

Abstract
Mu-opioid receptors (MORs) in the nucleus accumbens (NAc) can regulate reward-related behaviors that are dependent on mesolimbic dopamine, but the precise mechanism of this MOR regulation is unknown. We hypothesized that MORs within the NAc core regulate dopamine release. Specifically, we infused the MOR antagonist CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2) into the NAc core while dopamine release was evoked by electrical stimulation of the ventral tegmental area and measured by fast-scan cyclic voltammetry. We report that CTAP dose-dependently inhibited evoked dopamine release, with full blockade achieved with the 8 μg infusion. In contrast, evoked dopamine release increased after nomifensine infusion and was unchanged after vehicle infusion. These findings demonstrate profound local control of dopamine release by MORs within the NAc core, which has implications for regulation of reward processing.
Keywords: Mu opioid receptor, dopamine, accumbens, phasic, electrochemistry, local infusion
Graphical Abstract
The nucleus accumbens (NAc) is in the ventral extent of the striatum and constitutes a main input nucleus of the basal ganglia. Multiple neurotransmitter systems participate in the function of this nucleus; however, dopamine projections from the ventral tegmental area (VTA) are key modulators of the NAc.1–2 Mesolimbic dopamine is critical for reward learning and motivational processing,3 and its dysregulation is associated with disorders such as addiction and schizophrenia.e.g., 4–5
In the NAc, mu-opioid receptors (MORs) are located on both cholinergic and GABA interneurons and GABA medium spiny neurons.6–7 MOR activity affects reward-related behavior, as seen with direct infusion of opioid drugs to the NAc. For example, infusion of MOR agonists in the NAc promotes both hedonic taste reactions to sucrose8 and consumption of palatable foods, particularly those high in fat.9–14 Intriguingly, however, infusion of MOR antagonists into the NAc does not reliably reduce palatable food consumption, with some studies reporting reductions whereas others show no or smaller effects.15–19 In contrast, blockade of MORs in the NAc reduces dopamine-dependent appetitive behaviors in several paradigms, including progressive ratio for food reward,20 a runway task,21 operant responding during a Pavlovian-instrumental transfer test,22 cocaine-induced place preference and hyperactivity,23 and cued approach to a receptacle where cream reward is available.24 These results raise the hypothesis that endogenous opioids in the NAc promote reward-seeking behavior by increasing the release of dopamine from dopamine terminals. Consistent with this idea, NAc MOR activation can increase dopamine levels in the NAc as measured by microdialysis,25–29 perhaps by inhibition of GABA release onto cholinergic neurons that stimulate dopamine release via nicotinic receptor-mediated excitation of dopamine terminals.30–32
Although much evidence suggests that activation of MORs within the NAc is sufficient to enhance tonic dopamine, it is presently unclear whether endogenous opioids promote phasic dopamine release. We hypothesized that local infusion of the MOR antagonist CTAP (2, 4 and 8 μg) would decrease electrically evoked dopamine release in a dose-dependent fashion. To test that, we used fast scan cyclic voltammetry (FSCV), an electrochemical technique with a high anatomical and temporal resolution,33 to measure evoked dopamine transients before and after the local infusion of CTAP into the NAc. Unlike systemic administration, local infusion circumvents the possibility that the drug acts elsewhere in the brain or body, and specifically tests how local modulation of MORs influence dopamine dynamics.
To apply drugs directly to the electrochemical recording site in vivo, we employed an infusion technique adapted from previous studies.34–35 This method combined an infusion cannula with a guide cannula for a carbon-fiber microelectrode, such that the tip of the injector was positioned approximately 150 μm from the active surface of the electrode (Fig. 1A). Estimated electrode/injector placements for the CTAP infusions (described below) are depicted in Fig. 1B.
Figure 1.
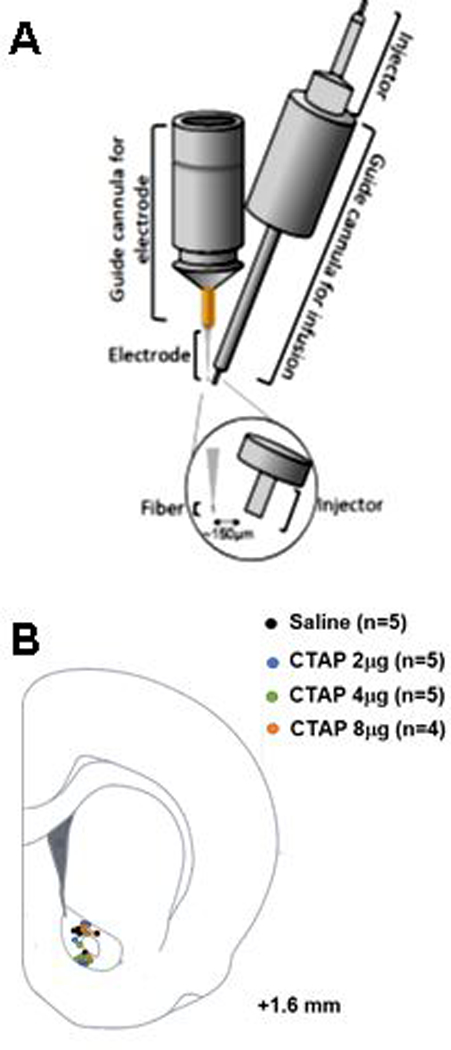
Schematic representation of the guide cannula ensemble and anatomical placements for simultaneous in vivo voltammetric measurement of evoked dopamine release and local application of a drug. (A) Guide cannula ensemble: The electrochemical measurements were performed at carbon-fiber microelectrodes lowered via the guide cannula on the left. The drug applications were performed via the injector inserted into the guide cannula on the right. Both cannulae were positioned and cemented together under a microscope prior to any experiment to ensure an approximate distance of 150 μm between the electrode and end of the injector. (B) Representations of electrode/injector placements within nucleus accumbens core.
