

 Telomerase participation causes microRNA circulation in a testing system, which reliably enhances the difference between cancer cells and normal cells.
Telomerase participation causes microRNA circulation in a testing system, which reliably enhances the difference between cancer cells and normal cells.
Abstract
Intracellular microRNA (miRNA) detection has attracted increasing attention, resulting in significant achievements. However, the development of an available tool that possesses a satisfactory signal-to-background ratio and high sensitivity for miRNA detection remains challenging. Herein, a class of telomerase-catalyzed FRET (fluorescence resonance energy transfer) ratioflares has been developed for the accurate sensing of low-abundance cancer-related miRNA both in a fluorescence assay and living cell imaging with signal amplification capacity. In this work, endogenous telomerase is led in a miRNA test system with signal amplification for the first time, wherein telomerase extends hexamer telomeric repeats (TTAGGG) using the 3′ end of the capture probe as the primer. The synergetic work of telomerase and the catalyst strand (CS) makes the target miRNA circulate in the system, resulting in high sensitivity and an enhanced FRET signal an improved detection limit of 2.27 × 10–15 M. Meanwhile, telomerase-catalyzed FRET ratioflares allow the difference between cancer cells and normal cells to be increased reliably. Furthermore, low false positive signals resulting from chemical interference and minimized system fluctuations are achieved through ratiometric measurements.
Introduction
In gene regulation, small interfering RNA (siRNA), microRNA (miRNA), and small hairpin RNA (shRNA) can modulate gene expression in different ways.1–3 Among these, microRNAs are sequence-specific single-strand RNA regulators (approximately 19–24 nucleotides) that function in the post-transcriptional regulation of gene expression.4–6 Increasing research has demonstrated that the occurrence and development of different cancers is closely associated with the dysregulated expression of specific miRNA.7–9 To better understand the functions and abundance differentiation of intracellular miRNA, fluorescence imaging of miRNAs in living cells has become increasingly important.10–13 In the last decade, miRNA has been detected in some reporter gene systems, such as real-time polymerase chain reaction (RT-PCR) technology, northern blotting, fluorescence in situ hybridization (FISH) technology, and DNA microarrays.14–17 However, these conventional methods are not suitable for monitoring miRNA in living cells, and the detection limits of around 0.7–5 pM are relatively low.18
Recently, miRNA detection has seen significant progress. An RNA aptamer sensor with co-expression of green fluorescent protein (GFP) was developed to reduce false positive signals when monitoring microRNA in living cells, but still suffers from limitations such as a sophisticated plasmid design process and insufficient sensitivity.19–21 Furthermore, rolling circle amplification (RCA) has been introduced to improve the detection sensitivity. Despite the high signal amplification efficiency, ionic strength, and hybrid temperature having a significant effect, the high background fluorescence still restricts the application of RCA.22,23 In particular, obtaining a satisfactory signal-to-background ratio and accurately sensing fluctuations in miRNA quantity within low concentration ranges in living cells remain challenging.
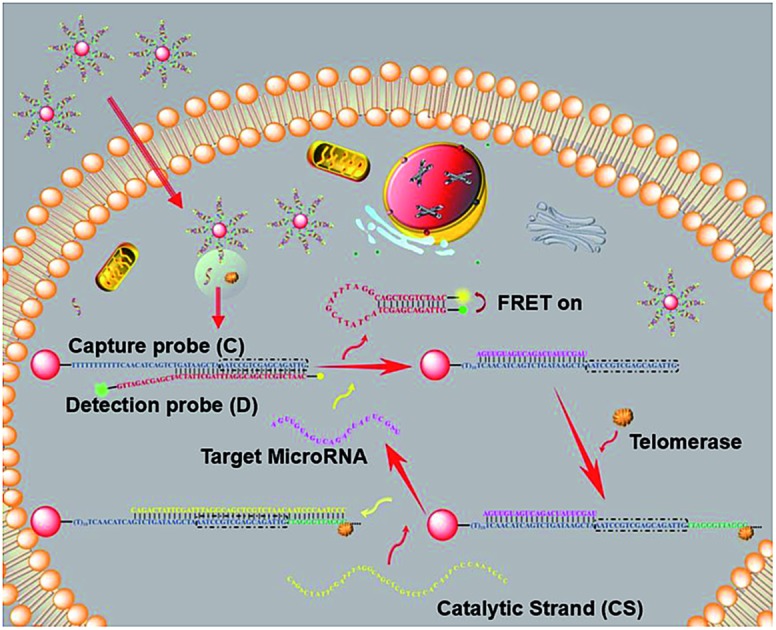
Herein, to address these issues, AuNP-based fluorescent-modified DNA ratioflares were designed to achieve intracellular miRNA recognition. In this study, a DNA detection probe was designed as a hairpin structure modified by FAM and TAMRA, leading to a fluorescence resonance energy transfer (FRET) effect. These elements led to AuNP-DNA ratioflares that meet the demands for minimizing the effect of system fluctuations through ratiometric measurement, and also reduced the false positive signal resulting from chemical interference compared with conventional single-dye nanoflares.24 Subsequently, to maximize the difference in fluorescence signals in cancer cells relative to normal cells and obtain a low detection limit, we led endogenous telomerase in a microRNA test system for the first time. Human telomerase is a ribonucleoprotein that can add repetitive nucleotide sequences (TTAGGG) onto the 3′ ends of telomeres through its intrinsic RNA template and reverse transcriptase to maintain telomere length.25–27 In normal cells, after each replication cycle, telomeres become increasingly short, leading to cell senescence and death.28 In contrast, the length of telomeres is maintained in most types of cancer cell (over 85%) due to a high expression level of telomerase, which makes cancer cells divide indefinitely.29 Typical telomerase concentrations in cancer cells (such as HeLa and MCF-7) are 10–12–10–11 IU level.30 Accordingly, telomerase will extend hexamer telomeric repeats (TTAGGG) using the 3′ end of the DNA capture probe as primer and endogenous deoxyribonucleoside triphosphate (dNTP) as the raw material. After catalytic strands are added, telomerase makes target microRNA circulate in the system, resulting in an enhanced FRET signal (Scheme 1) with an improved detection limit.
