

Summary
Obesity induces lipotoxic cardiomyopathy, a condition in which lipid accumulation in cardiomyocytes causes cardiac dysfunction. Here, we show that glycogen synthase kinase-3α (GSK-3α) mediates lipid accumulation in the heart. Fatty acids (FAs) upregulate GSK-3α, which phosphorylates PPARα at Ser280 in the ligand binding domain (LBD). This modification ligand-independently enhances transcription of a subset of PPARα targets, selectively stimulating FAs uptake and storage, but not oxidation, thereby promoting lipid accumulation. Constitutively-active GSK-3α, but not GSK-3β, was sufficient to drive PPARα signaling, while cardiac-specific knockdown of GSK-3α, but not GSK-3β, or replacement of PPARα Ser280 with Ala conferred resistance to lipotoxicity in the heart. Fibrates, PPARα ligands, inhibited phosphorylation of PPARα at Ser280 by inhibiting the interaction of GSK-3α with the LBD of PPARα, thereby reversing lipotoxic cardiomyopathy. These results suggest that GSK-3α promotes lipid anabolism through PPARα-Ser280 phosphorylation, which underlies the development of lipotoxic cardiomyopathy in the context of obesity.
Keywords: GSK-3α, PPARα, fatty acid metabolism, obesity, lipotoxic cardiomyopathy, fibrates
eTOC blurb
Nakamura et al. investigate the mechanisms underlying the development of lipotoxic cardiomyopathy. Fatty acids-induced upregulation of GSK-3α acts as a central regulator of cardiac fatty acid metabolism by stimulating a biased PPARα transcriptional response via Ser280-phosphorylation, favoring fatty acid uptake. A reversal of Ser280-phosphorylation attenuates lipotoxicity.
Graphical Abstract
Introduction
Fatty acids are major substrates for ATP generation in oxidative tissues, including the adult heart (Lopaschuk et al., 2010). Derangement in fatty acid metabolism has significant consequences for cellular functions, leading to organ failure. Decreases in the level of fatty acid oxidation (FAO) are a common feature of heart failure (Neubauer, 2007), a condition in which cardiac output is decreased due to dysfunction or death of individual cardiomyocytes (CMs). In lipotoxicity, an imbalance between uptake or synthesis of fatty acid and its consumption results in intracellular accumulation of lipid intermediates, which induces cellular dysfunction and death in non-adipose tissues, including the kidney, liver, skeletal muscle, and heart (Goldberg et al., 2012). Lipotoxic cardiomyopathy is the major mechanism through which patients with insulin resistance, diabetes and obesity develop cardiac hypertrophy and dysfunction (Schilling and Mann, 2012), and is an important cause of heart failure with preserved ejection fraction (HFpEF). However, how the balance of fatty acid metabolism is disrupted in obesity remains poorly understood.
Peroxisome proliferator-activated receptor α (PPARα) is a nuclear receptor transcription factor that plays a key role in regulating fatty acid metabolism in the heart (Vega and Kelly, 2017). Genetic overexpression of PPARα in the heart mimics the phenotype of lipotoxic cardiomyopathy (Finck et al., 2002) whereas knockdown of PPARα attenuates lipotoxic cardiomyopathy (Finck et al., 2003), suggesting that PPARα plays an important role in the pathogenesis of lipotoxic cardiomyopathy. However, how endogenous PPARα is functionally modulated in the presence of obesity and diabetes and how its modification contributes to lipotoxicity remain poorly understood. In particular, given that PPARα broadly controls expression of genes involved in fatty acid metabolism, including uptake, storage and oxidation, it remains unclear how changes in the level or the activity of PPARα alone can cause the imbalance between the various fatty acid metabolic mechanisms in turn leads to accumulation of lipid intermediates in CMs.
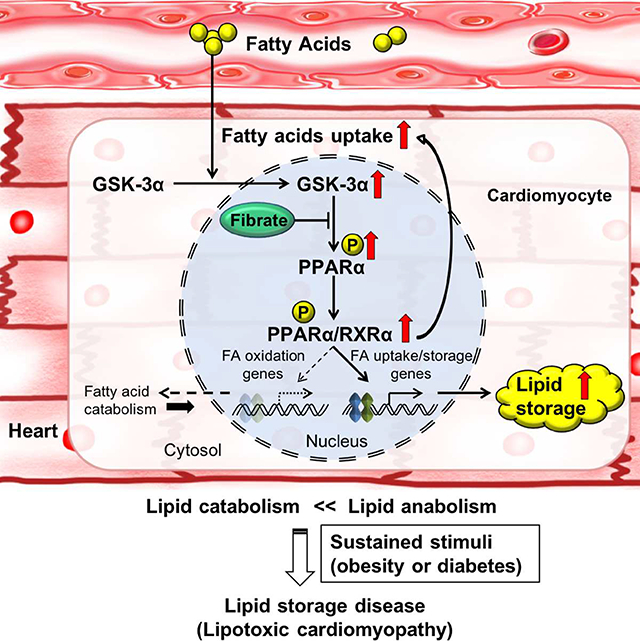
Through our extensive search for the molecular mechanism mediating lipotoxic cardiomyopathy in mouse models of obesity and diabetes induced by high-fat diet (HFD) consumption and genetic alteration, we found that activation of GSK-3α, an isoform of GSK-3, is essential for the induction of lipotoxic cardiomyopathy. Thus, the goal in this study was to elucidate the molecular mechanism by which GSK-3α controls lipid accumulation in CMs in the presence of metabolic syndrome. Our results show that fatty acids upregulate nuclear GSK-3α, which phosphorylates PPARα at Ser280 in the ligand binding domain (LBD). This modification induces transcription of genes involved in fatty acid uptake and storage, but not oxidation. Our data suggest that GSK-3α acts as a fatty acids sensor in CMs, stimulating lipid uptake and promoting its accumulation in CMs through biased activation of PPARα, and that this process underlies the development of lipotoxic cardiomyopathy in obesity and diabetes.
Results
Cardiac-specific haploinsufficiency of GSK-3α ameliorates HFD-induced cardiomyopathy
In order to elucidate changes in the signaling mechanism during the development of lipotoxic cardiomyopathy, we analyzed publicly available gene expression datasets from the hearts of obese (GSE16790) and diabetic mice (GSE36875). In order to distinguish the mechanism of cardiomyopathy caused by obesity or type II diabetes, on which we are focusing, from that caused by type I diabetes, we excluded gene sets upregulated in streptozotocin-induced diabetic hearts (GSE5606). Twenty-six genes were upregulated in the hearts of both obese and diabetic mice (Figure 1A). Consistent with the nature of obesity-related cardiomyopathy, analysis of gene ontology terms showed that the metabolic process (GO: 0008152), containing 13 genes, represented the largest biological process among the 26 overlapping genes, wherein GSK-3α was the only kinase (Figure 1B). We validated the upregulation of GSK-3α in a diet-induced obese model. Obesity induced by HFD (60% kcal from fat) consumption elicited lipotoxic cardiomyopathy with accumulation of intramyocardial lipid in the heart. In line with the paper by Bugger and Abel, we also identified insulin resistance in the heart, as evidenced by decreased Akt phosphorylation in response to insulin (Bugger and Abel, 2009) (Figure S1A). Since Akt, which negatively regulates GSK-3α, is downregulated and GSK-3α is translocated to the nucleus in response to stress (Azoulay-Alfaguter et al., 2011) (Figure S4A), we evaluated the activity of GSK-3α in the nucleus in response to HFD, using immune-complex in vitro kinase assays with recombinant β-catenin as a substrate. GSK-3α was activated by HFD consumption (Figures 1C, 1D). GSK-3α was similarly activated in genetically obese mouse (ob/ob) hearts (Figure S1B). These results suggest that the activity of GSK-3α is significantly increased in the hearts of mice fed a HFD and genetically obese mice.
Figure 1. Cardiac-specific GSK-3α haploinsufficiency ameliorates high-fat diet (HFD)-induced lipotoxic cardiomyopathy.

(A) Area-proportional Venn diagram representing the 26 genes significantly (p < 0.05) upregulated both in ob/ob (GSE16790) and db/db (GSE36875) mouse hearts, but not in streptozotocin-induced diabetic hearts (Type I DM) (GSE5606). (B) Pie chart illustrating the percent composition of Gene Ontology biological processes of the 26 common genes found in (A). Metabolic process (GO: 0008152) contains the largest gene set (13 genes), among which only GSK-3α is a kinase. (C) Immunoblots to evaluate nuclear GSK-3α activity in the hearts of wild-type (WT) mice fed a HFD or normal chow (NC) for the indicated periods. GSK-3α was immunoprecipitated from the nuclear fraction of heart lysates, followed by in vitro kinase assays with recombinant β-catenin. Recombinant GSK-3α protein was used as a positive control and immunoprecipitation with IgG was used as a negative control. (D) Quantification of the nuclear GSK-3α activity in (C) (n = 3). (E to L) GSK-3α cardiac-specific heterozygous knockout (GSK-3α cHKO) mice and heterozygous floxed (control) mice were fed a HFD or NC for 14 weeks. (E) Photograph of the hearts of control and GSK-3α cHKO mice fed a HFD or NC. (F) Left ventricular (LV) weight normalized by tibia length, a marker of cardiac hypertrophy (n = 8 (NC) and 22–24 (HFD)). (G and H) Diastolic function, as indicated by deceleration time (n = 8–15) (G), and the slope of the end-diastolic pressure-volume (PV) relation (EDPVR) (n = 5 (NC) and 9 (HFD)) (H). (I) Lipid accumulation in the hearts (Oil Red O staining, left). Scale bar, 100 μm. Inset scale bar, 20 μm. Quantification of myocardial lipid accumulation (right) (n = 6). (J) Palmitate oxidation in the hearts (n = 8–9 (NC) and 17 (HFD)). (K) Picric acid sirius red (PASR) staining, indicating cardiac fibrosis (left). Scale bar, 100 μm. Percentage of PASR positive areas (right) (n = 4). (L) mRNA expression related to cardiac metabolism, inflammation, and transcription factors in the hearts (n = 6). (M) Gene set enrichment analysis plot of Kyoto encyclopedia of genes and genomes (KEGG). PPAR signaling signatures in GSK-3α S21A knock-in (KI) and WT mice fed NC. NES denotes normalized enrichment score. FDR denotes false discovery rate. Error bars indicate s.e.m. * p<0.05, ** p<0.001. See also Figures S1 and S2 and Table S1.
In order to examine the functional significance of GSK-3α activation in the heart in response to HFD consumption, we fed cardiac-specific GSK-3α heterozygous knockout (GSK-3α cHKO) and heterozygous floxed (control) mice with HFD for up to 14 weeks (Figure S1C). Although there was no difference in body weight gain, food intake or systemic insulin resistance between control and GSK-3α cHKO mice (Figures S1D–F), there was a clear attenuation in HFD-induced cardiac and individual CM hypertrophy in GSK-3α cHKO mice (Figures 1E, 1F, S1G). Left ventricular (LV) systolic function was preserved in both groups (Figure S1H) after HFD consumption. However, HFD induced diastolic dysfunction in control mice, as evidenced by a longer deceleration time (DT) of the transmitral flow observed via Doppler echocardiography, a left upward shift of the slope of the end-diastolic pressure-volume (PV) relation loop (EDPVR), and an increase in Tau in PV loop analyses (Pacher et al., 2008) (Figures 1G, 1H, S1I–K). The HFD-induced diastolic dysfunction was significantly alleviated in GSK-3α cHKO mice. HFD consumption did not affect maximum LV pressure or stroke work in either control or GSK-3a cHKO mice (Figures S1L, S1M). These results indicate that HFD-mediated cardiac hypertrophy (lipotoxic cardiomyopathy) may not be induced by altered blood pressure within 3 months, and that suppression of hypertrophy does not lead to markedly elevated wall stress; thus, the overall function of the heart in GSK-3α cHKO mice is improved. Together, these results suggest that aberrant activation of GSK-3α plays a critical role in the development of cardiomyopathy in response to HFD consumption.
Intramyocardial lipid accumulation, as assessed by Oil Red O staining, and palmitic acid oxidation, assessed using 3H-palmitic acid, were increased in control mouse hearts in response to HFD, but were markedly attenuated in GSK-3α cHKO mice (Figures 1I, 1J). A Seahorse analysis indicated that the mitochondrial FAO rate was also increased in CMs isolated from the hearts of control mice fed HFD but was significantly suppressed in CMs isolated from the hearts of GSK-3α cHKO mice fed HFD (Figures S2A–D). In addition, picric acid Sirius red staining showed reduced cardiac fibrosis in GSK-3α cHKO mice compared to in control mice (Figure 1K). Glycogen storage in the heart was not significantly altered by GSK-3α suppression (Figures S2E, S2F). Notably, GSK-3β cHKO mice developed cardiac systolic dysfunction and more prominent myocardial lipid accumulation than control mice in response to HFD (Figures S2G–K), suggesting that the role of GSK-3 is isoform-dependent. HFD upregulated mRNA expression of genes involved in fatty acid metabolism in control mice, whereas these changes were attenuated in GSK-3α cHKO mice (Figure 1L). In contrast, gene expression of inflammatory cytokines and other transcription factors related to cardiac metabolism did not differ significantly between control and GSK-3α cHKO mice fed a HFD. These results indicate that alleviation of diet-induced cardiomyopathy and metabolic remodeling in GSK-3α cHKO mice may be mediated, at least in part, at the level of transcription of genes involved in fatty acid metabolism.
Since HFD activates GSK-3α, we hypothesized that activation of GSK-3α is sufficient to deregulate expression of fatty acid metabolism-related genes. We analyzed microarray data obtained from the hearts of GSK-3α S21A homozygous knockin (KI) mice harboring constitutively active GSK-3α and wild-type (WT) mice fed normal chow (NC) (Matsuda et al., 2008). Although there was no overt cardiac phenotype at the age of 3 months, the gene set enrichment analysis (GSEA) showed that, in the hearts of GSK-3α S21A KI mice, PPAR signaling is the most significantly enriched among the upregulated gene sets related to energy metabolism (Figure 1M, Table 1 and Table S1). In contrast, GSK-3β S9A homozygous KI mice, harboring constitutively active GSK-3β, displayed downregulation of genes involved in cardiac fatty acid metabolism (Figure S2L). One PPAR target gene, CD36, was significantly increased in GSK-3α S21A KI mice (Figure S2M). Consistent with the increased CD36 mRNA expression, both palmitate uptake into the heart and intramyocardial lipid accumulation were increased in GSK-3α S21A KI mice (Figures S2N, S2O). These results are also consistent with the absence of an increase in expression of CD36 in response to HFD consumption in GSK-3α cHKO mice (Figure 1L), which could partially explain the lower FAO, potentially resulting from less fatty acid import, in GSK-3α cHKO mice. Taken together, these results indicate that upregulation of GSK-3α, but not GSK-3β, positively regulates lipid metabolism through PPAR modification in the heart.
