

Abstract
Purpose
We previously identified an oxysterol, VP1-001 (also known as compound 29), that partially restores the transparency of lenses with cataracts. To understand the mechanism of VP1-001, we tested the ability of its enantiomer, ent-VP1-001, to bind and stabilize αB-crystallin (cryAB) in vitro and to produce a similar therapeutic effect in cryAB(R120G) mutant and aged wild-type mice with cataracts. VP1-001 and ent-VP1-001 have identical physicochemical properties. These experiments are designed to critically evaluate whether stereoselective binding to cryAB is required for activity.
Methods
We compared the binding of VP1-001 and ent-VP1-001 to cryAB using in silico docking, differential scanning fluorimetry (DSF), and microscale thermophoresis (MST). Compounds were delivered by six topical administrations to mouse eyes over 2 weeks, and the effects on cataracts and lens refractive measures in vivo were examined. Additionally, lens epithelial and fiber cell morphologies were assessed via transmission electron microscopy.
Results
Docking studies suggested greater binding of VP1-001 into a deep groove in the cryAB dimer compared with ent-VP1-001. Consistent with this prediction, DSF and MST experiments showed that VP1-001 bound cryAB, whereas ent-VP1-001 did not. Accordingly, topical treatment of lenses with ent-VP1-001 had no effect, whereas VP1-001 produced a statistically significant improvement in lens clarity and favorable changes in lens morphology.
Conclusions
The ability of VP1-001 to bind native cryAB dimers is important for its ability to reverse lens opacity in mouse models of cataracts.
Keywords: αB-crystallin, enantiomer, pharmacologic chaperone, biophysical assay, in vivo cataract model
A densely packed network of crystallin proteins provides the optical clarity and unique refractive index of lens tissue. In humans, the lens has limited capacity for new protein synthesis, requiring crystallins to remain soluble and stable for decades.1 This remarkable feature is facilitated in part by αB-crystallin (cryAB), a chaperone that helps to maintain protein stability and preserve lens transparency.2,3 The active form of cryAB consists of dimers and larger oligomers4 that prevent client proteins from aggregating via numerous, promiscuous, low-affinity amphipathic interactions.5,6
Structural or functional alterations in cryAB or its related isoform, cryAA, by mutation or posttranslational modification are associated with aggregation7 and early-onset hereditary cataracts.8–10 Mutant cryAB(R120G) is prone to aggregation in vitro11 and is associated with childhood cataracts.12 R120G disrupts two native salt bridges, which normally contribute to the dimerization of two cryAB protomers across an antiparallel β-sheet interface.13 Repeating units of cryAB dimers typically assemble into larger oligomers, but R120G disrupts overall assembly and function of the chaperone. We previously showed that the lenses of cryAB(R120G) knock-in mice contain a greater proportion of insoluble, aggregated cryAB and develop opacities at an early age as compared with wild-type (WT) mice.14 In addition, cryAB(R120G) oligomers in solution had a 2- to 3-fold higher average molecular mass than WT cryAB. Similar to cryAB(R120G), cryAA(R49C) knock-in mice develop lens opacity at an early age and exhibit a smaller proportion of water-soluble cryAA as compared with WT mice.8 Importantly, these mutations appear to damage the chaperone functions of crystallins. Interactions with client proteins, such as vimentin, are increased for cryAB(R120G) compared with WT in epithelial and fiber cells of mouse lenses.14 Along with aging and trauma to the lens, cryAB(R120G) can cause aggregate formation that leads to lens opacification and cataract formation.15,16 Recent studies identified sterols as pharmacologic chaperones that reverse crystallin aggregation in cataracts.17–19 Thus, one potential therapeutic approach for hereditary and/or age-associated cataracts is to bind and stabilize the native cryAB dimer.
We previously identified VP1-001, an oxysterol that limits cryAB aggregation in a thermal shift assay and restores transparency to the lenses of cryAB(R120G) heterozygous mice.17 We hypothesized that VP1-001 acts as a small-molecule pharmacologic chaperone20 by binding to a site within cryAB that stabilizes its native conformation. Indeed, nuclear magnetic resonance (NMR) studies suggested that a cryptic binding site for VP1-001 is located in a deep groove in the cryAB dimer. However, this compound may also act as a chemical chaperone, similar to glycerol or trimethylamine-N-oxide, to nonselectively protect cryAB from misfolding. In contrast to pharmacologic chaperones, chemical chaperones do not interact with a discrete binding site. A third possibility is that VP1-001 may partition into the lipid bilayers of lens cells, exerting its effect by influencing membrane fluidity. Further development of this therapeutic strategy requires clearer understanding of the mechanisms of action of VP1-001.
We measured binding of VP1-001 and its enantiomer, ent-VP1-001, to cryAB. Despite having physicochemical properties identical to VP1-001, the enantiomer does not engage the chiral cryAB pocket. Thus, ent-VP1-001 was a useful tool for determining whether these compounds act through binding cryAB or through an alternative mechanism. Indeed, only VP1-001 restored lens transparency in mouse models of hereditary and age-associated cataracts. These results strongly support selective binding of VP1-001 to cryAB with subsequent stabilization of the native cryAB conformation and partial restoration of crystallin networks. These findings clarify the mechanism of action of VP1-001 and support optimization of affinity for cryAB in the development of potential cataract treatments.
Methods
Docking
Crystallographic (Protein Data Bank [PDB] code: 2WJ721) and closed models of cryAB (receptor) were created using a standard docking protocol to prepare hot spots (spheres) and scoring grids (van der Waals, electrostatics, and ligand desolvation) as previously described,22 with one modification: qnifft23 was used in place of Delphi to prepare the electrostatic scoring grids. qnifft allows for the same calculation of Poisson-Bolzmann finite difference electrostatics but is available as an open-source software package.
Modeling was performed using UCSF (University of California, San Francisco, CA, USA) DOCK 3.624,25 as licensed by Blue Dolphin (San Francisco, CA, USA). The initial criteria for the model were a good overall fit of the ligand to receptor, favorable hydrophobic and polar complementarity, and minimal unfavorable interactions.26 To obtain closed receptor models, the crystallographic structure was modified as follows. In Chimera,27 one monomer was rotated toward the other along an axis determined by the centroids of the H-bonding atoms of the beta sheet through which the monomers were joined. In this approach, the promoters were considered rigid bodies. We created models in 4° increments, from 4° to 20°. In all cases, the hydroxyl of the ligand was constrained to engage both arginines (R120) at either end of the dimer interface.
Synthesis of ent-VP1-001
Compound synthesis was adapted from a previously described procedure.28 The full synthetic route can be found in Supplementary Figure S1, and the characterization of each intermediate is described in the Supplementary Methods.
