

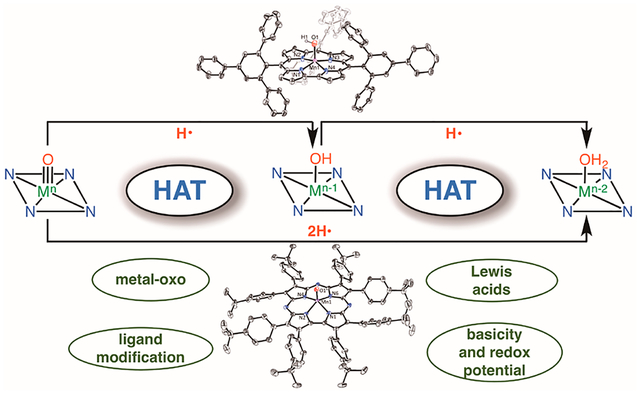
CONSPECTUS:
There has been considerable interest in hydrogen atom transfer (HAT) reactions mediated by metal/oxygen species because of their central role in metalloenzyme function as well as synthetic catalysts. This Account focuses on our progress in synthesizing high-valent metal–oxo and metal–hydroxo porphyrinoid complexes and determining their reactivities in a range of HAT processes. For these studies we have utilized corrolazine and corrole ligands, which are a ring-contracted subclass of porphyr inoid compounds designed to stabilize high-valent metal complexes. The high-valent manganese complex MnV(O)-(TBP8Cz) (TBP8Cz = octakis(4-tert-butylphenyl)corrolazine) provided an early example of a well-characterized low-potential oxidant that exhibits a redox potential indicating it to be a relatively weak oxidant yet can still be effective at abstracting H atoms from 20 certain C−H/O−H bonds. Approximating the thermodynamics of the HAT reactivity of the MnV(O) complex and related species with the help of a square scheme approach in which HAT can be formally separated into proton (pKa) and electron transfers (E°) indicates that affinity for the proton (i.e., the basicity) is a key factor in promoting HAT. Anionic axial ligands have a profound influence on the HAT reactivity of MnV(O)(TBP8Cz), supporting the conclusion that basicity is a critical parameter in determining the reactivity. The influence of Lewis acids on MnV(O)(TBP8Cz) was examined, and it was shown that both the electronic structure and reactivity toward HAT were significantly altered. High-valent Cr(O), Re(O), and Fe(O) corrolazines were prepared, and a range of HAT reactions were studied with these complexes. The chromium and manganese complexes form a rare pair of structurally characterized CrV(O) and MnV(O) species in identical ligand environments, allowing for a direct comparison of their HAT reactivities. Although CrV(O) was the better oxidant as measured by redox potentials, MnV(O) was significantly more reactive in HAT oxidations, pointing again to basicity as a key determinant of HAT reactivity. The iron complex, FeIV(O)(TBP8Cz+•), is an analogue of the heme enzyme Compound I intermediate and was found to be mildly reactive toward H atom abstraction from C–H bonds. In contrast, ReV(O)(TBP8Cz) was inert toward HAT, although one-electron oxidation to ReV(O)(TBP8Cz+•) led to some interesting reactivity mediated by the π-radical-cation ligand alone. Other ligand modifications, including peripheral substitution as well as novel alkylation of the meso position on the Cz core, were examined for their influence on HAT. A highly sterically encumbered corrole, tris(2,4,6-triphenylphenyl)corrole (ttppc), was employed for the isolation and structural characterization of the first MnIV(OH) complex in a porphyrinoid environment, MnIV(OH)(ttppc). This complex was highly reactive in HAT with O–H substrates and was found to be much more reactive than its higher-oxidation-state counterpart MnV(O)(ttppc), providing important mechanistic insights. These studies provided fundamental knowledge on the relationship between structure and function in high-valent M(O) and M(OH) models of heme enzyme reactivity.
Graphical Abstract
1. INTRODUCTION
Hydrogen atom transfer (HAT) is a fundamental chemical reaction involving the transfer of a combination of a proton and an electron from one group to another. This reaction is a key step in a variety of chemical and biological processes. These processes typically involve oxidation of substrate via a transition metal complex that has an oxidizing metal center to accept the electron and a basic ligand to accept the proton. One example, in particular, is the oxidation of organic substrates by high-valent metal–oxo species. In the heme monooxygenase cytochrome P450 (CYP), hydroxylation of a C–H bond involves an initial HAT to an iron(IV)–oxo π- radical cation, also known as Compound I (Cpd-I) followed by a faster radical recombination step (Scheme 1).1,2 However, alternative pathways have been observed for certain CYPs where, instead of radical recombination, the initial HAT step is followed by a second HAT to an FeIV(OH) intermediate, protonated Compound-II (Cpd-II), resulting in desaturation of the substrate instead of hydroxylation (Scheme 1).3 An analogous but reverse HAT reaction may be critical to water oxidation at the Mn-containing oxygen-evolving cluster (OEC) in Photosystem II (Scheme 1).4,5
Scheme 1.

Hydrogen Atom Transfers in CYP and the OECa
aReproduced from ref 33. Copyright 2018 American Chemical Society.
Our group has been interested in elucidating the fundamental structural and electronic factors that control HAT reactions involving high-valent metal–oxo complexes in biologically relevant ligand environments. A basic scheme showing HAT pathways is presented in Scheme 2.
Scheme 2.

