396 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Gordon JI, Duronio RJ, Rudnick DA, Adams SP & Gokel GW Protein N-myristoylation. J. Biol. Chem. 266, 8647–8650 (1991). [PubMed] [Google Scholar]
- 14.Berndt P, Fields GB & Tirrell M Synthetic lipidation of peptides and amino acids: Monolayer structure and properties. J. Am. Chem. Soc. 117, 9515–9522 (1995). [Google Scholar]
- 15.Hartgerink JD, Beniash E & Stupp SI Self-assembly and mineralization of peptideamphiphile nanofibers. Science 294, 1684–8 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Hamley IW Self-assembly of amphiphilic peptides. Soft Matter 7, 4122 (2011). [Google Scholar]
- 17.Cui H, Webber MJ & Stupp SI Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers 94, 1–18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silva GA et al. Selective differentiation of neural progenitor cells by high-epitope density nanofibers. Science 303, 1352–5 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Urry DW Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J. Phys. Chem. B 101, 11007–11028 (1997). [Google Scholar]
- 20.Roberts S, Dzuricky M & Chilkoti A Elastin-like polypeptides as models of intrinsically disordered proteins. FEBS Lett. 589, 2477–2486 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urry DW et al. Elastic protein-based polymers in soft tissue augmentation and generation. J. Biomater. Sci. Polym. Ed. 9, 1015–1048 (1998). [DOI] [PubMed] [Google Scholar]
- 22.MacEwan SR & Chilkoti A Elastin-like polypeptides: Biomedical applications of tunable biopolymers. Biopolymers 94, 60–77 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Amiram M, Luginbuhl KM, Li X, Feinglos MN & Chilkoti A Injectable proteaseoperated depots of glucagon-like peptide-1 provide extended and tunable glucose control. Proc. Natl. Acad. Sci. U. S. A. 110, 2792–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luginbuhl KM et al. One-week glucose control via zero-order release kinetics from an injectable depot of glucagon-like peptide-1 fused to a thermosensitive biopolymer. Nat. Biomed. Eng. 1, 78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McHale MK, Setton LA & Chilkoti A Synthesis and in vitro evaluation of enzymatically cross-linked elastin-like polypeptide gels for cartilaginous tissue repair. Tissue Eng. 11, 1768–1779 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Lim DW, Nettles DL, Setton L. a. & Chilkoti A In situ cross-linking of elastin-like polypeptide block copolymers for tissue repair. Biomacromolecules 9, 222–230 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacEwan SR & Chilkoti A Digital switching of local arginine density in a genetically encoded self-assembled polypeptide nanoparticle controls cellular uptake. Nano Lett. 12, 3322–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho Y et al. Effects of hofmeister anions on the phase transition temperature of elastin-like polypeptides. J. Phys. Chem. B 112, 13765–13771 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McDaniel JR, Radford DC & Chilkoti A A unified model for de novo design of elastin-like polypeptides with tunable inverse transition temperatures. Biomacromolecules 14, 2866–2872 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duronio RJ et al. Protein N-myristoylation in Escherichia coli: reconstitution of a eukaryotic protein modification in bacteria. Proc. Natl. Acad. Sci. U. S. A. 87, 1506–1510 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heal WP et al. Site-specific N-terminal labelling of proteinsin vitro and in vivo using N-myristoyl transferase and bioorthogonal ligation chemistry. Chem. Commun. 3, 480–482 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Kulkarni C, Kinzer-Ursem TL & Tirrell DA Selective functionalization of the protein N terminus with N-myristoyl transferase for bioconjugation in cell lysate. ChemBioChem 14, 1958–1962 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Kulkarni C, Lo M, Fraseur JG, Tirrell DA & Kinzer-Ursem TL Bioorthogonal Chemoenzymatic Functionalization of Calmodulin for Bioconjugation Applications. Bioconjug. Chem. 26, 2153–2160 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Ho SH & Tirrell DA Chemoenzymatic Labeling of Proteins for Imaging in Bacterial Cells. J. Am. Chem. Soc. 138, 15098–15101 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Eisenhaber F et al. Prediction of lipid posttranslational modifications and localization signals from protein sequences: Big-n, NMT and PTS1. Nucleic Acids Res. 31, 3631–3634 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paramonov SE, Jun HW & Hartgerink JD Self-assembly of peptide-amphiphile nanofibers: The roles of hydrogen bonding and amphiphilic packing. J. Am. Chem. Soc. 128, 7291–7298 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Ortony JH et al. Internal dynamics of a supramolecular nanofibre. Nat. Mater. 13, 1–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maurer-Stroh S, Eisenhaber B & Eisenhaber F N-terminal N-myristoylation of proteins: Refinement of the sequence motif and its taxon-specific differences. J. Mol. Biol. 317, 523–540 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Maurer-Stroh S, Eisenhaber B & Eisenhaber F N-terminal N-myristoylation of proteins: prediction of substrate proteins from amino acid sequence. J. Mol. Biol. 317, 541–557 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Römer L & Scheibel T The elaborate structure of spider silk: structure and function of a natural high performance fiber. Prion 2, 154–161 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu XD, Jin Y, Liu Y, Zhang XZ & Zhuo RX Self-assembly behavior of peptide amphiphiles (PAs) with different length of hydrophobic alkyl tails. Colloids Surf., B 81, 329–335 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Lee O-S, Stupp SI & Schatz GC Atomistic molecular dynamics simulations of peptide amphiphile self-assembly into cylindrical nanofibers. J. Am. Chem. Soc. 133, 3677–3683 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Smith CK, Withka JM & Regan L A thermodynamic scale for the beta-sheet forming tendencies of the amino acids. Biochemistry 33, 5510–7 (1994). [DOI] [PubMed] [Google Scholar]