Protein Purification
Three recombinant proteins were expressed and purified from Escherichia coli BL21(DE3) cells (Invitrogen, Thermo Fisher Scientific, Invitrogen, Waltham, MA, USA): a cryAB core domain, α-crystallin domain (ACD) mutant, WT ACD, and full-length cryAB(R120G). For the ACD mutant, the mutation E87C was induced by a mutagenesis kit (QuikChange; Agilent Technologies, Santa Clara, CA, USA) (forward primer: GGA TGT GAA GCA CTT TAG CCC GTG CGA ACT GAA AGT TAA GGT TCT GG; reverse primer: CCA GAA CCT TAA CTT TCA GTT CGC ACG GGC TAA AGT GCT TCA CAT CC), and the protein was expressed and purified as previously described.29 The WT ACD (amino acids 68–153) with an N-terminal hexahistidine (His6) tag followed by a tobacco etch virus (TEV) protease cleavage site was purified from E. coli expressing a pET28a plasmid (GenScript, Piscataway, NJ, USA) harboring the construct. The full-length cryAB(R120G) with an N-terminal His6 tag was purified from E. coli expressing a pD441 plasmid (ATUM) harboring the construct. Cultures were induced at an optical density of 3.0 by adding 1 mM isopropyl-β-d-thiogalactopyranoside to rich media (Terrific Broth; Thermo Fisher Scientific, Pittsburgh, PA, USA) and incubated at 25°C for 18 hours with agitation at 200 RPM. Pellets were centrifuged for 10 minutes at 7000g and flash frozen at −80°C for long-term storage. Frozen pellets were thawed and resuspended in 100 to 150 mL of lysis buffer (50 mM Tris, pH 8.0; 150 mM NaCl; 10 mM imidazole; 5% glycerol) or in lysis buffer containing 6 M urea and 0.5 mM [tris(2-carboxyethyl)phosphine] (TCEP) (for full-length cryAB) and homogenized by an homogenizing valve (EmulsiFlex-C3; Avestin, Inc., Ottowa, ON, Canada) with a target pressure of 1500 psi). The homogenates were clarified by centrifuging for 15 minutes at 46,000g. The supernatants were agitated with 15 mL Ni-NTA resin at 4°C for 2 hours. WT ACD or full-length cryAB(R120G) bound to resin was collected and washed on a glass filter with 150 mL of lysis buffer or lysis buffer with urea and TCEP, respectively, and then with 100 mL wash buffer or urea wash buffer (lysis buffers with 30 mM imidazole). Proteins were eluted in five 15-mL fractions with elution buffer or urea elution buffer (lysis buffers with 400 mM imidazole) or until eluants were colorless. The eluant containing WT ACD was agitated overnight at 4°C with TEV protease (purified in-house30 from ATCC plasmid pRK793 in E. coli BL21 CodonPlus(DE3)-RIL cells) to remove the His6 tag. The protein was centrifuged for 10 minutes at 4000g. The supernatant was concentrated to 5 mL and refolded, and buffer exchange was performed via size-exclusion chromatography (SEC) with SEC buffer (1× PBS [pH 7.5], 5% glycerol). The eluants containing full-length cryAB(R120G) were combined and concentrated to 5 mL and then quickly diluted in 95 mL 0.1× PBS with rapid stirring. The refolded protein was centrifuged for 10 minutes at 4000g, and the supernatant was dialyzed overnight into 4 L 0.1× PBS. All proteins were flash frozen and stored at −80°C. Full-length proteins were thawed and centrifuged at 10,000g for 3 minutes prior to plating assays.
Differential Scanning Fluorimetry (DSF)
DSF was performed on recombinant ACD in the presence of either the vehicle control (4% ethanol) or increasing concentrations of compounds in 4% ethanol. A sample of 2.4 μL of the compound stocks were thoroughly mixed with 51.6 μL of 70 μM recombinant cryAB ACD protein in DSF buffer (50 mM Tris-HCl [Invitrogen], 100 mM NaCl, 0.5 mM ZnCl2, 0.5 mM MgCl2, 0.5 mM CaCl2, pH 8.0) and 6 μL 100× orange protein gel stain (SYPRO; Invitrogen; 5000× stock. DSF buffer is diluent for intermediate stock). Then, 10 μL was added in quadruplicates to a 384-well reaction plate (MicroAmp Optical; Thermo Fisher Scientific, Applied Biosystems, Waltham, MA, USA). The plate was sealed with a MicroAmp Optical adhesive film (Applied Biosystems), centrifuged at 1,000g for 2 minutes, and loaded into a real-time PCR system (QuantStudio 6 Flex; Applied Biosystems). The DSF assay was performed by heating the plate from 30°C to 80°C with 0.5°C increments per cycle over 100 cycles. Each cycle consisted of a 90-second hold at the desired temperature and a 20-second hold at 25°C, during which the fluorescent signal (520 nm excitation/558 nm emission) was measured.
The DSF data was fitted using a Boltzmann model (Prism, version 7; GraphPad Software, San Diego, CA, USA) using a fitting range from 25°C to 3° to 5°C above the temperature at which the fluorescence signal was highest. The melting temperature (Tm) was determined to be the temperature at the midpoint between the highest and lowest fluorescent signals on the curve.
Microscale Thermophoresis (MST)
MST was performed on a mixture containing 10 μM full-length cryAB(R120G) and 100 nM of the ACD mutant labeled with a thiol-reactive dye (Alexa Fluor; Thermo Fisher Scientific, Invitrogen) 488 C5 maleimide (AF488-ACD). The compounds were first diluted to 10 mM in dimethylsulfoxide (DMSO) and then serially diluted 2-fold in DMSO before use. Samples containing 9 μL of MST-TP buffer (50 mM Tris, 150 mM NaCl, 10 mM MgCl2, 0.05% Tween-20, 0.1% polyethylene glycol-8000, pH 7.4), 1 μL of compound (VP1-001 or ent-VP1-001), and 10 μL cryAB mixture (for a final concentration of 5 μM cryAB in 5% DMSO) were incubated for 15 minutes at room temperature and then applied to capillaries (MO-K022) loaded into a single laser microscale thermophoresis instrument (Monolith NT.115; NanoTemper Technologies GmbH, München, Germany) for analysis (80% blue laser power, medium MST power, standard conditions). The response to ligand titration was determined at the 1.5-second threshold and fit to the KD model using NanoTemper analysis software. The fraction bound for ent-VP1-001 was normalized to VP1-001 due to the inability to saturate curves with the enantiomer.