HAT in Metal–Oxo Systems
We developed corrolazine (Cz) ligands, which are ring-contracted forms of tetraazaporphyrins (porphyrazines), to stabilize high-valent metal–oxo species.6 Corrolazines and the analogous ring-contracted corroles contain the typical tetrapyrrolic macrocyclic chelate of a porphyrinoid molecule with a trianionic charge upon full deprotonation of the internal pyrrole N–H groups. The corrolazine core has proven to be particularly stable toward oxidation, allowing us to isolate and fully characterize a range of high-valent metal–oxo complexes. This Account focuses on the HAT reactivity of a range of metal–oxo and metal–hydroxo species prepared by our laboratory with corrolazine and corrole ligands.
2. A MnV(O) COMPLEX THAT ABSTRACTS HYDROGEN ATOMS
Early work was conducted in our laboratory with an octa-tert butylphenyl-substituted corrolazine (TBP8Cz = octakis(4-tert butylphenyl)corrolazinato3−), which was synthesized via ring contraction of a porphyrazine precursor (Scheme 3).7 This ligand led to the synthesis and isolation of the high-valent manganese–oxo complex MnV(O)(TBP8Cz), which was stable enough for isolation.8,9 However, the stability of this complex varies in solution and is highly dependent on the identity of the solvent; it was not until more than 12 years later that we discovered the appropriate solvent system (toluene/acetonitrile) for growing single crystals of MnV(O)(TBP8Cz) for structural characterization by X-ray diffraction (XRD) (Figure 1).10 With the synthesis of MnV(O)(TBP8Cz) in hand, our lab was the first to be able to examine directly the reactivity of a MnV(O) porphyrinoid complex in HAT reactions.11 Reaction of the MnV(O) complex with a series of para-X-substituted phenols (4-X-2,6-tBu2C6H2OH, X = C(CH3)3, H, Me, OMe, CN) resulted in the formation of a MnIII complex and the corresponding phenoxyl radical (Figure 1), indicating that MnV(O)(TBP8Cz) was competent to extract hydrogen atoms from phenol O–H bonds. However, the immediate product from HAT should be a MnIV(OH) complex, but no evidence for this species could be obtained; instead, only the two- electron-reduced product MnIII(TBP8Cz) was observed.
Scheme 3.

Corrolazine Synthesis
Figure 1.

Reactivity of MnV(O)(TBP8Cz) with phenol derivatives, showing the postulated MnIV(OH) intermediate.
There are two possible mechanisms that could explain the absence of the MnIV(OH) complex, which are shown in Scheme 4. One possibility is that the MnIV(OH) complex is more reactive toward the phenol substrates than the starting MnV(O) complex, making the second HAT step fast in comparison with the initial slow HAT step (mechanism A). A second possibility is that the MnIV(OH) complex is unstable and rapidly disproportionates into MnIII and MnV(O) species (Scheme 4), with the MnV(O) complex being the sole oxidant in the system (mechanism B).
Scheme 4.

Possible Mechanisms of H-Atom Abstraction by a MnV(O) Complexa
aReproduced from ref 33. Copyright 2018 American Chemical Society.
The rate of conversion of MnV(O)(TBP8Cz) to MnIII(TBP8Cz) was monitored by UV–vis spectroscopy under pseudo-first-order conditions with excess phenol substrate. Isosbestic conversion of the MnV(O) complex to the MnIII complex was seen for all of the substituted phenol derivatives, leading to second-order rate constants ranging from 6.0 × 10−3 to 2.9 M−1 s−1. The modification of the para substituents in the phenol derivatives led to a linear correlation of rate constant versus the Hammett σ parameter with ρ = −1.26. A plot of the ArO–H bond dissociation energy, BDE(ArO–H), versus the logarithm of the second-order rate constant (k″) also showed a linear relationship with a slope of −0.39, with the relative rate decreasing as the O–H bond strength increases. Deuteration of the OH group in 2,4,6-tri-tert-butylphenol (TTBP) led to a kinetic isotope effect (KIE) of kH/kD = 5.9. All of these data provided compelling evidence for a concerted HAT process involving HAT from the phenol derivative to MnV(O)(TBP8Cz). We hypothesized that the MnV(O) complex would also be able to oxidize C–H bond substrates with BDE(C–H) values similar to the BDE(O–H) values of the phenol derivatives. Both cyclohexadiene (CHD) and dihydroanthracene (DHA), which have relatively weak C–H bonds (BDFECHD(C–H) = 67.8 kcal/mol (gas) and BDFEDHA(C–H) = 76.0 kcal/mol (DMSO))12 reacted with the MnV(O) complex to give the MnIII product, although at significantly lower rates (k″ ~ 10−5 M−1 s−1). Much lower reaction rates for C–H versus O–H bonds with metal complexes in HAT is expected.13
Although we were unable to isolate or even spectroscopically observe the MnIV intermediate during HAT reactions with MnV(O)(TBP8Cz), we were able to identify a nicely reversible wave in the cyclic voltammogram that could be assigned to one-electron reduction of MnV(O)(TBP8Cz).9 The electro-chemistry showed that the MnV(O) complex exhibited a low reduction potential (E½(MnV/MnIV) = 0.05 V vs SCE), which led to an intriguing hypothesis regarding HAT reactivity. Perhaps this “low-potential” oxidant was effective at abstracting H atoms from O–H/C–H bonds because the basicity of the incipient [MnIV(O)]− species was high enough to provide a sufficient overall thermodynamic driving force for HAT.
At about this time, similar observations were emerging regarding the reactivity of the high-valent iron–oxo intermediates in heme enzymes such as CYP and copropor phyrinogen oxidase (CPO). An important early contribution came from Green, Dawson, and Gray,14 who provided evidence for the idea that Cpd-II, the FeIV(O)(porph) species in heme enzymes, was far more basic than previously thought when the axial ligand was a cysteinate residue. This basicity led to protonation of Cpd-II to give FeIV(OH)(porph) under physiological conditions. Importantly, the high basicity also suggested that the redox potential for Cpd-I (FeIV(O)(π-cation-radical porph)) could remain relatively low and still allow for effective H-atom abstraction from inert C–H substrates because the basicity of Cpd-II would compensate and lead to a favorable driving force for HAT. The idea of a “low-potential” high-valent metal–oxo oxidant in CYP could help to explain a conundrum in the field: how could deleterious oxidation of the surrounding protein matrix by Cpd-I be avoided? We realized that we could examine these ideas in more detail by taking advantage of the Cz ligand and the access to well-characterized high-valent metal–oxo porphyrinoid species such as the MnV(O) complex. With such complexes in hand, we could determine the relative electronic and structural influences that are important for controlling the outcomes of HAT reactions.
3. BASICITY AS A FACTOR
Following the literature on HAT reactions, especially from Mayer,12,15 led us to consider breaking down the HAT step into the elementary proton transfer (PT) and electron transfer (ET) steps, as shown in Scheme 5. This “square scheme” approach was shown to be especially useful in transition-metal- mediated oxidations.
Scheme 5.