Table 1.
Top 10 upregulated KEGG gene sets in GSK-3α KI (vs WT) mouse heart.
| GS follow link to MSigDB | SIZE | ES | NES | NOM p-val | FDR q-val | |
|---|---|---|---|---|---|---|
| 1 | KEGG_COMPLEMENT_AND_COAGULATION_CASCADES | 45 | 0.76 | 2.52 | 0 | 0 |
| 2 | KEGG_PPAR_SIGNALING_PATHWAY | 60 | 0.66 | 2.31 | 0 | 0.001 |
| 3 | KEGG_GLYCINE_SERINE_AND THREONINE_METABOLISM | 28 | 0.74 | 2.22 | 0.001 | 0.001 |
| 4 | KEGG_CYSTEINE_AND_METHIONINE_METABOLISM | 29 | 0.72 | 2.15 | 0 | 0.002 |
| 5 | KEGG_SPLICEOSOME | 90 | 0.48 | 1.77 | 0 | 0.131 |
| 6 | KEGG_PHENYLALANINE_METABOLISM | 16 | 0.65 | 1.75 | 0.019 | 0.135 |
| 7 | KEGG_TRYPTOPHAN METABOLISM | 33 | 0.56 | 1.74 | 0.006 | 0.131 |
| 8 | KEGG_BASAL_TRANSCRIPTION_FACTORS | 30 | 0.57 | 1.72 | 0.012 | 0.131 |
| 9 | KEGG_PROTEASOME | 42 | 0.53 | 1.72 | 0.011 | 0.123 |
| 10 | KEGG_AMINO_SUGAR_AND_NUCLEOTIDE_SUGAR_METABOLISM | 42 | 0.54 | 1.71 | 0.001 | 0.115 |
The PPAR signaling pathway is most significantly enriched among energy metabolism-related gene sets in the hearts of GSK-3α knock-in (KI) mice compared to wild-type (WT) mice at three months of age, a time point before the development of cardiomyopathy in GSK-3α KI mice.
GSK-3α phosphorylates PPARα at Ser280, located in the ligand binding domain
Since the PPARα transcription factor plays a major role in the regulation of fatty acid metabolism in the heart, we examined the physical interaction between GSK-3α and PPARα. YFP-tagged PPARα pulled down endogenous GSK-3α, but not GSK-3β, in CMs (Figures 2A, S3A). Anti-FLAG immunoprecipitation assays showed that GSK-3α and PPARα interact with one another in mouse hearts overexpressing FLAG-tagged PPARα (Figure 2B). Co-immunoprecipitation assays also revealed that endogenous GSK-3α and endogenous PPARα interact with one another in CMs in vitro (Figure 2C). In addition, in vitro binding assays using recombinant GST-PPARα and recombinant GSK-3α or GSK-3β showed that GSK-3α, but not GSK-3β, directly interacts with PPARα (Figures 2D, S3B). We further evaluated which amino acids of PPARα are involved in the interaction with endogenous GSK-3α, using truncated recombinant GST-PPARα and CM lysates (Figures 2E, 2F). Although full-length (FL) and N-terminally truncated (T5) GST-PPARα were able to pull down endogenous GSK-3α, C-terminally truncated GST-PPARα (T1-T4) and GST alone failed to pull down GSK-3α, suggesting that the LBD of PPARα interacts with GSK-3α (Figure 2G).
Figure 2. GSK-3α physically interacts with and phosphorylates PPARα at Ser280 in cardiomyocytes (CMs) and in the heart.

(A and B) Immunoprecipitation assays to test the interaction between endogenous GSK-3α and exogenously expressed PPARα. YFP-tagged PPARα or FLAG-tagged PPARα was overexpressed in CMs using adenovirus (A) or in transgenic mouse hearts under the control of the αMHC-promoter (B), respectively. (C) Co-immunoprecipitation assays testing the interaction between endogenous GSK-3α and endogenous PPARα in CMs. (D) In vitro binding assays testing the direct interaction between recombinant (r) GSK-3α and rPPARα. (E to G) Immunoprecipitation assays to identify the amino acids in PPARα responsible for the interaction with endogenous GSK-3α. (E) Schematic representation of rGST-fused PPARα fragments. (F) Schema of the immunoprecipitation assays. rGST-fused-PPARα-full length (FL) or truncated PPARα (T1 to 5) was incubated with lysates extracted from cultured CMs, followed by pull-down with glutathione-sepharose and immunoblotting with anti-GSK-3α antibody. (G) Coomassie Brilliant Blue staining of rGST-PPARα-FL or truncated rGST-PPARα (T1 to T5) (left). Immunoblots testing the binding of endogenous GSK-3α to rGST-PPARα-FL or T1 to T5 (right). (H) Mass spectrometry analysis of the rGST-PPARα protein phosphorylated by GSK-3α in a kinase reaction. The MS/MS spectrum of the PPARα residue corresponding to Ser280 was increased at 80 Da, indicating phosphorylation. (I) Immunoblots showing Ser280 phosphorylation of endogenous PPARα in the hearts of WT mice fed a high-fat diet (HFD) or normal chow (NC) for 3 weeks. α-sarcomeric actinin was used as a loading control. (J) Immunoblots showing pPPARα (S280) in the hearts of control or GSK-3α cHKO mice fed a HFD for the indicated period. See also Figure S3.
We then hypothesized that GSK-3α phosphorylates PPARα. In vitro kinase assays showed that both GST-PPARα-FL and a PPARα fragment (amino acids 170–430) containing the LBD are phosphorylated by GSK-3α (Figure S3C). Furthermore, mass spectrometry analysis showed that Ser280 of PPARα, located in the LBD and highly conserved in PPARα across species, is phosphorylated by GSK-3α (Figures 2H, S3D). We therefore generated an antibody against PPARα phosphorylated at Ser280. This antibody effectively detected GST-tagged WT PPARα, but not GST-tagged PPARα-S280A, a non-phosphorylatable mutant, after in vitro kinase assays with GSK-3α (Figures S3E, S3F). Using this antibody, we confirmed that YFP-tagged PPARα-WT, but not PPARα-S280A, was phosphorylated in the presence of GSK-3α overexpression but not GSK-3α knockdown in CMs (Figure S3G). YFP-tagged PPARα-S280D, a phospho-mimetic mutant, was detected by the phospho-specific antibody even in cells in which GSK-3α was knocked down. Immunoblot analyses using this antibody showed that Ser280 phosphorylation of endogenous PPARα was increased in control mouse hearts in response to HFD consumption (Figures 2I, S3H), whereas there was significantly less Ser280 phosphorylation in GSK-3α cHKO mouse hearts (Figures 2J, S3I). Taken together, these data indicate that GSK-3α directly interacts with PPARα at the LBD and phosphorylates PPARα at Ser280 in the heart, and that Ser280 phosphorylation of PPARα is increased by HFD consumption through GSK-3α activation.
GSK-3α-mediated phosphorylation enhances PPARα transcriptional activity
We examined the functional role of PPARα-Ser280 phosphorylation. First, we investigated whether the phosphorylation status of endogenous PPARα is altered by fatty acids through GSK-3α in CMs in vitro. Bovine serum albumin (BSA)-conjugated palmitic acid, a major saturated fatty acid in mammals, increased the nuclear localization of GSK-3α in CMs within 1 hour, whereas GSK-3β was located diffusely in CMs, as shown by immunofluorescence staining (Figure S4A). The activity of GSK-3α in the nucleus was increased in response to palmitic acid, as determined by immune-complex in vitro kinase assays of the nuclear fraction of CMs (Figure S4B). In association with the enhanced GSK-3α activity, PPARα-Ser280 phosphorylation in the nuclear fraction was increased by palmitic acid in a dose-dependent manner (Figure 3A), an effect that was suppressed in the presence of adenovirus-mediated knockdown of GSK-3α (adenovirus (ad)-short-hairpin (sh)RNA-GSK-3α) (Figures 3B, S4C). Since saturated fatty acids, such as palmitic acid, monounsaturated fatty acids, such as oleic acid, and polyunsaturated fatty acids, such as linoleic acid, have distinct effects on metabolic diseases (Roberts et al., 2014), we examined the impact of unsaturated fatty acids and a fatty acids cocktail containing palmitic acid, oleic acid, and linoleic acid, on the GSK-3α-PPARα axis. Both oleic acid and the cocktail induced nuclear localization of GSK-3α and PPARα-Ser280 phosphorylation, an effect similar to that of palmitic acid. On the other hand, GSK-3α localization was unaffected and PPARα-Ser280 phosphorylation decreased in the presence of linoleic acid (Figures 3C, S4D–F). These results suggest that palmitic acid, oleic acid, and the fatty acids cocktail, but not linoleic acid, increase PPARα-Ser280 phosphorylation through GSK-3α in the CM nucleus in vitro.
Figure 3. Ser280 phosphorylation stimulates a subset of PPARα targets and promotes fatty acid uptake in cardiomyocytes.

(A) Immunoblots showing subcellular localization and expression of p-PPARα (Ser280) in cultured cardiomyocytes (CMs) treated with BSA-palmitic acid (PA) (0–500 μM) for 9 hours. (B) Immunoblots examining the involvement of GSK-3α in the BSA-PA-induced increase in PPARα phosphorylation at Ser280 in CMs. CMs transduced with adenovirus harboring shRNA-GSK-3α or scramble were treated with the indicated concentrations of PA, followed by nuclear extraction and immunoblotting. (C) Immunoblots showing the expression of p-PPARα (Ser280) in the nucleus of CMs treated with BSA-fatty acid cocktail for 9 hours. (D) Clustergram heat map of RNA-sequencing data. H9C2 cells were transduced with PPARα-wild type (WT), -S280A (SA) or -S280D (SD) mutant, or YFP alone as a control. Gene sets having 1) a fold difference of 1.5 or more between SA and SD and 2) a WT expression level located between SD and SA are shown in the heat map. (E) Gene set enrichment analysis plots of PPARα-SD (vs WT) showing upregulated signatures related to energy metabolism. Gene expression was determined by RNA-seq data. (F) qRT-PCR validation of expression of genes related to lipid metabolism and oxidative phosphorylation in CMs transduced with adenovirus harboring a YFP-PPARα-S280A or -S280D mutant or YFP alone as a control (n = 8–10). (G) Fatty acid uptake into CMs transduced with the indicated adenovirus. 3H-palmitate (left) and combined 3H-palmitate and 3H-oleate (right) incorporation into CMs was measured by scintillation counting (n = 4–5). (H) Relative oxygen consumption rate (OCR) of CMs transduced with the indicated adenovirus was measured in a 24-well Seahorse experiment in the presence of 500 μM of BSA-PA. Mitochondrial fatty acid oxidation (FAO) was evaluated by etomoxir-inhibitable OCR. Histograms show FAO rate (the ratio of FAO versus total OCR) (n = 5). (I) FAO rate in CMs transduced with the indicated adenovirus, measured in a 96-well Seahorse experiment in the presence of 500 μM of BSA-fatty acid cocktail (n = 8). (J and K) Oil Red O staining of CMs transduced with the indicated adenovirus in the presence or absence of BSA-PA (500 μM) (n = 5) (J) or in the presence of BSA-fatty acid cocktail (500 μM) (n = 6) (K). BSA alone was used as a control (PA 0 μM). Scale bars, 100 μm (upper panel) and 20 μm (lower panel). Error bars indicate s.e.m. * p < 0.05, ** p < 0.001. See also Figure S4 and Table S2.
Next, we examined the effect of Ser280-phosphorylation on PPARα transcriptional activity in response to palmitic acid in CMs. Knockdown of GSK-3α by ad-shRNA-GSK-3α abolished the palmitic acid-induced increase in PPARα activity (Figure S4G). The effect of PPARα-Ser280 phosphorylation on its activity was further elucidated using PPARα-WT, PPARα-S280A and PPARα-S280D. Whereas PPARα reporter activity dose-dependently increased in response to palmitic acid in PPARα-WT-transduced H9C2 cells, it was significantly attenuated in PPARα-S280A-transduced H9C2 cells (Figure S4H). Conversely, PPARα-S280D increased transcriptional activity independently of palmitic acid. These results suggest that GSK-3α positively regulates PPARα activity through Ser280 phosphorylation. Since PPARγ, another PPAR isoform, is phosphorylated at Ser273 by ERK/CDK5 in adipose tissue in obesity (Banks et al., 2015; Choi et al., 2010), we evaluated the phosphorylation status of PPARγ at Ser273. Neither palmitic acid nor changes in GSK-3α expression altered the PPARγ-Ser273 phosphorylation level in CMs (Figures S4I, S4J).
To demonstrate the functional role of PPARα phosphorylation, we performed an RNA-sequencing analysis of H9C2 cells transduced with PPARα-WT, PPARα-S280A, PPARα-S280D or YFP as a control. The results indicated that PPARα phosphorylation induces global changes in gene expression (Figure 3D). Ingenuity Pathway Analysis showed that phosphorylation of PPARα significantly alters energy metabolism (Figure S4K). Furthermore, GSEAs showed that fatty acid and glycerolipid metabolism-related gene sets are significantly enriched in PPARα-S280D-transduced cells relative to PPARα-WT (Figure 3E and Table S2). Gene expression changes were further examined in CMs by quantitative RT-PCR (Figure 3F). PPARα-S280D increased expression of genes related to fatty acid uptake and pyruvate dehydrogenase kinase 4 (PDK4), which inactivates the pyruvate dehydrogenase (PDH) complex and inhibits glucose oxidation, but genes related to β-oxidation and mitochondrial oxidative phosphorylation were mostly either unchanged or decreased (Figure 3F). These results suggest that PPARα-Ser280 phosphorylation upregulates expression of genes involved in fatty acid uptake and storage but not utilization.
We next investigated the functional significance of PPARα phosphorylation in CMs. Consistent with the gene expression patterns, transduction with PPARα-S280D markedly enhanced uptake of BSA-palmitic acid and BSA-fatty acids cocktail into CMs compared to transduction with either PPARα-WT or PPARα-S280A (Figure 3G). The oxygen consumption rate (OCR) inhibitable with etomoxir, a Cpt1 inhibitor, was evaluated as a measure of mitochondrial FAO in CMs using a Seahorse analyzer (Kim et al., 2013). Despite a significant increase in fatty acid uptake in CMs expressing the PPARα-S280D mutant compared to in those expressing PPARα-WT, FAO was not further increased in PPARα-S280D-transduced CMs compared to in PPARα-WT-transduced CMs (Figures 3H, 3I, S4L), suggesting that CMs expressing PPARα-S280D accumulate more lipids than those expressing PPARα-WT (Figures 3J, 3K, S4M). Taken together, these results suggest that Ser280-phosphorylation of PPARα may shift fatty acid metabolism to fatty acid uptake and storage, thereby promoting lipid accumulation.