Animals and Dosing
All animal procedures adhered to the ARVO Statement for Use of Animals in Ophthalmic and Vision Research. WT (C57BL/6J) mice (218–391 days old) and heterozygous cryAB(R120G) mice (on a C57BL/6J background; 187–362 days old) were used in this study. The Mouse Genetics Core at Washington University (St. Louis, MO, USA) was responsible for care, breeding, and genotyping of mice. Single nucleotide polymorphism services from DartMouse (Dartmouth Geisel School of Medicine, Lebanon, NY, USA) previously converted these mice to a C57BL/6J background using speed congenics. This process utilizes strain-specific polymorphic markers to speed up the conversion process from a mixed 129Sv/C57BL/6J background to >98% C57BL/6J background (Supplementary Fig. S2). These mice do not carry the CP49 mutation; thus, cataracts in these mice are unlikely to be due to the CP49 mutation present in the 129Sv strain.31
One eye of each animal was treated topically with either 0.04% ent-VP1-001 or 0.04% VP1-001 (1 mM in 8% cyclodextrin), and the other eye was treated with vehicle alone (8% cyclodextrin) three times per week (Monday, Wednesday, and Friday) for 2 weeks. Mouse cohorts were age matched between treatment groups (see Fig. 4).
Figure 4.

Treatment with VP1-001 significantly reduces cataract grade in cryAB(R120G) mutant mice, but ent-VP1-001 is inactive. (A) Cataract grading for cryAB(R120G) heterozygous mice treated with either compound. The vehicle group represents combined contralateral eyes from both treatment groups. (B) Age distribution of cryAB(R120G) heterozygous mice separated by treatment condition. Each point represents an individual mouse lens. The mean is shown by a dotted line, and the error bars represent the standard errors from the means. (C–H) Representative slit lamp images from a cryAB(R120G) heterozygous mouse showing the extent of lens opacity in vehicle-treated (C) and ent-VP1-001–treated eyes (D). Representative slit lamp images from a cryAB(R120G) heterozygous mouse showing the extent of lens opacity in vehicle-treated (E) and VP1-001–treated eyes (F). Representative slit lamp images from a cryAB(R120G) heterozygous mouse showing the extent of lens opacity in an untreated eye (G). Representative slit lamp images from an untreated normal, WT mouse showing a clear lens (H).
Slit Lamp Biomicroscopy
Mice were examined by slit lamp biomicroscopy (slit lamp model BG-2GN, serial number 941752; Topcon, Oakland, NJ, USA) 3 days following the final dosing. Prior to observation, the pupils were dilated with a 1% USP tropicamide ophthalmic solution and 10% USP phenylephrine hydrochloride solution in a 9:1 ratio. The slit was placed orthogonal to the mouse for best viewing of the lens opacities. Each eye was recorded on a video camera in the night mode (DCR-DVD403 3MP DVD Handycam Camcorder with 10× optical zoom with Carl-Zeiss Vario-Sonnar Super Steady Shot; Sony, Tokyo, Japan). Still frames were extracted from the videos using software (Topaz Moment; free of charge from Topaz Labs, Dallas, TX, USA) or Adapter (Adapter; Macroplant, McKinney, TX, USA). Improvements in lens transparency were scored using a system we previously modeled after LOCSIII but adapted for mice.14,17 Specifically, an independent observer who did not apply the topical treatments scored each eye from 0 to 4 using the following system: stage 0, clear lens; stage 1, loss of normal lens appearance in the anterior, posterior, and nuclear regions of the lens; stage 2, distinct nuclear opacity accompanied by discrete anterior lens changes; stage 3, increased opacity in nuclear and cortical regions, discrete punctate opacities, and overall opacities covering approximately two-thirds of the lens; stage 4, complete loss of transparency and opacities involving most of the cortex and nucleus.
Gel Permeation Chromatography (GPC) of Water-Soluble Lens Crystallins
The treated mouse eyes were enucleated, and the lenses were excised and placed in individual tubes for analyses. The lenses were homogenized with disposable grinders (Axygen, Union City, CA, USA) in 1.5-mL Eppendorf tubes containing 400 μL PBS and centrifuged for 30 minutes at 4°C and 10,286g. The supernatants were filtered through a 0.22-μm filter, transferred to a vial, and analyzed by GPC as follows. Proteins (100 μL aliquots of the supernatants) were separated in succession on SEC columns (G3000 XL and G5000 XL; Tosoh Bioscience LLC, Prussia, PA, USA) in line with a TDA302 detector that measured the refractive index (RI), right-angle light scattering (RALS), viscosity, and UV absorbance (Malvern PANalytical Products, Westborough, MA, USA).
Transmission Electron Microscopy of Treated Lenses
For ultrastructural analyses, the lenses were fixed in 2% formaldehyde–2.5% glutaraldehyde (Polysciences, Inc., Warrington, PA, USA) in 100 mM sodium cacodylate buffer (pH 7.2) for 2 hours at room temperature and then overnight at 4°C. The samples were washed in sodium cacodylate buffer at room temperature and postfixed in 1% osmium tetroxide (Polysciences, Inc.) for 1 hour. The samples were then rinsed extensively in deionized water prior to en bloc staining with 1% aqueous uranyl acetate (Ted Pella Inc., Redding, CA, USA) for 1 hour. Following several rinses in deionized water, the samples were dehydrated in a graded ethanol series and embedded in resin (Eponate 12; Ted Pella, Inc.). Sections 95-nm thick were cut with an ultramicrotome (Leica Ultracut UCT; Leica Microsystems Inc., Bannockburn, IL, USA), stained with uranyl acetate and lead citrate, and viewed on a transmission electron microscope (JEOL 1200 EX; JEOL USA Inc., Peabody, MA, USA) equipped with an 8-megapixel digital camera and software (Image Capture Engine V602; Advanced Microscopy Techniques, Woburn, MA, USA). Multiple images of the lens epithelial and lens fiber cells were captured in the equatorial region. Trained staff at the Molecular Microbiology Imaging Facility at Washington University performed all processing, plastic embedding, and ultrastructural analysis by electron microscopy (EM).
Morphologic damage to the lens epithelium and lens fiber cells was documented in EM images of the equatorial region of the lens. Morphologic damage to the lens epithelium included loss of cell-cell contacts, presence of vacuoles, and loss of contacts at the epithelial-capsule interface. Morphologic damage to the lens fiber cells included nonuniform cell size, irregular packing, and limited membrane blebbing. The numbers of damaged cells were determined in each image, and a t test was used to determine statistical significance. EM analysis of WT, untreated mouse lenses can be found in Supplementary Figure S3.
Results
VP1-001 is an oxysterol that contains eight chiral centers. To determine if binding to cryAB is an important aspect of compound activity, we synthesized the enantiomer, ent-VP1-001 (Fig. 1A; Supplementary Fig. S1). Importantly, VP1-001 and ent-VP1-001 have identical physicochemical properties (e.g., molecular mass and calculated logP) but mirror-image three-dimensional configurations (Fig. 1B). Thus, we predicted that only VP1-001 would bind the chiral site in cryAB.
Figure 1.

VP1-001 and ent-VP1-001 have the same physical properties but distinct shapes. (A) Chemical structures and physicochemical properties of the oxysterol VP1-001 and its enantiomer ent-VP1-001. Total polar surface area is the sum of the surface contributions of polar atoms. Calculated logP is a measure of hydrophilicity and is defined as the partition coefficient of a compound in mixtures of n-octanol and water. (B) Three-dimensional comparison of the structures of VP1-001 and its enantiomer in different orientations, highlighting the distinct shapes. These structural differences were predicted to hinder binding of ent-VP1-001 to cryAB by misaligning the key hydroxyl groups (red).