Square Scheme for HAT to MnV(O)(TBP8Cz)
The driving force (ΔG°) for the HAT reaction (diagonal) can be equated to the sum of the ΔG° values for the individual ET (horizontal) and PT (vertical) steps, as shown in Scheme 5. This analysis leads to eq 1, which was initially provided by Bordwell et al.16 for estimating homolytic bond dissociation free energies (BDFEs) in solution from equilibrium acidities and electrochemical data:
| (1) |
The strength of the O–H bond formed in the Mn−1(OH)species can be related to the physical parameters of redox potential (E°) and basicity (pKa) of the relevant metal–oxo species in the scheme. The larger the bond strength (we will use BDFE in place of BDE because the Bordwell equation strictly refers to free energies; see Warren et al.12) of the O–H bond formed, the greater the driving force for HAT. In addition, the square scheme shows explicitly that if E° for the MV(O)/MIV(O) reduction is relatively low, the driving force for HAT can be maintained by a relatively high pKa, i.e., a high basicity for MIV(O). The reactivity of MnV(O)(TBP8Cz) with the phenol and C–H derivatives suggested that HAT was thermodynamically favored for these substrates. With the assumption that the reaction is thermodynamically favored, the BDFE of the putative MnIV(O–H) bond should be greater than that of the X–H bond being cleaved in the substrate, i.e., ~80 kcal/mol. This estimate of the BDFE for the [MnIV(OH)]− species combined with E½(MnV(O)/[MnIV(O)]−) = 0.02 V vs SCE in PhCN (−0.38 V vs Fc+/Fc)9 and use of the Bordwell equation with a CG value of 54.9 (in CH3CN)12 leads to a predicted pKa of ≥24.7 for [MnIV(OH)]−. This estimate is valid provided that the assumption that the CG value in CH3CN is a reasonable approximation of the CG value for PhCN. This value can be compared to that for the basic Cpd-II in CYP, which has pKa = 12 for the ferryl heme.17 We reasoned that the comparison of the pKa value of [MnIV(OH)]− estimated in organic solvent with that of the pKa value for Cpd-II in aqueous solution was reasonable because the Cpd-II active site is buried in a highly hydrophobic pocket, and therefore, the local environment is much closer to an organic solvent than it is to an aqueous medium. Basicity appeared to be a key factor in promoting HAT for MnV(O)(TBP8Cz), and further studies supported this conclusion (vide infra).
The electron transfer properties of MnV(O)(TBP8Cz) were next examined and provided further insights into overall oxidative reactivity, including HAT, and the related process of hydride transfer.18 Reaction of the MnV(O) complex with decamethylferrocene (Fc*) led to the two-electron-reduced MnIII product (Figure 2a). Spectral titrations confirmed the 2:1 reductant:Mn complex stoichiometry (Figure 2b). As in the HAT chemistry, a MnIV intermediate was not observed, and it was postulated that rapid protonation of the putative basic MnIV(O)− species occurred from exogenous water, giving a transient MnIV(OH) which was then rapidly reduced by a second equivalent of reductant. The data were consistent with the mechanism in Scheme 6, which shows a proton-coupled electron transfer (PCET) mechanism for the reduction of MnV(O) by ferrocene derivatives.
Figure 2.

(a) UV–vis changes for the reaction between Fc* and MnV(O)(TBP8Cz) in PhCN. (b) Spectral titration showing a 2:1 stoichiometry of electron transfer.
Scheme 6.

Mechanism of Electron Transfer from [Fe(C5H4Me)2] to MnV(O)(TBP8Cz)
The reactions of MnV(O)(TBP8Cz) with NADH analogues as potential hydride donors were examined. Addition of the acridine derivatives shown in Scheme 7 to the MnV(O) complex caused the rapid formation of [MnIII(OH)-(TBP8Cz)]− and acridinium (AcrH+) products. Kinetic analyses, including a KIE of (Figure 3), indicated that the rate-determining step involves C–H bond cleavage. A significant decrease in rate was also seen for the ethyl-substituted acridine (Figure 3), which would be expected if an initial HAT step in Scheme 7 were the rate-determining step, as opposed to hydride transfer.
Scheme 7.

Mechanism of Hydride Transfer from NADH Analogues (AcrHR) to MnV(O)(TBP8Cz)
Figure 3.

Plots of the observed pseudo-first-order rate constants (kobs) for the reaction of NADH analogues with MnV(O)(TBP8Cz).
4. AXIAL LIGAND EFFECT
The heme enzymes CPO and CYP are unique in that they contain rare thiolate ligation at the axial position, in contrast to the more common His ligation in other enzymes (e.g., peroxidases, heme dioxygenases). Studies of CPO and CYP suggested that the anionic axial ligand is linked to the increase in basicity exhibited by Cpd-II, which in turn facilitates HAT (vide supra). The addition of thiols to MnV(O)(TBP8Cz) as potential axial donors was problematic because of the direct oxidation of sulfur donors to disulfide. However, addition of more inert anionic axial ligands, such as F− and CN−, led to the in situ formation of the six-coordinate MnV(O)(F−)(TBP8Cz) and MnV(O)(CN−)(TBP8Cz) complexes.19 Spectroscopic data indicated weak binding of the anionic donors to the position trans to the terminal oxo group.
The reactivity of the six-coordinate species was dramatically different from that of the starting five-coordinate complex. The fluoride-ligated complex reacted with the C–H substrate DHA with a second-order rate constant that was ~2100-fold greater than that measured for the five-coordinate complex. A large KIE of 10.5 for DHA versus [D4]DHA was also obtained, supporting a rate-determining HAT step. An even more dramatic rate enhancement was observed when the anionic donor was changed from F− to the more electron-donating CN–, with a rate enhancement of ~16000-fold. Similar trends in reaction rate were seen with other C–H substrates, including CHD, indene, xanthene, and triphenylmethane, as well as for the HAT reactivity of non-heme iron(IV)–oxo complexes.20
Density functional theory calculations were performed on the reactions of MnV(O)(TBP8Cz) and the six-coordinate F−- and CN−-ligated derivatives (1, 1-F, and 1-CN in Figure 4) with DHA. The potential energy profiles for all three complexes show a similar reaction mechanism (Figure 4) that proceeds via two HAT steps, with the first step being rate- determining. The energy profiles show a dramatic lowering of the initial, rate-determining HAT barrier upon coordination of F− or CN−. Thus, the calculations are in satisfying agreement with the trend in reactivity that was observed experimentally. Additional calculations suggested that the reactivity could be linked to increased basicity of the oxo group. It is interesting to note that the addition of the axial ligand has a greater impact on the increase in basicity of the metal–oxo group, compared with the lowering of the redox potential, which would have lowered the driving force for HAT and attenuated the reactivity. It should also be noted that no “multistate reactivity” was implicated for the MnV(O) corrolazines. Recent CASSCF calculations confirmed the singlet ground states for the five-coordinate and axially ligated MnV(O) complexes.21
Figure 4.