Phosphorylation of PPARα at Ser280 increases both interaction between PPARα and RXRα and specific DNA binding
We investigated the mechanism by which Ser280-phosphorylation enhances PPARα activity. YFP-tagged PPARα-S280A and -S280D mutants were both found primarily in the nucleus (Figure S5A), indicating that Ser280 phosphorylation does not affect the nuclear localization of PPARα. PPARα heterodimerizes with retinoid X receptor (RXR) and binds to the PPAR response element (PPRE) to activate transcription (Evans and Mangelsdorf, 2014). Immunoprecipitation assays showed that heterodimerization with RXRα occurs more efficiently in PPARα-S280D than in PPARα-WT or PPARα-S280A (Figure 4A). Although PPARα activity can be altered by recruitment of Sirt1 to PPARα (Oka et al., 2011), interaction with Sirt1 was phosphorylation-independent (Figure S5B). According to the three-dimensional structure of PPARα previously analyzed by X-ray crystallography (Xu et al., 2002), Ser280 is located on helix 3 near His440 and Lys448/449, basic residues on helix 11 across the ligand binding pocket (Figure S5C). Thus, we predict that Ser280-phosphorylation shortens the distance between Ser280 and His440 or Lys448/449 through increased electrostatic interaction, which could allow the activating function-2 (AF-2) on helix 12 to adopt a more stable and active conformation in an agonist-independent manner (Figures S5D). In fact, in vitro PPARα reporter assays showed that combined mutations replacing His440 and Lys448/449 with uncharged amino acids suppressed the activity of PPARα-S280D to a level similar to that of PPARα-WT (Figures 4B, S5E), consistent with the aforementioned hypothesis.
Figure 4. PPARα-Ser280 phosphorylation enhances both interaction with RXRα and PPRE binding.

(A) Representative immunoblots showing the interaction between YFP-PPARα-wild type (WT), -S280D (SD) or -S280A (SA) mutant and RXRα in cardiomyocytes (CMs) in vitro. YFP alone was used as a control. (B) PPRE-luciferase reporter assay using a series of alanine mutations to evaluate the effect of the indicated basic residues on the activity of PPARα-S280D (n = 12). ## p<0.001 compared to PPARα-S280D. (C) Chromatin immunoprecipitation (ChIP) assays using CMs transduced with adenovirus (Ad)-YFP-PPARα-WT, -SD, -SA, or YFP alone as a control. DNA was amplified by PCR with specific primers flanking the promoter of the indicated genes containing the PPARα-binding motif. PCR using input DNA as template served as an internal control. The data shown are representative of three independent experiments. (D) ChIP assays using specific primers flanking the promoter of the indicated genes containing the PPARα-binding motif. The data shown are representative of three independent experiments. (E) Double-stranded oligo pull-down assays, using biotinylated oligos containing the specific PPRE sequences in the indicated gene promoters. Recombinant GST-PPARα-WT or -SA was subjected to in vitro kinase assays using recombinant GSK-3α prior to the oligo pull-down assays. An oligo containing a PPRE with mutations in four base pairs was used as a negative control. (F) Venn diagram showing the number of genes in the rat genome containing the specific PPRE/DR1 motif (shown in Figure S5F) in their promoters. The numbers of overlapping genes in the Venn diagram (red circle) were significantly different (p=0.0002, Fisher’s exact test). See also Figure S5.
Next, we asked whether increased association of PPARα-S280D with RXRα enhances DNA binding. We compared the chromatin association of the PPARα-WT, PPARα-S280A, and PPARα-S280D alleles using chromatin immunoprecipitation assays. PPARα-S280D exhibited enhanced chromatin association with the promoters of genes involved in fatty acid uptake and transport in CMs compared to PPARα-WT, whereas PPARα-S280A exhibited less chromatin association than PPARα-WT (Figures 4C, S5F). However, DNA binding of PPARα-S280D was unchanged or decreased compared to PPARα-WT and PPARα-S280A when we used primers for the promoters of genes encoding mitochondrial FAO proteins (Figures 4D, S5F).
We further investigated the underlying mechanisms by which Ser280-phosphorylated PPARα preferentially stimulates fatty acid uptake-related genes rather than oxidation-related genes. First, we evaluated whether the binding affinity of PPARα to the PPRE is altered by the phosphorylation, using a double-stranded oligo pull-down assay. The oligos containing the PPRE/DR1 sequences in the uptake-related gene promoters were able to pull down phosphorylated recombinant GST-PPARα to a greater extent than those containing the PPRE/DR1 sequences in the oxidation-related gene promoters (Figure 4E). On the other hand, the binding ability of PPARα to the PPRE sequences of the oxidation-related genes was higher when PPARα was not phosphorylated at Ser280 (recombinant GST-PPARα-S280A). These results suggest that phosphorylation at Ser280 is sufficient to change the DNA binding preference of PPARα, even in the absence of other proteins.
In order to test whether differences in the PPRE/DR1 sequence confer a preference for recruiting either Ser280-phosphorylated or unphosphorylated PPARα and, consequently, selective control of fatty acid uptake vs oxidation, we first inspected PPRE/DR1 sequences in the promoters of genes upregulated by PPARα-S280D vs S280A to generate PPRE/DR1 motifs (Figure S5G). The genes identified by the PPARα-S280D-related PPRE/DR1 motif were more frequently observed in the gene set identified by the fatty acid uptake-related PPRE/DR1 motif than in that associated with the oxidation-related PPRE/DR1 motif (Figure 4F). These results indicate that the selective control of fatty acid uptake vs oxidation by PPARα-Ser280 phosphorylation is modulated at least in part by the PPRE/DR1 sequence. Together, these findings suggest that GSK-3α-mediated phosphorylation of PPARα at Ser280 increases the interaction between PPARα and RXRα and the binding of PPARα to specific PPREs, thereby stimulating a subset of PPARα target genes favoring fatty acid uptake and storage (Figure S5H).
PPARα phosphorylation is critical for HFD-induced lipid derangement in the heart
In order to demonstrate the functional significance of PPARα-Ser280 phosphorylation in the heart in vivo in response to HFD, we generated KI mice in which the Ser280 residue in PPARα was replaced with Ala (PPARα-S280A KI mice) on the C57BL/6J background, using homologous recombination in ES cells (Figures S6A–D). Since homozygous KI mice were embryonic lethal, we used heterozygous KI (het KI) mice for further characterization. The cardiac phenotype of the het KI mice was normal at 18 weeks of age. We first evaluated how the lack of PPARα-Ser280 phosphorylation affects metabolites in the heart at baseline by GC/MS-based metabolomic analysis. Glucose metabolites and branched-chain amino acids were similar in WT and het KI mice, whereas myocardial levels of free fatty acids were decreased in het KI mice, accompanied by decreases in fumaric acid and malic acid (Figures 5A, 5B, and Table S3). In order to test whether PPARα-Ser280 phosphorylation mediates HFD-induced lipotoxicity in the heart, het KI and WT mice were subjected to HFD feeding for 8 weeks. Although HFD induced Ser280 phosphorylation of PPARα in WT mice, the phosphorylation was reduced in het KI mice (Figure S6E). Cardiac hypertrophy induced by HFD was ameliorated in het KI mice (Figure 5C). Echocardiography showed a preserved ejection fraction in both WT and het KI mice in both the presence and absence of HFD (Figure S6F), while the HFD-induced diastolic dysfunction observed in WT mice was significantly attenuated in het KI mice (Figures 5D, S6G–J). Lipid accumulation was also significantly attenuated in het KI mice (Figure 5E). These results suggest that a HFD-induced increase in PPARα-Ser280 phosphorylation plays a critical role in the development of lipotoxic cardiomyopathy.
Figure 5. Phosphorylation of PPARα at Ser280 is critical for the development of lipotoxic cardiomyopathy.

(A) Heatmap of metabolites associated with fatty acids/glycerol, glycolysis and BCAA metabolism in the hearts of wild-type (WT) and heterozygous PPARα-S280A knock-in (KI) mice at baseline, measured by metabolomics. (B) Histograms showing myocardial free fatty acids in the hearts of WT and heterozygous PPARα-S280A KI mice (n = 5). (C to E) WT and heterozygous PPARα-S280A KI mice were fed a high-fat diet (HFD) or normal chow (NC) for 8 weeks. (C) Left ventricular (LV) weight normalized by tibia length (n = 5). (D) The slope of the end-diastolic pressure-volume (PV) relation (EDPVR), a marker of diastolic function (n = 5–8). (E) Oil Red O staining of the heart sections (left). Scale bar, 50 μm. Quantification of myocardial lipid accumulation (right) (n = 6). (F to M) Either PPARα-WT or PPARα-S280D (SD) mutant was expressed in the hearts of WT mice (C57BL/6J background) fed NC for 8 weeks via adeno-associated virus (AAV)-mediated gene delivery. AAV-empty injection (Emp or Empty) was performed as a control. (F) Representative immunoblots showing the expressions of p-PPARα (Ser280), PPARα-S280D and total PPARα in the heart and skeletal muscle. Histogram indicates the expression levels relative to Histone H3 (n = 8). (G) LV weight normalized by tibia length (n = 5–7). (H) LV posterior wall thickness on diastole (LVPWd), a marker of hypertrophy (n = 5–6). (I) Deceleration time, a marker of diastolic function (n = 5–6). (J) The slopes of EDPVR (left) and Tau (right), both markers of diastolic function (n = 4–6). (K) Fatty acid oxidation (FAO) rate in CMs isolated from the hearts of mice transduced with the indicated AAV (n = 5). (L) Oil Red O staining of the heart sections (left). Scale bars, 50 μm. Quantification of myocardial lipid accumulation (right) (n = 4–6). (M) Picric acid sirius red (PASR) staining indicating cardiac fibrosis in the heart sections (left) and the percentage of PASR positive areas (right). Scale bars, 200 μm (n = 4–7). Error bars indicate s.e.m. * p<0.05, ** p<0.001. See also Figure S6 and Table S3.
Next, we asked whether an increase in PPARα-Ser280 phosphorylation is sufficient to induce cardiac lipotoxicity. Either PPARα-WT or PPARα-S280D was expressed in the hearts of WT mice (C57BL/6J background) fed NC for 8 weeks using adeno-associated virus (AAV)-DJ/8-mediated gene delivery (Figure 5F). AAV-empty injection was performed as a control. Although no significant difference in body weight was observed among the groups (Figure S6K), PPARα-S280D-expressing mice exhibited cardiac hypertrophy and diastolic dysfunction with preserved ejection fraction (Figures 5G–J, S6L–N). Lung weight/body weight was increased in PPARα-S280D-expressing mice, suggesting that PPARα phosphorylation leads to the development of HFpEF (Figure S6K). Despite an increase in FAO rate (Figures 5K, S6O), PPARα-S280D induced cardiac lipid accumulation to a markedly greater degree than PPARα-WT or AAV-empty injection, accompanied by increased cardiac fibrosis (Figures 5L, 5M). These results suggest that PPARα phosphorylation at Ser280 is sufficient to induce lipid accumulation and cardiomyopathy.
PPARα ligands inhibit GSK-3α-mediated phosphorylation of PPARα
Fenofibrate is a US Food and Drug Administration (FDA)-approved PPARα agonist. Given that GSK-3α interacts with and phosphorylates PPARα in the LBD, we asked whether fenofibrate alters the status of Ser280 phosphorylation. Interestingly, fenofibrate inhibited PPARα-Ser280 phosphorylation in a dose-dependent manner in CMs in the presence of palmitic acid or the fatty acids cocktail (Figures 6A, S7A, S7B). Another PPARα agonist, WY-14643, also inhibited GSK-3α-mediated PPARα-Ser280 phosphorylation in vitro, as well as palmitic acid-induced Ser280 phosphorylation in cultured CMs (Figures S7C, S7D).
Figure 6. Fenofibrate, a PPARα ligand, inhibits GSK-3α-mediated PPARα-Ser280 phosphorylation, thereby ameliorating lipotoxic cardiomyopathy.

(A) Immunoblots examining the effect of fenofibrate, a PPARα agonist, on PPARα-Ser280 phosphorylation in cardiomyocytes (CMs) in the presence or absence of 500 μM of BSA-palmitic acid (PA). (B to E) Wild-type (WT) mice were fed a high-fat diet (HFD) in the presence or absence of fenofibrate (Feno) for the indicated periods, as shown in Figure S7C. (B) Immunoblots examining the effect of fenofibrate on PPARα-Ser280 phosphorylation in the hearts. (C) Left ventricular (LV) weight normalized by tibia length (n = 8–12). (D) The slope of the end-diastolic pressure-volume (PV) relation (EDPVR), a marker of diastolic function (n = 5–11). (E) Oil Red O staining of heart sections after 14 weeks of HFD in the presence or absence of fenofibrate (left). Scale bar, 100 μm. Quantification of myocardial lipid accumulation (right) (n = 5). (F) The differential effect of fenofibrate on PPRE-luciferase reporter activity in H9C2 cells transduced with PPARα-WT or PPARα-S280D mutant in the presence of a high concentration of fatty acid (500 μM of PA) or BSA control. YFP alone was used for background extraction (n = 6). (G) Fatty acid oxidation (FAO) rate in CMs in the presence of 50 μM or 500 μM of PA. Total oxygen consumption rate (OCR) in CMs transduced with the indicated adenoviruses was measured by 96-well Seahorse experiment and mitochondrial FAO was evaluated by etomoxir-inhibitable OCR (n = 18–23 (WT) and 6–7 (S280D)). (H) Oil Red O staining of rat neonatal CMs transduced with the indicated adenovirus in the presence or absence of fenofibrate with 50 μM or 500 μM of BSA-PA (left). Scale bar, 50 μm. Quantification of myocardial lipid accumulation (right) (n = 6). (I and J) Immunoprecipitation assays showing the interaction between endogenous GSK-3α and YFP-PPARα or YFP alone as a control in CMs treated with 100 μM or 500 μM of BSA-PA in the presence or absence of 10 μM of fenofibrate (I) and quantification of the data (n = 4) (J). (K) Proximity Ligation Assay (PLA) showing the in situ interaction between endogenous GSK-3α and YFP-PPARα in CMs treated with 100 μM or 500 μM of PA in the presence or absence of 10 μM of fenofibrate. PLA was performed using anti-GSK-3α and anti-GFP-YFP antibodies. Red color indicates localization in close proximity. Scale bar, 10 μm. (L) Immunoblots examining GSK-3α activity in failing human hearts in the presence (n = 7) or absence (n = 14) of diabetes. See also Figure S7 and Table S4.