To characterize this technique, we first infused nomifensine (NOM), a dopamine transporter inhibitor with well characterized effects on extracellular dopamine: it enhances evoked dopamine release and slows subsequent clearance.36 Each rat was anesthetized and secured in a stereotaxic frame, and the pre-assembled guide cannula ensemble was secured on the skull to target the NAc core. Next, a carbon-fiber microelectrode was lowered into the NAc via a manipulator inserted in the guide cannula; each turn of the manipulator wheel pushed the electrode 300 μm into the tissue. The electrode was lowered to a predetermined position near the infusion cannula. There, a range of potentials was applied to the electrode using a triangle waveform (−0.4V to 1.3V to −0.4V, 400V/s, 10Hz) while dopamine neurons were periodically activated via electrical stimulation to the VTA (125nA, 24 pulses at 60Hz, biphasic, 2ms/phase). Dopamine release time-locked to the electrical stimulation was electrochemically confirmed via the background-subtracted cyclic voltammogram containing oxidative and reductive current peaks characteristic for catecholamines. Next, the infusion injector was slowly inserted and evoked dopamine was again confirmed.
Locally applied NOM (40 μM, 0.5 μl over 2 minutes) enhanced VTA-evoked dopamine release in the NAc core of anesthetized rats (n=4). Representative dopamine signals obtained before and after NOM application in an individual rat are shown in Fig. 2A. The color plots show current (color) at each applied potential (y-axis) over a 10-second scan (x-axis). Current at the peak oxidation potential of dopamine, converted to dopamine concentration using in vitro electrode calibrations, is depicted in the line graph above the color plot. Composite data (Fig. 2B) show that NOM significantly increased evoked dopamine within 5 minutes after the start of infusion (one-way RM ANOVA: F3,12=3.9, p<0.001). Pre-infusion, evoked [DA]max was approximately 65 nM, and this increased 5-fold at 15 min post-infusion. Post-hoc analysis revealed that [DA]max was significantly higher at each time point post-infusion (all t’s > 2.3, all p’s < 0.03). These results, along with those from the saline group, indicate that drugs delivered via this infusion technique could quickly alter local dopamine release.
Figure 2.
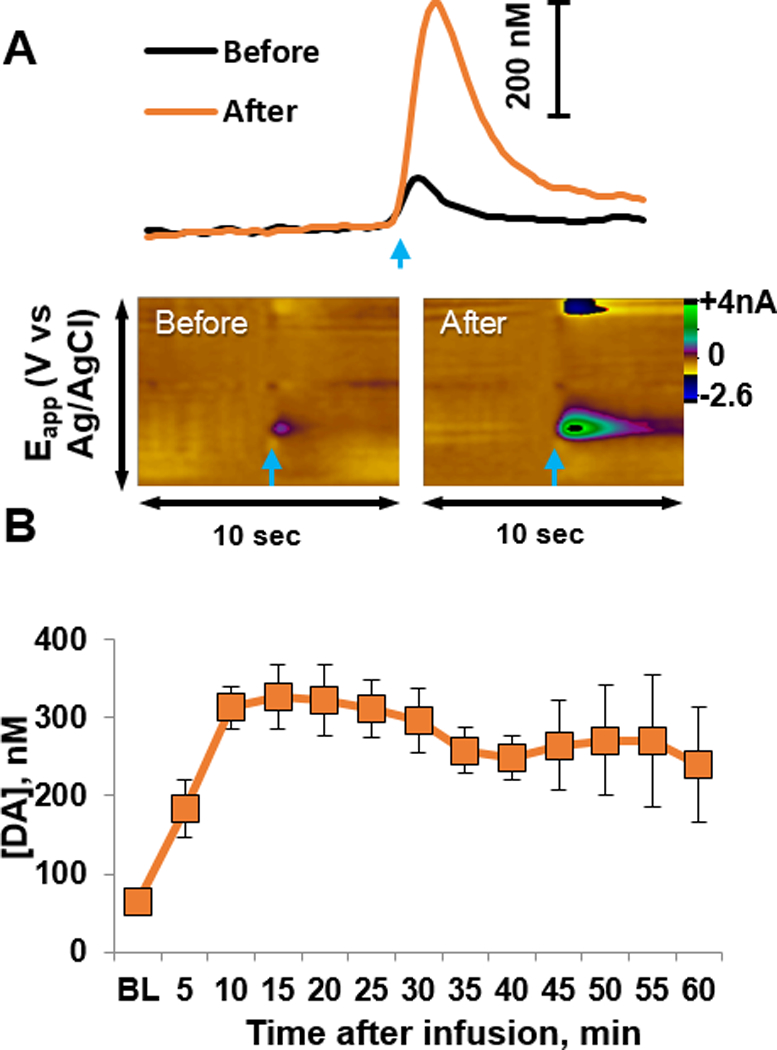
Locally applied nomifensine enhanced evoked dopamine transients. (A) Current versus time traces (top) and color plots (bottom) of evoked dopamine release from an individual rat before and after nomifensine infusion. The time of electrical stimulation is indicated by blue arrows. In the color plots, current (color) is depicted at the different applied potentials (y-axis) over time (x-axis). Dopamine oxidation is evident as positive current at ~ 0.65 V and reduction is evident as negative current at ~ −0.25 V. (B) Composite data (n=4 rats) show that nomifensine enhanced evoked [DA]max within 5 minutes, with a peak response at 15 minutes. * different from baseline (BL), p<0.03.