(left) Calculated potential energy profiles for reactions of singlet-state [MnV(O)(TBP8Cz)(X)] (X = none (1), F (1-F), CN (1-CN)) with DHA; TS = transition state; Int = intermediate; Prod = product). (right) Transition state structures for the initial HAT step, with selected bond distances in angstroms, bond angles in degrees, and imaginary frequencies in wavenumbers. (bottom) Mechanism of HAT with DHA as the substrate. Adapted with permission from ref 19. Copyright 2010 John Wiley & Sons.
5. ELECTRONIC STRUCTURE
With the elucidation of the X-ray structure of the oxygen- evolving complex of Photosystem II showing a Lewis acidic Ca2+ ion closely associated with the catalytic center in a Mn4CaO5 cluster, there has been increased interest in understanding the potential influence of redox-inactive metal ions on high-valent Mn(O) species.22–26
We found that the Lewis acid Zn2+ stabilizes a valence tautomer of MnV(O)(TBP8Cz), formulated as MnIV(O–Zn)(TBP8Cz+•) (Scheme 8).27 The paramagnetic 1H NMR spectrum, Evans’ method measurement (μeff = 4.11μB), and EPR silence were consistent with an integer-spin ground state for the Zn2+ adduct. Subsequent studies found that the valence tautomer was stabilized by the nonmetallic Lewis acid B(C6F5)323 as well as the Bronsted acids24 trifluoroacetic acid (TFA) and HBArF (Scheme 8). The Zn2+ and B(C6F5)3 adducts were characterized by X-ray absorption spectroscopy, revealing elongated Mn–O bonds consistent with binding of the Lewis acid to the oxo ligand. The latter species abstracted H atoms from the O–H bonds in 2,4-di-tert-butylphenol (DTBP) and 2,4,6-tri-tert-butylphenol (TTBP), acting as one-electron oxidants to give MnIV complexes, in contrast with the MnV(O) tautomer, which acts as a two-electron oxidant (Scheme 9). An increased rate of HAT reactivity up to ~100- fold was seen for the MnIV(O–LA)(TBP8Cz+•) complexes with the O–H donors (Table 1).
Scheme 8.

Reversible Binding of Lewis/Bronsted Acids Stabilizing MnIV(O–LA)(TBP8Cz+•)a
aReproduced from ref 24. Copyright 2016 American Chemical Society.
Scheme 9.

HAT Reactivities of MnV(O)(TBP8Cz), MnV(O)(tpfc), and Lewis Acid Adducts
Table 1.
Rate Constants for Different Lewis Acids Ranked by Acceptor Number
| k2 (M−1 s−1) | ||
|---|---|---|
| Lewis acid (A.N.)a | DTBP | xanthene |
| MnV(O)(TBP8Cz) | ||
| none | 2.9 ± 0.1 | (1.8 ± 0.2) × 10−3 |
| TFA (40) | – | (9 ± 1) × 10−4 |
| Zn2+ (68) | 17 ± 1 | (8.1 ± 0.4) × 10−3 |
| HBArF (102) | – | (1.9 ± 0.2) × 10−2 |
| B(C6F5)3 (82) | 107 ± 8 | (5.5 ± 0.3) × 10−2 |
| MnV(O)(tpfc) | ||
| none | 45 ± 0.3 | – |
| TFA | 32.6 ± 0.5 | – |
| Zn2+ | 943 ± 26 | – |
| Sc3+ | 27.3 ± 0.7 | – |
| Yb3+ | 42 ± 2 | – |
| Ca2+ | 1386 ± 5 | – |
| B(C6F5)3 | 271 ± 6 | – |
A.N. = acceptor number, which is proportional to the Lewis acid strength (see ref 21).
In 2015, a study by Abu-Omar on Mn(O) in a corrole ligand revealed different results. MnV(O)(tpfc) (tpfc = tris- (pentafluorophenyl)corrole) forms the valence tautomer MnIV(O)(tpfc+•) upon addition of TFA. Reaction of MnIV(O)-(tpfc+•) with 2,4-DTBP gives the MnIII complex as the product (Scheme 9), a two-electron reduction process. In addition, slower HAT reactivity for MnIV(O)(tpfc+•) compared with the MnV(O) tautomer was seen.25 A follow-up study by our lab showed that MnIV(O–LA)(TBP8Cz+•) (LA = Zn2+, B(C6F5)3, TFA, HBArF) acted as a one-electron oxidant with the C–H donors xanthene, DHA, and CHD.24 The stoichiometry suggests that the MnIV product following HAT is more stable in the Cz than in the corrole. As can be seen in Table 1, Zn2+, B(C6F5)3, and HBArF adducts lead to rate enhancements of HAT in comparison to the naked MnV(O), while a slightly lower k2 was observed for reactivity of the TFA adduct, similar to what was found for MnIV(O)(tpfc+•) with TFA. These results suggest that TFA is too weak an acid under the reaction conditions to increase the rate of HAT.
Abu-Omar also explored HAT reactivity with O–H substrates of MnV(O)(tpfc) and Zn2+, Ca2+, Sc3+, Yb3+, and B(C6F5)3. The Ca2+ adduct, which was slowest to form, was found to be most reactive toward HAT, while Sc3+ and Yb3+, similar to TFA, showed fast kinetics of formation but slower HAT reactivity. It is interesting to note that the Ca2+ adduct exhibited superior activity given that calcium is involved in the Mn-containing OEC of Photosystem II. However, the underlying factors (e.g., steric or electronic properties) that influenced the HAT rate, and in particular what makes Ca2+ adduct the most reactive, could not be determined.26
6. METAL SUBSTITUTION
6.1. Chromium versus Manganese
The isolation and structural characterization of CrV(O)-(TBP8Cz) (Figure 5a), which is isomorphous with MnV(O)-(TBP8Cz), provided a unique opportunity to compare their HAT reactivities.10
Figure 5.