- 44.Minor DL & Kim PS Measurement of the [beta]-sheet-forming propensities of amino acids. Nature 367, 660–663 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Quiroz FG & Chilkoti A Sequence heuristics to encode phase behaviour in intrinsically disordered protein polymers. Nat. Mater. 14, 1164–1171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hassouneh W, Christensen T & Chilkoti A Elastin-like polypeptides as a purification tag for recombinant Proteins. Curr. Protoc. Protein Sci. 6.11.1–6.11.16 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyer DE & Chilkoti A Genetically Encoded Synthesis of Protein-Based Polymers with Precisely Specified Molecular Weight and Sequence by Recursive Directional Ligation: Examples from the Elastin-like Polypeptide System. Biomacromolecules 3, 357–367 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Serrano V, Liu W & Franzen S An infrared spectroscopic study of the conformational transition of elastin-like polypeptides. Biophys. J. 93, 2429–2435 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang H, Guler MO & Stupp SI The internal structure of self-assembled peptide amphiphiles nanofibers. Soft Matter 3, 454 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Cui H, Cheetham AG, Pashuck ET & Stupp SI Amino acid sequence in constitutionally isomeric tetrapeptide amphiphiles dictates architecture of one-dimensional nanostructures. J. Am. Chem. Soc. 136, 12461–12468 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.LeVine H Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–10 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stsiapura VI et al. Thioflavin T as a Molecular Rotor: Fluorescent Properties of Thioflavin T in Solvents with Different Viscosity. J. Phys. Chem. B 112, 15893–15902 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Sulatskaya AI, Maskevich AA, Kuznetsova IM, Uversky VN & Turoverov KK Fluorescence Quantum Yield of Thioflavin T in Rigid Isotropic Solution and Incorporated into the Amyloid Fibrils. PLoS One 5, e15385 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newcomb CJ, Moyer TJ, Lee SS & Stupp SI Advances in cryogenic transmission electron microscopy for the characterization of dynamic self-assembling nanostructures. Curr. Opin. Colloid Interface Sci. 17, 350–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mcdaniel JR et al. Noncanonical self-assembly of highly asymmetric genetically encoded polypeptide amphiphiles into cylindrical micelles. Nano Lett. 14, 6590–6598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simon JR, Carroll NJ, Rubinstein M, Chilkoti A & Lopez GP Programming molecular self-assembly of intrinsically disordered proteins containing sequences of low complexity. Nat. Chem. 9, 509–515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muiznieks LD & Keeley FW Proline periodicity modulates the self-assembly properties of elastin-like polypeptides. J. Biol. Chem. 285, 39779–39789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muiznieks LD et al. Modulated growth, stability and interactions of liquid-like coacervate assemblies of elastin. Matrix Biol. 36, 39–50 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Reichheld SE, Muiznieks LD, Keeley FW & Sharpe S Direct observation of structure and dynamics during phase separation of an elastomeric protein. Proc. Natl. Acad. Sci. 114, E4408–E4415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albertazzi L et al. Probing Exchange Pathways in One-Dimensional Aggregates with Super-Resolution Microscopy. Science 344, 491–495 (2014). [DOI] [PubMed] [Google Scholar]
- 61.da Silva RMP et al. Super-resolution microscopy reveals structural diversity in molecular exchange among peptide amphiphile nanofibres. Nat. Commun. 7, 11561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aluri S, Pastuszka MK, Moses AS & MacKay JA Elastin-like peptide amphiphiles form nanofibers with tunable length. Biomacromolecules 13, 2645–2654 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mejuch T & Waldmann H Synthesis of lipidated proteins. Bioconjug. Chem. 27, 1771–1783 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Triola G, Waldmann H & Hedberg C Chemical Biology of Lipidated Proteins. ACS Chem. Biol. 7, 87–99 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Inostroza-Brito KE et al. Co-assembly, spatiotemporal control and morphogenesis of a hybrid protein-peptide system. Nat. Chem. 7, 897–904 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the main data supporting the findings of this study are available within the article and its Supplementary Information.