With this validated infusion technique, we next aimed to explore the local effect of MOR antagonism on evoked DA release. In separate groups of rats (n=4–5 rats per dose), CTAP (2, 4 or 8 μg in 0.5 μl) was infused over 2 minutes and compared to vehicle infusions (0.5 μl saline). Data from individual rats (Fig. 3A, as described for Fig. 2A) illustrate that saline did not alter evoked dopamine release, while CTAP diminished it. The left panels illustrate dopamine release after electrical stimulation (at 5 seconds) under baseline conditions; note the individual variability in release due to factors such as the stimulating electrode placement and carbon-fiber length. The right panels show evoked dopamine release in the same rats 30 min after saline, 4 or 8 μg CTAP, and CTAP reduced release while saline did not. The dose-dependent effect of CTAP across rats is depicted in Fig. 3B, as the change in dopamine release from baseline across all doses is presented over time. While saline and 2 μg CTAP minimally affected evoked dopamine release, both 4 and 8 μg CTAP reduced the dopamine signal. Statistics were calculated on the average change in dopamine over 60 minutes post-infusion (Fig. 3C). A one-way ANOVA revealed a main effect of group (F3,15=11.7, p<0.001) and Holm-Sidak post-hoc comparisons found that both 4 and 8 μg CTAP significantly reduced evoked dopamine release compared to saline (both t’s>4.6, p’s≤0.002), while the 8 μg dose was also different from the 2 μg dose (t=3.3, p<0.02).
Figure 3.
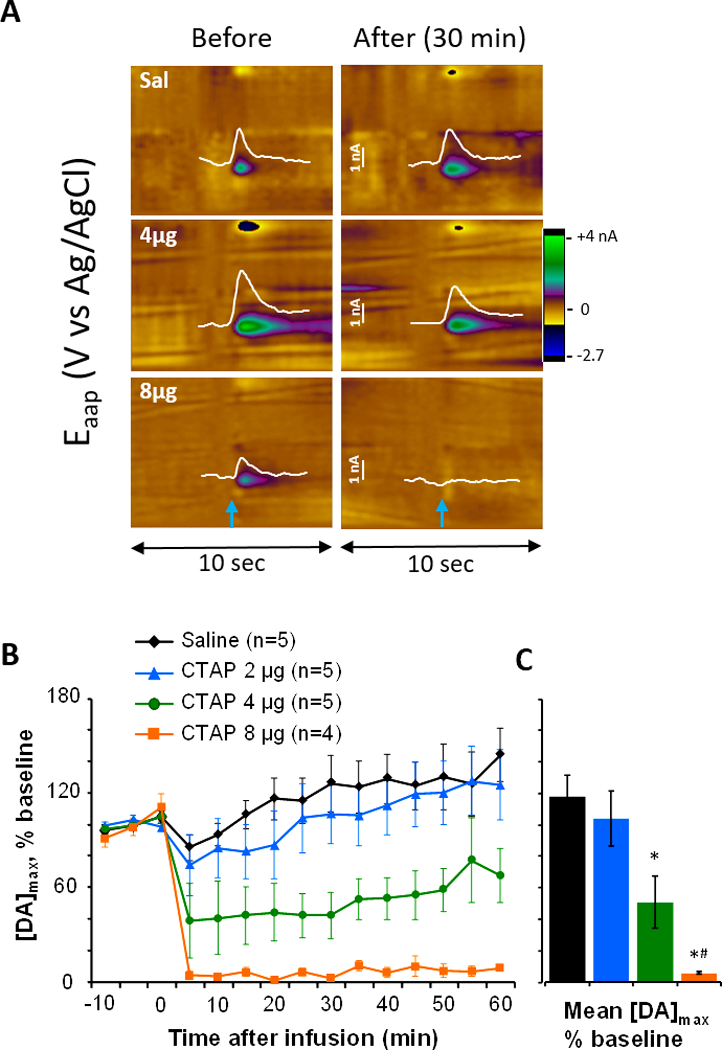
Infusion of CTAP to the nucleus accumbens core dose-dependently reduces evoked dopamine release. The selective MOR antagonist CTAP (2, 4, and 8 μg) or saline was infused to the area of dopamine measurement via an infusion cannula approximately 150 μm from the voltammetric electrode. (A) Current-versus-time traces at the oxidation potential of dopamine (white, 7 seconds) are overlaid on color plots of evoked dopamine release from individual rats before and after saline, 4 μg and 8 μg CTAP infusion. The time of electrical stimulation is indicated by blue arrows. Infusion of 4 μg CTAP partially blunted electrically-evoked dopamine release while 8 μg CTAP blocked it. (B) and (C): Composite data show the dose-dependent effects of CTAP on evoked dopamine release over time (B) and averaged across post-infusion time points (C). Electrically-evoked dopamine was unchanged by saline and 2 μg CTAP, but reduced by 4 and 8 μg CTAP. Statistics were calculated on the data in panel C: * different from Saline, p<0.05; # different from 2 μg CTAP, p<0.05.