(a) Crystal structure of CrV(O)(TBP8Cz). (b) Cyclic voltammogram of CrV(O)(TBP8Cz) in CH2Cl2.
While MnV(O) was reactive with a range of HAT substrates, the CrV(O) species was reactive only with TEMPOH (Table 2 and Figure 6). An E½ of −0.43 V vs Fc+/Fc is seen for the CrV/CrIV redox potential (Figure 5b), which is ~100 mV more positive than that for the MnV/MnIV couple. Since the reactivity of the CrV(O) complex (Table 2) implies a weaker CrIV(O–H) bond compared with MnIV(O–H), eq 1 suggests that the reduced [CrIV(O)−] is 8 orders of magnitude less basic than [MnIV(O)−]. The dramatic difference in reactivity therefore can be assigned to the different basicities of the metal–oxo units.
Table 2.
Reactivities of CrV(O) and MnV(O) toward HAT Substrates
| substrate | BDFE (kcal/mol) | MnV(O) | CrV(O) |
|---|---|---|---|
| HMBa | 83 | no | no |
| 2,4,6-TTBP | 80 | yes | no |
| DHA | 77 | yes | no |
| xanthene | 73 | yes | no |
| TEMPOH | 67 | yes | yes |
HMB = hexamethylbenzene.
Figure 6.

(a) Time-resolved UV–vis spectral changes for the reaction of CrV(O)(TBP8Cz) with excess TEMPOH. (b) Plot of the absorbance at 653 nm vs time for the reaction in (a). Inset: dependence of the first-order rate constant on [TEMPOH] and the best-fit line.
6.2. Iron
The iron complex FeIII(TBP8Cz) functioned as a catalyst for oxidation of alkenes in the presence of pentafluoroiodosylbenzene (C6F5IO, PFIB),28 and spectroscopic evidence suggested the formation of FeIV(O)(TBP8Cz+•), a Cpd-I analogue, during catalysis.29 Quantitative oxidation of FeIII(TBP8Cz) to FeIV(O)(TBP Cz+•) was initially accomplished by addition of PFIB at −78 °C (Scheme 10). The use of the oxidant m - chloroperoxybenzoic acid (mCPBA) in place of PFIB led to more efficient formation of the Cpd-I analogue.
Scheme 10.

Generation of FeIV(O)(TBP8Cz+•) Using Different Oxidants
Examination of the C–H activation capacity of FeIV(O)-(TBP8Cz+•) was carried out with a series of substrates with a range of BDE(C–H) values. A plot of the second-order rate constants versus the BDE(C–H) values for these substrates revealed a slope of −0.42 (Figure 7a). This plot was similar to what was seen for ([FeIV(O)(p-X-PyO)(tmp+•)]+)30 (tmp = meso-tetramesitylporphyrin) and MnV(O)(TBP8Cz). A KIE of 5.7 was also found for xanthene (Figure 7b). These results support a HAT mechanism for the rate-determining step of FeIV(O)(TBP8Cz+•) in reaction with C–H substrates.
Figure 7.

(a) Plot of log k versus BDE for the reaction of FeIV(O)(TBP8Cz+•) with C–H substrates. (b) Second-order plots for xanthene (green circles) and 9,10-d2-xanthene (blue diamonds).
The reactivity of [FeIV(O)(p-X-PyO)(tmp+•)]+ with xanthene is approximately 50–225 times higher than that of FeIV(O)(TBP8Cz+•). This difference in reactivity was initially thought to be attributed to the extra negative charge on the corrolazine ligand, which should reduce the electrophilicity of FeIV(O)(TBP8Cz+•) in comparison with the porphyrin system. However, we have shown that addition of negatively charged axial ligands to the neutral MnV(O)(TBP8Cz) greatly increases the reactivity toward C–H substrates, which suggests that a difference in ligand charge cannot by itself explain the reactivity trends observed.
6.3. Rhenium
A rhenium(V)–oxo corrolazine was the first metallocorrolazine containing a third-row metal ion.31 While its isoelectronic analogue MnV(O)(TBP8Cz) showed both oxygen atom transfer and HAT reactivity, ReV(O)(TBP8Cz) was found to be unreactive in both processes. However, the ReV(O) complex showed a reversible wave in cyclic voltammetry at +1.04 V vs SCE, indicative of Cz ring oxidation.10 Oxidizing the ReV(O) complex with [(4-BrC6H4)3N+•][SbCl6−] (Ered = +1.16 V vs SCE) gave the monocationic complex ReV(O)-(TBP8Cz+•), and reaction of this species with DHA resulted in quantitative conversion to the [ReV(O)(Cz(H))]+ complex, in which a meso-N atom was the presumed site of protonation. Anthracene was also formed in good yield (90%). However, further experiments revealed that instead of HAT to the ReV(O)(TBP8Cz+•) complex, the mechanism involved an electron back-transfer between ReV(O)(TBP8Cz+•) and (4-BrC6H4)3N, and it was the amminium radical cation that oxidized DHA directly. To avoid electron back-transfer between the reduced oxidant and the oxidized rhenium complex, the more powerful oxidant CeIV(NH4)2(NO3)6 (Ered = +1.33 V vs SCE) was used to generate ReV(O)-(TBP8Cz+•). In this case, direct HAT between the Re complex and DHA was seen (Scheme 11).
Scheme 11.