Molecular Docking Suggests That VP1-001, But Not Its Enantiomer, Binds a Deep Groove in the Native cryAB Dimer
In silico docking studies were performed to predict how VP1-001 and ent-VP1-001 bind cryAB. There are multiple, nonequivalent structures of the cryAB ACD solved by crystallography and NMR.21,32–34 Most of the differences among these structures occur at the dimer interface, where an extended β-sheet that is either flat and coplanar or curved and V-shaped exists between the two protomers. The β-sheets that form the dimer interface were crystalized in three different registers.33 This structural information suggests that the dimer has conformational flexibility. Accordingly, we started with one representative structure (PDB: 2WJ721) to create several models by varying the angle of the two protomers relative to one another. These motions influenced the size and shape of the binding pocket for VP1-001.17 Six of these models were considered, and the angle between protomers was increased from 0° to 20° in 4°-increments. Docking of VP1-001 and ent-VP1-001 revealed that the 16° closed structure was the last in the series to accommodate VP1-001. In the remaining four models, including the 8° example (Fig. 2), VP1-001 fit well into the known binding pocket. Specifically, the sterol rings made contacts with hydrophobic side chains on either side of the groove. By contrast, the terminal hydroxyl groups were positioned to make hydrogen bonds with residues on either end of the groove (Fig. 2B, green lines). Notably, when VP1-00 hydroxyls were positioned to interact with R120 residue, a key residue mutated in some cases of hereditary cataracts, we observed an additional potential hydrogen bond with the R116 residue. Next, we found that ent-VP1-001 did not fit well into the groove due to misalignment of possible hydrogen bond partners and steric clashes with hydrophobic residues in the groove. Thus, even the best models of ent-VP1-001 showed that much of the compound projected out of the groove. These docking studies suggested that only VP1-001 would bind cryAB.
Figure 2.

VP1-001 binds to the ACD, but ent-VP1-001 does not. (A) Electrostatic surface model of the ACD dimer in complex with docked VP1-001 (top) or ent-VP1-001 (bottom). Note the relatively poor ability of ent-VP1-001 to fit into the pocket. (B) Ribbon views of the ACD pocket with bound VP1-001 (top) or ent-VP1-001 (bottom). Green lines (top) indicate predicted hydrogen bonds (distance 4 Å or less). The protein coordinates are from the x-ray structure 2WJ7. However, the angle between protomers (shown in white and gray) was reduced by 8° by a rigid body movement (as described in Methods).
VP1-001, But Not Its Enantiomer, Binds and Stabilizes cryAB
To test our computational and structural predictions, we measured binding of VP1-001 and ent-VP1-001 to cryAB in vitro using two different methods. DSF was used to calculate an apparent Tm for solutions of purified, recombinant cryAB ACD.35,36 In this platform, binding of a ligand to cryAB ACD was predicted to shift the apparent Tm to a higher temperature.37,38 Consistent with this prediction, we found that VP1-001, but not ent-VP1-001, dose-dependently (Fig. 3A; n = 3, P < 0.0001 2-way ANOVA 0 μM versus 100 μM VP1-001) increased stability of cryAB ACD (Fig. 3A; n = 3, P < 0.0001 2-way ANOVA 100 μM ent-VP1-001 versus 100 μM VP1-001).
Figure 3.

VP1-001 binds to the ACD and full-length α-crystallin, but ent-VP1-001 does not. (A) Measured Tm of cryAB ACD (WT) calculated from Boltzmann fits of DSF traces (n = 3). The shaded gray area represents three standard deviations from the DMSO control. ent-VP1-001 does not shift the thermal stability of the ACD, whereas VP1-001 does. (B) Normalized binding (n = 3) to full-length cryAB (R120G) measured by MST. The shaded gray region represents three standard deviations from the DMSO control. Calculated KD values: VP1-001 = 56 ± 7.0 μM; ent-VP1-001 = 370 ± 30 μM. Thus, VP1-001 binds tighter to cryAB than ent-VP1-001 does, consistent with the increased number of predicted contacts in the docking studies. Note that the ent-VP1-001 curve does not fully saturate, so the calculated KD value is only approximate.
Next, we used an orthogonal method, MST, to verify this result using cryAB oligomers. MST relies on tracking the movement of a fluorescently tagged protein through a thermal gradient.39,40 We engineered a unique cysteine residue (E87C) into cryAB ACD and selectively labeled it with a cysteine-reactive Alexa Fluor dye to generate AF488-cryAB(E87C) ACD. Because cryAB protomers undergo subunit exchange, we mixed AF488-cryAB(E87C) ACD into full-length oligomers to reconstitute a fluorescently labeled protein sample for MST. Binding affinity was calculated from a fit of the dose-responsive MST signal change using the law of mass action.41 Consistent with docking and DSF studies, we found that VP1-001 bound cryAB 7-fold more strongly than ent-VP1-001, with VP1-001 KD = 56 ± 7 μM versus ent-VP1-001 KD = 370 ± 30 μM (Fig. 3B, n = 3). Importantly, the ent-VP1-001 binding curve did not saturate at 250 μM, so the KD value for that compound is only an estimate.
VP1-001, But Not ent-VP1-001, Partially Restores Transparency in Cataract Mouse Models
The cryAB(R120G) mice harbor a mutation that causes lens opacities to develop as early as 8 weeks at a frequency 3-fold higher than that in WT mice.14 We previously found that topical application of VP1-001 partially reverses cataract formation in these mice even when they are more than 1 year of age.17 Based on biophysical studies and the hypothesis that binding to cryAB is important, we predicted that only VP1-001 would be active in this animal model. One eye of each animal was treated topically with ent-VP1-001 or VP1-001 (1 mM in 8% cyclodextrin), and the other eye was treated with vehicle alone. Treatments were given three times per week for 2 weeks. Lens transparency was measured by slit lamp biomicroscopy and graded using a previously published cataract scoring system (see Methods). Mouse cohorts ranged in age from 187 to 362 days and were age matched between treatment groups (Fig. 4). In 9 of 13 cryAB(R120G) heterozygous mice that received ent-VP1-001 treatment, there was either no change or an increase in cataract lens grade relative to vehicle controls. The remaining four mice exhibited improvements not exceeding one grade (posttreatment lens opacity grade, 2.6 ± 0.50, P = 0.22; Fig. 4A). Representative slit lamp images are shown comparing ent-VP1-001 to the vehicle-treated, contralateral eye (Fig. 4). By contrast, and in agreement with previous work, the lenses of animals treated with VP1-001 were significantly improved (lens opacity grade, 1.2 ± 0.43, P < 0.0001, Fig. 4). Strikingly, the magnitude of the improvement was at least two grades in 9 of 13 mice and at least one grade in the other four mice. The VP1-001 lens closely resembled a normal, 196-day-old, WT untreated lens (Fig. 4).