One potential explanation for the reduction in evoked dopamine signal after CTAP application is that CTAP fouled the electrode, reducing its sensitivity for dopamine. To test this, fresh, unused electrodes (n=8) were calibrated with 1 μM dopamine before and after immersion in 16 μg/μl CTAP solution (the concentration used for 8 μg infusions) for 10 minutes. The average current from dopamine oxidation was initially 14±3 nA. Next, electrodes were immersed in the CTAP solution while we applied the same potentials to the carbon-fiber electrode as used in vivo. The 10-minute period was chosen to ensure any potential CTAP effect; however, during experiments the infusion occurred over a 2-minute period followed by drug diffusion and subsequent decrease in CTAP concentration around the electrode. The average current from dopamine oxidation obtained after CTAP application was unchanged, at 12±2 nA (paired t-test, t7=1.4, p=0.207).
Together, these data clearly demonstrate that MOR antagonism can reduce evoked dopamine release, indicating a role for endogenous opioids in the NAc to regulate mesolimbic dopamine. This finding is consistent with studies reporting that NAc MOR activation increases dopamine levels in the NAc.25, 29–32 As dopamine terminals express kappa-opioid receptors and delta-opioid receptors, but not MORs,37–39 CTAP is not acting directly on dopamine terminals. However, MORs are expressed on GABA interneurons,37 cholinergic interneurons46 and medium spiny neurons expressing dopamine D1 receptors (D1-MSNs),38 and together these cells appear to modulate neuronal activity of the whole striatal network.40 Thus, CTAP would act on several different targets into the NAc changing local and circuital dynamics, and the observations reported here likely result from a combination of actions. For example, it is well known that acetylcholine (ACh) locally regulates dopamine release via nicotinic ACh receptors.39–42 In this way, nicotinic ACh receptor activation on dopamine terminals can act as a low-frequency pass filter, enhancing initial dopamine release probability but reducing phasic transients, while inactivation of nicotinic ACh receptors – either by reduction of ACh release or desensitization of the receptors – acts as a high-frequency pass filter to blunt low-frequency stimulated dopamine release but augment burst-like stimulated dopamine release. Thus, CTAP, by blocking the inhibitory effects of MORs on ACh release, might enhance these effects of ACh on dopamine terminals. On the other hand, GABA interneurons also express MORs, and by the same reasoning, CTAP antagonism at those receptors would enhance GABA inhibition of cholinergic interneurons and subsequent inhibition of ACh release. Also, other players involved in local activity include M2/M4 metabotropic autoreceptors that modulate the release of ACh,43–44 GABA-B receptors expressed on dopamine terminals that inhibit dopamine release,45 and glutamatergic terminals that are regulated by ACh via nicotinic ACh receptors and in turn stimulate dopamine terminals via ionotropic glutamate receptors.46 Thus, decreased dopamine release after CTAP could be the result of multiple, different interactions in the NAc.
Of course, CTAP effects on striato-tegmental circuits may have contributed to the present results. MORs are present on direct-pathway D1-MSNs, and there is evidence that MSNs form monosynaptic connections with dopamine neurons in the VTA.47–49 In this case, if CTAP binds to MORs expressed on these D1-MSNs, GABA release would increase and, consequently, inhibit VTA dopamine neurons. On the other hand, electrophysiological evidence indicates that the majority of MSN input from the NAc to the VTA is to GABA neurons, including interneurons.50–52 In that case, one would predict the opposite effect of CTAP in the NAc, as enhanced GABA release would inhibit VTA interneurons and disinhibit dopamine neurons.51 However, by locally infusing CTAP into the NAc and delivering current to stimulate the dopamine neurons in the VTA, it is less likely that circuit-based pharmacological effects were the main contributor to the findings reported here.
As several different variables are involved in regulating dopamine dynamics, the complexity of the variables makes it difficult to give more weight to one particular factor over the others. In fact, the results we are presenting here could arise from a complex interaction between different variables (as mentioned) and possibly others not included. The nature of such interaction remains to be determined. At present we cannot rule out the possibility that CTAP had non-specific effects on receptors other than MOR receptors, although we are confident that our results are not due to fouling of the carbon-fiber electrodes.