Scheme 1. (A) Working principle of telomerase-catalyzed FRET ratioflares with signal amplification based on specific sequence responsive. (B) Design of telomerase-catalyzed FRET ratioflares. (C) Transmission electron microscopy (TEM) of telomerase-catalyzed FRET ratioflares.

Results and discussion
Working principle of telomerase-catalyzed FRET ratioflares
As shown in Scheme 1, newly designed AuNP nanostructures, termed telomerase-catalyzed FRET ratioflares, were constructed using a DNA frame, and their structures showed ratio signals. Briefly, two strands of DNA chains were designed and the AuNP functionalized with a 5′-thiolated DNA (capture probe, C), consisting of recognition sequences and telomerase primer at the 3′ end, was hybridized to the detection probe (D). The detection probes were designed as hairpin structures and fluorescently labelled at their 5′ and 3′ termini with acceptors (TAMRA) and donors (FAM), respectively. In the absence of targets, D was captured by binding with the complementary sequences in C, separating the fluorescent donor and acceptor, and inducing low FRET efficiency. In this state, only the fluorescence of FAM could be detected. In contrast, in the presence of target miRNAs, D was gradually displaced from C by the target miRNA, forming hairpin structures that brought the FAM and TAMRA into close proximity and resulted in high FRET efficiency, such that the fluorescence of TAMRA could be detected. Furthermore, the telomerase used endogenous deoxyribonucleoside triphosphate (dNTP) as the raw material, which led to the extension of hexamer telomeric repeats (TTAGGG) following the telomerase primer of C. After the catalyst strand (CS) was added, CS hybridized with C, which displaced and released targets miRNA and made them circulate in the test system. Therefore, the amplified fluorescence emission ratio of TAMRA to FAM (FA/FD) was used as a signal for quantitation of the target miRNA.
Construction and characterization of telomerase-catalyzed FRET ratioflares
To prepare the telomerase-catalyzed FRET ratioflares, 13 ± 2 nm AuNPs (Fig. S1†) were prepared and functionalized with thiol-terminated DNA capture probes (C) via gold–thiol bond formation. This was confirmed by the bathochromic shift of the gold absorbance band from 519 to 524 nm in the UV-vis spectrum (Fig. S2†). These nanoconjugates were then purified by cleaning and centrifuging repeatedly and allowed to hybridize with D. Quantification of the DNA surface loading by fluorescence showed that each AuNP contains ∼63 FRET pairings (Fig. S3†).31 The morphology of the telomerase-catalyzed FRET ratioflares was also investigated. As shown in Scheme 1C, transmission electron microscopy (TEM) observation indicated that the ratioflares were formed with average diameters of 17 ± 2 nm and possessed a globular structure. The above results illustrated that telomerase-catalyzed FRET ratioflares were successful prepared and able to execute miRNA detection in this study.
Fluorescence assay of target microRNA using telomerase-catalyzed FRET ratioflares
MicroRNA miR-21 was selected as the target template in our experiments. MiR-21, among the most important biomarkers, is considered an oncogene and is upregulated in various cancer cells.32 First, to test the target-sensing behavior of the telomerase-catalyzed FRET ratioflares, we examined the response of the probes to a synthetic DNA template (DNA-21), instead of the target miRNA (miR-21). Furthermore, to realize signal amplification, we tested the FRET signal in the absence of telomerase as contrast. The ratioflare concentration was fixed at 2 nM in the subsequent spectrum test at 37 °C. The results in Fig. 1A show excellent FRET signal changes at different target concentrations when the telomerase was present (final concentration, 0.5 IU per L). Furthermore, the telomerase activity was quantified using the standard curve obtained from enzyme-linked immunosorbent assay (ELISA) kit analysis (Fig. S5†). The ratioflares corresponded to a favorable linear relationship at low concentration, leading to an improved detection limit of 2.27 × 10–15 M (Fig. 1B). Meanwhile, Fig. S4† showed a fairly low FA/FD ratio signal for the system without telomerase. These results demonstrated that the telomerase-catalyzed FRET ratioflares were able to signal the low-abundance target (∼5-fold increase in FA/FD signal when concentration of DNA-21 was 10 nM) and gave a lower limit of detection when telomerase was added (∼104-fold increase compared to the system without telomerase).
Fig. 1. (A) Fluorescence spectra determination at various DNA template concentrations from 50 fM to 10 nM. (B) Linear relationship between FA/FD signal and log concentration (Log(C)) of the DNA template. (C) Fluorescence spectra determination with or without each component of the testing system when DNA template was presence or absent. Ratioflare concentration was fixed at 2 nM in each group. (D) Distinguishing middle-mismatched templates (mismatch-a and mismatch-b) and terminal-mismatched templates (mismatch-c and mismatch-d) from complementary target DNA-21. No template was added in the control group. Error bars show standard deviations of three replicates. λex = 492 nm, λem(FAM) = 520 nm, λem(TAMRA) = 580 nm.