HAT Reactivity of ReV(O)(TBP8Cz+•) with DHA
Thus, the inertness of the Re–O group allowed us to demonstrate that a π-radical-cation porphyrinoid ligand isolated from its metal–oxo core was capable of HAT reactivity.
7. LIGAND MODIFICATION
The influence of the Cz ligand on HAT was examined by peripheral modification of the aryl substituents. The p-tBu groups on the eight phenyl rings were substituted with p-OMe groups. This new ligand was used to generate MnV(O)-(MeOP8Cz).7 The redox potential of the MnV/MnIV couple (−0.57 V vs Fc+/Fc) was shifted in the negative direction by 40 mV, and that of the Cz ring oxidation (+0.45 V vs Fc+/Fc) was shifted by 60 mV in the same direction. The reactivity of MnV(O)(MeOP8Cz) was examined with C–H substrates, and rate constants ranging from 6.8(5) × 10−5 to 1.70(2) × 10−1 M−1 s−1 were obtained, similar to what was seen for the p-tBu derivative. While there was little change in the reaction rates for HAT reactivity with the substrates xanthene and acridine (kMeO/kTBP ratios of 1.3 and 1.0, respectively), a 2-fold increase in rate was observed for the 1,4-CHD substrate. The lower redox potential of the p-OMe derivative might be expected to lead to a slightly weaker driving force for HAT. However, since the HAT rates for the p-OMe derivative were seen to be slightly greater than those for the p-tBu derivative, it is reasonable to conclude that the basicity of the unseen [MnIV(O)]− species is enhanced and compensates for the lower redox potential to increase the driving force for HAT.
Another modification of the Cz ligand that led to changes in reactivity is alkylation of the meso-N position. We devised a synthetic strategy for the selective alkylation of a single meso-N atom of ReV(O)(TBP8Cz), leading to the cationic complex [ReV(O)(N-MeTBP8Cz)]+.32 This complex was capable of abstracting hydrogen atoms from weak O–H and N–H bonds, leading to a rare, air-stable 19-π-electron porphyrinoid complex, [ReV(O)(N-MeTBP8Cz)]•, in which the alkylated ligand was reduced by one electron. Kinetic analysis with TEMPOH as the substrate gave k2 = 0.76(2) M−1 s−1 and a KIE of 1.4. On the basis of the relatively small KIE, it was suggested that a PCET process occurs in which a weakly basic meso-N atom of the Cz ring is the initial proton acceptor but is then deprotonated by OTf−. Scheme 12 shows a summary of the influence of various ligand modifications on the reactivity of ReV(O)(TBP8Cz).
Scheme 12.

Influence of Ligand Modification on the HAT Reactivity of ReV(O)(TBP8Cz)
8. MISSING HAT INTERMEDIATE
We were able to synthesize an Fe(OH) complex with a formal +4 oxidation state by employing a large, sterically encumbered corrole ligand, tris(2,4,6-triphenylphenyl)corrole (ttppc).33 We hypothesized that the ttppc ligand might allow us entry into the analogous yet elusive MnIV(OH) species, which is the missing intermediate in many HAT reactions with the MnV(O) starting complex.33 The desired MnIV(OH)(ttpcc) complex was successfully synthesized and characterized by XRD (Figure 8). We also prepared and structurally characterized the related MnV(O)(ttppc) and MnIII(H2O)(ttppc) complexes.34 Thus, the series of well-characterized Mn complexes shown in Figure 8 provided an opportunity to study the nature of the interconversion of these species through HAT.
Figure 8.

(top) HAT steps relating Mn(ttppc) complexes and (bottom) their crystal structures.
Reaction of MnV(O)(ttppc) with excess 2,4-DTBP showed complete isosbestic conversion to the MnIII–aquo complex in minutes, with kH = 17.4(1) M−1 s−1 and a KIE of 1.05 (Figure 9a–c). Product analysis showed the formation of the bis(phenol) dimer (91% by GC-FID), supporting HAT with a MnV(O):DTBP stoichiometry of 1:2. This stoichiometry is the same as that seen for MnV(O)(TBP8Cz) and MnV (O)-(tpfc) (vide supra). The expected product after initial HAT is the MnIV(OH) species, but this species was not observed. Either of the two possible mechanisms in Scheme 4 may apply. However, with the isolation of the MnIV(OH)(ttppc) complex, we were able to directly examine this mechanistic question for the first time.
Figure 9.

(a) Reaction of MnV(O)(ttppc) and 2,4-DTBP. (b) Time-resolved UV–vis spectral changes for the reaction between MnV(O)(ttppc) and 2,4-DTBP. Inset: change in absorbance vs time for the growth of MnIII(ttppc) (660 nm) (green circles). (c) Plots of kobs versus [2,4-DTBP-OH] (black circles) and [2,4-DTBP-OD] (red squares). (d) Reaction of MnIV(OH)(ttppc) and 2,4-DTBP. (e) Time-resolved UV–vis spectral changes for the reaction between MnIV(OH)(ttppc) and 2,4-DTBP. Inset: change in absorbance vs time for the growth of MnIII(ttppc) (660 nm) (green circles). (f) Plots of kobs versus [2,4-DTBP-OH] (black circles) and [2,4-DTBP-OD] (red squares). Reproduced from ref 33. Copyright 2018 American Chemical Society.
Addition of excess of 2,4-DTBP to MnIV(OH)(ttppc) resulted in rapid conversion to MnIII(H2O)(ttppc) with k2 =2.73(12) × 104 M− s−1 (Figure 9d–f). This study clearly showed that the MnIV(OH) complex reacts much more rapidly with 2,4-DTBP than the MnV(O) complex does. The >1500-fold rate enhancement is consistent with mechanism A in Scheme 4. No evidence of disproportionation of MnIV(OH)-(ttppc) in solution over many hours was observed, which further rules out mechanism B. This settles the debate between the two suggested mechanisms in favor of mechanism A. Further kinetic studies were performed on a series of para- substituted 2,6-di-tert-butylphenol substrates (4-X-2,6-DTBP, X = OMe, Me, tBu, H). A Hammett plot for these substrates gave ρ+ = −1.4(2), and a Marcus analysis of the rate constants gave a weak slope of −0.12(1) (Figure 10). This latter slope is much smaller than expected for a rate-limiting ET reaction (−0.5) and is comparable to the slope observed for the reaction of cumylperoxyl radical and phenols (−0.05), which is a clear example of a concerted HAT.35 Taken together, the data show that the MnIV(OH) complex abstracts hydrogen atoms fashion. in a concerted
Figure 10.