In conclusion, this study determined that the MOR antagonist CTAP dose-dependently reduced evoked DA release in the NAc via local mechanisms. Specifically, we found that while 2 μg CTAP did not alter dopamine release, 4 and 8 μg CTAP immediately reduced [DA]max following infusion. These findings provide a reliable method to measure effects of a drug directly infused at the electrode and further the understanding of the role of μ-opioid receptor antagonism on dopamine regulation in the NAc. Our results offer support for behavioral studies that showed reduction of dopamine-dependent appetitive behaviors20 after blockade of MORs in the NAc, suggesting that the reported behavioral effects are due to reductions in dopamine release. However, the mechanism underlying the observed effects is still unknown and additional studies are necessary to completely understand those mechanisms.
Methods
Animals
Adult, male Long-Evans rats were purchased from Envigo Laboratories (Indianapolis, IN) at 225–250g; we chose these rats as male Long Evan rats were also used in studies demonstrating that CTAP infused into the NAc reduced cue-evoked reward seeking.24 All animals were pair-housed in a facility with a 12-hr light, 12-hr dark cycle with rat chow and water available ad libitum. All experiments were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals with procedures approved by the Institutional Animal Care and Use Committee of the University of North Carolina.
Fast scan cyclic voltammetric measurements
Urethane (50% w/w solution in saline, 1.5g/kg, IP) was used to anesthetize the rats throughout the experiment. During surgery, a reference electrode was placed in the left hemisphere and secured with a stainless steel screw and dental acrylic. Then, a stimulating electrode was placed in the VTA at the coordinates (from bregma) AP −5.2mm, ML +1.0mm and DV −8.0mm; the DV coordinate was adjusted in each rat to elicit an evoked dopamine signal with S:N > 30. Finally, an assembled guide cannula ensemble was placed in the NAc core with coordinates AP +1.6mm and ML +1.7mm. A schematic of the guide cannula ensemble is shown in Fig. 1. The ensemble consisted of a guide cannula for the carbon-fiber electrode (left) and a guide cannula for the injector (right) used to deliver the drug during the experiment. As demonstrated in the figure, the two cannulae were arranged to achieve an approximate distance of ~150 μm between the carbon-fiber and end of the injector. The assembly of the two guides was performed prior to the experiments and was done under the microscope to ensure the distance between the electrode and site of drug infusion.
FSCV at carbon-fiber electrodes (cylinders, 6–7μm diameter, 86±13μm active length, insulated in glass) was used to measure electrically evoked DA release in the NAc core as previously described.53–54 Measurements were taken every 100 ms with an applied potential from −0.4V to +1.3V and back to −0.4V at a rate of 400 V/s versus the Ag/AgCl reference electrode. In order to mimic phasic dopamine release, 24 pulses of 125–300μA (60 Hz, biphasic, 2 ms/phase) were delivered to the VTA using a bipolar stimulating electrode (Plastics One, Inc., Roanoke, VA). TarHeel CV (UNC Department of Chemistry) was used to collect and analyze the electrochemical data. The current associated with dopamine oxidation was used to evaluate dopamine release.
During each experiment, the carbon-fiber electrode was lowered into the NAc using a micromanipulator to the predetermined depth for that cannula ensemble (approximately DV −6.5 to −7.0mm from bregma), and electrically-evoked dopamine release was measured with FSCV. The stimulating electrode was lowered at increments of 0.2 mm until an evoked dopamine signal at appropriate signal:noise was found. Next, the injection apparatus was set up, the injector was filled with a drug or saline solution, and the injector was inserted into the guide cannula. At least 15 minutes elapsed to allow the tissue to adjust to the injector insertion. Three basal level measurements and twelve post-drug infusion measurements of electrically evoked dopamine release were collected with 5 minutes between each stimulation.
Drugs and Injection Procedure
NOM and CTAP were purchased from Sigma-Aldrich (St. Louis, MO, USA). All drugs were dissolved in saline, which was used as a vehicle control solution. Drugs were injected in a volume of 0.5 μl over a period of 2 minutes, and at least 2 additional minutes were allowed for diffusion before the next electrical stimulation. Independent groups of rats received different doses of CTAP (0, 2, 4 and 8 μg). Measurements continued for up to 60 minutes post-infusion with 5 minutes between stimulations.
In vitro calibration
Electrodes were calibrated in vitro after each experiment. A flow cell was used such that the carbon fiber electrode and reference electrode were constantly submerged in flowing TRIS buffer (32.5 mM KCl, 12 mM CaCl2, 12 mM MgCl2, 20mM Na2SO4, 12.5 mM NaH2PO4, 1.45 M NaCl, 150 MM TRIS). A ValveLink 8.2 controller (AutoMate Scientific, Berkeley, CA) was used to switch from buffer to 1 μM dopamine in buffer for a period of 5 seconds, then back to buffer.