Compared with conventional miRNA probes, telomerase-catalyzed FRET ratioflares were expected to possess an excellent capability for avoiding false positive signals. First, all or some components were added into the test system to remove their influences with a fixed ratioflare concentration of 2 nM. As shown in Fig. 1C, no FA/FD signal was observed in the groups without DNA-21 (500 pM), showing almost no background signal for the ratioflares themselves. Furthermore, a weaker FA/FD signal was observed in groups lacking one of the active components (telomerase, dNTPs, and CS) but with DNA-21 present. Notably, CS barely generated the FA/FD signal when telomerase was absent, but the signal was very close in proximity to the group with target only. Furthermore, compared with the synergetic effect of telomerase and CS, which makes target miRNA circulate in the system, the influence of the separate parts is negligible. We believe that, except for the last 12 bases in 5′ of CS, which are used to recognize the extending hexamer telomeric repeats following the telomerase primer of C, CS receives the same base sequence with detection probe D. Before D becomes a hairpin structure, it might be incompletely out of C or reintegrated with C, which greatly reduces the replacing ability of CS and results in a failure to turn on the fluorescence of TAMRA. Once the telomerase extends the terminal primer of C, the extra 12 bases provide a greater opportunity for CS to release the target miRNA. Furthermore, only telomerase with dNTPs and DNA-21 was added to the test system without the CS. There was no obvious signal enhancement, indicating that telomerase and CS played irreplaceable roles in the cyclic detection system and were both indispensable.
Within a microRNA family, the difference can only be only one or two bases, which might be another factor resulting in false positive signals. Therefore, we synthesized two strands of DNA templates with one-base middle-mismatches, mismatch-a and mismatch-b, bearing single mismatches differentiated from the sequence at the 7th and 15th base, respectively. To fully reflect the specificity of our ratioflares, two strands with a one-base terminal-mismatched template (mismatch-c) and two-base terminal-mismatched template (mismatch-d) were also tested. FRET ratioflares were added to DNA-21 (1 nM) and the mismatched templates (100 equiv.), with Fig. 1D showing the weak increase observed with those mismatched targets. However, compared with the FA/FD signal of the complementary target, these disturbances can be considered negligible. These results suggested that the telomerase-catalyzed FRET ratioflares were efficient at monitoring the presence of a specific target and could also be used to distinguish targets from single-base mismatches.
False positive signals might also be caused by several types of biological disruptor in vivo, such as sulfhydryl-containing substances, protein, and Dnase I. Therefore, interferents (500 equiv. relative to the target control) were added to the test system, as shown in Fig. 2A. Only the DNA-21 template caused a dramatic increase in the FA/FD signal, with a clear change during 6 h, while no signal was observed in other groups. Next, the biological stability of this system was verified by incubating the probes with a large excess (5 mM, 500 equiv.) of reductants (tris(2-carboxyethyl)phosphine, TCEP), oxidants (hydrogen peroxide, H2O2) and thiol nucleophiles (such as cysteine, mercaptoethanol, and glutathione). After incubation for 12 h, the system provided a nearly full fluorogenic response when 10 nM DNA-21 (final concentration) was added compared with the untreated group (Fig. 2B). This result indicated that our probes had outstanding specificity and avoid being destroyed by these bioactive compounds in vivo.
Fig. 2. (A) FRET fluorescence signal toward various analytes. 10 nM for DNA-21 template and 5 mM (500 equiv.) for all other analytes. Each bar represents relative responses at 0 h (black), 0.5 h (red), 1 h (blue), 2 h (pink), 3 h (green), 4 h (orange), and 6 h (purple) after adding the appropriate analytes. (B) Normalized fluorescence emission signal of telomerase-catalyzed FRET ratioflares (2 nM) after 12 h of incubation with different reactive additives, showing the stability of the probes with respect to the reactive additives. Error bars show standard deviations of three replicates. λex = 492 nm, λem(FAM) = 520 nm, λem(TAMRA) = 580 nm.

MicroRNA analysis in living cells using telomerase-catalyzed FRET ratioflares
After establishing the discerning capacity of target miRNA using our ratioflares through spectroscopy, we attempted to use our ratioflares to detect endogenous miRNA in living cells. Breast cancer cells (MCF-7) and human cervical carcinoma cells (HeLa) were selected for comparison with the mouse embryonic fibroblast cell (NIH-3T3). A lower miR-21 expression has been shown to occur in normal cells compared with cancer cells.33 Prior to microscopy, the cells were transfected with catalyst strand (1 μg) using Lipofectamine 3000 (1.8 μL) in Opti-MEM (250 μL). Initially, an MTT experiment was conducted, with the results showing that telomerase-catalyzed FRET ratioflares displayed low cytotoxicity at both low (0.04 nM, 95–99% cell viability) and high concentrations (10 nM, 82–86% cell viability) after incubation for 24 h (Fig. S6†). Next, to determine the ratioflare incubation time, we incubated HeLa cells with ratioflares for different periods ranging from 0 to 240 min, after which images of the cells were obtained by one-photon fluorescence confocal microscopy, with the results shown in Fig. S7.† After 120 min, the TAMRA fluorescence was brilliant, and the intensities did not increase with further increasing incubation times in HeLa cells.