(a) Hammett and (b) Marcus plots for the reaction of MnIV(OH)(ttppc) and 4-X-2,6-DTBP (X = OMe, Me, tBu, H). Reproduced from ref 33. Copyright 2018 American Chemical Society.
SUMMARY
High-valent metal–oxo species of corrolazine and corrole ligands were synthesized and employed to examine factors that can influence HAT reactivity. The ability of MnV(O)(TBP8Cz) to abstract H atoms from various substrates initially showed that a low-potential oxidant can still function as an effective H- atom abstractor and supported the idea that metal–oxo basicity is an important part of the driving force for HAT. These ideas were strengthened by the findings on the effects of axial ligands on MnV(O)(TBP8Cz) as well as the effects of peripheral substitution (e.g., p-OMe vs p-tBu) and metal ion identity (MnV(O) vs CrV(O)). Lewis acids were found to have a significant influence on HAT reactivity as well. Although the ReV(O)(TBP8Cz) analogue was found to be unreactive toward HAT, the inert Re–O group allowed for the opportunity to study the isolated HAT reactivity of the corrolazine ligand itself. The isolation and structural characterization of a series of Mn(corrole) complexes with different oxidation and proto nation states allowed for the study of the interconversion of these species through HAT and settled a debate regarding the mechanism of HAT for previously reported Mn–oxo porphyrinoid systems. This work showed that the lower-valent MnIV(OH) complex is a much more reactive H-atom abstractor than the higher-valent MnV(O) analogue. The results in this Account show that an interplay of a variety of factors (e.g., metal identity, ligand modifications, electronic structure) can greatly influence the H-atom reactivity of metal–oxo/hydroxo species. Analysis of these factors for the different porphyrinoid complexes presented here has provided knowledge about the thermodynamic forces that drive HAT as well as mechanistic insights into HAT processes relevant to enzyme systems.
ACKNOWLEDGMENTS
This work was supported by the NIH (Grant GM101153 to D.P.G.).
Biographies
Jireh Joy D. Sacramento received her B.S. degree in Biochemistry from the University of the Philippines Manila in 2010. She is currently a Ph.D. candidate at the Johns Hopkins University working with Professor David P. Goldberg.
David P. Goldberg received his B.A. degree from Williams College in 1989 and a Ph.D. degree from M.I.T in 1995. After completing a postdoctoral fellowship at Northwestern University, he moved to Johns Hopkins University in 1998, where he is currently a Professor of Chemistry.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Poulos TL Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C–H Bond Activation Kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- (3).Grant JL; Mitchell ME; Makris TM Catalytic strategy for carbon-carbon bond scission by the cytochrome P450 OleT. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 10049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Umena Y; Kawakami K; Shen JR; Kamiya N Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 A. Nature 2011, 473, 55–60. [DOI] [PubMed] [Google Scholar]
- (5).Cox N; Retegan M; Neese F; Pantazis DA; Boussac A; Lubitz W Electronic structure of the oxygen- evolving complex in photosystem II prior to O-O bond formation. Science 2014, 345, 804–808. [DOI] [PubMed] [Google Scholar]
- (6).Ramdhanie B; Stern CL; Goldberg DP Synthesis of the First Corrolazine: A New Member of the Porphyrinoid Family. J. Am. Chem. Soc. 2001, 123, 9447–9448. [DOI] [PubMed] [Google Scholar]
- (7).Joslin EE; Zaragoza JP; Baglia RA; Siegler MA; Goldberg DP The Influence of Peripheral Substituent Modification on P(V), Mn(III), and Mn(V)(O) Corrolazines: X-ray Crystallography, Electrochemical and Spectroscopic Properties, and HAT and OAT Reactivities. Inorg. Chem. 2016, 55, 8646–8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mandimutsira BS; Ramdhanie B; Todd RC; Wang H; Zareba AA; Czernuszewicz RS; Goldberg DP A Stable Manganese(V)–Oxo Corrolazine Complex. J. Am. Chem. Soc. 2002, 124, 15170–15171. [DOI] [PubMed] [Google Scholar]
- (9).Lansky DE; Mandimutsira BS; Ramdhanie B; Clausen M; Penner-Hahn J; Zvyagin SA; Telser J; Krzystek J; Zhan R; Ou Z; Kadish KM; Zakharov LN; Rheingold AL; Goldberg DP Synthesis, Characterization, and Physicochemical Properties of Manganese(III) and Manganese(V)–Oxo Corrolazines. Inorg. Chem. 2005, 44, 4485–4498. [DOI] [PubMed] [Google Scholar]
- (10).Baglia RA; Prokop-Prigge KA; Neu HM; Siegler MA; Goldberg DP Mn(V)(O) versus Cr(V)(O) Porphyrinoid Complexes: Structural Characterization and Implications for Basicity Controlling H-Atom Abstraction. J. Am. Chem. Soc. 2015, 137, 10874–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lansky DE; Goldberg DP Hydrogen Atom Abstraction by a High-Valent Manganese(V)–Oxo Corrolazine. Inorg. Chem. 2006, 45, 5119–5125. [DOI] [PubMed] [Google Scholar]