We also tested whether fenofibrate inhibits PPARα-Ser280 phosphorylation in the heart in vivo and whether it ameliorates HFD-induced lipid dysregulation and cardiac dysfunction. Mice were fed ad libitum with HFD in the presence or absence of 0.2% (wt/wt) fenofibrate (Haemmerle et al., 2011) for the indicated periods (Figure S7E). Consistent with the in vitro results, fenofibrate treatment suppressed HFD-induced PPARα-Ser280 phosphorylation in the heart (Figures 6B, S7F). Systolic function was preserved in both the control and fenofibrate groups (Figure S7G), but fenofibrate significantly attenuated cardiac hypertrophy and diastolic dysfunction (Figures 6C–D, S7H–K). Although fenofibrate decreased palmitate oxidation in the hearts of mice fed a HFD (Figure S7L), it significantly suppressed HFD-induced lipid accumulation in the heart (Figure 6E).
In order to test whether fenofibrate directly affects lipid metabolism in CMs, in vitro experiments were conducted. Fenofibrate enhanced the transcriptional activity of PPARα in both the absence and presence of low concentrations of palmitic acid in CMs (Figure S7M), confirming the role of fenofibrate as a PPARα agonist. However, fenofibrate decreased fatty acid-induced PPARα activation in the presence of high concentrations of palmitic acid (Figure S7M). Although fenofibrate suppressed PPARα reporter gene activity in response to a high concentration of palmitic acid in H9C2 cells transduced with PPARα-WT, it failed to suppress the PPARα activity in those transduced with PPARα-S280D (Figure 6F), suggesting that fenofibrate-induced suppression of PPARα activity in the presence of a high concentration of palmitic acid is mediated through suppression of PPARα-Ser280 phosphorylation. To further demonstrate the functional alterations in lipid metabolism in response to fenofibrate, CMs transduced with either PPARα-WT or PPARα-S280D were treated with either a low (50 μM) or high (500 μM) concentration of palmitic acid or the fatty acid cocktail. Seahorse analyses showed that fenofibrate suppressed the FAO rate at high, but not low, concentrations of fatty acids in CMs expressing PPARα-WT. However, fenofibrate did not affect the mitochondrial FAO rate at either low or high concentrations of fatty acids in CMs expressing PPARα-S280D (Figure 6G, data using the cocktail not shown). Similarly, although intracellular lipid accumulation, evaluated with Oil Red O staining, induced by a high concentration of palmitic acid was suppressed by fenofibrate in CMs expressing PPARα-WT, it was not affected by fenofibrate in CMs expressing PPARα-Ser280D (Figure 6H). These results indicate that fenofibrate acts as a PPARα agonist at low concentrations of fatty acids but suppresses fatty acid metabolism in the presence of high concentrations of fatty acids, by inhibiting PPARα-Ser280 phosphorylation (Figure S7N).
Next, we examined the mechanisms by which fenofibrate decreases Ser280 phosphorylation. GSK-3α and YFP-tagged PPARα physically interact with one another in a palmitic acid concentration-dependent manner, as evaluated with pull-down assays (Figures 6I, 6J) and in situ proximity ligation assays (Figure 6K). Fenofibrate inhibited the interaction between endogenous GSK-3α and YFP-tagged PPARα in CMs. Taken together, these results suggest that fenofibrate negatively regulates PPARα phosphorylation in CMs by inhibiting the interaction between GSK-3α and PPARα, thereby suppressing PPARα activity and normalizing lipid derangement in the heart in the presence of high concentrations of fatty acid.
Finally, we examined GSK-3α activity in human failing hearts. Diabetic patients showed a reduction in the ratio of Ser21-phosphorylated versus total GSK-3α (Figures 6L, S7O and Table S4), indicating enhanced GSK-3α activity in the presence of diabetes in human hearts.
Discussion
In this study, we show that GSK-3α acts as a key kinase controlling lipid uptake and storage through PPARα phosphorylation. Fatty acids upregulate nuclear GSK-3α, which phosphorylates PPARα at Ser280, selectively stimulating transcription of genes involved in fatty acid uptake and storage without affecting those involved in FAO. The resultant imbalance between lipid uptake and consumption leads to intracellular lipid accumulation in metabolic syndrome. We propose that the GSK-3α-PPARα pathway is an essential mediator of fatty acid uptake/storage.
Ligand-independent PPARα activation through Ser280 phosphorylation
Our results suggest that Ser280 phosphorylation stimulates PPARα function independently of ligands. The Ser280 residue of PPARα is located in helix 3 (H3) within the LBD (Zoete et al., 2007). Based on the crystal structure of the PPARα LBD (Xu et al., 2002) and PPRE luciferase reporter assays, we predict that phosphorylation of PPARα-Ser280 enhances its electrostatic interaction with His440 and Lys448/449, basic residues on helix 11 (H11), thereby shortening the distance between H3 and H11 and allowing the adjacent helix 12 (H12), containing AF-2, to fold up against the LBD core, creating a lid over the ligand-binding pocket even in the absence of endogenous ligands. PPARα-Ser280 phosphorylation also enhances the binding of PPARα to the PPRE in the promoters of genes involved in fatty acid uptake but not mitochondrial FAO. Thus, it is likely that Ser280 phosphorylation affects the function of PPARα through multiple mechanisms by allosterically affecting the structure of PPARα. This, in turn, allows Ser280-phosphorylated PPARα to selectively activate transcription of genes involved in fatty acid uptake and storage but not utilization; this selectivity is modulated at least in part by the PPRE sequence.
Previous investigations have shown that PPARγ is phosphorylated by ERK/CDK5 at Ser273 in adipose tissues in a model of obesity (Banks et al., 2015; Choi et al., 2010). Although both Ser280 in PPARα and Ser273 in PPARγ are located in the LBD, there is no amino acid sequence homology between the regions adjacent to PPARα Ser280 and PPARγ Ser273. Consistently, neither fatty acids nor changes in GSK-3α activity alter the level of PPARγ-Ser273 phosphorylation in CMs. Furthermore, although fenofibrate inhibits phosphorylation of PPARα at Ser280 by competing with GSK-3α, it does not inhibit phosphorylation of PPARγ at Ser273 (Figure S7P). More importantly, phosphorylation of PPARα at Ser280 and that of PPARγ at Ser273 affect distinct sets of downstream genes and exhibit distinct functional consequences, namely promoting fatty acid uptake and lipotoxicity in the heart and lipid-independent insulin resistance in adipose tissue, respectively.
GSK-3 in metabolism
GSK-3α and GSK-3β share 98% sequence similarity in their catalytic domains and phosphorylate some common targets in vitro. However, genetic deletion of GSK-3β in mice causes embryonic lethality, whereas deletion of GSK-3α does not, suggesting that GSK-3α and GSK-3β have distinct biological targets (Hoeflich et al., 2000). The differences in the amino acid sequences at the N- and C-termini confer distinct substrate specificities, and the difference in subcellular localization between GSK-3α and GSK-3β allows them to phosphorylate distinct substrates as well. Here, only GSK-3α, but not GSK-3β, translocates to the nucleus and interacts with PPARα in CMs in response to fatty acids. Interestingly, although downregulation of GSK-3α in CMs inhibited the development of cardiomyopathy in response to HFD consumption, downregulation of GSK-3β exacerbated HFD-induced cardiomyopathy. Constitutively active GSK-3α and GSK-3β regulate expression of genes involved in fatty acid uptake and transport in diametrically opposite directions. Elucidating the molecular mechanisms allowing GSK-3α and GSK-3β to exert opposite functions is important because none of the currently available small molecule inhibitors for GSK-3 are isoform specific and, thus, the beneficial effect of GSK-3α inhibition upon lipotoxic cardiomyopathy could be diminished or overwhelmed by the detrimental effect of GSK-3β inhibition. In theory, only the unique actions of GSK-3α ought to be targeted for the treatment of lipotoxic cardiomyopathy.
GSK-3 has recently been shown to act as a glucose sensor in B cells in the germinal center (Jellusova et al., 2017): GSK-3 is inactivated in the presence of abundant glucose, promoting cell growth and proliferation, whereas GSK-3 is activated under conditions of glucose deprivation, increasing glycolysis and energy production. Glucose and fatty acid metabolism are often reciprocally regulated. According to the Randle hypothesis, fatty acid suppresses glucose oxidation and induces insulin resistance in muscles through an increase in intracellular citrate concentration (Randle et al., 1963). Alternatively, fatty acid negatively regulates glucose oxidation through DAG-mediated activation of PKCθ and IRS-1 and the subsequent inhibition of PI3K and GLUT4 translocation (Samuel and Shulman, 2012; Shulman, 2014). Our results represent another important mechanism in the heart by which fatty acids negatively regulate glucose oxidation: increases in fatty acids activate GSK-3α, which in turn upregulates PDK4 through PPARα phosphorylation. We propose that glucose and fatty acid metabolism may be coordinately regulated via GSK-3α in the heart and that the GSK-3α-PPARα axis is a major mechanism in the induction of lipotoxic cardiomyopathy in obesity. Since expression of GSK-3α is increased in the presence of fatty acids, GSK-3α and PPARα may constitute a feed-forward mechanism that not only contributes to myocardial energy production in the intact heart, such as during fasting, but also facilitates the progression of lipotoxic cardiomyopathy in metabolic syndrome. It is tempting to speculate that mammals, including humans, have developed this mechanism to allow the heart to promote fatty acid uptake and storage without stimulating FAO or consequent production of reactive oxygen species when fat is abundant in preparation for future use as a backup when fat is less available.
Bidirectional role of fibrates in cardiac lipid metabolism
Fenofibrate is a synthetic PPARα agonist that has been shown to stimulate fatty acid metabolism in the liver and skeletal muscle. However, the pharmacological action of fenofibrate in the heart remains elusive because fenofibrate fails to alter myocardial fatty acid metabolism in healthy human volunteers and, conversely, decreases fatty acid metabolism in the hearts of mice fed a HFD (Sarma et al., 2012). Here, we demonstrate that fenofibrate inhibits PPARα activity by blocking GSK-3α-mediated PPARα phosphorylation in the presence of high concentrations of fatty acids in CMs. GSK-3α is translocated into the nucleus in the presence of high concentrations of fatty acids, where it phosphorylates PPARα. Since GSK-3α and fenofibrate both bind to the PPARα LBD, fenofibrate can competitively inhibit the access of GSK-3α to the LBD of PPARα and inhibit Ser280 phosphorylation.
It should be noted that the effectiveness of fenofibrate for treatment of diabetic patients remains controversial. Although a recent meta analysis showed that fibrates have an additive cardioprotective effect upon statin therapy in humans (Silverman et al., 2016), the ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial had previously showed little or no effect of combination therapy with fenofibrate and simvastatin compared to simvastatin alone in patients with diabetes (Group et al., 2010). Given the aforementioned difficulty in developing an isoform-specific inhibitor for GSK-3α and the absence of an approved drug for lipotoxic cardiomyopathy in the clinical setting, interventions directly and selectively suppressing PPARα-phosphorylation at Ser280 may be more effective as a treatment for lipotoxic cardiomyopathy than either GSK-3 inhibitors or fenofibrate. PPARα has cell-type specific roles. For example, enhanced PPARα activity in muscle or liver is protective against diet-induced obesity or diabetes (Finck et al., 2005; Kersten et al., 1999). Furthermore, transcription may be regulated in a cell-type specific manner (Gosselin et al., 2014; Heinz et al., 2015). Thus, whether PPARα-Ser280 phosphorylation also contributes to fatty acid metabolism in other cell types remains to be elucidated.
Increasing evidence suggests that the fatty acid composition of fats, including monounsaturated or polyunsaturated fatty acids and even-chain or odd-chain saturated fatty acids, the total and individual concentrations of fatty acids, and the ratio of saturated to unsaturated fatty acids all have distinct effects on cellular metabolism, thereby either promoting or preventing metabolic syndrome and cardiovascular diseases. Thus, it is challenging to conduct an in vitro study that faithfully mimics the fatty acid composition observed in HFD models in vivo. The palmitic acid concentration in human plasma ranges from 25 to 2500 μM, and the level of PPARα phosphorylation at Ser280 in CMs in vitro in response to the fatty acid concentrations we used is comparable to those in the hearts of mice fed either a HFD or a control diet in vivo. Therefore, the experiments conducted using a range of 0 to 500 μM of BSA-conjugated fatty acids in CMs in vitro closely resemble human lipotoxic cardiomyopathy in terms of lipid accumulation and altered cellular metabolism. It would be interesting to investigate the impact of additional single fatty acids or multiple fatty acids in combination with different ratios on the Ser280-phosphorylation level in the future.
Limitations of the study
The HFD we used for the in vivo study contained lard-based fats. It would be important to identify the key components of the fat or the ratio of each fatty acid in the blood that is critical for the induction of PPARα phosphorylation and the development of lipotoxic cardiomyopathy in vivo. In addition, the specific combination of fatty acids that would faithfully replicate in vivo conditions of HFD consumption for in vitro studies remains to be established. Although our results suggest that the PPRE/DR1 sequence plays a critical role in mediating the effect of Ser280 PPARα phosphorylation on the biased control of fatty acid metabolism, the possibility remains that the effect of Ser280 PPARα phosphorylation is mediated through either a DNA binding motif-independent mechanism or indirect regulation of fatty acid metabolism.
In summary, we demonstrate that GSK-3α acts as a central regulator of cardiac fatty acid metabolism by stimulating a biased PPARα transcriptional response through Ser280 phosphorylation and consequent changes in the structure of PPARα. Activation of GSK-3α under conditions similar to metabolic syndrome contributes to the development of lipotoxic cardiomyopathy by inducing an imbalance between fatty acid uptake/storage and catabolism in CMs. We show that a reversal of Ser280 phosphorylation of PPARα normalizes lipotoxicity and cardiac dysfunction, representing a promising therapeutic intervention to combat lipotoxic cardiomyopathy in patients with metabolic syndrome.
STAR Methods
• Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Junichi Sadoshima (sadoshju@njms.rutgers.edu).