To evaluate a potential fouling effect of CTAP application on the sensitivity of electrodes to dopamine, an in vitro experiment was performed. Freshly made carbon-fiber electrodes with lengths of 75±20 μm were soaked in isopropyl alcohol for 20 minutes,55 and then calibrated as described. After initial calibration, electrodes were placed in 8 μg CTAP solution along with a reference electrode with the FSCV waveform continuously applied at 10 Hz for a period of 10 minutes. Immediately afterward, the electrodes were recalibrated in the flow cell using the same method as described above.
Acknowledgments
Funding sources: This research was funded by a Klarman Family Foundation grant to SMN and by the UNC Bowles Center for Alcohol Studies. ELB was supported by a Summer Undergraduate Research Fellowship from the Office for Undergraduate Research at the University of North Carolina at Chapel Hill.
Abbreviations
- ACh
acetylcholine
- CTAP
D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2, MOR antagonist
- FSCV
fast scan cyclic voltammetry
- MOR
mu opioid receptor
- NAc
nucleus accumbens
- nAChR
nicotinic acetylcholine receptor
- NOM
nomifensine, dopamine transporter blocker
Footnotes
Conflict of interest: The authors declare no real or perceived conflict of interest associated with this research.
References
- 1.Nicola SM, The nucleus accumbens as part of a basal ganglia action selection circuit. Psychopharmacology 2007, 191 (3), 521–50. [DOI] [PubMed] [Google Scholar]
- 2.Da Cunha C; Boschen SL; Gomez AA; Ross EK; Gibson WS; Min HK; Lee KH; Blaha CD, Toward sophisticated basal ganglia neuromodulation: Review on basal ganglia deep brain stimulation. Neuroscience and biobehavioral reviews 2015, 58, 186–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berke JD, What does dopamine mean? Nature neuroscience 2018, 21 (6), 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seeman MV; Seeman P, Is schizophrenia a dopamine supersensitivity psychotic reaction? Progress in neuro-psychopharmacology & biological psychiatry 2014, 48, 155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Volkow ND; Wise RA; Baler R, The dopamine motive system: implications for drug and food addiction. Nature reviews 2017, 18 (12), 741–752. [DOI] [PubMed] [Google Scholar]
- 6.Britt JP; McGehee DS, Presynaptic opioid and nicotinic receptor modulation of dopamine overflow in the nucleus accumbens. J Neurosci 2008, 28 (7), 1672–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma YY; Cepeda C; Chatta P; Franklin L; Evans CJ; Levine MS, Regional and cell-type-specific effects of DAMGO on striatal D1 and D2 dopamine receptor-expressing medium-sized spiny neurons. ASN Neuro 2012, 4 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pecina S; Berridge KC, Opioid site in nucleus accumbens shell mediates eating and hedonic ‘liking’ for food: map based on microinjection Fos plumes. Brain research 2000, 863 (1–2), 71–86. [DOI] [PubMed] [Google Scholar]
- 9.Taha SA, Preference or fat? Revisiting opioid effects on food intake. Physiology & behavior 2010, 100 (5), 429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woolley JD; Lee BS; Taha SA; Fields HL, Nucleus accumbens opioid signaling conditions short-term flavor preferences. Neuroscience 2007, 146 (1), 19–30. [DOI] [PubMed] [Google Scholar]
- 11.Zhang M; Kelley AE, Opiate agonists microinjected into the nucleus accumbens enhance sucrose drinking in rats. Psychopharmacology 1997, 132 (4), 350–60. [DOI] [PubMed] [Google Scholar]
- 12.Zhang M; Gosnell BA; Kelley AE, Intake of high-fat food is selectively enhanced by mu opioid receptor stimulation within the nucleus accumbens. The Journal of pharmacology and experimental therapeutics 1998, 285 (2), 908–14. [PubMed] [Google Scholar]
- 13.Bakshi VP; Kelley AE, Feeding induced by opioid stimulation of the ventral striatum: role of opiate receptor subtypes. The Journal of pharmacology and experimental therapeutics 1993, 265 (3), 1253–60. [PubMed] [Google Scholar]
- 14.Mucha RF; Iversen SD, Increased food intake after opioid microinjections into nucleus accumbens and ventral tegmental area of rat. Brain research 1986, 397 (2), 214–24. [DOI] [PubMed] [Google Scholar]
- 15.Lardeux S; Kim JJ; Nicola SM, Intermittent-access binge consumption of sweet high-fat liquid does not require opioid or dopamine receptors in the nucleus accumbens. Behavioural brain research 2015, 292, 194–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacDonald AF; Billington CJ; Levine AS, Effects of the opioid antagonist naltrexone on feeding induced by DAMGO in the ventral tegmental area and in the nucleus accumbens shell region in the rat. American journal of physiology 2003, 285 (5), R999–R1004. [DOI] [PubMed] [Google Scholar]