- (12).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Mayer JM Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Green MT; Dawson JH; Gray HB Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. [DOI] [PubMed] [Google Scholar]
- (15).Waidmann CR; Miller AJM; Ng C-WA; Scheuermann ML; Porter TR; Tronic TA; Mayer JM Using combinations of oxidants and bases as PCET reactants: thermochemical and practical considerations. Energy Environ. Sci. 2012, 5, 7771–7780. [Google Scholar]
- (16).Bordwell FG; Cheng J-P; Harrelson JA Homolytic Bond Dissociation Energies in Solution from Equilibrium Acidity and Electrochemical Data. J. Am. Chem. Soc. 1988, 110, 1229–1231. [Google Scholar]
- (17).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C–H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Fukuzumi S; Kotani H; Prokop KA; Goldberg DP Electron- and hydride-transfer reactivity of an isolable manganese-(V)–oxo complex. J. Am. Chem. Soc. 2011, 133, 1859–1869. [DOI] [PubMed] [Google Scholar]
- (19).Prokop KA; de Visser SP; Goldberg DP Unprecedented rate enhancements of hydrogen-atom transfer to a manganese(V)–oxo corrolazine complex. Angew. Chem., Int. Ed. 2010, 49, 5091–5095. [DOI] [PubMed] [Google Scholar]
- (20).Sastri CV; Lee J; Oh K; Lee YJ; Lee J; Jackson TA; Ray K; Hirao H; Shin W; Halfen JA; Kim J; Que L Jr.; Shaik S; Nam W Axial ligand tuning of a nonheme iron(IV)–oxo unit for hydrogen atom abstraction. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19181–19186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Yang T; Quesne MG; Neu HM; Cantu Reinhard FG; Goldberg DP; de Visser SP Singlet versus Triplet Reactivity in an Mn(V)–Oxo Species: Testing Theoretical Predictions against Experimental Evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen J; Lee YM; Davis KM; Wu X; Seo MS; Cho KB; Yoon H; Park YJ; Fukuzumi S; Pushkar YN; Nam W A mononuclear non-heme manganese(IV)–oxo complex binding redox- inactive metal ions. J. Am. Chem. Soc. 2013, 135, 6388–6391. [DOI] [PubMed] [Google Scholar]
- (23).Baglia RA; Durr M; Ivanovic-Burmazovic I; Goldberg DP Activation of a high-valent manganese–oxo complex by a nonmetallic Lewis acid. Inorg. Chem. 2014, 53, 5893–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Baglia RA; Krest CM; Yang T; Leeladee P; Goldberg DP High-Valent Manganese–Oxo Valence Tautomers and the Influence of Lewis/Bronsted Acids on C–H Bond Cleavage. Inorg. Chem. 2016, 55, 10800–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bougher CJ; Liu S; Hicks SD; Abu-Omar MM Valence Tautomerization of High-Valent Manganese(V)–Oxo Corrole Induced by Protonation of the Oxo Ligand. J. Am. Chem. Soc. 2015, 137, 14481–14487. [DOI] [PubMed] [Google Scholar]
- (26).Bougher CJ; Abu-Omar MM Lewis-Acid-Assisted Hydrogen Atom Transfer to Manganese(V)–Oxo Corrole through Valence Tautomerization. ChemistryOpen 2016, 5, 522–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Leeladee P; Baglia RA; Prokop KA; Latifi R; de Visser SP; Goldberg DP Valence tautomerism in a high-valent manganese–oxo porphyrinoid complex induced by a Lewis acid. J. Am. Chem. Soc. [DOI] [PubMed] [Google Scholar]
- (28).Mcgown AJ; Kerber WD; Fujii H; Goldberg DP Catalytic Reactivity of a Meso-N-Substituted Corrole and Evidence for a High-Valent Iron–Oxo Species. J. Am. Chem. Soc. 2009, 131, 8040–8048. [DOI] [PubMed] [Google Scholar]
- (29).Cho K; Leeladee P; McGown AJ; DeBeer S; Goldberg DP A high-valent iron–oxo corrolazine activates C–H bonds via hydrogen-atom transfer. J. Am. Chem. Soc. 2012, 134, 7392–7399. [DOI] [PubMed] [Google Scholar]
- (30).Kang Y; Chen H; Jeong YJ; Lai W; Bae EH; Shaik S; Nam W Enhanced reactivities of iron(IV)–oxo porphyrin pi-cation radicals in oxygenation reactions by electron-donating axial ligands. Chem. - Eur. J 2009, 15, 10039–10046. [DOI] [PubMed] [Google Scholar]
- (31).Zaragoza JP; Siegler MA; Goldberg DP Rhenium(V)–oxo corrolazines: isolating redox-active ligand reactivity. Chem. Commun. 2016, 52, 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Joslin EE; Zaragoza JP; Siegler MA; Goldberg DP meso-N-Methylation of a porphyrinoid complex: activating the H-atom transfer capability of an inert ReV(O) corrolazine. Chem. Commun. 2017, 53, 1961–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zaragoza JPT; Siegler MA; Goldberg DP A Reactive Manganese(IV)–Hydroxide Complex: A Missing Intermediate in Hydrogen Atom Transfer by High-Valent Metal–Oxo Porphyrinoid Compounds. J. Am. Chem. Soc. 2018, 140, 4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liu H-Y; Yam F; Xie Y-T; Li X-Y; Chang CK A Bulky Bis-Pocket Manganese(V)–Oxo Corrole Complex: Observation of Oxygen Atom Transfer between Triply Bonded Mn(V)O and Alkene. J. Am. Chem. Soc. 2009, 131, 12890–12891. [DOI] [PubMed] [Google Scholar]
- (35).Osako T; Ohkubo K; Taki M; Tachi Y; Fukuzumi S; Itoh S Oxidation Mechanism of Phenols by Dicopper-Dioxygen (Cu2/O2) Complexes. J. Am. Chem. Soc. 2003, 125, 11027–11033. [DOI] [PubMed] [Google Scholar]