Age-associated cataracts are far more prevalent in humans than hereditary forms. Therefore, we compared the efficacy of VP1-001 and ent-VP1-001 in aged WT C57BL/6J mice that had spontaneously occurring, age-related cataracts. Using a similar dosing scheme as described above, treatment of WT mice with ent-VP1-001 did not significantly affect cataract grade (posttreatment lens opacity grade, 2.6 ± 1.1; n = 4; P = 0.61), whereas VP1-001 did (lens opacity grade, 1.0 ± 0.44; n = 10; P = 0.0013; Fig. 5A). VP1-001 partially restored transparency in both hereditary and age-associated murine models.
Figure 5.
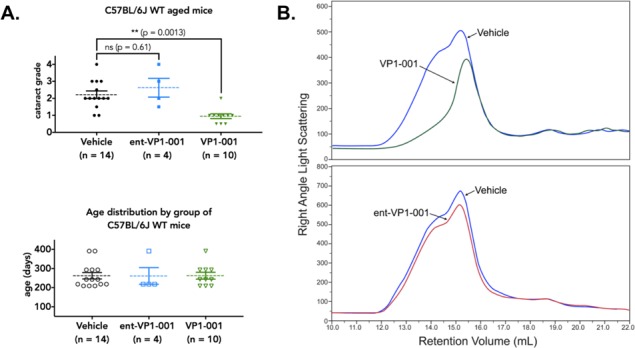
Treatment with VP1-001 significantly reduces cataract grade and refractive measures in aged WT C57BL/6J mice, but ent-VP1-001 is inactive. (A, top) Cataract grading for aged WT C57BL/6J mice treated with either compound. Vehicle group represents contralateral eyes from both treatment groups. (A, bottom) Age distribution of WT C57BL/6J mice separated by treatment condition. Each point represents an individual mouse lens. The mean is shown by a dotted line, and the error bars represent the standard errors from the means. (B) Comparison of GPC RALS of water-soluble fractions from homogenized lens tissues of VP1-001 or ent-VP1-001–treated and contralateral vehicle-treated eyes.
VP1-001, But Not ent-VP1-001, Increases the Proportion of Water-Soluble Protein in cryAB(R120G) Mouse Lenses
To determine the effects of these compounds on crystallin aggregation and water solubility, we analyzed the RI and average molecular weight of cryAB in mouse lenses treated with ent-VP1-001 or VP1-001. RI measurements were conducted on water-soluble lens proteins and generated the expected peaks for α-, β-, and γ-crystallins.8 Here, as well as in previous reports,17 VP1-001 appears to retain protein in the soluble fraction (67% increase in protein solubility, n = 7). Moreover, the RI measurements showed larger peak areas for α-, β-, and γ-crystallins in the VP1-001–treated samples (Fig. 6A). VPI-001 increased the relative amount of total α-crystallin and β-crystallins with less of an effect on γ-crystallin. This result suggests that VP1-001 increases the water-soluble proportion of not only cryAB but also other lens crystallins, perhaps by restoring chaperone function to cryAB in vivo. These results are consistent with the reported association of α-crystallin with β-crystallin and γ-crystallin.5 In parallel sets of experiments, we found that ent-VP1-001 did not improve the protein water-soluble content compared with vehicle (Fig. 6C).
Figure 6.

Treatment with VP1-001 significantly increases protein solubility in lenses of cryAB(R120G) mutant mice, but ent-VP1-001 is inactive. Homogenized tissue from cryAB(R120G) heterozygous mice treated with vehicle, VP1-001 (A), or ent-VP1-001 (C) were separated by GPC and measured by RI to reveal peaks for α--, β-, and γ-crystallins. Alternatively, samples were measured for RALS to study the molecular mass of soluble α-crystallin (B, D). Treatment with VP1-001 (A), but not ent-VP1-001 (C), increased the amount of soluble lens protein compared with vehicle. In all chromatography traces, the treatments were compared with vehicles taken from the contralateral eye of the same mouse.
Among the lens crystallins, α-crystallins are the major source of light scattering.8 Therefore, we used RALS measurements to estimate the molecular weight of the α-crystallin heterooligomer. Treatment with ent-VP1-001 did not change the molecular weight distribution of α-crystallins compared with that of vehicle-treated contralateral eyes, whereas VP1-001 treatment showed a robust response (Table). The average molecular weight of the oligomer decreased by 50% with VP1-001 treatment (n = 5), whereas treatment with ent-VP1-001 increased the average molecular weight by 8% (n = 5). Furthermore, the RALS peak shape was not altered by ent-VP1-001 treatment (Fig. 6D), whereas VP1-001 shifted the peak to the right (Fig. 6B). The decreased molecular weight of α-crystallin in lenses treated with VP1-001 suggests that VP1-001 affects the self-association of αB-crystallin such that the protein is less aggregated. These findings suggest that VP1-001 treatment may be beneficial to the protective functions of α-crystallins.
Table.
Differences in Molecular Weight and Water-Soluble α-Crystallin Concentration From cryAB(R120G) Heterozygous Mouse Lenses
|
Mouse |
Lens Treatment |
Avg. Molecular Weight |
Δ Protein Concentration |
| 864-3 | Vehicle | 1270 | |
| ent-VP1-001 | 1238 | −0.14 | |
| 983-4 | Vehicle | 1036 | |
| VP1-001 | 561 | 1.2 |
The water-soluble supernatants from homogenized lenses obtained from aged WT (C57BL/6J) mice were also tested by GPC. There was a large difference in the RALS peak shape of the VP1-001 and ent-VP1-001 groups (Fig. 5B). The vehicle-treated samples had broad peaks consistent with aggregated material, whereas treatment with VP1-001 eliminated a large portion of the higher-molecular-weight α-crystallin oligomer peak. The broad peak also shifted the retention volume by 3 mL with VP1-001 treatment (Fig. 5B). In contrast, ent-VP1-001 had no effect.
Effect of VP1-001 and ent-VP1-001 Treatment on Lens Epithelial Cell-Cell Contacts and Lens Fiber Cell Morphologies in cryAB(R120G) Mice
EM was performed 3 days after treatment ended to assess epithelial and lens fiber architecture in treated cryAB(R120G) mice. We investigated the effect of VP1-001 and ent-VP1-001 on newly formed lens epithelial and fiber cells during the treatment period. Lens epithelial sections display the cells just adjacent to the lens capsule in the equatorial region (Fig. 7). Fiber cell sections show the cross-sectional view of cortical fiber cells that have most recently differentiated in these adult mice. Both eyes (vehicle and compound) were imaged from cryAB(R120G) heterozygous mice treated with VP1-001 (10 images/eye; n = 8 mice) or ent-VP1-001 (7–13 images /eye; n = 6 mice). The average numbers of epithelial or fiber cell images with morphologic damage were counted, and differences between treated and untreated (vehicle) eyes were assessed by t test.
