

Abstract
The combination of chronic dietary exposure to the fungal toxin, aflatoxin B1 (AFB1), and hepatitis B viral (HBV) infection is associated with an increased risk for early onset hepatocellular carcinomas (HCCs). An in-depth knowledge of the mechanisms driving carcinogenesis is critical for the identification of genetic risk factors affecting the susceptibility of individuals who are HBV infected and AFB1 exposed. AFB1-induced mutagenesis is characterized by G to T transversions. Hence, the DNA repair pathways that function on AFB1-induced DNA adducts or base damage from HBV-induced inflammation are anticipated to have a strong role in limiting carcinogenesis. These pathways define the mutagenic burden in the target tissues and ultimately limit cellular progression to cancer. Murine data have demonstrated that NEIL1 in the DNA base excision repair pathway was significantly more important than nucleotide excision repair relative to elevated risk for induction of HCCs. These data suggest that deficiencies in NEIL1 could contribute to the initiation of HCCs in humans. To investigate this hypothesis, publicly-available data on variant alleles of NEIL1 were analyzed and compared with genome sequencing data from HCC tissues derived from individuals residing in Qidong County (China). Three variant alleles were identified and the corresponding A51V, P68H, and G245R enzymes were characterized for glycosylase activity on genomic DNA containing a spectrum of oxidatively-induced base damage and an oligodeoxynucleotide containing a site-specific AFB1-formamidopyrimidine guanine adduct. Although the efficiency of the P68H variant was modestly decreased, the A51V and G245R variants showed nearly wild-type activities. Consistent with biochemical findings, molecular modeling of these variants demonstrated only slight local structural alterations. However, A51V was highly temperature sensitive suggesting that its biological activity would be greatly reduced. Overall, these studies have direct human health relevance pertaining to genetic risk factors and biochemical pathways previously not recognized as germane to induction of HCCs.
Keywords: base excision repair, formamidopyrimidines, thymine glycol, aflatoxin, hepatocellular carcinoma
1. Introduction
There exist a number of barriers to understanding the fundamental genetic drivers that modulate susceptibility to the development of early onset hepatocellular carcinomas (HCCs) in individuals who are not only chronically exposed to the food-borne fungal toxin, aflatoxin B1 (AFB1), but also infected with hepatitis B virus (HBV) (reviewed in (1) and (2)). Many extrinsic factors are known to be well-correlated with individuals who develop early-onset HCCs, including AFB1, HBV and other factors which promote chronic inflammation. However, these environmental and lifestyle conditions are so ubiquitously distributed among at-risk populations in portions of East Asia, sub-Saharan Africa, Mexico, Central, and South America, that they are insufficient to predict individual susceptibility to the development of HCCs. Although it was anticipated that variants or altered gene expression of bioactivation and detoxication pathways or deficiencies in DNA nucleotide excision repair (NER) might contribute to individual susceptibility to the major AFB1 DNA adducts, i.e. AFB1-N7-guanine (AFB1-N7-Gua) and two diastereomers of the imidazole ring-opened 8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxyaflatoxin B1 (AFB1-FapyGua) (3–8), these hypotheses have generally not been supported by genome analyses except for rare homozygotic inactivating mutations in xeroderma pigmentosum (XP) complementation groups C and D (XPC and XPD) NER enzymes (9, 10). In addition, studies demonstrating that XP complementation group A (XPA) knockout mice only exhibited a 1.2 risk ratio for the development of HCCs following maximally tolerated AFB1 exposures cast doubt on the exclusive role of NER in the correction of AFB1-induced DNA adducts (11). These results, in combination with a paucity of data correlating specific allelic variants with increased susceptibility, suggest that the current understanding of the drivers in AFB1- and inflammation-associated HCCs is inadequate to account for early onset disease.
Germane to these issues, we have discovered an additional DNA repair pathway for the removal of AFB1-induced DNA damage which is of critical importance in limiting HCC formation. Specifically, we have demonstrated that NEIL1, a base excision repair (BER) DNA glycosylase/abasic site lyase, plays a key role in repair of AFB1-FapyGua and that mice deficient in this enzyme are significantly more susceptible to AFB1-induced HCCs (7). These data raise the possibility that allelic variants of NEIL1 which compromise catalytic activity or intracellular stability may modulate individual susceptibility to HCC formation. Furthermore, NEIL1 is also one of the key DNA glycosylases responsible for the removal of several oxidatively-induced DNA base lesions which are produced as a result of chronic inflammation. The major NEIL1 substrates include 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua), 4,6-diamino-5-formamidopyrimidine (FapyAde) and thymine glycol (ThyGly) (reviewed in (12)). Collectively, these data identified NEIL1 as an essential enzyme for genome maintenance under conditions of both AFB1 exposure and chronic inflammation.
We had demonstrated that the G83D and C136R variants of NEIL1 (Fig. 1) were glycosylase inactive (13), and suggested that catalytically or otherwise compromised NEIL1 variants could add to overall DNA adduct burdens sustained by hepatocytes. Relevant to this conclusion, recent data demonstrated that expression of the G83D variant of NEIL1 resulted in an oncogenic phenotype in MCF10A immortal human breast epithelial cells (14). Expression of this variant, in the presence of WT NEIL1, induced replication stress, genomic instability, and cellular transformation through a mechanism postulated to involve the masking of DNA adducts at replication forks that are substrates for NEIL1. These data suggest that repair of AFB1- and oxidatively-induced DNA damage could be blocked by catalytically-compromised, but correctly folded, variants of NEIL1 that are capable of binding to specific DNA base lesions. Additionally, we have previously demonstrated a significant phenotype in Neil1+/− mice that showed intermediate (relative to WT and knockout) manifestations of the metabolic syndrome, including obesity, fatty liver disease, and elevated circulating leptin levels (15). These data have significant implications for human health by linking NEIL1 functional heterozygosity with increased susceptibility to HCCs caused by AFB1 or oxidatively-induced DNA damage.
Fig. 1. Location of amino acid residues in NEIL1 that are changed in the variant alleles.