• Experimental Model and Subject Details
Mice
GSK-3α heterozygous floxed mice (C57BL/6 background) were a kind gift from Dr. P.S. Klein (University of Pennsylvania). GSK-3β floxed mice (C57BL/6 background) were a kind gift from Dr. C.J. Phiel (He et al., 2010). GSK-3α and β knock-in mice were a kind gift from Dr. D.R. Alessi (University of Dundee). Cardiomyocyte-specific deletion of GSK-3α or GSK-3β was obtained by crossing the mice with α-myosin heavy chain promoter-driven heterozygous Cre mice (a kind gift from Dr. M.D. Schneider). Cardiomyocyte-specific Tg-FLAG-PPARα (α-myosin heavy chain promoter) mice were a kind gift from Dr. D. Kelly. Male C57BL/6J ob/ob mice and male C57BL/6J wild-type mice were purchased from Jackson Labs at 5–6 weeks of age. For diet-induced obesity, male 5 to 6-week-old animals were fed a high-fat (60%) diet (Research Diets, D12492). The PPARα ligand, fenofibrate (Sigma-Aldrich), was provided via ad libitum feeding with a high-fat diet containing 0.2% (wt/wt) fenofibrate (custom diet purchased from Research Diets) for the indicated periods (Haemmerle et al., 2011). For cardiac insulin resistance tests, mice were fasted for 4 hours, followed by intraperitoneal injection of 0.75 U/kg body weight insulin. Fifteen minutes after insulin injection, the hearts were harvested to evaluate phosphorylation of Akt by immunoblot. For intraperitoneal glucose tolerance tests in HFD-fed mice, mice were fasted for six hours and body weight and fasting glucose level from a small tail clip were measured. Glucose (1 mg/g body weight) was injected intraperitoneally and blood glucose values were obtained at 15, 30, 45, 60, 90, and 120 mins. Mice were housed in a temperature-controlled environment within a range of 21–23 °C with 12-hour light/dark cycles and were fed an indicated diet. We used age-matched male mice in all animal experiments. All protocols concerning the use of animals were approved by the Institutional Animal Care and Use Committee at New Jersey Medical School, Rutgers University.
Gene targeting mouse
PPARα genomic DNA was isolated from BAC clone (RP24–545F24) to construct the PPARα S280A KI targeting vector. PCR-based site-directed mutagenesis was performed to introduce a single mutation of T to G in codon 280 in exon 8 of PPARα to change codon 280 from Ser to Ala. A pGK neo cassette flanked by two FRT sites (a kind gift from Dr. Takeda, Osaka University) was inserted into intron 8 for selection of the targeted allele. Diphtheria Toxin A fragment was used for negative selection. The DNA sequences used for constructing the targeting vector, including introns 7 and 8 and exon 8, were confirmed by DNA sequencing. The targeting vectors were linearized with PmeI and subsequently electroporated into ES cells. G418-resistant ES clones were screened for homologous recombination by long range PCR using the primers listed in Table S5 with the Expand Long Template PCR system (Roche #11681842001). Three positive clones were identified out of 288 clones. Homologous recombinant ES clones were microinjected into blastocysts from C57BL/6J mice and transferred into pseudo-pregnant recipients to generate male chimeras. The chimeric male mice resulting from the microinjection were bred with C57BL/6J female mice to generate germline-transmitted heterozygous S280A KI mice. PCR analysis was performed on tail DNA from offspring. Sequence analysis was performed on PCR products to verify the presence of the mutations using standard procedures. The mutant offspring were backcrossed into the C57BL/6J background.
GSK-3α flox/flox mice were generated in Dr. Peter S. Klein’s laboratory (University of Pennsylvania, Philadelphia). Briefly, a conventional targeting vector with neomycin resistance and TK was generated to insert loxP sites on either side of exon 2 in the Gsk3a gene. The neomycin resistance cassette, inserted downstream of exon 2, was also flanked by FRT sites. The construct was electroporated into ES cells derived from C57BL/6 mice and positive and negative selection was used to isolate positive clones, which were then screened by PCR for the correct insertion and confirmed by Southern blot. Cre-mediated excision removes exon 2, which encodes an essential portion of the catalytic domain, to generate a null allele.
Human samples from explanted hearts
The samples from explanted hearts used in this study were obtained from 14 non-diabetic (mean age 52.0 ± 11.8 years; 13 males) and 7 diabetic (mean age 50.4 ± 11.6 years; 6 males) patients who had received heart transplants at the Taipei Veterans General Hospital. The study was approved by the Ethics Committee of Taipei Veterans General Hospital, and all patients or their families expressed their willingness to participate through an informed consent form. Myocardial samples from near the mitral annulus were obtained at the time of therapeutic transplantation. Immediately after tissue procurement, the samples for biochemical studies were stored in liquid nitrogen and kept at −80°C.
Cell line
H9C2 cells were maintained at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium/Nutrient Mixture F-12 supplemented with 10% fetal bovine serum. Information on the sex is not available.
Primary Rat Neonatal Cardiomyocytes
Primary cultures of ventricular CMs were prepared from 1-day-old Crl:(WI)BR-Wistar rats (both sexes) (Harlan Laboratories, Somerville) and maintained in culture. A cardiomyocyte-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. CMs were cultured in complete medium containing Dulbecco’s modified Eagle’s medium/F-12 supplemented with 5% horse serum, 4 μg/ml transferrin, 0.7 ng/ml sodium selenite, 2 g/l bovine serum albumin (fraction V), 3 mM pyruvate, 15 mM Hepes pH 7.1, 100 μM ascorbate, 100 mg/l ampicillin, 5 mg/l linoleic acid, and 100 μM 5-bromo-2’-deoxyuridine (Sigma). Culture dishes were coated with 0.3% gelatin or 2% gelatin for immunofluorescence staining on chamber slides.
• Method Details
Antibodies and reagents
The following commercial antibodies were used at the indicated dilutions: phospho-GSK-3α (Ser21) (36E9) (1:1,000), total GSK-3α (D80E6, for WB) (1:2,000), total GSK-3α (D80D1, for IF) (1:100), total GSK-3α/β (D75D3) (1:4,000), phospho-GSK-3α/β (1:2,000), histone H3 (1:5,000), GAPDH (14C10) (1:5,000), GFP (D5.1, for IF) (1:500), and secondary antibodies (anti-mouse or rabbit IgG) conjugated with horseradish peroxidase (1:4,000) (Cell Signaling); secondary antibodies (anti-mouse or rabbit IgG) conjugated with Alexa Fluor 488 or 555 (1:100) (Life Technologies); GFP-magnetic beads (Fisher/MBL); total PPARα (1:3,000) (Cayman Chemical); RXRα (D-20) (1:4,000) (Santa Cruz); α-actinin (1:4,000) (sarcomeric) (Sigma-Aldrich). For detection of phosphorylation of PPARα at Ser280, a polyclonal phosphorylation-specific antibody was generated by immunizing rabbits with a phospho-peptide corresponding to residues surrounding Ser280 of PPARα (1:1,000). Antibodies were diluted in either 5% (w/v) BSA or 5% (w/v) non-fat dry milk in 1xTBS/0.5%Tween 20, depending on the level of background intensity. The following reagents were used: WY-14643 and fenofibrate (R & D Systems, Tocris for in vitro experiments); Palmitic acid, Duolink In Situ PLA, and Etomoxir (Sigma-Aldrich).
Adenovirus constructs
Recombinant adenovirus vectors for overexpression and short hairpin RNA-mediated gene silencing were constructed, propagated and titered as previously described (Oka et al., 2011). pBHGloxΔE1,3Cre plasmid was co-transfected with the pDC316 shuttle vector (Microbix) or pDCSilencer (Microbix) containing YFP-PPARα-WT, YFP-PPARα-mutants, or GSK-3α targeting sequences into HEK293 cells using Lipofectamine 2000 (Life Technologies). The cDNA of mouse PPARα was amplified using pDC316-PPARα (Oka et al., 2011) as a template and ligated into pDC316-YFP-N terminal vector (Maejima et al., 2013). The shuttle vector for the short hairpin sequence of rat GSK-3α was generated by insertion of a complementary hairpin sequence of rat GSK-3α (see key resources table) into pDCSilencer (Microbix). The luciferase reporter plasmid (pPPRE-tk-luc) was a kind gift from Dr. Ronald Evans at the Salk Institute. Mutations were performed by site directed mutagenesis. Adenovirus vectors harboring LacZ (Ad-LacZ) and sh-Scramble (Ad-shScr) were used as controls. The total MOI of adenovirus was kept constant using LacZ or Scramble virus.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-rabbit monoclonal phospho-GSK-3α (Ser21) (36E9) | Cell Signaling | 9316 |
| Anti-rabbit monoclonal GSK-3α (D80E6) | Cell Signaling | 4337 |
| Anti-rabbit monoclonal GSK-3α (D80D1) XP | Cell Signaling | 4818 |
| Anti-rabbit monoclonal GSK-3α/β (D75D3) XP | Cell Signaling | 5676 |
| Anti-rabbit polyclonal phospho-GSK-3α/β (Ser21/9) | Cell Signaling | 9331 |
| Anti-rabbit monoclonal GSK-3β (27C10) | Cell Signaling | 9315 |
| Anti-mouse monoclonal GSK-3α (H-12) | Santa Cruz | sc-5264 |
| Anti-rabbit polyclonal GSK-3β (H-76) | Santa Cruz | sc-9166 |
| Anti-rabbit polyclonal PPARα | Cayman Chemical | 101710 |
| Anti-rabbit polyclonal PPARα | Abcam | ab24509 |
| Anti-rabbit polyclonal RXRα (D-20) | Santa Cruz | sc-553 |
| Anti-rabbit polyclonal RXRα/β/γ (ΔN197) | Santa Cruz | sc-774X |
| Anti-rabbit monoclonal β-Catenin (6B3) | Cell Signaling | 9582 |
| Anti-rabbit polyclonal phospho-β-catenin (Ser33/37/T41) | Cell Signaling | 9561 |
| Anti-rabbit polyclonal Akt | Cell Signaling | 9272 |
| Anti-rabbit polyclonal phospho-Akt (Ser473) | Cell Signaling | 9271 |
| Anti-rabbit polyclonal Histone H3 | Cell Signaling | 9715 |
| Anti-rabbit monoclonal GAPDH (14C10) | Cell Signaling | 2118 |
| Anti-rabbit monoclonal GFP (D5.1) XP | Cell Signaling | 2956 |
| Anti-rabbit polyclonal GST | Cell Signaling | 2622 |
| Anti-mouse monoclonal α-actinin (Sarcomeric) | Sigma-Aldrich | A7811 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling | 7076 |
| Anti-rabbit IgG, HRP-linked antibody | Cell Signaling | 7074 |
| Anti-rabbit polyclonal Troponin T antibody | Abcam | 45932 |
| Goat anti-mouse IgG secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | A-11029 |
| Goat anti-rabbit IgG secondary antibody, Alexa Fluor 488 | Thermo Fisher Scientific | A-11034 |
| Goat anti-rabbit IgG secondary antibody, Alexa Fluor 594 | Thermo Fisher Scientific | A-11037 |
| Clean-Blot IP Detection Reagent (HRP) | Thermo Fisher Scientific | 21230 |
| Anti-rabbit polyclonal phospho-specific PPARα (Ser280) | GeneScript | This paper |
| Bacterial and Virus Strains | ||
| Subcloning Efficiency DH5α Competent Cells | Thermo Fisher Scientific | 18265017 |
| One Shot BL21(DE3) Chemically Competent E. coli | Thermo Fisher Scientific | C600003 |
| BAC clone | BACPAC | RP24–545F24 |
| Biological Samples | ||
| Human heart samples | Taipei Veterans General Hospital | This paper |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Anti-GFP mAb-Magnetic beads | MBL | D153–11 |
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | A2220 |
| Protein A/G PLUS-Agarose | Santa Cruz | sc-2003 |
| Glutathione Sepharose 4B | VWR/GE | 17–0756-01 |
| GSK3α protein, active | Millipore | 14–492 |
| GSK3β protein, active | Millipore | 14–306 |
| Recombinant GST-PPARa (full-length and truncated) | This paper | N/A |
| Recombinant GST-β-catenin | This paper | N/A |
| PPARα LBD (human recombinant) | Cayman Chemical | 10009088 |
| Recombinant GSK-3α | This paper | N/A |
| IPTG | Invitrogen | 15529–019 |
| FuGENE 6 Reagent | VWR/Promega | E2691 |
| LipofectamineTM 2000 Transfection Reagent | Invitrogen | 11668–019 |
| Palmitic acid | Sigma-Aldrich | P0500 |
| Oleic acid | Sigma-Aldrich | O1008 |
| Linoleic acid | Sigma-Aldrich | L1376 |
| Bovine Serum Albumin | Boston BioProducts | P753 |
| Palmitic Acid, [9,10–3H(N)]-, 5mCi (185MBq) | Perkin Elmer | NET043005MC |
| Oleic Acid, [9,10–3H(N)]-, 1mCi | Perkin Elmer | NET289001MC |
| Etomoxir sodium salt hydrate | Sigma-Aldrich | E1905 |
| 2-Deoxy-D-glucose | Sigma-Aldrich | D8375 |
| L-Carnitine hydrochloride | Sigma-Aldrich | C0283 |
| Sodium Pyruvate | Sigma-Aldrich | P4562 |
| Fenofibrate | Sigma-Aldrich | F6020 |
| WY-14643 | R&D Systems, Tocris | 1312 |
| Rodent Diet with 60% kcal% fat | Research Diets | D12492 |
| Rodent Diet with 10% kcal% fat | Research Diets | D12450B |
| High-fat diet containing 0.2% (wt/wt) fenofibrate | Research Diets | This paper |
| High-fat diet without Fenofibrate | Research Diets | This paper |
| Insulin solution human | Sigma-Aldrich | I9278 |
| 2,2,2-Tribromoethanol | Sigma-Aldrich | T48402 |
| Oil Red O solution in 0.5% in isopropanol | Sigma-Aldrich | O1391 |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Aldrich | P0044 |
| Protease Inhibitor Cocktail | Sigma-Aldrich | P8340 |
| Maxima SYBR Green/ROX qPCR Master Mix (2x) | Thermo/Fermentas | K0223 |
| TRIzol Reagent (Ambion) | Life Technologies | 15596–018 |
| Adenosine 5’-diphosphate sodium salt | Sigma-Aldrich | A2754 |
| Collagenase Type 2 CLS2 | Worthington | LS004177 |
| Percoll | GE Healthcare | 17–0891-01 |
| Biotinylated Thrombin | EMD Millipore | 69672–3 |
| Streptavidin Agarose | EMD Millipore | 69203–3 |