Figure 7.

VP1-001, but not ent-VP1-001, improves lens morphology in cryAB(R120G)-mutant mice. Electron micrographs of lens epithelium and lens fiber cell sections from four mice are grouped horizontally (for example, images A1, B1, C1, and D1 are from one mouse). Scale bars: 2 μm, white. Red arrows indicate vacuoles, cyan arrows indicate an abnormal nuclear shape, yellow arrows indicate irregular cell size, and white arrows indicate abnormal cell protrusions. (Left) Lens epithelial sections from treated and untreated eyes of cryAB(R120G) heterozygous mice. For each treatment condition, two replicate mice are displayed. (Right) Lens fiber sections from the same four mice. The images are representative of a majority of the sections from mice. In contrast to VP1-001, the enantiomer did not reverse morphologic changes in cryAB(R120G) heterozygous mice. (Bottom panel) Low-magnification image of a lens treated with VP1-001. The equatorial region in which the EM images were taken is enclosed by a box. Scale bar: 0.5 mm, black.
The vehicle-treated lenses displayed disorganization (Fig. 7), similar to that of sections previously imaged by immunofluorescence microscopy.14 Large intercellular gaps in lens epithelium were observed (red arrows). Disruption of contacts between the lens capsule and epithelium were also observed in vehicle-treated lenses (left red arrow in Fig. 7A2). In the equatorial fiber cells, there was normal fiber cell elongation, but cell packing was not uniform, and membranes appear blebbed and irregular. The morphologies of lenses treated with ent-VP1-001 were similar to those treated with vehicle with disordered epithelial and fiber cell layers (Fig. 7; images represent a majority, 75%, of sections). Conversely, VP1-001 had a striking effect on epithelial cell-cell contacts and fiber cell packing, with the hexagonal architecture resembling healthy tissue (Fig. 7, top; images represent a majority, 88%, of sections). We measured a statistically significant difference in morphologic damage in lens sections treated with VP1-001 versus vehicle (or enantiomer) (t test, P < 0.001).
Effect of VP1-001 and ent-VP1-001 on Cell-Cell Contacts in Aged WT Mice
We also analyzed electron micrographs of lens sections from aged WT mice to determine if they showed abnormal lens morphologies that improved after treatment with VP1-001 (32 images/eye; n = 4 mice) or ent-VP1-001 (15 images/eye; n = 2 mice). The lens epithelium of vehicle-treated lenses from aged WT mice exhibited disruption of the cell-cell interface near the capsule (Fig. 8A) similar to cryAB(R120G) heterozygous mice (Fig. 7, left). These abnormalities were eliminated in lenses treated with VP1-001, but not ent-VP1-001. In these experiments, we noted that fiber cell morphology in aged mice was less disorganized than in the mutant model; nevertheless, we observed nonuniform cell sizes, irregular packing, and limited membrane blebbing. The fiber cells of aged mouse lenses showed slight improvement with ent-VP1-001 treatment, with the appearance of the expected hexagonal shape in some cells. The improvement in morphology was more pronounced with VP1-001 treatment (Fig. 8B, top right).
Figure 8.

VP1-001, but not ent-VP1-001, improves lens morphology in aged WT C57BL/6J mice. Electron micrographs of lens epithelium and lens fiber cell sections from three mice. Scale bars: 2 μm, white. Mouse 3 was imaged at a slightly lower magnification. (A) Lens epithelial sections from contralateral treated and untreated eyes of two WT C57BL/6J mice. Red arrows indicate the gaps between adjacent cells. (B) Lens fiber sections from two mice. Yellow arrows indicate irregular cell size. White arrow indicates a vacuole. The appearance of normal cell-cell associations in the lens epithelium of VP1-001–treated mice suggests that VP1-001 improves morphologic changes associated with aging of the lens epithelium.
To better understand the prominent gaps at the capsular interface in the aged mice, we captured higher magnification images of cell-cell contact surfaces in the epithelium (Fig. 9). In the vehicle-treated lenses, gaps were interspersed among normal cell-cell boundaries, suggesting local areas of weakened interactions. Notably, these gaps were absent from VP1-001–treated lenses. To our knowledge, these are the first observations of large gaps at the cell-cell interfaces of epithelial cells in cataract models. Previous immunofluorescence studies of cataract lenses in 12-week-old mice did not report these gaps.14 However, we attribute this difference to the more advanced age of the mice in the current study (aged 31–35 weeks).
Figure 9.

VP1-001 protects cell-cell contacts in the lens epithelia of aged WT C57BL/6J mice. Electron micrographs of lens epithelium sections from contralateral treated and untreated eyes of WT mice. White arrows indicate typical healthy cell-cell contacts, whereas red arrows indicate areas of weak adhesion. Scale bars: 0.5 μm, black. (Left panels) Lens epithelial cell sections from two mice treated with vehicle only. (Right panels) Lens epithelial cell sections from the contralateral eyes of the same mice treated with 1 mM VP1-001.
Discussion
Stable interactions between small heat shock proteins and their substrates do not necessarily prevent protein aggregation.42 However, some small heat shock proteins do hinder aggregation through transient interactions.42 Thus, binding and aggregation are two separately controlled processes. In a cellular complex, both transient and stable interactions are likely to contribute to these processes. CryAB activity limits aggregation by maintaining protein solubility in the lens, preventing cataract formation. In this work, we probed how a small-molecule ligand for cryAB, VP1-001, might maintain its function. A classical and rigorous approach is to characterize the activity of the enantiomer because interactions with chiral proteins will only occur with one enantiomer. For example, direct interaction of the anticancer compound, YK-4-279, with its target was confirmed using its enantiomer.43 This approach is powerful, because the enantiomers of active compounds have identical physical properties. Thus, the enantiomer can be used to rule out alternative models, such as chemical chaperone activity or interactions with membranes. By comparing the activities of VP1-001 to ent-VP1-001, we aimed to address several important mechanistic questions.
We found that VP1-001 was predicted to bind into a deep groove in the cryAB ACD dimer. In this bound state, key hydrogen bond contacts were predicted at both extremes of the groove at the dimer interface (see Fig. 2). However, ent-VP1-001 did not make these same contacts and was predicted to have weaker affinity. Indeed, these predictions were experimentally confirmed with two biophysical methods, DSF and MST. VP1-001 bound better to cryAB than did ent-VP1-001. Thus, we were able to use these compounds to rigorously probe whether direct binding to cryAB was required to improve lens transparency and protein solubility in mouse models. More explicitly, if we found that ent-VP1-001 was bioactive in vivo, then we would conclude that alternative, nonstereospecific mechanisms, such as membrane interactions, may contribute to the activity of VP1-001. In this way, the results would have a profound impact on the future development of anticataract compounds. If binding to cryAB is important (e.g., VP1-001 is active, but ent-VP1-001 is inactive), then further optimization would likely require increasing affinity for cryAB. In contrast, if alternative mechanisms existed (e.g., both compounds were active), then one would envision ways of improving activity by promoting lipophilicity and membrane binding. As discussed in greater detail below, we found that VP1-001, but not ent-VP1-001, was active in a range of biological cataract assays. Thus, one major conclusion of these studies is that direct binding to cryAB is required for the observed biological activity of VP1-001.