- 17.Kelley AE; Bless EP; Swanson CJ, Investigation of the effects of opiate antagonists infused into the nucleus accumbens on feeding and sucrose drinking in rats. The Journal of pharmacology and experimental therapeutics 1996, 278 (3), 1499–507. [PubMed] [Google Scholar]
- 18.Bodnar RJ; Glass MJ; Ragnauth A; Cooper ML, General, mu and kappa opioid antagonists in the nucleus accumbens alter food intake under deprivation, glucoprivic and palatable conditions. Brain research 1995, 700 (1–2), 205–12. [DOI] [PubMed] [Google Scholar]
- 19.Ward HG; Nicklous DM; Aloyo VJ; Simansky KJ, Mu-opioid receptor cellular function in the nucleus accumbens is essential for hedonically driven eating. Eur J Neurosci 2006, 23 (6), 1605–13. [DOI] [PubMed] [Google Scholar]
- 20.Zhang M; Balmadrid C; Kelley AE, Nucleus accumbens opioid, GABaergic, and dopaminergic modulation of palatable food motivation: contrasting effects revealed by a progressive ratio study in the rat. Behavioral neuroscience 2003, 117 (2), 202–11. [DOI] [PubMed] [Google Scholar]
- 21.Shin AC; Pistell PJ; Phifer CB; Berthoud HR, Reversible suppression of food reward behavior by chronic mu-opioid receptor antagonism in the nucleus accumbens. Neuroscience 2010, 170 (2), 580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pecina S; Berridge KC, Dopamine or opioid stimulation of nucleus accumbens similarly amplify cue-triggered ‘wanting’ for reward: entire core and medial shell mapped as substrates for PIT enhancement. Eur J Neurosci 2013, 37 (9), 1529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soderman AR; Unterwald EM, Cocaine reward and hyperactivity in the rat: sites of mu opioid receptor modulation. Neuroscience 2008, 154 (4), 1506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caref K; Nicola SM, Endogenous opioids in the nucleus accumbens promote approach to high-fat food in the absence of caloric need. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshida Y; Koide S; Hirose N; Takada K; Tomiyama K; Koshikawa N; Cools AR, Fentanyl increases dopamine release in rat nucleus accumbens: involvement of mesolimbic mu- and delta-2-opioid receptors. Neuroscience 1999, 92 (4), 1357–65. [DOI] [PubMed] [Google Scholar]
- 26.Okutsu H; Watanabe S; Takahashi I; Aono Y; Saigusa T; Koshikawa N; Cools AR, Endomorphin-2 and endomorphin-1 promote the extracellular amount of accumbal dopamine via nonopioid and mu-opioid receptors, respectively. Neuropsychopharmacology 2006, 31 (2), 375–83. [DOI] [PubMed] [Google Scholar]
- 27.Hirose N; Murakawa K; Takada K; Oi Y; Suzuki T; Nagase H; Cools AR; Koshikawa N, Interactions among mu- and delta-opioid receptors, especially putative delta1- and delta2-opioid receptors, promote dopamine release in the nucleus accumbens. Neuroscience 2005, 135 (1), 213–25. [DOI] [PubMed] [Google Scholar]
- 28.Borg PJ; Taylor DA, Involvement of mu- and delta-opioid receptors in the effects of systemic and locally perfused morphine on extracellular levels of dopamine, DOPAC and HVA in the nucleus accumbens of the halothane-anaesthetized rat. Naunyn Schmiedebergs Arch Pharmacol 1997, 355 (5), 582–8. [DOI] [PubMed] [Google Scholar]
- 29.Hipolito L; Sanchez-Catalan MJ; Zanolini I; Polache A; Granero L, Shell/core differences in mu- and delta-opioid receptor modulation of dopamine efflux in nucleus accumbens. Neuropharmacology 2008, 55 (2), 183–9. [DOI] [PubMed] [Google Scholar]
- 30.Cachope R; Mateo Y; Mathur BN; Irving J; Wang HL; Morales M; Lovinger DM; Cheer JF, Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep 2012, 2 (1), 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fields HL; Margolis EB, Understanding opioid reward. Trends in neurosciences 2015, 38 (4), 217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wenzel JM; Cheer JF, Endocannabinoid Regulation of Reward and Reinforcement through Interaction with Dopamine and Endogenous Opioid Signaling. Neuropsychopharmacology 2018, 43 (1), 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson DL; Venton BJ; Heien ML; Wightman RM, Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin.Chem. 2003, 49 (10), 1763–1773. [DOI] [PubMed] [Google Scholar]
- 34.Spanos M; Gras-Najjar J; Letchworth JM; Sanford AL; Toups JV; Sombers LA, Quantitation of hydrogen peroxide fluctuations and their modulation of dopamine dynamics in the rat dorsal striatum using fast-scan cyclic voltammetry. ACS Chem Neurosci 2013, 4 (5), 782–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson LR; Panda S; Schmidt AC; Sombers LA, Selective and Mechanically Robust Sensors for Electrochemical Measurements of Real-Time Hydrogen Peroxide Dynamics in Vivo. Analytical chemistry 2018, 90 (1), 888–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garris PA; Budygin EA; Phillips PE; Venton BJ; Robinson DL; Bergstrom BP; Rebec GV; Wightman RM, A role for presynaptic mechanisms in the actions of nomifensine and haloperidol. Neuroscience 2003, 118 (3), 819–829. [DOI] [PubMed] [Google Scholar]