To study the lysosomal escape of telomerase-catalyzed FRET ratioflares, we stained the HeLa and MCF-7 cells with LysoTracker Blue and monitored the cells using confocal fluorescence images. As shown in Fig. S8 and S9,† after incubation for 30 min with ratioflares, FAM and LysoTracker Blue co-localized in the two types of cells, indicating that the telomerase-catalyzed FRET ratioflares were located in the lysosomes. After incubation for 180 min, nearly all the ratioflares were localized in the cytoplasm, while only a very small fraction were colocalized with the lysosome tracker. We then quantified the Pearson correlation coefficient of ratioflares with lysosomes over incubation time in HeLa cells (Fig. S10†). The result was consistent with the observation in cells. Therefore, we believe that the telomerase-catalyzed FRET ratioflares can escape from the lysosomes into the cytoplasm for target microRNA detection.34,35
The cells were then imaged after incubation for 120 min in the following experiments. From the imaging of three types of cell (Fig. 3A), strong fluorescent staining of TAMRA was observed in the tumor cells (MCF-7 and HeLa), and much less TAMRA staining was observed in normal cells (NIH-3T3). The average fluorescence intensity is shown in Fig. 3B, with enhancements of about seven-fold (MCF-7) and five-fold (HeLa) in the FA/FD signal of cancer cells compared with normal cells. Furthermore, to verify that the observed differences were not caused by the probe uptake capacity between different cell lines, cell-derived lysates were tested using our ratioflares. After incubation for 1.5 h at 37 °C, compared with the FA/FD signal from NIH-3T3 cells, increases in the FA/FD signal of about 4.8- and 2.5-fold were observed from the lysates of MCF-7 and HeLa, respectively (Fig. 3C). Therefore, confocal imaging in living cells and cell lysates testing showed an identical tendency. Furthermore, detecting miRNA in cell-derived lysates of telomerase-catalyzed FRET ratioflares is distinctly advantageous, making our ratioflares more convenient than conventional applicable technologies, which require careful RNA extraction before detection.
Fig. 3. (A) One-photon confocal fluorescence imaging of miR-21 in MCF-7, HeLa, and NIH-3T3 cells after incubation with ratioflares for 120 min. λex = 488 nm, λem(FAM) = 500–540 nm, λem(TAMRA) = 560–600 nm. Scale bar = 20 μm. (B) Average fluorescence intensity ratio between FAM channel and TAMRA channel after incubation for 120 min with ratioflares in three types of living cell. (C) FA/FD signal from cell-derived lysates of MCF-7, HeLa, and NIH-3T3 cells. Only telomerase-catalyzed FRET ratioflares were added to the control group.

Telomerase and CS play irreplaceable roles in the cyclic detection system. Firstly, the effect of telomerase level was explored at different concentrations of telomerase inhibition reagent (epigallocatechin gallate, EGCG) added to HeLa cells. Before and after treatment, the telomerase activity was quantified by ELISA kit analysis (Fig. S5†). Originally, an MTT experiment was conducted, with the results showing that EGCG had low cytotoxicity at both low (30 μg mL–1, 96–99% cell viability) and high concentrations (360 μg mL–1, 78–69% cell viability) after incubation for 48 h (Fig. S11†). HeLa cells were then treated with different concentrations of EGCG for 48 h. As shown in Fig. S14,† 120 μg mL–1 of EGCG led to a reduction of nearly 70% (from 0.26 to 0.08) in the telomerase activity of HeLa cells, which was similar to the telomerase activity in MCF-7 and other less active cells. As shown in Fig. S12 and S13,† the telomerase activity had little influence on the cyclic efficiency (Fig. S14†) when the amount of EGCG added was low. This demonstrated that our ratioflares can maintain catalytic function and ignore small differences in the expression level of telomerase in different cancer cells. When the EGCG concentration was increased up to 260 μg mL–1, nearly no telomerase activity was observed by ELISA kit analysis, which showed a similar activity to normal cells.
Next, all cell types were treated with EGCG for comparison (Fig. 4A(d–f)). After treatment with 260 μg mL–1 of EGCG for 48 h, telomerase-catalyzed FRET ratioflares were added and the fluorescent intensity was obtained using confocal imaging. Another group of three types of cell were then incubated with ratioflares without CS (Fig. 4A(g–i)), using the same probe concentration and other test conditions as the control group (Fig. 4A(a–c)). Both outcomes retained the ability to distinguish cancer cells from normal cells, but showed lower signals and smaller differences (only two-fold increase for MCF-7 and one-fold increase for HeLa) with NIH-3T3 (Fig. 4A, S15, and S16†). These results suggested that the catalytic disassembly of ratioflares proceeded in living cells and the synergetic effect of telomerase and CS can enhance the differentiation between cancer cells and normal cells. In particular, ratioflares barely cause a background signal alone and can be used for high-sensitive imaging of low-abundance miRNA biomarkers.
Fig. 4. One-photon confocal fluorescence imaging of miR-21 in HeLa cells after incubation with telomerase-catalyzed FRET ratioflares for 120 min. (A) group (a–c) was a control group, group (d–f) was treated with EGCG to inhibit the telomerase activity, group (g–i) did not contain catalyst strand. (B) group (j–l) was a control group, group (m–o) was treated with anti-miR-21 sequence to inhibit the miR-21 concentration, and group (p–r) was treated with contrastive probes. (C) Average fluorescence intensity ratio between FAM channel and TAMRA channel after incubating with EGCG and without CS. (D) Average fluorescence intensity ratio between FAM channel and TAMRA channel after incubating with anti-miR-21 sequence and contrastive probes. λex = 488 nm, λem(FAM) = 500–540 nm, λem(TAMRA) = 560–600 nm. Scale bar = 20 μm.