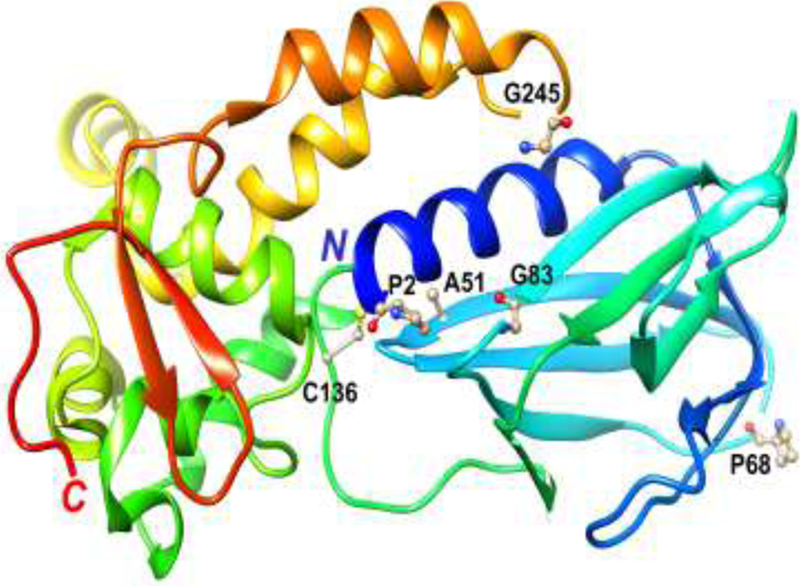
Structure of NEIL1 (truncated form lacking 95 amino acids at the C-terminal domain (20)) with the protein depicted as a ribbon cartoon and in rainbow coloring from blue, N-terminus, to red, C-terminus. The key catalytic residue (P2) and the relevant residues that are changed in the variant alleles (G83, C136, A51, P68, and G245) are highlighted in ball-and-stick mode and labeled.
Given the sum of biochemical and rodent data demonstrating the ability of NEIL1 to efficiently remove oxidatively-induced DNA base lesions and chemically stable AFB1-FapyGua adducts, and considering its critical role in protecting against AFB1-induced HCCs in mice, this investigation was designed to identify and characterize the major NEIL1 variants in East Asia.
2. Materials and Methods
2.1. Expression constructs
The variant alleles coding for full-length NEIL1 enzymes were constructed using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs). The primer oligodeoxynucleotides (Integrated DNA Technologies) had the following sequences, with the variant nucleotide sites underlined: GCTTCAGTCCGCGGCAAGGA (A51V forward), TGA GAT GCG GTA GGC ACT GCT (A51V reverse); CCCAGCACCAACAGGAGCC (P68H forward); CCCC AGG CAG AGG GCT CAG (P68H reverse); GGCTACAGGTCAGAGAGCGGG (G245R forward); CCTGCCCCCCAACTGGACCA (G245R reverse). All designed substitutions were confirmed by DNA sequence analysis of the open reading frame for NEIL1 with a C-terminal 6-His-tag (DNA Sequencing Core, Vollum Institute, Oregon Health & Science University).
2.2. Expression and purification of NEIL1 enzymes
The expression vectors described above were introduced into BL21(DE3) Escherichia coli (New England Biolabs) and NEIL1 enzymes were purified as previously described (13). Specifically, an overnight culture was prepared from a single colony in lysogeny broth supplemented with 50 µg/mL ampicillin at 37 °C, the culture was diluted 100-fold and continued to grow under these conditions to an optical density (OD600) of 0.6. Expression of NEIL1 was induced by addition of isopropyl 1-thio-β-D-galactopyranoside to 0.5 mmol/L at 20 °C for 3.5 h. Cells were harvested and resuspended in 50 mmol/L sodium phosphate, pH 7.5, 300 mmol/L sodium chloride, 50 mmol/L imidazole (binding buffer) with EDTA-free complete protease inhibitor cocktail (Roche). Single-cell suspensions were passed through a French pressure cell at 8,300 kPa. Cellular debris was removed by centrifugation at 10,000 g for 20 min and the supernatant loaded onto a Ni-NTA agarose column (Qiagen), pre-equilibrated with binding buffer. The matrix was washed with ≈ 20 times the column volume of binding buffer, and proteins were eluted with a 50 mmol/L to 500 mmol/L imidazole gradient in a total volume of 12-times the column volume and 2–3 mL fractions collected. The NEIL1-containing fractions were combined and dialyzed against 20 mmol/L Tris-HCl (pH 7.4), 100 mmol/L KCl, 1 mmol/L β-mercaptoethanol, 0.1 mmol/L EDTA, 15 % (v/v) glycerol.
2.3. Site-specifically modified DNA substrates
The double-stranded ThyGly-containing DNA substrates conjugated with the carboxytetramethylrhodamine (TAMRA) fluorescent reporter were prepared from commercially produced oligodeoxynucleotides (Integrated DNA Technologies) according to the previously described protocol (16). The adducted 17-mer oligodeoxynucleotide (5′-TCACCGXCGTACGACTC-3′, where X shows the location of ThyGly) had the TAMRA moiety on its 5′ terminus. In assays using a plate reader for monitoring DNA incisions, the complement oligodeoxynucleotide (5′-GAGTCGTACGACGGTGA-3′) contained the Black Hole Quencher 2 (BHQ2) on its 3′ terminus. In the gel-based analyses, the complement strand had no modifications.
The 24-mer oligodeoxynucleotide containing a site-specific AFB1-FapyGua was synthesized as reported (17). The sequence of modified DNA was 5′-ACCACTACTATXATTCATAACAAC-3′, where X corresponds to the adducted site. The adducted oligodeoxynucleotides were labeled at the 5′ termini using [γ-32P]ATP (PerkinElmer Life Sciences) and T4 polynucleotide kinase (New England Biolabs) and annealed with the complement strand (5′-GTTGTTATGAATCATAGTAGTGGT-3′) as described (7).