| Critical Commercial Assays | ||
| XF24 Extracellular Flux Analyzer | Seahorse Bioscience | https://www.agilent.com/en/products/cell-analysis-(seahorse)/seahorse-analyzers |
| XF96 Extracellular Flux Analyzer | Seahorse Bioscience | https://www.agilent.com/en/products/cell-analysis-(seahorse)/seahorse-analyzers |
| BriteLite Plus | Perkin Elmer | 6066761 |
| Luciferase assay system | VWR/Promega | E1501 |
| Duolink In Situ PLA probe anti-Mouse MINUS | Sigma-Aldrich | 92004–0030 |
| Duolink In Situ PLA probe anti-Rabbit PLUS | Sigma-Aldrich | 92002–0030 |
| TnT T7 Coupled Reticulocyte Lysate System | VWR/Promega | L4610 |
| QIAGEN HiSpeed Plasmid Maxi Kit | QIAGEN | 12663 |
| QIAGEN HiSpeed Plasmid Midi Kit | QIAGEN | 12643 |
| QIAprep Spin Miniprep Kit | QIAGEN | 27104 |
| QIAquick PCR Purification Kit | QIAGEN | 28106 |
| QIAquick Gel Extraction Kit | QIAGEN | 28706 |
| Deposited Data | ||
| GSK-3α/β knock-in mice microarray data | Matsuda, T. et al., 2008 | GSE112160 |
| PPARα mutants RNaseq data | This paper | GSE112309 |
| Experimental Models: Cell Lines | ||
| H9C2 | Oka, S. et al., 2011 | N/A |
| HEK293 | Maejima, Y. et al., 2013 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: GSK-3α heterozygous floxed | Gift from Dr. P.S. Klein (University of Pennsylvania) | N/A |
| Mouse: GSK-3β floxed | Gift from Dr. C.J. Phiel (Nationwide Children’s Hospital) | He, F. et al., 2010. |
| Mouse: GSK-3α-Ser21Ala knock-in | Gift from Dr. D.R. Alessi (University of Dundee) | N/A |
| Mouse: GSK-3β-Ser9Ala knock-in | Gift from Dr. D.R. Alessi (University of Dundee) | N/A |
| Mouse: α-myosin heavy chain promoter-driven heterozygous Cre transgenic | Gift from Dr. M.D. Schneider (Imperial College London) | N/A |
| Mouse: α-myosin heavy chain promoter-driven FLAG-PPARα transgenic | Gift from Dr. D. Kelly (University of Pennsylvania) | N/A |
| Mouse: C57BL/6J | Jackson Laboratory | JAX 000664 |
| Mouse: ob/ob: B6.Cg-Lepob/J | Jackson Laboratory | JAX 000632 |
| Mouse: PPARα-Ser280Ala knock-in | This paper | N/A |
| Rat: Primary cultured neonatal ventricular cardiomyocytes | Harlan | 1-day-Crl:(WI)BR-Wistar rats |
| Oligonucleotides | ||
| Oligonucleotides used in this study are provided in Table S5 | This paper | NCBI PRIMER-BLAST |
| Recombinant DNA | ||
| pDC316 shuttle vector | Microbix | N/A |
| pDC316-YFP shuttle vector | Sadoshima lab | Maejima, Y. et al., 2013 |
| pDC316-FLAG shuttle vector | Sadoshima lab | Maejima, Y. et al., 2013 |
| pDCSilencer | Microbix | N/A |
| pBHGloxΔE1.3 Cre | Microbix | N/A |
| pCold-GST | Takara/Clontech | 3372 |
| CTNNB1 (untagged)-Human catenin, beta 1 | Origene | SC107921 |
| GSK3 alpha pMT2 | Addgene | 15896 |
| pPPRE-tk-luciferase reporter | Gift from Dr. Ronald Evans (Salk Institute) | N/A |
| pCITE-GSK-3α | This paper | N/A |
| pL451 | Gift from Dr. Takeda (Osaka University) | N/A |
| pMCS-DTA | Gift from Dr. Takeda (Osaka University) | N/A |
| pMCS-Neo-PPARα-S280A-DTA | This paper | N/A |
| pCold-GST-PPARα-full length | This paper | N/A |
| pCold-GST-PPARα-truncate 1 | This paper | N/A |
| pCold-GST-PPARα-truncate 2 | This paper | N/A |
| pCold-GST-PPARα-truncate 3 | This paper | N/A |
| pCold-GST-PPARα-truncate 4 | This paper | N/A |
| pCold-GST-PPARα-truncate 5 | This paper | N/A |
| pDC316-YFP-PPARα-wild type | This paper | N/A |
| pDC316-YFP-PPARα-S280A | This paper | N/A |
| pDC316-YFP-PPARα-S280D | This paper | N/A |
| pDC316-YFP-PPARα-S280E | This paper | N/A |
| pDC316-YFP-PPARα-WT-H440A | This paper | N/A |
| pDC316-YFP-PPARα-WT-H440Q | This paper | N/A |
| pDC316-YFP-PPARα-WT-K448A | This paper | N/A |
| pDC316-YFP-PPARα-WT-K448N | This paper | N/A |
| pDC316-YFP-PPARα-WT-K449A | This paper | N/A |
| pDC316-YFP-PPARα-WT-K449N | This paper | N/A |
| pDC316-YFP-PPARα-WT-K448A/K449A | This paper | N/A |
| pDC316-YFP-PPARα-WT-K448N/K449N | This paper | N/A |
| pDC316-YFP-PPARα-WT-H440A/K448A/K449A | This paper | N/A |
| pDC316-YFP-PPARα-WT-H440Q/K448N/K449N | This paper | N/A |
| pDC316-YFP-PPARα-WT-H440A | This paper | N/A |
| pDC316-YFP-PPARα-S280D-H440Q | This paper | N/A |
| pDC316-YFP-PPARα-S280D-K448A | This paper | N/A |
| pDC316-YFP-PPARα-S280D-K448N | This paper | N/A |
| pDC316-YFP-PPARα-S280D-K449A | This paper | N/A |
| pDC316-YFP-PPARα-S280D-K449N | This paper | N/A |
| pDC316-YFP-PPARα-S280D-K448A/K449A | This paper | N/A |
| pDC316-YFP-PPARα-S280D-K448N/K449N | This paper | N/A |
| pDC316-YFP-PPARα-S280D-H440A/K448A/K449A | This paper | N/A |
| pDC316-YFP-PPARα-S280D-H440Q/K448N/K449N | This paper | N/A |
| pDC316-GSK-3α | This paper | N/A |
| shRNA-GSK-3α #1 | This paper | N/A |
| shRNA-GSK-3α #2 | This paper | N/A |
| shRNA-GSK-3α #3 | This paper | N/A |
| pBluescript II | Maejima, Y. et al., 2013 | N/A |
| AAV-DJ/8-CMV | UPenn Vector Core | N/A |
| AAV-DJ/8-CMV-PPARα-wild type | This paper | N/A |
| AAV-DJ/8-CMV-PPARα-S280D | This paper | N/A |
| Software and Algorithms | ||
| R version 3.3.1 | The R Project | https://www.r-project.org/ |
| CLC Genomics Workbench 8.5 version | Qiagen | http://www.clcbio.com/products/clc-genomics-workbench/ |
| GSEA ver 2.0 | Broad institute of MIT and Harvard | http://software.broadinstitute.org/gsea/ |
| IPA pathway analysis software | Qiagen | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| MEME Suite ver 5.0.2 | The MEME SUITE | http://meme-suite.org/ |
| UCSC Genome Browser | UCSC | http://genome.ucsc.edu/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism version 6 | GraphPad | https://www.graphpad.com |
| Metaboanalyst ver 3.0 | MetaboAnalyst | http://www.metaboanalyst.ca/ |
| MultiExperiment Viewer ver4.9.0 | MeV | http://mev.tm4.org/#/welcome |
| Other | ||
Adeno-associated virus
The recombinant adeno-associated virus (AAV) vectors used to generate AAV-DJ/8-PPARα-WT or S280D were constructed by cloning the cDNA of PPARα-WT or S280D (mouse) from the pDC316-YFP-PPARα-WT or S280D vector into the pAAV-MCS expression vector (Cell Biolabs, Inc, #VPK-410) downstream of the CMV promoter with BamHI and SalI. 293AAVcells (Cell Biolabs, Inc.) were co-transfected with the recombinant AAV vectors, pAAV-DJ/8 vector, and helper plasmid in a 1:1:1 ratio using polyethylenimine (PEI) at the AAV core, Department of Cell Biology and Molecular Medicine, New Jersey Medical School, Rutgers University, Newark, NJ (Grimm et al., 2008). The recombinant AAV produced was purified by the iodixanol gradient/ultra-centrifugation method, and the AAV fraction was concentrated using a VIVASPIN 20 concentrator (100 kDa cut-off, Sartorius, Germany). The virus titer was determined using the Cell Biolabs AAV quantitation kit (Cat. # VPK-145). To administer recombinant AAVs, doses of 2×1011 vector genome per mouse were injected intravenously via the jugular vein of C57BL/6J wild-type mice.
Recombinant proteins
The bacterial expression vectors for GST-fused PPARα-full length and -truncates (T1–5) were generated by insertion of mouse PPARα cDNA amplified by PCR into the pCold-GST-vector. The BL21 E. coli strain was transformed with pCold-GST-PPARα-full length or truncates. The E. coli was grown in 3 ml LB medium containing ampicillin overnight at 37°C, and then transferred to 250 ml LB medium containing ampicillin. Protein expression was induced by addition of 1 mM isopropylthio-β-galactoside. After overnight culture at 15°C, the E. coli were lysed in lysis buffer (1% Triton X-100 and 1 mM DTT in PBS) with sonication. The lysate was incubated with 0.5 ml Glutathione-sepharose 4B (GE Healthcare) for 1 hour at 4°C. The sepharose was washed 3 times with 5 ml lysis buffer, and then suspended with 1 ml cleavage buffer (20 mM Tris pH 7, 150 mM NaCl, 1 mM DTT).
In vitro kinase assay
Recombinant active GSK-3α was purchased from Millipore. Recombinant PPARα-LBD (human) was purchased from Cayman Chemical. GST-tagged full-length PPARα protein was generated using the pCold-GST-vector. Recombinant active GSK-3α (10 ng) was incubated with recombinant GST-full length-PPARα (1 μg) or PPARα-LBD (1 μg) in a kinase buffer (50 mM HEPES (pH 7.4), 15 mM MgCl2 and 200 μM sodium vanadate containing 100 μM ATP in the presence or absence of 10 μCi [γ−32P]ATP per reaction) at 30°C for 15 minutes. Phosphorylated proteins were separated by SDS-PAGE and analyzed by autoradiography or immunoblot.
Mass spectrometry
A kinase reaction was performed using recombinant GST-tagged PPARα and GSK-3α proteins. Phosphorylated proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue. The gel band of interest was excised for in-gel trypsin digestion. The resulting peptides were subjected to LC-MS/MS analysis on an Ultimate 3000 (Dionex, Sunnyvale, CA, USA) LC system coupled with an Orbitrap Velos tandem mass spectrometry instrument (Thermo Fisher Scientific). The MS spectra were acquired in positive mode with a spray voltage of 2 kV and a capillary temperature of 275ºC. The Lock Mass feature was used for accurate mass measurement. The automatic gain control (AGC) target was set to 1.0E+6 for full scan in the Orbitrap mass analyzer and 3.0E+4 for MS/MS scans in the ion trap mass analyzer. The precursor was selected within a 2 m/z isolation window and fragmented using collision-induced dissociation (CID) fragmentation. The MS/MS spectra were searched against the Swissprot mouse database with an MS error tolerance of 10 ppm and an MS/MS error tolerance of 0.5 Da. Methionine oxidation, cysteine carbamidomethylation (IAM) and serine/threonine phosphorylation were set as variable modifications. One % false discovery rate (FDR) was used for both protein and peptide identification.
RNA-Seq library preparation, sequencing and data analysis
Total RNA was isolated from H9C2 cells transduced with the indicated plasmid using TRIzol (Invitrogen). Isolated RNA was first checked for integrity on an Agilent Bioanalyzer 2100; samples with RNA integrity number (RIN) >7.0 were used for subsequent processing. Total RNA was subjected to two rounds of poly(A) selection using oligo-d(T)25 magnetic beads (New England Biolabs). A paired-end (strand specific) cDNA library was prepared using the NEB Next Ultra-directional RNA-Seq protocol. Briefly, poly(A)+ RNA was fragmented by heating at 94°C for 10 minutes followed by reverse transcription and second strand cDNA synthesis using the reagents provided in the NEB Next kit. End-repaired cDNA was then ligated with double stranded DNA adapters, followed by purification of ligated DNA with AmpureXP beads. cDNA was then amplified by PCR for 15 cycles with a universal forward primer and a reverse primer with bar code. The sequencing of the cDNA libraries was performed on the Illumina HiSeq 2500 platform (Illumina, San Diego, CA) using the single-read 1×50 cycles configuration. The raw reads files have been deposited in the NCBI Gene Expression Omnibus.
CLC Genomics Workbench version 8.5 (http://www.clcbio.com/products/clc-genomics-workbench/; Qiagen) was used for RNA-seq analysis. De-multiplexed fastq files from RNA-Seq libraries were imported into the CLC software. Adaptor sequences and bases with low quality were trimmed and reads were mapped to a reference genome, Rattus norvegicus (assembly Rnor_6.0). Read mapping was carried out with the RNA-Seq Analysis Tool of CLC Genomics Workbench. RPKM values were calculated for each gene to quantify absolute expression. Statistical analysis of differentially expressed genes was carried out with the Empirical analysis of Digital Gene Expression data tool in CLC Genomic Workbench. Functional Pathway analysis of PPARα-Ser280 phosphorylation-regulated genes (SD vs SA) was performed using IPA pathway analysis software (QIAGEN). Expression patterns of regulated genes were graphically represented in a heat map. Hierarchical clustering was performed to group genes with similar features in the expression prolife. The clustering and visualization were carried out using the MultiExperiment Viewer (MeV).
The following publicly available microarray datasets were used: OB/OB (GSE16790), DB/DB (GSE36875), and streptozotocin-induced type I diabetic model (GSE5606). Raw intensity values were normalized using the Robust Multi-array Average (RMA) method on R. A p value less than 0.05 in the empirical analysis was considered statistically significant. Normalized expression data were also analyzed with GSEA v2.0.13 software using the JAVA program (Broad Institute, Cambridge, MA). All gene sets were obtained from the Molecular Signatures Database v4.0 distributed on the GSEA Web site.
Quantitative RT-PCR
Total RNA was prepared from mouse hearts and CMs using TRIzol (Invitrogen). cDNA was generated using 300 ng total RNA and SuperScript III Reverse Transcriptase (ThermoFisher). Using Maxima SYBR Green qPCR master mix (Fermentas), real-time RT-PCR was performed under the following conditions: 94°C for 10 minutes; 40 cycles of 94°C for 15 seconds, 58°C for 30 seconds, 72°C for 30 seconds; and a final elongation at 72°C for 15 minutes. Relative mRNA expression was determined by the ΔΔ-Ct method normalized to the ribosomal RNA (18S) level. Primer sequences are shown in Table S5.