Furthermore, to verify the specificity of the FRET signal for miR-21 in living cells, three types of cell were transfected in advance with anti-miR-21 sequence to reduce the miR-21 concentration. As shown in Fig. 4B(m–o), S15, and S16,† the TAMRA fluorescence intensity was lower and the FAM fluorescence intensity was higher in the anti-miR-21 treated cells compared with those in untreated cells (Fig. 4B(j–l)), which led to subdued FA/FD signals when the miR-21 expression level was inhibited. A contrastive probe containing a three-base mismatched recognition sequence was then prepared. The three types of cell were incubated with telomerase-catalyzed FRET ratioflares and contrastive probes (Fig. 4B(p–r), S15, and S16†) at consistent concentrations and ambient conditions, and then imaged using one-photon confocal microscopy. The results showed that cells treated with telomerase-catalyzed FRET ratioflares had high FA/FD signals compared with those treated with contrastive probes. Furthermore, these results indicated that the ratioflares showed specific sensing of miR-21 that correlated well with the miR-21 expression level. This further demonstrated that our ratioflares possessed the capacity to monitor changes in miRNA expression levels in living cells.
Conclusions
We have developed a new tool, named telomerase-catalyzed FRET ratioflares, for the accurate sensing of low-abundance miRNA molecules both in fluorescence assays and living cells. Compared with conventional microRNA probes, the telomerase-catalyzed FRET ratioflares possessed various merits. Firstly, the participation of telomerase causes the target miRNA to circulate in the test system, which provides high sensitivity and the ability to amplify FRET signals with an improved detection limit of 2.27 × 10–15 M for miRNA detection. Secondly, the synergetic effect of telomerase and catalyst strand (CS) allows the difference between cancer cells and normal cells to be increased reliably. Furthermore, low false positive signals from chemical interference and minimized system fluctuations were achieved through ratiometric measurements. This method is more convenient than conventional applicable technologies, which require careful RNA extraction before detection, because it is suitable for detecting miRNA in cell-derived lysates. Telomerase-catalyzed FRET ratioflares provide a widely applicable approach for visualizing low-abundance nucleic acids, providing a valuable means for elucidating their functions in living tumor cells and facilitating clinical diagnostics.
Experimental
Reagents and apparatus
All reagents used were obtained from commercial suppliers and used without further purification, unless otherwise stated. Solvents used were purified using standard methods. Human telomerase ELISA kit was purchased from Nanjing SenBeiJia Biological Technology Co., Ltd (Nanjing, China). HSA (human serum albumin), BSA (bovine serum albumin), Hcy (homocysteine), Cys (cysteine), GSH (glutathione), MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide), trisodium citrate, and HAuCl4·3H2O (hydrogen tetrachloroaurate trihydrate) were purchased from Energy Chemical Reagent Company. Mercaptoethanol, EGCG (epigallocatechin gallate) and TCEP·HCl (tris(2-carboxyethyl)phosphine hydrochloride) were purchased from Solarbio Science & Technology Co., Ltd. Lipofectamine 3000 transfection reagent, Lipofectamine-RNAiMAX transfection reagent, and DNase I endonuclease were purchased from Life Technologies Co. (USA). dNTP (deoxyribonucleoside triphosphate) was obtained from TaKaRa Biotechnology Co., Ltd (Dalian, China). All other reagents were of analytical grade. Twice-distilled purified water obtained from Milli-Q systems was used in all experiments. The morphological features of AuNPs were obtained by transmission electron microscopy (TEM, T20) at 300 kV. Fluorescence spectra were recorded on a Thermo Scientific NanoDrop 3300 Fluorospectrometer and VAEIAN CARY Eclipse fluorescence spectrophotometer (serial no. FL0812-M018), with slit widths modified to adjust the fluorescence intensity to a suitable range.
Dulbecco's modified Eagle's medium (DMEM), pancreatin, phosphate buffer saline (PBS), and penicillin/streptomycin were purchased from HyClone (Logan City, USA). Fetal bovine serum (FBS) was purchased from Zhejiang Tianhang Biological Technology Stock Co., Ltd. (Zhejiang, China). Gibco Opti-ME (reduced serum media) was purchased from Thermos Scientific (USA). Human breast cancer cells (MCF-7), human cervical carcinoma cells (HeLa), and mouse embryonic fibroblast cells (NIH-3T3) were purchased from the Institute of Basic Medical Sciences (IBMS) of the Chinese Academy of Medical Sciences. One-photon confocal fluorescent images were acquired on an Olympus FV1000 confocal inverted microscope (Olympus, Tokyo, Japan) with a 60 × objective lens.
All oligonucleotides were synthesized and HPLC-purified by TaKaRa Biotechnology Co., Ltd (Dalian, China). The sequences of the involved oligonucleotides are listed below (from 5′ to 3′):
Capture probe (C): SH-T10T CAA CAT CAG TCT GAT AAG CTA AAT CCG TCG AGC AGA TTG;
Contrast probe (non-recognition sequence): SH-T10T CTA CAT CAG TTT GAT AAG CTT AAT CCG TCG AGC AGA TTG;
Detection probe (D): TAMRA – CAA TCT GCT CGA CGG ATT TAG CTT ATC A TCG AGC AGA TTG – FAM;
Catalytic strand (CS): CCC TAA CCC TAA CAA TCT GCT CGA CGG ATT TAG CTT ATC AGA C;
MiR-21: UAG CUU AUC AGA CUG AUG UUG A;
DNA template: TAG CTT ATC AGA CTG ATG TTG A;
Mismatch-a template (one-base middle-mismatched): TAG CTT CTC AGA CTG ATG TTG A;
Mismatch-b template (one-base middle-mismatched): TAG CTT ATC AGA CTC ATG TTG A;
Mismatch-c template (two-base terminal-mismatched): AAG CTT ATC AGA CTG ATG TTG T;
Mismatch-d template (one-base terminal-mismatched): AAG CTT ATC AGA CTG ATG TTG A;
MiR-21 inhibitor (anti-miR-21): UCA ACA UCA GUC UGA UAA GCU A.