2.4. DNA Glycosylase assays using site-specifically modified DNA substrates
The NEIL1-catalyzed reactions were conducted in 20 mmol/L Tris-HCl, pH 7.4, 100 mmol/L KCl, 100 μg/mL BSA, and 0.01 % Tween 20. The detailed description of the experimental approach using TAMRA-conjugated DNA substrates and detection of the TAMRA fluorescent signal by a plate reader to monitor for DNA incisions was previously reported (16). Briefly, DNA substrate with the TAMRA moiety on the 5′ terminus of ThyGly-containing strand and the BHQ2 on the 3′ terminus of the complement strand was aliquoted into wells of a black 384-well plate, and reactions were initiated by the addition of NEIL1 enzymes. The volume of a standard reaction was 20 μL, DNA and enzyme were present at equimolar concentrations (50 nmol/L). Plates were incubated at 37 °C in the Infinite M200 plate reader (Tecan Group Ltd.). The result of DNA incision with subsequent dissociation of the TAMRA-conjugated product from the BHQ2-containing strand was an increase in fluorescent signal that was detected using a 525/9 nm excitation filter and a 598/20 nm emission filter. The data were recorded in arbitrary fluorescence units. In a subset of these experiments, formation of the incision products was validated using gel-based assays described below. These analyses revealed that there was a consistent, but slight delay, in the build-up of the fluorescence signal relative to DNA incision. However, the fluorescence plateau values correspond to complete incision of the substrate DNAs. Thus, the results of assays using a plate reader are presented in arbitrary fluorescence units, as opposed to percentages of product formed.
In gel-based assays, products were resolved through a 15% polyacrylamide gel under denaturing conditions (gel containing 8 mol/L urea and DNA samples containing 63 % (v/v) formamide). For TAMRA-conjugated DNA, gel images were captured with the FluorChem M system (Protein Simple) using a 534 nm LED light source and 593 nm emission filter. The intensities of DNA bands were measured using Image Studio™ Lite version 4.0 software (LI-COR). The 32P-labeled DNA was visualized using a Personal Molecular Imager™ System (Bio-Rad) and quantified from a phosphor screen image by the Quantity One Software (Bio-Rad).
The rate constants for NEIL1-catalyzed incisions were measured under single turnover conditions as previously described (7). Following pre-incubation at 37 °C for 2 min, the 32P-labeled AFB1-FapyGua substrate and enzyme were combined, incubated at 37 °C, and the aliquots removed during the course of reaction. The standard reaction contained 20 nmol/L double-stranded DNA substrate, with enzyme being present at 20- or 25-fold excess molar concentration. The aliquots were removed in the course of reactions, and in order to analyze glycosylase activity independently of lyase activity, DNA was incubated in the presence of 250 mmol/L NaOH at 90 °C for 5 min. Following gel electrophoresis and determination of band intensities, the product formation (P) was plotted as a function of time (t), and rate constant (kobs), non-specific product (Pns), and extrapolated amplitude of substrate used (S) were obtained from the best fit of the data to equation P = Pns + S(1 – e−kobs*t) using KaleidaGraph software (Synergy Software). The average values with standard deviations were calculated from three independent experiments.
2.5. Stabilities of NEIL1 enzymes at 37 °C
The NEIL1 enzymes were diluted in the reaction buffer described above to 100 nmol/L and incubated at 37 °C for 20 min. Incubations were monitored at 5 min intervals, tubes were placed on ice, and aliquots were collected and tested for activity using TAMRA/BHQ2-conjugated ThyGly substrate at equimolar enzyme to DNA concentrations (50 nmol/L). The production of fluorescent signal was monitored in a plate reader for 20 min and the rates were calculated from the linear phase of reactions (first 5 min). To assess the initial glycosylase activity, similar reactions also were conducted prior to incubation of enzyme at 37 °C. The rates obtained from the latter reactions were used as a reference to calculate the fraction of activity that remained at the given time point (A(t)). The average A(t) values from at least three independent experiments were plotted as a function of time (t), and rates of reduction in activity (λ) were obtained from the best fit of the data to equation A(t) = e−λ*t; the half-life was calculated using t1/2 = ln(2)/λ.
2.6. Preparation of high-molecular weight DNA substrate for glycosylase reactions
Commercially available high-molecular weight calf thymus DNA (Sigma-Aldrich, St. Louis, MO) was dissolved in phosphate buffer (pH 7.4) at a concentration of 0.3 mg/mL at 4 °C. DNA solution was saturated with N2O for 1 h and then irradiated in a 60Co γ-ray source at a dose of 5 Gy, followed by dialysis against water for 18 h at 4 °C. Water outside the dialysis tubes was replaced twice. Aliquots of 50 µg DNA were dried in a Speedvac under vacuum and kept at 4 °C until use.