- 37.Ferezou I; Hill EL; Cauli B; Gibelin N; Kaneko T; Rossier J; Lambolez B, Extensive overlap of mu-opioid and nicotinic sensitivity in cortical interneurons. Cereb Cortex 2007, 17 (8), 1948–57. [DOI] [PubMed] [Google Scholar]
- 38.Cui Y; Ostlund SB; James AS; Park CS; Ge W; Roberts KW; Mittal N; Murphy NP; Cepeda C; Kieffer BL; Levine MS; Jentsch JD; Walwyn WM; Sun YE; Evans CJ; Maidment NT; Yang XW, Targeted expression of mu-opioid receptors in a subset of striatal direct-pathway neurons restores opiate reward. Nature neuroscience 2014, 17 (2), 254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rice ME; Cragg SJ, Nicotine amplifies reward-related dopamine signals in striatum. Nature neuroscience 2004, 7 (6), 583–4. [DOI] [PubMed] [Google Scholar]
- 40.Goutier W; Lowry JP; McCreary AC; O’Connor JJ, Frequency-Dependent Modulation of Dopamine Release by Nicotine and Dopamine D1 Receptor Ligands: An In Vitro Fast Cyclic Voltammetry Study in Rat Striatum. Neurochem Res 2016, 41 (5), 945–50. [DOI] [PubMed] [Google Scholar]
- 41.Threlfell S; Lalic T; Platt NJ; Jennings KA; Deisseroth K; Cragg SJ, Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 2012, 75 (1), 58–64. [DOI] [PubMed] [Google Scholar]
- 42.Threlfell S; Cragg SJ, Dopamine signaling in dorsal versus ventral striatum: the dynamic role of cholinergic interneurons. Front Syst Neurosci 2011, 5, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yan Z; Surmeier DJ, Muscarinic (m2/m4) receptors reduce N- and P-type Ca2+ currents in rat neostriatal cholinergic interneurons through a fast, membrane-delimited, G-protein pathway. J Neurosci 1996, 16 (8), 2592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W; Basile AS; Gomeza J; Volpicelli LA; Levey AI; Wess J, Characterization of central inhibitory muscarinic autoreceptors by the use of muscarinic acetylcholine receptor knock-out mice. J Neurosci 2002, 22 (5), 1709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pitman KA; Puil E; Borgland SL, GABA(B) modulation of dopamine release in the nucleus accumbens core. Eur J Neurosci 2014, 40 (10), 3472–80. [DOI] [PubMed] [Google Scholar]
- 46.Kaiser S; Wonnacott S, alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Molecular pharmacology 2000, 58 (2), 312–8. [DOI] [PubMed] [Google Scholar]
- 47.Watabe-Uchida M; Zhu L; Ogawa SK; Vamanrao A; Uchida N, Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 2012, 74 (5), 858–73. [DOI] [PubMed] [Google Scholar]
- 48.Beier KT; Steinberg EE; DeLoach KE; Xie S; Miyamichi K; Schwarz L; Gao XJ; Kremer EJ; Malenka RC; Luo L, Circuit Architecture of VTA Dopamine Neurons Revealed by Systematic Input-Output Mapping. Cell 2015, 162 (3), 622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faget L; Osakada F; Duan J; Ressler R; Johnson AB; Proudfoot JA; Yoo JH; Callaway EM; Hnasko TS, Afferent Inputs to Neurotransmitter-Defined Cell Types in the Ventral Tegmental Area. Cell Rep 2016, 15 (12), 2796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morales M; Margolis EB, Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nature reviews 2017, 18 (2), 73–85. [DOI] [PubMed] [Google Scholar]
- 51.Bocklisch C; Pascoli V; Wong JC; House DR; Yvon C; de Roo M; Tan KR; Luscher C, Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 2013, 341 (6153), 1521–5. [DOI] [PubMed] [Google Scholar]
- 52.Xia Y; Driscoll JR; Wilbrecht L; Margolis EB; Fields HL; Hjelmstad GO, Nucleus accumbens medium spiny neurons target non-dopaminergic neurons in the ventral tegmental area. J Neurosci 2011, 31 (21), 7811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson DL; Volz TJ; Schenk JO; Wightman RM, Acute ethanol decreases dopamine transporter velocity in rat striatum: in vivo and in vitro electrochemical measurements. Alcoholism, clinical and experimental research 2005, 29 (5), 746–755. [DOI] [PubMed] [Google Scholar]
- 54.Shnitko TA; Taylor SC; Stringfield SJ; Zandy SL; Cofresi RU; Doherty JM; Lynch WB; Boettiger CA; Gonzales RA; Robinson DL, Acute phenylalanine/tyrosine depletion of phasic dopamine in the rat brain. Psychopharmacology 2016, 233 (11), 2045–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bath BD; Michael DJ; Trafton BJ; Joseph JD; Runnels PL; Wightman RM, Subsecond adsorption and desorption of dopamine at carbon-fiber microelectrodes. Anal.Chem. 2000, 72 (24), 5994–6002. [DOI] [PubMed] [Google Scholar]