Preparation of telomerase-catalyzed FRET ratioflares
AuNPs were prepared by sodium citrate reduction of HAuCl4, as described in the literature.36 Transmission electron microscopy (TEM) images indicated particle sizes of 13 ± 2 nm (Fig. S1†). Following the “ageing process” proposed by the Mirkin group,37 functionalized ratioflares were obtained. For purification from non-grafted oligonucleotides, the solution containing the FRET ratioflares was centrifuged (13 000 rpm, 30 min) three times and then resuspended in PBS for the following experiments. The particle concentration was determined by measuring their extinction at 524 nm (ε = 2.7 × 108 L mol–1 cm–1).38
Cell culture conditions and telomerase extraction
MCF-7 (human breast adenocarcinoma) cells, HeLa (human cervical carcinoma) cells, and NIH-3T3 (mouse embryonic fibroblast) cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, and were then seeded in 25 cm2 culture flasks at 37 °C under 5% CO2 and 95% humidity. The cells were resuspended in ice-cold CHAPS lysis buffer (200 μL, 10 mM Tris–HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.5% (w/v) CHAPS, 10% (v/v) glycerol, 0.1 mM PMSF). Telomerase was then extracted as described in the literature.39
A standard addition method was used to quantify telomerase activity in HeLa cell extract using the ELISA kit. The standard curve was obtained and is shown in Fig. S5.† The activity of telomerase extracted from HeLa cells earlier was calculated to be 11.5 IU per L.
Fluorescence assay of microRNA template turnover detection experiments
For microRNA detection, telomerase-catalyzed FRET ratioflares were diluted to a concentration of 2 nM in PBS buffer and treated with increasing concentrations of the DNA targets (0, 50 fM, 100 fM, 1 pM, 10 pM, 100 pM, 500 pM, 1 nM, and 10 nM). When telomerase was present, the telomerase activity was 0.5 IU per L with dNTPs (10 mM each) and 200 nM catalyst strand added. After incubation for 90 min at 37 °C, the fluorescence was monitored at appropriate excitation wavelengths. As contrast, only DNA targets and telomerase-catalyzed FRET ratioflares were added into the comparison group, while retaining the same probe concentration and other test conditions (Fig. S4†). The fluorescence was recorded on a Thermo Scientific NanoDrop 3300 Fluorospectrometer with excitation at 492 nm and measuring emissions from 500 to 650 nm in 1 nm increments. Means and standard deviations were obtained from three parallel experiments in each trial.
Calculation of detection limit
The fluorescence intensity of FRET ratioflares was measured three times and the standard deviation of the blank measurement was obtained. The detection limit was calculated using the following equation:Detection limit = 3σ/kwhere σ is the standard deviation of the blank measurement, σ = 0.00931, and k is the slope of the fluorescence intensity vs. miRNA template concentration growth (k = 0.07836).
Cell viability experiment
The cell viability was measured by the reduction of MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) to formazan crystals using mitochondrial dehydrogenases. MCF-7, HeLa, and NIH-3T3 cells were seeded in 96-well microplates at a density of 1 × 105 cells per mL in 100 μL of medium containing 10% FBS buffer. After cell attachment for 24 h, the plates were washed with PBS (100 μL per well). To test the cytotoxicity of telomerase-catalyzed FRET ratioflares, the cells were cultured in medium with 0.04, 0.08, 0.16, 0.31, 0.625, 1.25, 2.5, 5, or 10 nM of ratioflares for 24 h. Cells in culture medium without ratioflares were used as control (Fig. S6†). To test the cytotoxicity of EGCG, the cells were cultured in medium with 30, 60, 90, 120, 180, 260, 300, and 340 μg mL–1 of EGCG for 48 h, respectively. Cells in culture medium without EGCG were used as the control (Fig. S11†). Six replicate wells were used for each control and test concentration. A total of 10 μL of MTT (5 mg mL–1) prepared in PBS was added to each well, and the plates were incubated at 37 °C for another 4 h in a 5% CO2 humidified incubator. The medium was then carefully removed and the purple crystals were lysed in DMSO (100 μL). The optical density was determined on a microplate reader (Thermo Fisher Scientific) at 570 nm with subtraction of the absorbance of the cell-free blank volume at 630 nm. Cell viability was expressed as a percentage of the control culture value, and calculated using the following equation:Cell viability (%) = (ODdye – ODKdye)/(ODcontrol – ODKcontrol) × 100
Live-cell detection of telomerase-catalyzed FRET ratioflares by microscopy
For imaging experiments, the cells were seeded into a glass-bottomed dish (35.0 mm dish with 20.0 mm bottom well) with culture medium (2.0 mL) in the incubator until the cell density was approximately 80%. To transfect the cells, we incubated Lipofectamine 3000 (1.8 μL) in Opti-MEM (125 μL) in one centrifuge tube (tube A), and incubated p3000 (1.2 μL) in Opti-MEM containing 1 μg of catalyst strand (125 μL) in another centrifuge tube (tube B). The contents of tube B was then added into tube A, mixed homogeneously, and then incubated for 5 min at RT to allow the Lipofectamine-oligo complex to form. The cell media was then replaced with the newly prepared Opti-MEM solution containing the complex (500 μL) and incubated with FRET ratioflares (1 nM) for 120 min at 37 °C with 5% CO2 and 95% humidity. The cells were then washed twice with PBS. Fluorescence imaging was performed using an Olympus FV-1000 inverted fluorescence microscope with a 60 × objective lens. Under the confocal fluorescence microscope, the probe was excited at 488 nm and emission was collected at 500–540 nm (green channel) for FAM imaging and collected at 560–600 nm (red channel) for TAMRA imaging. All parameters remained the same throughout the cell experiments.