Previous studies demonstrated that expression of WT α-crystallin protects the lens epithelium from a variety of stress conditions and mutations.44 Our in vivo studies in mutant mice with cataracts revealed that VP1-001, but not ent-VP1-001, partially corrected lens transparency, as assessed by the cataract grading scale and decreased average molecular weight of cryAB oligomers. We also tested VP1-001 and ent-VP1-001 in WT aged mice, which is critical because cataracts in old mice are expected to arise from a more heterogeneous set of circumstances. Indeed, mass spectrometry studies have shown that aged lens proteins harbor many posttranslational modifications that accumulate over time, including oxidation,45 glycation,46 deamidation,47 racemization, and isomerization.48 The ability of VP1-001 to lower the cataract grade and the appearance of nearly normal lens morphology in the newly synthesized epithelial and fiber cells in the aged mice are particularly interesting. Despite a heterogeneous collection of molecular defects, VP1-001 was able to compensate for damage to cryAB in the lens epithelial and fiber cells of the equatorial region of the lens and improve lens epithelial cell morphology (see Figs. 7–9) and fiber cell packing (see Figs. 7, 8). Adult mouse lenses add approximately 100 to 200 lens epithelial cells per day. No cell death is observed in the adult mouse lens epithelium, indicating that 100 to 200 lens epithelial cells differentiate per day into fiber cells.49 Our data suggest that the newly differentiated lens fiber cells, which emerged during the course of VP1-001 treatment, adopted a normal structure and are overlaid on the deformed cells. The appearance of normal cell-cell contacts in the lens epithelium of VP1-001–treated mice suggests that binding of VP1-001 to cryAB improves the morphology of the lens epithelium.
Our EM studies suggested that normal cell-cell interactions in the lens epithelium may contribute to the effects of VP1-001. In addition, we observed concurrent enhancement in the soluble fraction of β- and γ-crystallin (Figs. 5A, 6B), which does not directly bind to VP1-001.17 Taken together, these observations suggest that binding of VP1-001 does not disrupt cryAB normal function. Indeed, we speculate that compound-bound cryAB may still function as a chaperone and that this activity may contribute to the potency of VP1-001 in these models. Future studies using in vitro chaperone assays would corroborate this speculation.
Supplementary Material
Acknowledgments
The authors thank John Irwin, PhD (Blue Dolphin) for in silico docking. We also thank Wandy Beatty, PhD, at Washington University Department of Molecular Microbiology for electron microscopy studies.
Supported by the National Institutes of Health (Grants EY05681-32, EY02687, and 1R43EY026474-01), The Taylor Family Institute for Innovative Psychiatric Research, and Research to Prevent Blindness, Inc.
Disclosure: K.S. Molnar, ViewPoint Therapeutics (E); B.M. Dunyak, ViewPoint Therapeutics (E); B. Su, ViewPoint Therapeutics (E); Y. Izrayelit, None; B. McGlasson-Naumann, None; P.D. Hamilton, None; M. Qian, None; D.F. Covey, None; J.E. Gestwicki, ViewPoint Therapeutics (S), P; L.N. Makley, ViewPoint Therapeutics (E), P; U.P. Andley, ViewPoint Therapeutics (F, S)
References
- 1.Bloemendal H, de Jong W, Jaenicke R, Lubsen NH, Slingsby C, Tardieu A. Ageing and vision: structure, stability and function of lens crystallins. Prog Biophys Mol Biol. 2004;86:407–485. doi: 10.1016/j.pbiomolbio.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopal P, Tse E, Borst AJ, et al. A conserved histidine modulates HSPB5 structure to trigger chaperone activity in response to stress-related acidosis. Elife. 2015;4:e07304. doi: 10.7554/eLife.07304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci U S A. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aquilina JA, Benesch JL, Bateman OA, Slingsby C, Robinson CV. Polydispersity of a mammalian chaperone: mass spectrometry reveals the population of oligomers in alphaB-crystallin. Proc Natl Acad Sci U S A. 2003;100:10611–10616. doi: 10.1073/pnas.1932958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andley UP, Malone JP, Townsend RR. In vivo substrates of the lens molecular chaperones alphaA-crystallin and alphaB-crystallin. PLoS One. 2014;9:e95507. doi: 10.1371/journal.pone.0095507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derham BK, Harding JJ. Alpha-crystallin as a molecular chaperone. Prog Retin Eye Res. 1999;18:463–509. doi: 10.1016/s1350-9462(98)00030-5. [DOI] [PubMed] [Google Scholar]
- 7.Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76:145–153. doi: 10.1016/s0014-4835(02)00278-6. [DOI] [PubMed] [Google Scholar]
- 8.Andley UP, Hamilton PD, Ravi N. Mechanism of insolubilization by a single-point mutation in alphaA-crystallin linked with hereditary human cataracts. Biochemistry. 2008;47:9697–9706. doi: 10.1021/bi800594t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng C, Xia CH, Huang Q, Ding L, Horwitz J, Gong X. Altered chaperone-like activity of alpha-crystallins promotes cataractogenesis. J Biol Chem. 2010;285:41187–41193. doi: 10.1074/jbc.M110.154534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perng MD, Cairns L, van den IP, Prescott A, Hutcheson AM, Quinlan RA. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. 1999;112(pt 13):2099–2112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- 11.Bova MP, Yaron O, Huang Q, et al. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A. 1999;96:6137–6142. doi: 10.1073/pnas.96.11.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 13.Sacconi S, Feasson L, Antoine JC, et al. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Disord. 2012;22:66–72. doi: 10.1016/j.nmd.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Andley UP, Hamilton PD, Ravi N, Weihl CC. A knock-in mouse model for the R120G mutation of alphaB-crystallin recapitulates human hereditary myopathy and cataracts. PLoS One. 2011;6:e17671. doi: 10.1371/journal.pone.0017671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Truscott RJ, Friedrich MG. The etiology of human age-related cataract. Proteins don't last forever. Biochim Biophys Acta. 2016;1860:192–198. doi: 10.1016/j.bbagen.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Truscott RJ, Augusteyn RC. Changes in human lens proteins during nuclear cataract formation. Exp Eye Res. 1977;24:159–170. doi: 10.1016/0014-4835(77)90256-1. [DOI] [PubMed] [Google Scholar]