2.7. NEIL1-catalyzed reactions using high-molecular weight DNA substrate and detection of excised DNA base lesions by gas chromatography-tandem mass spectrometry (GC-MS/MS)
For NEIL1-catalyzed reactions, aliquots of γ-irradiated DNA (50 μg) were supplemented with stable isotope-labeled internal standards, i.e., FapyAde-13C,15N2, FapyGua-13C,15N2, 8-OH-Ade-13C,15N2, 8-OH-Gua-15N5, 5-OH-Cyt-13C,15N2, 5-OH-Ura-13C4,15N2, ThyGly-2H4, 5-OH-5-MeHyd-13C,15N2 and 5,6-diOH-Ura-13C,15N2 (isodialuric acid-13C,15N2), which are a part of the NIST Standard Reference Material 2396 Oxidative DNA Damage Mass Spectrometry Standards (for details see http://www.nist.gov/srm/index.cfm and https://wwws.nist.gov/srmors/view_detail.cfm?srm=2396). The samples were then dissolved in 50 μL of 50 mmol/L phosphate buffer (pH 7.4), 100 mmol/L KCl, 1 mmol/L EDTA and 0.1 mmol/L dithiotreitol. Samples were incubated for 1 h at 37 °C using enzyme concentrations that are indicated in the figure legend. In addition to reactions in the presence of NEIL1 enzymes, control reactions were performed in parallel that had no enzyme or contained a combination of the E. coli formamidopyrimidine glycosylase (Fpg) (R&D Systems, Inc. Minneapolis, MN) and endonuclease III (Nth) (Prospec-Tany TechnoGene Ltd., Ness Ziona, Israel). Following precipitation of DNA with ethanol, the supernatant fractions were collected. After removal of ethanol in a Speedvac, the samples were lyophilized and trimethylsilylated. Specifically, 60 μL of a mixture of nitrogen-bubbled bis(trimethylsilyl)trifluoroacetic acid [containing trimethylchlorosilane (1 %; v/v)] (BSTFA) (Pierce, Rockford, Ill.) and pyridine (Sigma-Aldrich, St. Louis, MO) (1:1, v/v) was added to each lyophilized supernatant fraction. The samples were vortexed and purged individually with ultra-high-purity nitrogen, sealed with Teflon-coated septa, and heated at 120 °C for 30 min. After cooling, the clear samples were removed and placed in vials used for injection onto the GC-column. Aliquots (4 µL) of derivatized samples were analyzed by GC-MS/MS under the conditions described previously (18). On the basis of the known mass spectra of trimethylsilyl derivatives of DNA base lesions (reviewed in (19)), the following mass transitions were used for identification and quantification: m/z 369 → m/z 280 and m/z 372 → m/z 283 for FapyAde and FapyAde-13C,15N2 respectively; m/z 457 → m/z 368 and m/z 460 → m/z 371 for FapyGua and FapyGua-13C,15N2, respectively; m/z 367 → m/z 352 and m/z 370 → m/z 355 for 8-OH-Ade and 8-OH-Ade-13C,15N2, respectively; m/z 455 → m/z 440 and m/z 460 → m/z 445 for 8-OH-Gua and 8-OH-Gua-15N5, respectively; m/z 343 → m/z 342, and m/z 346 → m/z 345 for 5-OH-Cyt and 5-OH-Cyt-13C,15N2, respectively; m/z 344 → m/z 343, and m/z 350 → m/z 349 for 5-OH-Ura and 5-OH-Ura-13C4,15N2, respectively; m/z 448 → m/z 259 and m/z 452 → m/z 262 for ThyGly and ThyGly-2H4, respectively; m/z 331 → m/z 316 and m/z 334 → m/z 319 for 5-OH-5-MeHyd and 5-OH-5-MeHyd-13C,15N2, respectively; m/z 432 → m/z 417 and m/z 435 → m/z 420 for 5,6-diOH-Ura and 5,6-diOH-Ura-13C,15N2, respectively. The quantification was achieved using integrated areas of the signals of the mass transitions of the monitored DNA base lesions and their corresponding stable isotope-labeled analogues. The measured levels were expressed as number of lesions per 106 DNA bases. The mean values with standard deviations were calculated from three independent measurements. Significant excision of FapyGua, FapyAde and ThyGly from DNA was observed, whereas the other DNA lesions measured in this work were not significantly excised by NEIL1 enzymes over control levels.
2.8. Modeling the structural consequences of NEIL1 A51V, P68H and G245R allelic variants
The crystallographic coordinates for C-terminally truncated NEIL1 (Δ95-CTD) (20) were retrieved from the Protein Data Bank (www.rcsb.org; PDB ID 5ITQ). Amino acids A51, P68 and G245 were swapped to V, H, and R, respectively, in UCSF Chimera (21). The unfavorable cis conformation of H68 following mutation from proline was converted to trans using the program Coot (22), followed by real-space refinement inside the confines of the final electron density for the NEIL1 crystal structure. The mutated protein structure was then energy minimized with Amber 14 (23) using steepest descent and conjugate gradient cycles until convergence.
3. Results and Discussion
3.1. NEIL1 allelic variants in East Asian populations
Previous data from our laboratories have revealed that in addition to NER being important in the repair of AFB1-induced DNA adducts, the BER pathway is of critical importance in limiting HCC formation by the removal of AFB1-induced DNA damage (reviewed in (2)). These data raise the possibility that allelic variants of NEIL1 with compromised catalytic activity or intracellular stability may modulate individual susceptibility to AFB1-induced HCC. Consequently, we have hypothesized that genetic variation in NEIL1 may modulate HCC susceptibility arising from both AFB1 exposure and high frequency HBV infection.
To investigate this hypothesis, we have carried out systematic analyses of publicly-available DNA sequence data bases for NEIL1 variant allele frequencies specifically using the Genome Aggregation Database (gnomAD) (24) and the 1000 Genomes (25). The gnomAD contains ~300,000 alleles representing 6 global and 8 sub-continental ancestries. The previously characterized glycosylase-incompetent NEIL1 variants were not found in East Asian populations; G83D is associated with European, Ashkenazi Jewish, and Latino ancestries and C136R is found in African populations and at extremely low frequencies. When singletons were excluded, a total of 20 missense variants in the exomes of NEIL1 were identified in East Asian populations, with 3 of these, A51V (rs768187848), P68H (rs187873972), and G245R (rs140982397), present at > 0.1 % frequencies. The positions of these residues within the NEIL1 structure are shown in Fig. 1. Subsequent analyses of the 1000 Genomes database (25) revealed that the most common East-Asian variant, P68H, which presents in this region at an overall frequency of ≈ 1.4 %, is overrepresented in some ethnic groups. Specifically, 8 and 6 alleles for this variant were found in exomes of 190 individuals from Vietnam (Kinh in Ho Chi Minh City) and 180 individuals from China (Chinese Dai in Xishuangbanna), respectively.
Our hypothesis may also be supported by exome sequencing data of HCC patients who reside in a region of Southeast China with known AFB1 exposure (26). In this data set, each of the three NEIL1 variant alleles (A51V, P68H, and G245R) was identified once in the 49-patient cohort. These findings are notable and unanticipated because individually, these allelic variants are only present in East Asia at ≈ 0.17 %, 1.4 %, and 0.65 %, respectively. Further population-based studies are necessary to test the hypothesis that there is an association between rare genetic variations in the NEIL1 gene and susceptibility to HCC.