Cell lysate preparation and detection
All types of cell were counted and followed by centrifugation. RIPA buffer was used to resuspend the cells to obtain 1.5 × 106 cells per mL. RNaseOUT (400 U mL–1) was added to slow RNA degradation. Cells were kept on ice with intermittent vortexing for 15 min. The debris was removed by centrifugation and the supernatant was collected. The telomerase-catalyzed FRET ratioflares were added to the supernatant at a final concentration of 2 nM. The solution was then incubated at 37 °C for 2 h before measuring the fluorescence on a Thermo Scientific NanoDrop 3300 fluorospectrometer.
Cell imaging with inhibited telomerase activity
After treatment with 0, 60, 120, 180, and 260 μg mL–1 of EGCG for 48 h, HeLa cells were transfected with catalyst strand and incubated with FRET ratioflares for 120 min. The cell was then imaged by confocal microscopy (Fig. S12†) and the average fluorescence intensity ratio between the FAM channel and TAMRA channel was obtained, as shown in Fig. S13.† The telomerase activity was then obtained by ELISA kit analysis (Fig. S14†). The other two types of cell were treated with 260 μg mL–1 of EGCG for 48 h, and then both channels of fluorescence intensity with the ratioflare were detected using confocal imaging (Fig. S15 for MCF-7 and S16† for NIH-3T3).
Cell imaging with inhibited miR-21 content
The cells (2 × 105 cells) were transfected with synthetic anti-miR-21. The transfection experiments were conducted using Lipofectamine-RNAiMAX Transfection Reagent (60 μL) in Opti-MEM (2 mL) containing 10 pM anti-miR-21, which were replaced with cell medium, followed by incubation of the three types of cell for 24 h. The two channel of fluorescence intensity with the ratioflares were then detected using confocal imaging (Fig. S15 for MCF-7 and S16† for NIH-3T3).
Conflicts of interest
The authors declare no competing financial interests.
Supplementary Material
Acknowledgments
This work was financially supported by the National Science Foundation of China (Project No. 21421005, 21576037, and U1608222).
Footnotes
†Electronic supplementary information (ESI) available: Additional documentation (12 figures) including: characterization of AuNPs and DNA-AuNPs; absorption spectra of AuNPs and DNA functionalized AuNPs; standard linear calibration curve of fluorescence signal against concentration of TAMRA-labeled detection probes; fluorescence spectra when telomerase is absent; standard curve obtained from ELISA kit standard method; MTT experiments of HeLa, MCF-7, and NIH-3T3 cells with telomerase-catalyzed FRET ratioflares for 24 h; endosome/lysosome escape experiment; MTT experiment of HeLa, MCF-7, and NIH-3T3 cells with EGCG for 48 h; time-dependent fluorescence intensity of ratioflares in HeLa cells; one-photon confocal fluorescence imaging of miR-21 in HeLa cells after incubation with different concentrations of EGCG for 48 h; average fluorescence intensity ratio between FAM channel and TAMRA channel after incubating with EGCG; absorbance of telomerase activity in HeLa cells after incubation with different concentrations of EGCG for 48 h through ELISA kit analysis; one-photon confocal fluorescence imaging of miR-21 in MCF-7 and NIH-3T3 cells after incubation with anti-miR-21 sequence, contrast probes, and EGCG, and without CS. See DOI: 10.1039/c9sc02301a
References
- Xia Y. Q., Zhang R. L., Wang Z. L., Tian J., Chen X. Y. Chem. Soc. Rev. 2017;46:2824. doi: 10.1039/c6cs00675b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M. I., Reis R. M., Calin G. A. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2011;717:1. doi: 10.1016/j.mrfmmm.2011.03.009. [DOI] [PubMed] [Google Scholar]
- Lu J., Getz G., Miska E. A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B. L., Mak R. H., Ferrando A. A., Downing J. R., Jacks T., Horvitz H. R., Golub T. R. Nature. 2005;435:834. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- He L., Thomson J. M., Hemann M. T., Hernando-Monge E., Mu D., Goodson S., Powers S., Cordon-Cardo C., Lowe S. W., Hannon G. J., Hammond S. M. Nature. 2005;435:828. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. Cell. 2004;116:281. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Ambros V. Nature. 2004;431:350. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S., Saini H. K., van Dongen S., Enright A. J. Nucleic Acids Res. 2008;36:D154. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill J., Deiters A. J. Am. Chem. Soc. 2013;135:10512. doi: 10.1021/ja404350s. [DOI] [PubMed] [Google Scholar]
- Xu L. G., Gao Y. F., Kuang H., Liz-Marzan L. M., Xu C. L. Angew. Chem., Int. Ed. 2018;57:10544. doi: 10.1002/anie.201805640. [DOI] [PubMed] [Google Scholar]