- 17.Makley LN, McMenimen KA, DeVree BT, et al. Pharmacological chaperone for alpha-crystallin partially restores transparency in cataract models. Science. 2015;350:674–677. doi: 10.1126/science.aac9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Chen XJ, Yang Z, et al. Synthesis, evaluation, and structure-activity relationship study of lanosterol derivatives to reverse mutant-crystallin-induced protein aggregation. J Med Chem. 2018;61:8693–8706. doi: 10.1021/acs.jmedchem.8b00705. [DOI] [PubMed] [Google Scholar]
- 19.Chemerovski-Glikman M, Mimouni M, Dagan Y, et al. Rosmarinic acid restores complete transparency of sonicated human cataract ex vivo and delays cataract formation in vivo. Sci Rep. 2018;8:9341. doi: 10.1038/s41598-018-27516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Convertino M, Das J, Dokholyan NV. Pharmacological chaperones: design and development of new therapeutic strategies for the treatment of conformational diseases. ACS Chem Biol. 2016;11:1471–1489. doi: 10.1021/acschembio.6b00195. [DOI] [PubMed] [Google Scholar]
- 21.Bagneris C, Bateman OA, Naylor CE, et al. Crystal structures of alpha-crystallin domain dimers of alphaB-crystallin and Hsp20. J Mol Biol. 2009;392:1242–1252. doi: 10.1016/j.jmb.2009.07.069. [DOI] [PubMed] [Google Scholar]
- 22.Irwin JJ, Shoichet BK, Mysinger MM, et al. Automated docking screens: a feasibility study. J Med Chem. 2009;52:5712–5720. doi: 10.1021/jm9006966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp KA, Friedman RA, Misra V, Hecht J, Honig B. Salt effects on polyelectrolyte-ligand binding: comparison of Poisson-Boltzmann, and limiting law/counterion binding models. Biopolymers. 1995;36:245–262. doi: 10.1002/bip.360360211. [DOI] [PubMed] [Google Scholar]
- 24.Lorber DM, Shoichet BK. Flexible ligand docking using conformational ensembles. Protein Sci. 1998;7:938–950. doi: 10.1002/pro.5560070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem. 2005;5:739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bissantz C, Kuhn B, Stahl M. A medicinal chemist's guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang CC, Couch GS, Pettersen EF, Ferrin TE. Chimera: an extensible molecular modeling application constructed using standard components. Pac Symp Biocomput. 1996;1:724. [Google Scholar]
- 28.Westover EJ, Covey DF. Synthesis of ent-25-hydroxycholesterol. Steroids. 2006;71:484–488. doi: 10.1016/j.steroids.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Jehle S, van Rossum B, Stout JR, et al. alphaB-crystallin: a hybrid solid-state/solution-state NMR investigation reveals structural aspects of the heterogeneous oligomer. J Mol Biol. 2009;385:1481–1497. doi: 10.1016/j.jmb.2008.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kapust RB, Tozser J, Fox JD, et al. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 31.Alizadeh A, Clark J, Seeberger T, Hess J, Blankenship T, FitzGerald PG. Characterization of a mutation in the lens-specific CP49 in the 129 strain of mouse. Invest Ophthalmol Vis Sci. 2004;45:884–891. doi: 10.1167/iovs.03-0677. [DOI] [PubMed] [Google Scholar]
- 32.Jehle S, Rajagopal P, Bardiaux B, et al. Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nat Struct Mol Biol. 2010;17:1037–1042. doi: 10.1038/nsmb.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hochberg GK, Ecroyd H, Liu C, et al. The structured core domain of alphaB-crystallin can prevent amyloid fibrillation and associated toxicity. Proc Natl Acad Sci U S A. 2014;111:E1562–E1570. doi: 10.1073/pnas.1322673111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laganowsky A, Benesch JL, Landau M, et al. Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 2010;19:1031–1043. doi: 10.1002/pro.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 36.Cummings MD, Farnum MA, Nelen MI. Universal screening methods and applications of ThermoFluor. J Biomol Screen. 2006;11:854–863. doi: 10.1177/1087057106292746. [DOI] [PubMed] [Google Scholar]
- 37.Shih MH, Lim HS. Screening methods for identifying pharmacological chaperones. Mol BioSyst. 2017;13:638–647. doi: 10.1039/c6mb00866f. [DOI] [PubMed] [Google Scholar]
- 38.Burns JN, Orwig SD, Harris JL, Watkins JD, Vollrath D, Lieberman RL. Rescue of glaucoma-causing mutant myocilin thermal stability by chemical chaperones. ACS Chem Biol. 2010;5:477–487. doi: 10.1021/cb900282e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rainard JM, Pandarakalam GC, McElroy SP. Using microscale thermophoresis to characterize hits from high-throughput screening: a European lead factory perspective. SLAS Discov. 2018;23:225–241. doi: 10.1177/2472555217744728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seidel SA, Dijkman PM, Lea WA, et al. Microscale thermophoresis quantifies biomolecular interactions under previously challenging conditions. Methods. 2013;59:301–315. doi: 10.1016/j.ymeth.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waage P, Gulberg CM. Studies concerning affinity. J Chem Educ. 1986;63 [Google Scholar]
- 42.Mymrikov EV, Daake M, Richter B, Haslbeck M, Buchner J. The chaperone activity and substrate spectrum of human small heat shock proteins. J Biol Chem. 2017;292:672–684. doi: 10.1074/jbc.M116.760413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barber-Rotenberg JS, Selvanathan SP, Kong Y, et al. Single enantiomer of YK-4-279 demonstrates specificity in targeting the oncogene EWS-FLI1. Oncotarget. 2012;3:172–182. doi: 10.18632/oncotarget.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andley UP. The lens epithelium: focus on the expression and function of the alpha-crystallin chaperones. Int J Biochem Cell Biol. 2008;40:317–323. doi: 10.1016/j.biocel.2007.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Truscott RJ. Age-related nuclear cataract-oxidation is the key. Exp Eye Res. 2005;80:709–725. doi: 10.1016/j.exer.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 46.van Boekel MA, Hoenders HJ. Glycation of crystallins in lenses from aging and diabetic individuals. FEBS Lett. 1992;314:1–4. doi: 10.1016/0014-5793(92)81446-s. [DOI] [PubMed] [Google Scholar]
- 47.Lampi KJ, Wilmarth PA, Murray MR, David LL. Lens beta-crystallins: the role of deamidation and related modifications in aging and cataract. Prog Biophys Mol Biol. 2014;115:21–31. doi: 10.1016/j.pbiomolbio.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakaue H, Takata T, Fujii N, Sasaki H, Fujii N, Alpha B- and betaA3-crystallins containing d-aspartic acids exist in a monomeric state. Biochim Biophys Acta. 2015;1854:1–9. doi: 10.1016/j.bbapap.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 49.Sikic H, Shi Y, Lubura S, Bassnett S. A full lifespan model of vertebrate lens growth. R Soc Open Sci. 2017;4:160695. doi: 10.1098/rsos.160695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.