3.2. Substrate specificity of WT and allelic variants of NEIL1
To express proteins encoded by NEIL1 nonsynonymous allelic variants (A51V, P68H, and G245R), the full-length coding sequence for each variant was engineered into an expression plasmid for K242R NEIL1. The K242R version of NEIL1 results from editing of pre-mRNA by adenosine deaminase, ADAR1 that converts the central adenosine in an AAA codon to inosine, thus altering the translation code from K to R (27). The initial characterization of human NEIL1 (28–30), as well as the majority of subsequent studies on NEIL1 biochemical properties, structure, and modulation of its activity by amino acid changes has been done using the edited version. Thus, WT NEIL1 and its variants investigated here were represented by the edited, K242R forms. All plasmids were sequence verified prior to protein expression in E. coli, and each was purified using Ni-agarose chromatography. Protein preparations were determined to be 90 % to 95 % pure, with no non-specific nicking activities on control DNA.
Preliminary kinetic analyses were carried out on fluorescently-labelled synthetic oligodeoxynucleotides in which a 5’ TAMRA-labelled 17-mer, containing a ThyGly base at position 7, was annealed with a complementary strand containing a 3’ BHQ2 moiety (16). Reactions were initiated by addition of WT or variant proteins to DNA substrate and analyzed in real-time in a plate reader. Since the site of DNA modification was 6 nucleotides from the 5’ TAMRA label, incision of substrate resulted in the melting of the 6-mer and an enhancement of fluorescence. As shown in Fig. 2, all three variant enzymes could cleave the ThyGly-containing oligodeoxynucleotide. The rate of the fluorescent signal production in the presence of A51V (Fig. 2A) was comparable with that of WT during the early phase of reaction (≈ first 10 min), but significantly slowed soon thereafter. This kinetics contrasted with a parallel reaction with WT, in which the fluorescent signal continued to increase throughout the time course. The reaction kinetics observed for P68H (Fig. 2B) and G245R (Fig. 2C) were indistinguishable relative to that of WT. These results demonstrating the abilities of A51V, P68H, and G245R to cleave ThyGly-containing oligodeoxynucleotide were corroborated using gel-based assays (data not shown) and in relation to G245R, agreed with results of a previous study (31). Thus, all three NEIL1 variants possess glycosylase activity for ThyGly and retain lyase function.
Fig. 2. Initial characterization of the NEIL1 variants using fluorescently-labeled ThyGly-containing oligodeoxynucleotides.

The activities of the A51V (A), P68H (B), and G245R (C) variants were tested along with activity of WT NEIL1 using TAMRA/BHQ2-conjugated double-stranded oligodeoxynucleotides that contained a site-specific ThyGly. The reactions were conducted with equimolar DNA and enzyme concentrations (50 nmol/L) at 37 °C. The TAMRA emission was recorded using an Infinite M200 plate reader.
Although the analyses of incision of ThyGly-containing synthetic oligodeoxynucleotides provided initial insights into the activity of these enzymes, the substrate specificity of NEIL1 is more broad and includes FapyGua and FapyAde as the primary base lesions excised from genomic DNAs containing a spectrum of oxidatively-induced DNA damages (reviewed in (12)). Thus, purified WT and each of the allelic variants of NEIL1 were incubated with calf thymus DNA that had been previously exposed to a 5 Gy γ-irradiation dose. Control reactions (no enzyme treatment) and treatment with a combination of the E. coli Fpg and Nth were also performed. Fpg has been previously characterized to release FapyGua, FapyAde and 8-hydroxyguanine with similar excision kinetics (32–34), while Nth has a broad substrate specificity for pyrimidine-derived lesions, including ThyGly, and also removes FapyAde (35, 36). Relative to the WT NEIL1, the efficiency of P68H was reduced on both FapyGua and FapyAde, while the efficiency of G245R was only reduced relative to the release of FapyGua (Fig. 3A and B). Consistent with the results of experiments with site-specifically modified oligodeoxynucleotides, P68H and G245R released ThyGly from genomic DNA with the same efficiently as WT NEIL1, while A51V showed a modest increase in the efficiency of ThyGly incision (Fig. 3C). These data on the excision of ThyGly from genomic DNAs emphasize that this damage is only poorly recognized by edited NEIL1 (37) relative to the total ThyGly in the sample as revealed by the combined Nth/Fpg treatment. The poor excision of ThyGly was in stark contrast to the efficient excision of both FapyGua and FapyAde by NEIL1 enzymes; these base lesions were released at levels comparable to that observed in Fpg/Nth-catalyzed reactions (Fig. 3A and B).
Fig. 3. Removal of oxidatively-induced DNA base lesions from high-molecular weight DNA by WT and variants of NEIL1.

The γ-irradiated calf thymus DNA (50 μg) was incubated for 1 h at 37 °C in the presence of NEIL1 enzymes (2 μg), combination of Fpg and Nth (1 μg each), or in reaction buffer. The released FapyGua (A), FapyAde (B), and ThyGly (C) were measured by GC-MS/MS using their stable isotope-labeled analogues as internal standards. The statistically significant differences are only shown for variant enzyme versus WT (* = p < 0.05; ** = p < 0.01). The differences between the non-specific base hydrolysis and the base release in the presence of enzymes were statistically significant in all cases (p < 0.05). The uncertainties are standard deviations.