- Yao Q. C., Wu D. E., Ma R. Z., Xia M. J. Organomet. Chem. 2013;743:1. [Google Scholar]
- Wei Q. M., Huang J., Li J., Wang J. L., Yang X. H., Liu J. B., Wang K. M. Chem. Sci. 2018;9:7802. doi: 10.1039/c8sc02943a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Jiang J., Yuan R., Zhuo Y., Chai Y. Q. J. Am. Chem. Soc. 2018;140:9361. doi: 10.1021/jacs.8b04648. [DOI] [PubMed] [Google Scholar]
- He X. W., Zeng T., Li Z., Wang G. L., Ma N. Angew. Chem., Int. Ed. 2016;128:3125. [Google Scholar]
- Huang J., Wang H., Yang X. H., Quan K., Yang Y. J., Ying L., Xie N. L., Ou M., Wang K. M. Chem. Sci. 2016;7:3829. doi: 10.1039/c6sc00377j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W., Sun M. Z., Fu P., Li S., Xu L. G., Kuang H., Xu C. L. Adv. Mater. 2017;29:1703410. doi: 10.1002/adma.201703410. [DOI] [PubMed] [Google Scholar]
- Pritchard C., Cheng H., Tewari M. Nat. Rev. Genet. 2012;13:358. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Shuai Z. H., Zhou H., Luo Z. M., Liu B., Zhang Y. N., Zhang L., Chen S. F., Chao J., Weng L. X., Fan Q. L., Fan C. H., Huang W., Wang L. H. J. Am. Chem. Soc. 2018;140:3988. doi: 10.1021/jacs.7b12772. [DOI] [PubMed] [Google Scholar]
- Sun Y. Y., Tian H., Liu C. H., Sun Y. Y., Li Z. P. Chem. Commun. 2017;53:11040. doi: 10.1039/c7cc06140d. [DOI] [PubMed] [Google Scholar]
- Huang Z., Liu C., Fu Q. Q., Li J., Zou J. M., Xie S. T., Qiu L. P. Chin. J. Appl. Chem. 2018;35:28. [Google Scholar]
- Quan K., Huang J., Yang X. H., Yang Y. J., Ying L., Wang H., He Y., Wang K. M. Chem. Commun. 2015;51:937. doi: 10.1039/c4cc08488h. [DOI] [PubMed] [Google Scholar]
- Karunanayake Mudiyanselage A. P. K. K., Yu Q. K., Leon-Duque M. A., Zhao B., Wu R., You M. X. J. Am. Chem. Soc. 2018;140:8739. doi: 10.1021/jacs.8b03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Liu M. H., Zhang C. Y. Chem. Sci. 2018;9:4258. doi: 10.1039/c8sc00832a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Feng J., Liu H. Y., Li Q. L., Tong L. L., Tang B. Chem. Sci. 2016;7:1940. doi: 10.1039/c5sc03909f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. J., Huang J., Yang X. H., Quan K., Wang H., Ying L., Xie N. L., Ou M., Wang K. M. J. Am. Chem. Soc. 2015;137:8340. doi: 10.1021/jacs.5b04007. [DOI] [PubMed] [Google Scholar]
- Qian R. C., Ding L., Yan L. W., Lin M. F., Ju H. X. J. Am. Chem. Soc. 2014;136:8205. doi: 10.1021/ja5042995. [DOI] [PubMed] [Google Scholar]
- Cohen S. B., Graham M. E., Lovrecz G. O., Bache N., Robinson P. J., Reddel R. R. Science. 2007;315:1850. doi: 10.1126/science.1138596. [DOI] [PubMed] [Google Scholar]
- Ma F., Wei S. H., Leng J. H., Tang B., Zhang C. Y. Chem. Commun. 2018;54:2483. doi: 10.1039/c8cc00093j. [DOI] [PubMed] [Google Scholar]
- Chadeneau C., Hay K., Hirte H. W., Gallinger S., Bacchetti S. Cancer Res. 1995;55:2533. [PubMed] [Google Scholar]
- Zhou X. M., Xing D., Zhu D. B., Jia L. Anal. Chem. 2009;81:255. doi: 10.1021/ac801914b. [DOI] [PubMed] [Google Scholar]
- Ma W., Fu P., Sun M. Z., Xu L. G., Kuang H., Xu C. L. J. Am. Chem. Soc. 2017;139:11752. doi: 10.1021/jacs.7b03617. [DOI] [PubMed] [Google Scholar]
- Haiss W., Thanh N. T. K., Aveyard J., Fernig D. G. Anal. Chem. 2007;79:4215. doi: 10.1021/ac0702084. [DOI] [PubMed] [Google Scholar]
- Ying Z. M., Wu Z., Tu B., Tan W. H., Jiang J. H. J. Am. Chem. Soc. 2017;139:9779. doi: 10.1021/jacs.7b04527. [DOI] [PubMed] [Google Scholar]
- Li L., Feng J., Liu H. Y., Li Q. L., Tong L. L., Tang B. Chem. Sci. 2016;7:1940. doi: 10.1039/c5sc03909f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R., Chen J., Wang J. H., Rao J. M., Du X. C., Liu Y. M., Zhang L. S., Qiu L., Liu B., Zhao Y. D., Jiang P. J., Chen C. Y., Li Y. Q. Small. 2018;14:1802745. doi: 10.1002/smll.201802745. [DOI] [PubMed] [Google Scholar]
- Lei Y. F., Tang L. X., Xie Y. Z., Xianyu Y. L., Zhang L. M., Wang P., Hamada Y., Jiang K., Zheng W. F., Jiang X. Y. Nat. Commun. 2017;8:15130. doi: 10.1038/ncomms15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabar K. C., Freeman R. G., Hommer M. B., Natan M. J. Anal. Chem. 1995;67:735. [Google Scholar]
- Demers L. M., Mirkin C. A., Mucic R. C., Reynolds R. A., Letsinger R. L., Letsinger R., Viswanadham G. A. Anal. Chem. 2000;72:5535. doi: 10.1021/ac0006627. [DOI] [PubMed] [Google Scholar]
- Cao Y. W., Jin R. C., Mirkin C. A. J. Am. Chem. Soc. 2001;123:7961. doi: 10.1021/ja011342n. [DOI] [PubMed] [Google Scholar]
- Wang W. J., Li J. J., Rui K. P., Gai P., Zhang J. R., Zhu J. J. Anal. Chem. 2015;87:3019. doi: 10.1021/ac504652e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.