Metabolomics analysis
Myocardial tissue samples were harvested from heterozygous PPARα-S280A KI and littermate control mice at the age of 18 to 20 weeks (n=5). Untargeted screening of the metabolome in ventricular tissue was performed using GC/MS at the University of Utah Metabolomics Core Facility. The freeze-clamped tissue samples (~15 mg) were homogenized in the extraction solution (methanol/water (8:1) with amino-acid standards and D4 succinate) using a ceramic bead tube kit (MO BIO Laboratories, Inc., Carlsbad, CA) and Bead Ruptor (Omni International, Kennesaw, GA). After incubation for 2 hours at −20°C followed by centrifugation at 13,000g at 4°C for 10 minutes, the extraction samples were dried using a Speed-vac (MiVac Duo, Barnstead/Genevac Inc., Gardiner, NY) overnight and suspended in 40 μl of 40 mg/ml O-methoxylamine hydrochloride (MOX) in pyridine. GC/MS analysis was performed with a Waters GCT Premier mass spectrometer fitted with an Agilent 6890 gas chromatograph and a Gerstel MPS2 autosampler. A 30 m Phenomenex ZB5–5 MSi column with a 5 m long guard column was employed for chromatographic separation. Data were collected by MassLynx 4.1. Initial analysis of known metabolites was performed using QuanLynx with data transfer to Excel. Peak picking was performed using MarkerLynx with data mining performed using SIMCA-P ver. 12.0.1. The values were normalized by wet tissue weight. Bioinformatic analysis of the metabolomic data was carried out using Metaboanalyst 3.0 software, including the generation of heat maps. Differences in the abundance of individual metabolites were determined using a Student’s t-test (2 tails, unpaired comparison). A value of p<0.05 was considered statistically significant. Data are given as mean ± s.e.m.
Immunoblotting
CM lysates and heart homogenates were prepared in RIPA buffer containing protease and phosphatase inhibitors (Sigma-Aldrich). Lysates were centrifuged at 13,200 r.p.m. at 4°C for 15 minutes. Total protein lysates (10–30 μg) were incubated with SDS sample buffer (final concentration: 100 mM Tris pH 6.8, 2% SDS, 5% glycerol, 2.5% 2-mercaptoethanol, and 0.05% bromophenol blue) at 95°C for 5 minutes. The denatured protein samples were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes by wet electrotransfer, and probed with primary antibodies.
Subcellular fractionation
Cultured neonatal rat CMs were washed with PBS and collected with ice-cold PBS, followed by centrifugation at 600g for 5 minutes. CMs were then resuspended in hypotonic lysis buffer (10 mM K-HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.1 mM EGTA, 0.1 mM EDTA, 1% IGEPAL, 1% Phosphatase Inhibitor Cocktail, and 1% Protease Inhibitor Cocktail) and were incubated for 15 minutes on ice with intermittent pipetting. Whole-cell lysates were centrifuged at 1200g for 5 minutes. The supernatant was collected for the cytosolic fraction, and the pellets were resuspended in lysis buffer (20 mM K-HEPES, 25% Glycerol, 0.45 M NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1 mM EDTA, 1% Phosphatase Inhibitor Cocktail, and 1% Protease Inhibitor Cocktail) and were incubated for 15 minutes on ice with intermittent pipetting. The total homogenate was centrifuged at 13,000rpm for 10 minutes to collect the nuclear fraction. The pelleted nuclei were resuspended in lysis buffer and protein content was determined for all fractions. The nuclear fraction from mouse hearts was prepared with NE-PER Extraction Reagent (Thermo Scientific).
Immunoprecipitation
CMs or heart samples were lysed with lysis buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton-X 100, 1% Sodium Deoxycholate, Protease Inhibitor Cocktail (Sigma), and Phosphatase Inhibitor Cocktail (Sigma). Primary antibody was covalently immobilized on protein A/G agarose using the Pierce Crosslink Immunoprecipitation Kit according to the manufacturer’s instructions (Thermo Scientific). Samples were incubated with immobilized antibody beads for at least 2 hours at 4°C. After immunoprecipitation, the samples were washed with TBS five times. They were then eluted with glycine-HCl (0.1 M, pH 3.5) and the immunoprecipitates were subjected to immunoblotting using specific primary antibodies and a conformation-specific secondary antibody that recognizes only native IgG (Cell Signaling).
Immune complex in vitro kinase assay
The nuclear fraction was obtained as described in the section on subcellular fractionation. GSK-3α was immunoprecipitated as described in the section on immunoprecipitation. Immunoprecipitated endogenous GSK-3α protein or recombinant GSK-3α protein was incubated with recombinant GST-β-catenin or GST alone as a control in kinase buffer as described in the section on in vitro kinase assays, then incubated with 5xSDS sample buffer at 95°C for 5 minutes, followed by SDS-PAGE. Proteins were detected by immunoblot using pβ-catenin antibody.
In vitro binding assays
Recombinant GST-fused proteins in a slurry of glutathione sepharose were incubated with CM lysates, recombinant GSK-3α, or recombinant GSK-3β in lysis buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton-X 100, 1% Sodium Deoxycholate, Protease Inhibitor Cocktail (Sigma), and Phosphatase Inhibitor Cocktail (Sigma) with rotation for 1 hour at 4°C, followed by pull-down with glutathione-sepharose. After washing five times with PBS, proteins were eluted with 5xSDS sample buffer, followed by SDS-PAGE and immunoblots.
Reporter gene assay
PPRE reporter gene activity in rat neonatal CMs was measured with a luciferase assay system (Promega). Cardiomyocytes were transfected with 3xPPRE luciferase reporter plasmids (0.2 μg) overnight (24 well plate) using LipofectAmine 2000 (Invitrogen). The PPRE reporter gene assay was performed after 9 hours of palmitic acid treatment. CMs were lysed with 50 μl Reporter lysis buffer (24 wells each). The luminescence reaction was started by adding 5 μl lysate to 50 μl Reaction buffer, and luminescence was measured for 10 seconds using an OPTOCOMP I luminometer (MGM Instruments, Inc.). The luminescence was normalized by protein content measured by protein assay kit (BioRad). H9C2 cells were transfected in bulk with 7 μg of total DNA consisting of 6.4 μg of 3xPPRE-luciferase reporter and 0.6 μg of PPARα in a pDC316 vector using FuGene 6 (Roche) at a lipid to DNA ratio of 3:1. Following 18 hours of transfection, cells were trypsinized and replated into 96 well plates (Perkin Elmer) and incubated for 4 hours at 37°C and 5% CO2. Palmitic acid and PPARα ligands were added and the cells were further incubated for 9 hours. Luciferase levels were assayed using BriteLite Plus (Perkin Elmer) and read using a multi-label plate reader (Perkin Elmer).
Chromatin immunoprecipitation (ChIP) assay
CMs were transduced with Ad-YFP-PPARα-WT or -mutants or Ad-YFP as a control (10 cm dish). Two days after transduction, CMs were fixed to cross-link protein-DNA complexes for 10 minutes with 1% formaldehyde in complete medium containing Dulbecco’s modified Eagle’s medium/F-12 supplemented with 5% horse serum, 4 μg/ml transferrin, 0.7 ng/ml sodium selenite, 2 g/l bovine serum albumin (fraction V), 3 mM pyruvate, 15 mM Hepes pH 7.1, 100 μM ascorbate, 100 mg/l ampicillin, 5 mg/l linoleic acid, and 100 μM 5-bromo-2’-deoxyuridine (Sigma) at room temperature. Cross-linking was stopped by addition of 1 M Glycine to a final concentration of 125 mM for 5 minutes. CMs were washed twice with ice-cold PBS and the nuclear fraction was collected as described in the section on subcellular fractionation. The nuclear fractions were subjected to ultrasonic treatment with a Diagenode Bioruptor to generate 200-to 1000-bp DNA fragments and were then diluted 10-fold with dilution buffer [16.7 mM Tris-HCl pH 8.1, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton X-100, protease inhibitor cocktail]. After the addition of EDTA to a final concentration of 0.1 mM, the digested sample was centrifuged at 15,000g for 10 min at 4°C and the resulting supernatant was incubated with rotation at 4°C overnight with anti-GFP-YFP conjugated magnetic beads. The immunoprecipitated chromatin was washed five times with low salt buffer [20 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 1% Triton X, and 0.1% SDS] and high salt buffer [20 mM Tris-HCl, 500 mM NaCl, 2 mM EDTA, 1% Triton X, and 0.1% SDS], followed by elution with elution buffer [0.1 M NaHCO3 and 1% SDS]. Protein-DNA cross-links were reversed by incubation overnight at 65°C with 1% SDS in Tris-EDTA buffer, followed by incubation at 45°C for 1 hour with proteinase K and RNase A. DNA fragments were purified with a Qiaquick PCR Purification kit (QIAGEN). The DNA was then used as a template for RT-PCR analysis. The PCR primer sequences are listed in Table S5.
In vitro oligo pull-down assay
Recombinant Glutathione S-transferase (GST)-fused PPARα-WT or PPARα-S280A proteins were purified from the BL21 E. coli strain transformed with pCold-GST backbone plasmids. The recombinant proteins were subjected to in vitro kinase assays using recombinant GSK-3α protein, followed by purification with Glutathione-sepharose 4B (GE Healthcare), washing with binding buffer 3 times and resuspension with 150 μl binding buffer [10 mM Hepes pH 7.9, 2.5 mM MgCl2, 50 mM KCl, 150 mM NaCl, 5% glycerol, 1 mM DTT, 0.1% IGEPAL CA-630]. The recombinant proteins were incubated with biotin-labeled double-stranded DNAs with streptavidin-beads at 4°C for 2 hours. The recovered proteins were washed 3 times and resolved by SDS-PAGE. The sequences of the double-stranded DNAs are listed in Table 5.
Generation of the PPRE/DR1 sequence logos
We searched the PPRE/DR1 sequences in the promoters of genes upregulated in PPARα-S280D- or-S280A-transduced H9C2 cells (SD vs SA, fold change > 1.5 using the RNAseq data) and in the promoters of fatty acid uptake- or oxidation-related genes using the MEME software (Multiple EM for Motif Elicitation Version 5.0.2, The MEME SUITE). The sequence of the PPRE element containing direct repeats of the AGGTCA half-site separated by one base pair (canonical PPRE/DR1) together with the upstream and downstream three base pairs (nnnAGGTCAnAGGTCAnnn) was used as an input motif for matches to the DR1 sequences in the promoters with a threshold of p < 1.0E-4. Based on previously published reports, the following genes were assigned for fatty acid uptake/transport or oxidation-related genes in the heart: Slc27a1, Lpl, Acsl1, Cd36, Fabp3, and Fabp4 for uptake/transport; Acadvl, Acadm, Acadl, Acads, Acadsb, Hadha, Hadhb, Echs1, and Hadh for oxidation. Using the identified sequences, we generated the PPRE/DR1 sequence logos with the MEME software. Finally, we identified rat genes containing each PPRE/DR1 motif in their promoters with a threshold of p < 1.0E-5 using MEME. The 5000 bp promoter sequences in the rat genome were obtained using the UCSC Genome Browser (RGSC 6.0/m6). Genes containing both fatty acid uptake- and oxidation-related motifs were excluded. Genes containing both PPARα-S280D- and -S280A-related motif were excluded as well. The remaining genes were plotted in a Venn Diagram using Bioinformatics & Evolutionary Genomics website.
Echocardiography
Mice were anesthetized using 12 μl/g body weight of 2.5% avertin (Sigma-Aldrich), and echocardiography was performed using ultrasound (Vivid 7, GE Healthcare). A 13-MHz linear ultrasound transducer was used. Mice were subjected to 2-dimension guided M-mode measurements of LV internal diameter to measure systolic function and wall thickness, and transmitral inflow Doppler echocardiography in apical 4-chamber view to measure deceleration time of early filling of mitral inflow. Both were taken from at least three beats and averaged. LV ejection fraction was calculated as follows: Ejection fraction = [(LVEDD)3 – (LVESD)3]/(LVEDD)3 × 100. For all mouse experiments, data analysis was conducted in a blinded manner.
Pressure-volume (PV) loop analysis
Mice were anesthetized with pentobarbital (60 mg/kg, intraperitoneal injection) and subjected to PV loop analyses to measure diastolic and systolic cardiac function. The right common carotid artery was surgically isolated and clamped proximally and distally. A small incision (5–10 mm) was made on the carotid artery, and a 1.4-French pressure-conductance catheter (Millar Instruments) was inserted in the vessel and introduced into the left ventricle. The PV relation and hemodynamics were continuously recorded. After stabilization, the end-systolic PV relation and its slope and the end-diastolic PV relation and its slope were obtained by intermittent occlusion of the inferior vena cava.
Metabolic assay
The oxygen consumption rate (OCR, pmol/min) in cultured CMs was determined using a Seahorse XF24 or XF96 Extracellular Flux Analyzer (Seahorse Bioscience) according to the Installation and Operation Manual from Seahorse Bioscience. CMs were plated at a density of 200,000 cells/well in 24-well Seahorse assay plates or at a density of 50,000 cells/well in 96-well Seahorse assay plates, followed by transduction with Ad-PPARα-WT or Ad-PPARα-mutants for 34 hours and additional incubation with 500 μM BSA-conjugated fatty acids for 14 hours prior to measurement. One hour prior to the beginning of measurements, the medium was replaced with XF assay medium containing unbuffered DMEM, 5.5 mM glucose, and 0.5 mM carnitine, and CMs were incubated for 1 hour in a 37°C incubator without CO2. Etomoxir (100 μM), a specific Cpt-1 inhibitor, was added to assess the degree of FAO (Kim et al., 2013).
Adult CMs isolation and metabolic assay
Adult CMs were isolated as described previously with a modification (Ackers-Johnson et al., 2016). Briefly, the heart of a male mouse was perfused with 12 ml EDTA buffer [130 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 10 mM HEPES, 10 mM Glucose, 10 mM BDM, 10 mM Taurine, 5 mM EDTA] to stop the beating of the heart. Digestion was achieved using 30 ml perfusion buffer [130 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 10 mM HEPES, 10 mM Glucose, 10 mM BDM, 10 mM Taurine, 1 mM MgCl2] containing Collagenase type II (0.5mg/ml) and Protease XIV (0.05mg/ml). Cellular dissociation was stopped by addition of 5 ml Perfusion buffer containing 5% FBS and 500 μM BSA-conjugated fatty acid cocktail (palmitic acid: oleic acid: linoleic acid = 2:1:1 and BSA: fatty acid = 1:5). CMs and non-CMs were separated by 4 sequential rounds of gravity settling with calcium reintroduction medium containing 500 μM BSA-conjugated fatty acid cocktail. Adult CMs were plated at a density of 200 cells/well in 96-well Seahorse assay plates with XF assay medium, containing 5.5 mM glucose, 500 μM BSA-conjugated fatty acid cocktail, 10 mM BDM and 0.5 mM carnitine. Adult CMs were incubated in a 37°C incubator with CO2 for 30 minutes and without CO2 for 30 minutes prior to measurement. The FAO rate was determined by Etomoxir (250 μM)-inhibitable OCR using a Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience).