3.3. Temperature sensitivity of the A51V NEIL1 variant
The kinetics of A51V-catalyzed reaction with ThyGly-containing oligodeoxynucleotides (Fig. 2A) suggested that incision was nearly complete ≈ 10 min into the reaction. One possible explanation is that the A51V allelic variant is temperature sensitive and that during the reaction, there was an appreciable loss in catalytic activity. To test this hypothesis, WT and each of the variants were incubated at 37 °C for increasing times in reaction buffer that did not contain the DNA substrate and the activities were evaluated from the linear phase of reaction following the addition of damage-containing oligodeoxynucleotides. The fraction of activity remaining relative to the initial activity was plotted as a function of pre-incubation time (Fig. 4) and the experimental points were fit to an exponential equation to obtain rates of reduction in activity. Consistent with the hypothesis of A51V being temperature sensitive, the half-life at 37 °C was estimated to be ≈ 5 min versus ≈ 30 min estimated for WT (Fig. 4A). The gel-based analyses confirmed a rapid inactivation of A51V at 37 °C (data not shown). The A51V enzyme that had been inactivated for 20 min at 37 °C was kept in ice for 1 h and 24 h, and its activity was retested. No evidence for the activity being restored was found (data not shown), suggesting that inactivation of this variant under physiological temperature conditions is irreversible. In contrast, P68H and G245R displayed no enhanced temperature inactivation relative to that of the WT enzyme (Fig. 4B and C, respectively).
Fig. 4. Kinetics of temperature inactivation of WT and variants of NEIL1.

WT and variants of NEIL1 A51V (A), P68H (B), and G245R (C) were pre-incubated at 37 °C and the activities were tested with equimolar DNA and enzyme concentrations (50 nmol/L) using TAMRA/BHQ2-conjugated double-stranded ThyGly substrate. The reactions were conducted at 37 °C and monitored by an Infinite M200 plate reader. The rates were calculated from the linear phase of reactions. The fraction of activity remaining at the given time point (A(t)) was calculated relative to the initial enzyme activity. The average A(t) values from at least three independent experiments were plotted as function of time (t). The uncertainties are standard deviations.
Considering a poor recognition of ThyGly by the edited NEIL1 enzymes (Fig. 3C), an experiment was designed to validate thermal instability of A51V using the preferred NEIL1 substrates, FapyGua and FapyAde. The A51V variant was pre-incubated at 37 °C for 30 min, and then reacted with γ-irradiated calf thymus DNA. The levels of the released base lesions were assessed by GC-MS/MS (Fig. 5). The data confirmed enzyme inactivation under physiological temperature conditions. The removal of FapyGua and FapyAde was reduced 7-fold and 5-fold, respectively (Fig. 5A and B) and consistent with the results of experiments with synthetic oligodeoxynucleotides (Fig. 4A), no residual activity was detected with regard to excision of ThyGly (Fig. 5C).
Fig. 5. Removal of oxidatively-induced DNA base lesions by A51V following pre-incubation of enzyme at 37 °C.

The γ-irradiated calf thymus DNA (50 μg) was incubated for 1 h at 37 °C in the presence of A51V enzyme (2 μg). The aliquot of A51V was pre-incubated at 37 °C for 30 min. Subsequently, DNA was added to the solution which was then incubated for another 30 min. The released FapyGua (A), FapyAde (B), and ThyGly (C) were measured by GC-MS/MS using their stable isotope-labeled analogues as internal standards (* = p < 0.05; ** = p < 0.01; *** = p < 0.001). The uncertainties are standard deviations.
3.4. Incision of synthetic oligodeoxynucleotides containing AFB1-FapyGua
In addition to characterizing the activities of these allelic variants on DNAs containing oxidatively-induced base damages, we also characterized their efficiencies in the incision of DNA containing a site-specific AFB1-FapyGua base adduct (Table 1 and Fig. 6). As shown in Table 1, P68H showed reduced rates of incision relative to WT, with the p < 0.05, while the rate of G245R-catalyzed reaction was comparable to that of WT. The analysis of A51V under single-turnover conditions was not practical due to incompatibility of its temperature sensitivity with conditions under which the single-turnover experiments were conducted. Thus, the ability of A51V to incise AFB1-FapyGua was tested in short, 5-min reactions and the incision product was observed (Fig. 6). However, given a short half-life of this variant at physiologically relevant temperature, the biological significance of this observation can be questioned.
Table 1.
Single-turnover kinetics of WT and variants of NEIL1 on AFB1-FapyGua-containing oligodeoxynucleotides. The uncertainties are standard deviations.
| Enzyme | Incision rates (kobs, min−1) |
|---|---|
| Wild type | 0.18 ± 0.01 |
| A51V | ND |
| P68H | 0.13 ± 0.01 |
| G245R | 0.16 ± 0.02 |
Fig. 6. Incision of AFB1-FapyGua from site-specifically modified oligodeoxynucleotides by WT and A51V NEIL1.

The reactions were conducted with 500 nmol/L WT or A51V NEIL1 at 37 ○C for 5 min using 32P-labeled 24-mer double-stranded DNA (20 nmol/L) that contained a centrally-located AFB1-FapyGua. Representative gel image (A) and percent of product formation (average ± standard deviation) calculated from three independent experiments (B) are shown (* = p < 0.05; ** = p < 0.01; *** = p < 0.001). The uncertainties are standard deviations.
3.5. Modeling the structural consequences of NEIL1 A51V, P68H and G245R allelic variants
The conformational changes as a result of amino acid substitutions are quite limited as per the computational model of A51V-P68H-G245R NEIL1 (Fig. 7). In the WT protein, amino acids P68 and G245 map to loop regions on the surface whereas A51 lies in the interior between a β-strand and an α-helix (Fig. 7A). In the latter case, only slight shifts are necessary to accommodate the bulkier valine side chain (Fig. 7B). The change from cis-amide proline to trans-amide histidine at position 68 only alters the conformation locally and main and side chain atoms in the WT and mutated NEIL1 are superimposable at the neighboring Q67 and Q70 (Fig. 7C). Likewise, the large arginine side chain in the loop at position 245 juts away from the protein surface and can be accommodated by slight adjustments in the preceding Y244 and following S246 and the neighboring residues F13 and Q86 (Fig. 7D).
Fig. 7. NEIL1 crystal structure and conformational effects of allelic variants.
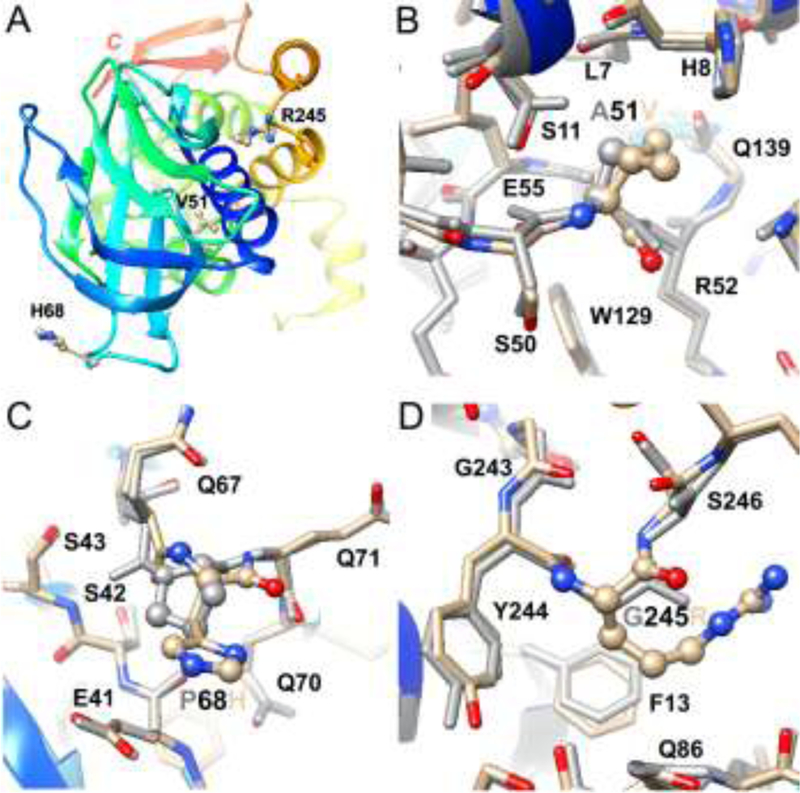
Overall structure of NEIL1 (truncated form lacking 95 amino acids at the C-terminal domain (20)) with the protein depicted as a ribbon cartoon and in rainbow coloring from blue, N-terminus, to red, C-terminus, and variant amino acids highlighted in ball-and-stick mode and labeled (A). Overlay of the WT and variant structures viewed in the regions around A/V 51 (B), P/H 68 (C), and G/R 245 (D). Residues in the variant and energy-minimized structure are colored by atom, with carbon, oxygen and nitrogen atoms shown in tan, red and blue, respectively, and the WT structure is uniformly colored in gray. Variant residues 51, 68 and 245 are highlighted in ball-and-stick mode and selected neighboring residues are labeled.
The observed correlations between the locations of substituted amino acids, the structural changes as inferred from computational modeling, and the phenotypes of NEIL1 sequence variants follow general trends found in protein structure-function relationships. Thus, amino acid changes near the periphery are typically less deleterious than changes mapping to the active site or near the ligand and/or substrate (38). In 3-methyladenine DNA glycosylase, mutations in surface loops, if not involved in DNA binding, are tolerated better relative to those buried in the protein’s exterior, especially when present in β sheets (39). Consistent with these prior observations, substitutions in the surface loop regions, at P68 and G245, imposed only small local changes, with no or minor phenotypical consequences. Deviations from the WT structure in A51V were also minimal, but occurred at the interface of a β-strand and an α-helix. We hypothesize that these local changes are sufficient to facilitate enzyme misfolding, which is manifested in thermal instability. The structural basis for the severely affected phenotype of the G83D variant had previously been addressed. G83D was predicted to affect the conformation of the β4/5 loop by introducing an unfavorable negative charge in an otherwise hydrophobic local environment (40). Indeed, the displacement of residues of the β4/5 loop was observed in the crystal structure of the corresponding G86D variant in Mimivirus Nei1 (37).
Conclusion
The goal of this investigation was to determine whether rare allelic variants of NEIL1 that are geographically clustered in East Asia display alterations in kinetics of excision, substrate specificities, and stabilities relative to the WT enzyme. Our data demonstrated that the P68H allelic variant had a modest reduction in catalytic activity on the release of FapyGua, FapyAde, and AFB1-FapyGua. The substrate specificity and catalytic efficiency of G245R was undistinguishable from WT, except for a reduced efficiency in excision of FapyGua. Additionally, the A51V variant was unstable under physiological temperature conditions, suggesting that it may have no biological activity. Although the significance of these changes is yet to be determined in humans, the presence of these rare allelic variants in HCC patients from Qidong County is suggestive that there could be adverse health outcomes. These studies provide the framework from which larger, disease-based molecular epidemiology studies will be conducted.
Highlights.
Hepatocellular carcinomas (HCCs) occur with highest frequency in East Asia
HCCs are associated with aflatoxin exposures and chronic inflammation
NEIL1 initiates repair of aflatoxin- and inflammation-induced DNA base damage
Variants of NEIL1 that occur in East Asian populations are characterized
A subset of NEIL1 variants are expected to have human health implications
Acknowledgments
Certain commercial equipment or materials are identified in this paper in order to specify adequately the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
This study was supported by the National Institutes of Health Grants R01 CA055678, R01 ES029357, and R56 ES027632. The work of Oskar K. Linde was partially supported by the Summer Internship Program (Oregon Institute of Occupational Health Sciences, Oregon Health & Science University).
Funding Source
All sources of funding should also be acknowledged and you should declare any involvement of study sponsors in the study design; collection, analysis and interpretation of data; the writing of the manuscript; the decision to submit the manuscript for publication. If the study sponsors had no such involvement, this should be stated.