Fatty acid oxidation
Fatty acid β-oxidation was measured using heart homogenates as described previously (Watanabe et al., 2000). The left ventricle (LV) of a fresh heart was washed with PBS, weighed and homogenized in four volumes of homogenate buffer [0.25 M Sucrose, 1 mM EDTA] per LV weight (mg). Forty μl of the heart homogenate was dispensed per tube and centrifuged at 800g for 1.5 minutes and the precipitates were incubated with 200 μl of Reaction buffer [10 mM Hepes pH 7.4, 150 mM KCl, 0.1 mM EDTA, 1 mM K2HPO4, 10 mM MgCl2, 1 mM Malate, 10 mM Glucose, 1 μCi [9,10-3H] palmitic acid and 50 μM of unlabeled palmitic acid, both bound to BSA at the molecular ratio of 1:3.7 (BSA : fatty acid)] at 37°C for 10 minutes. The reaction was stopped by the addition of 1 ml chloroform/methanol (2:1) and the samples were mixed well. The samples were kept at −20°C for 4 hours and centrifuged at 12,000g at room temperature for 10 minutes. The supernatants containing 3H2O were mixed with 4 ml scintillation fluid and the radioactivity was measured with a scintillation counter.
Fatty acid uptake in vitro
Rat neonatal CMs were transduced with adenovirus harboring PPARα-WT, PPARα-S280A, PPARα-S280D or YFP alone. Two days after transduction, CMs were incubated in CM culture medium containing 500 μM unlabeled BSA-conjugated palmitic acid or 500 μM unlabeled BSA-conjugated fatty acid cocktail (palmitic acid, oleic acid, and linoleic acid (2:1:1 ratio)) with 1 μCi/ml of [9,10-3H(N)]-palmitic acid alone or 1 μCi/ml of combined [9,10-3H(N)]-palmitic acid and [9,10-3H(N)]-oleic acid and 1.25% BSA for 10 minutes (Liu et al., 2011). After washing CMs with ice-cold PBS four times, CMs were harvested with trypsin-EDTA and centrifuged at 800g at room temperature for 3 minutes. The pellet was resuspended with 1 ml PBS and centrifuged at 800g at room temperature for 3 minutes again. CMs were lysed with 1 ml of 0.5 M NaOH and neutralized with 6 M HCl. The radioactivity was measured with a scintillation counter.
Palmitate uptake into the heart in vivo
In vivo palmitic acid uptake was measured as previously described with modifications (Khan et al., 2013). Palmitic acid [9, 10-3H(N)]- 5 mCi (PerkinElmer, NET043005MC) was complexed with 6% BSA at 37°C. Male GSK-3α S21A KI and control mice were fasted for 2 hours and were intravenously administered the labeled BSA-conjugated palmitic acid and 400 μM of unlabeled BSA-conjugated palmitic acid via the femoral vein with ~106 counts and euthanized 5 minutes post-injection. Blood samples collected 2 minutes after the injection were used for normalization. The heart was quickly washed with PBS to remove the blood from the ventricles and weighed. The heart was lysed with 1 ml of 0.5 M NaOH and neutralized with 6 M HCl. Radioactivity was determined in 20 μl of blood and a whole heart on an LS 6500 multipurpose scintillation counter (Beckman-Coulter). Palmitic acid uptake into the heart was normalized by the radioactivity of the blood and the heart weight.
Oil Red O staining
Neonatal CMs were cultured on coverslips and fixed in 4% paraformaldehyde. The left ventricle of the heart was frozen with O.C.T. compound and Cryomold Biopsy (Tissue-Tek). The heart tissue was cut into 10 μm thick sections on a glass slide. Frozen sections were air-dried for 10 minutes at room temperature and fixed with 10% formalin for 5 minutes. After washing with PBS, the sections were dipped into 60% isopropyl alcohol three times and then put into Oil Red O working solution [0.5% Oil Red O (Sigma) dissolved in isopropyl alcohol (W/V)] for 15 minutes. The sections were washed with 60% isopropyl alcohol three times and then water for 30 minutes. Cover glasses were mounted with mounting medium. Images were obtained in a blinded manner and analyzed with ImageJ software (NIH).
Immunohistochemistry
The heart tissue was washed with PBS, fixed in 4% paraformaldehyde overnight, embedded in paraffin, and sectioned at 10-μm thickness onto a glass slide. After de-paraffinization, sections were stained with Picric acid sirius red (PASR) or Periodic acid-Schiff for evaluation of fibrosis or glycogen accumulation, respectively. Images were obtained in a blinded manner. The cross-sectional area of CMs was measured after wheat germ agglutinin (WGA) staining using ImageJ software (NIH).
Immunofluorescence staining
Rat neonatal CMs were cultured on coverslips, transduced with adenoviruses for 2 to 4 days, washed with PBS, and fixed in 4% paraformaldehyde for 10 minutes. Samples were permeabilized with PBST (0.5% Triton-X in PBS) for 15 minutes and blocked in 5% BSA, 5% goat serum in PBST for 30 minutes at 37°C. Samples were incubated with specific antibodies at 4°C overnight, washed, and incubated with secondary antibody conjugated with Alexa Fluor 488, 555, or 594 dye (Life Technologies). Samples were washed with PBS and mounted on glass slides with mounting medium containing DAPI to stain nuclei (VECTASHIELD, Vector Laboratories). Cells were observed under a fluorescence microscope. Images were obtained in an unblinded manner.
Proximity Ligation Assay
The interaction between GSK-3α and PPARα was examined in situ using Duolink. Rat neonatal CMs were transduced with adenovirus harboring YFP-PPARα, followed by incubation with 100 μM or 500 μM of BSA-conjugated palmitic acid in the presence or absence of fenofibrate. After fixation, slides were incubated for 10 min at 95°C in antigen retrieval buffer (100 mM Tris, 5% urea, pH 9.5). Slides were then blocked for 4 hours at 4°C with PBS containing 5% normal goat serum. Anti-GSK-3α and anti-GFP-YFP primary antibodies were added (diluted 1:100 in blocking buffer) and slides were incubated in a humidity chamber overnight at 4°C. Slides were further blocked at 37°C in Duolink blocking buffer for 1 hour in a humidity chamber. Slides were then washed in Duolink Wash Buffer A twice at room temperature. PLA PLUS Anti-mouse and PLA MINUS Anti-rabbit probes were then incubated with slides in a preheated humidity chamber for 1 hour at 37°C. After washing in Wash Buffer A twice for 5 minutes, slides were incubated with Duolink Ligation Stock for 30 minutes at 37°C. After that, slides were washed in Wash Buffer A and incubated with Duolink Polymerase in Duolink Amplification Stock for 100 minutes at 37°C. After two washes with Wash Buffer B, slides were mounted with Duolink Mounting Medium with DAPI. Images were obtained in an unblinded manner.
• Quantification and Statistical Analysis
All values for continuous data are expressed as mean ± s.e.m. Statistical analyses were carried out by 2-tailed unpaired Student t test for 2 groups or 1-way ANOVA followed by the Newman-Keuls post-hoc analysis for 3 groups or more, assuming independent variables, normal distribution, and equal variance of samples. Categorical variables were compared using Fisher’s exact test. A p value of less than 0.05 was considered significant. Statistical parameters can be found in the figure legends. Statistical analyses were performed using GraphPad Prism (GraphPad Software).
• Data and Software Availability
The RNA sequencing data on H9C2 cell lines transduced with PPARα mutants have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE112309. The microarray data on the hearts of GSK-3α and GSK-3β knock-in mice have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE112160.
Supplementary Material
Highlights.
Fatty acids upregulate GSK-3α, which, in turn, phosphorylates PPARα at Ser280.
Ser280-phosphorylation stimulates biased gene expression favoring fatty acid uptake.
PPARα-Ser280 phosphorylation promotes the development of lipotoxic cardiomyopathy.
Fibrates inhibit Ser280-phosphorylation, thereby reversing cardiac lipotoxicity.
Acknowledgments
We thank Dr. Peter S. Klein for providing us with GSK-3α heterozygous floxed mice. We thank Daniela Zablocki for critical reading of the manuscript and Mayumi Nakamura for the graphics. We thank Dr. Richard Kitsis for his help in generating PPARα-S280A KI mice. The authors thank Dr. Shinichi Oka for technical suggestions. This work was supported in part by U.S. Public Health Service grants HL67724, HL91469, HL102738, HL112330 and AG23039 (to J.S.). This work was also supported by the Leducq Foundation Transatlantic Network of Excellence (to J.S.), American Heart Association Founders Affiliate Postdoctoral Fellowship (14POST18870094) and Scientist Development Grant (17SDG33660358) (to M.N.), and DRC at Washington University Grant (No. 5 P30 DK020579) (to J.S.W.). The mass spectrometry data were obtained from an Orbitrap instrument funded in part by an NIH grant (NS046593) for the support of the Rutgers University Neuroproteomics Core Facility.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackers-Johnson M, Li PY, Holmes AP, O’Brien SM, Pavlovic D, and Foo RS (2016). A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circulation research 119, 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Alfaguter I, Yaffe Y, Licht-Murava A, Urbanska M, Jaworski J, Pietrokovski S, Hirschberg K, and Eldar-Finkelman H (2011). Distinct molecular regulation of glycogen synthase kinase-3alpha isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. The Journal of biological chemistry 286, 13470–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks AS, McAllister FE, Camporez JP, Zushin PJ, Jurczak MJ, Laznik-Bogoslavski D, Shulman GI, Gygi SP, and Spiegelman BM (2015). An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature 517, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugger H, and Abel ED (2009). Rodent models of diabetic cardiomyopathy. Disease models & mechanisms 2, 454–466. [DOI] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Bostrom P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Bluher M, et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, and Mangelsdorf DJ (2014). Nuclear Receptors, RXR, and the Big Bang. Cell 157, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Bernal-Mizrachi C, Han DH, Coleman T, Sambandam N, LaRiviere LL, Holloszy JO, Semenkovich CF, and Kelly DP (2005). A potential link between muscle peroxisome proliferator-activated receptor-alpha signaling and obesity-related diabetes. Cell metabolism 1, 133–144. [DOI] [PubMed] [Google Scholar]
- Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, and Kelly DP (2003). A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A 100, 1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, et al. (2002). The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 109, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg IJ, Trent CM, and Schulze PC (2012). Lipid metabolism and toxicity in the heart. Cell metabolism 15, 805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, et al. (2014). Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 159, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, and Kay MA (2008). In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. Journal of virology 82, 5887–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group AS, Ginsberg HN, Elam MB, Lovato LC, Crouse JR 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, et al. (2010). Effects of combination lipid therapy in type 2 diabetes mellitus. The New England journal of medicine 362, 1563–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, et al. (2011). ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med 17, 1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Popkie AP, Xiong W, Li L, Wang Y, Phiel CJ, and Chen Y (2010). Gsk3beta is required in the epithelium for palatal elevation in mice. Developmental dynamics : an official publication of the American Association of Anatomists 239, 3235–3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nature reviews Molecular cell biology 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, and Woodgett JR (2000). Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406, 86–90. [DOI] [PubMed] [Google Scholar]
- Jellusova J, Cato MH, Apgar JR, Ramezani-Rad P, Leung CR, Chen C, Richardson AD, Conner EM, Benschop RJ, Woodgett JR, et al. (2017). Gsk3 is a metabolic checkpoint regulator in B cells. Nature immunology 18, 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, and Wahli W (1999). Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. The Journal of clinical investigation 103, 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan RS, Lin Y, Hu Y, Son NH, Bharadwaj KG, Palacios C, Chokshi A, Ji R, Yu S, Homma S, et al. (2013). Rescue of heart lipoprotein lipase-knockout mice confirms a role for triglyceride in optimal heart metabolism and function. American journal of physiology Endocrinology and metabolism 305, E1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, et al. (2013). Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature 494, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Yu S, Khan RS, Ables GP, Bharadwaj KG, Hu Y, Huggins LA, Eriksson JW, Buckett LK, Turnbull AV, et al. (2011). DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. Journal of lipid research 52, 732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, and Stanley WC (2010). Myocardial fatty acid metabolism in health and disease. Physiological reviews 90, 207–258. [DOI] [PubMed] [Google Scholar]
- Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, Sciarretta S, Del Re DP, Zablocki DK, Hsu CP, et al. (2013). Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 19, 1478–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S, et al. (2008). Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proceedings of the National Academy of Sciences of the United States of America 105, 20900–20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer S (2007). The failing heart--an engine out of fuel. N Engl J Med 356, 1140–1151. [DOI] [PubMed] [Google Scholar]
- Oka S, Alcendor R, Zhai P, Park JY, Shao D, Cho J, Yamamoto T, Tian B, and Sadoshima J (2011). PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell metabolism 14, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Nagayama T, Mukhopadhyay P, Batkai S, and Kass DA (2008). Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nature protocols 3, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle PJ, Garland PB, Hales CN, and Newsholme EA (1963). The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789. [DOI] [PubMed] [Google Scholar]
- Roberts LD, Koulman A, and Griffin JL (2014). Towards metabolic biomarkers of insulin resistance and type 2 diabetes: progress from the metabolome. The lancet Diabetes & endocrinology 2, 65–75. [DOI] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2012). Mechanisms for insulin resistance: common threads and missing links. Cell 148, 852–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma S, Ardehali H, and Gheorghiade M (2012). Enhancing the metabolic substrate: PPAR-alpha agonists in heart failure. Heart Fail Rev 17, 35–43. [DOI] [PubMed] [Google Scholar]
- Schilling JD, and Mann DL (2012). Diabetic cardiomyopathy: bench to bedside. Heart Fail Clin 8, 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI (2014). Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. The New England journal of medicine 371, 1131–1141. [DOI] [PubMed] [Google Scholar]
- Silverman MG, Ference BA, Im K, Wiviott SD, Giugliano RP, Grundy SM, Braunwald E, and Sabatine MS (2016). Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions: A Systematic Review and Meta-analysis. Jama 316, 1289–1297. [DOI] [PubMed] [Google Scholar]
- Vega RB, and Kelly DP (2017). Cardiac nuclear receptors: architects of mitochondrial structure and function. The Journal of clinical investigation 127, 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, et al. (2000). Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor alpha associated with age-dependent cardiac toxicity. The Journal of biological chemistry 275, 22293–22299. [DOI] [PubMed] [Google Scholar]
- Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, et al. (2002). Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 415, 813–817. [DOI] [PubMed] [Google Scholar]
- Zoete V, Grosdidier A, and Michielin O (2007). Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochimica et biophysica acta 1771, 915–925. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.