Abbreviations:
- HCC
hepatocellular carcinoma
- AFB1
aflatoxin B1
- HBV
hepatitis B virus/viral
- XP
xeroderma pigmentosum
- NER
nucleotide excision repair
- BER
base excision repair
- AFB1-FapyGua
8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-dihydropyrimid-5-yl-formamido)-9-hydroxyaflatoxin B1
- FapyGua
2,6-diamino-4-hydroxy-5-formamidopyrimidine
- FapyAde
4,6-diamino-5-formamidopyrimidine
- ThyGly
thymine glycol
- TAMRA
carboxytetramethylrhodamine
- BHQ2
Black Hole Quencher 2
- GS-MS/MS
gas chromatography-tandem mass spectrometry
- Fpg
E. coli formamidopyrimidine glycosylase
- Nth
E. coli endonuclease III
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None declared.
References
- (1).Fedeles BI and Essigmann JM (2018) Impact of DNA lesion repair, replication and formation on the mutational spectra of environmental carcinogens: Aflatoxin B1 as a case study. DNA Repair (Amst) 71, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McCullough AK and Lloyd RS (2019) Mechanisms underlying aflatoxin-associated mutagenesis - Implications in carcinogenesis. DNA Repair (Amst) 77, 76–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Essigmann JM, Croy RG, Nadzan AM, Busby WF Jr., Reinhold VN, Buchi G and Wogan GN (1977) Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc Natl Acad Sci U S A 74, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Croy RG, Essigmann JM, Reinhold VN and Wogan GN (1978) Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver. Proc Natl Acad Sci U S A 75, 1745–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Croy RG and Wogan GN (1981) Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1. Cancer Res 41, 197–203. [PubMed] [Google Scholar]
- (6).Woo LL, Egner PA, Belanger CL, Wattanawaraporn R, Trudel LJ, Croy RG, Groopman JD, Essigmann JM, Wogan GN and Bouhenguel JT (2011) Aflatoxin B1-DNA adduct formation and mutagenicity in livers of neonatal male and female B6C3F1 mice. Toxicol Sci 122, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Vartanian V, Minko IG, Chawanthayatham S, Egner PA, Lin YC, Earley LF, Makar R, Eng JR, Camp MT, Li L, Stone MP, Lasarev MR, Groopman JD, Croy RG, Essigmann JM, McCullough AK and Lloyd RS (2017) NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc Natl Acad Sci U S A 114, 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Coskun E, Jaruga P, Vartanian V, Erdem O, Egner PA, Groopman JD, Lloyd RS and Dizdaroglu M (2019) Aflatoxin-guanine DNA adducts and oxidatively-induced DNA damage in aflatoxin-treated mice in vivo as measured by liquid chromatography-tandem mass spectrometry with isotope-dilution. Chem Res Toxicol 32, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Long XD, Ma Y, Zhou YF, Ma AM and Fu GH (2010) Polymorphism in xeroderma pigmentosum complementation group C codon 939 and aflatoxin B1-related hepatocellular carcinoma in the Guangxi population. Hepatology 52, 1301–1309. [DOI] [PubMed] [Google Scholar]
- (10).Long XD, Ma Y, Zhou YF, Yao JG, Ban FZ, Huang YZ and Huang BC (2009) XPD codon 312 and 751 polymorphisms, and AFB1 exposure, and hepatocellular carcinoma risk. BMC Cancer 9, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Takahashi Y, Nakatsuru Y, Zhang S, Shimizu Y, Kume H, Tanaka K, Ide F and Ishikawa T (2002) Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene-deficient mice. Carcinogenesis 23, 627–633. [DOI] [PubMed] [Google Scholar]
- (12).Dizdaroglu M, Coskun E and Jaruga P (2017) Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutat Res 771, 99–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M and Lloyd RS (2007) Human polymorphic variants of the NEIL1 DNA glycosylase. J Biol Chem 282, 15790–15798. [DOI] [PubMed] [Google Scholar]
- (14).Galick HA, Marsden CG, Kathe S, Dragon JA, Volk L, Nemec AA, Wallace SS, Prakash A, Doublie S and Sweasy JB (2017) The NEIL1 G83D germline DNA glycosylase variant induces genomic instability and cellular transformation. Oncotarget 8, 85883–85895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S, Ballinger SW, Corless CL, McCullough AK and Lloyd RS (2006) The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci U S A 103, 1864–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jacobs AC, Calkins MJ, Jadhav A, Dorjsuren D, Maloney D, Simeonov A, Jaruga P, Dizdaroglu M, McCullough AK and Lloyd RS (2013) Inhibition of DNA glycosylases via small molecule purine analogs. PLoS One 8, e81667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Banerjee S, Brown KL, Egli M and Stone MP (2011) Bypass of aflatoxin B1 adducts by the Sulfolobus solfataricus DNA polymerase IV. J Am Chem Soc 133, 12556–12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jaruga P, Coskun E, Kimbrough K, Jacob A, Johnson WE and Dizdaroglu M (2017) Biomarkers of oxidatively induced DNA damage in dreissenid mussels: A genotoxicity assessment tool for the Laurentian Great Lakes. Environ Toxicol 32, 2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dizdaroglu M, Coskun E and Jaruga P (2015) Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic Res 49, 525–548. [DOI] [PubMed] [Google Scholar]
- (20).Zhu C, Lu L, Zhang J, Yue Z, Song J, Zong S, Liu M, Stovicek O, Gao YQ and Yi C (2016) Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc Natl Acad Sci U S A 113, 7792–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Petterson EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC and Ferrin TE (2004) UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 25 1605–1612. [DOI] [PubMed] [Google Scholar]
- (22).Emsley P, Lohkamp B, Scott WG and Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham I,T. E., Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walk-er RC, Wang J, Wolf RM, Wu X and Kollman PA (2014) AMBER14, University of California, San Francisco. [Google Scholar]
- (24).Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ and MacArthur DG (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210.
- (25).Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and Abecasis GR (2015) A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang W, He H, Zang M, Wu Q, Zhao H, Lu LL, Ma P, Zheng H, Wang N, Zhang Y, He S, Chen X, Wu Z, Wang X, Cai J, Liu Z, Sun Z, Zeng YX, Qu C and Jiao Y (2017) Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterology 153, 249–262 e242. [DOI] [PubMed] [Google Scholar]
- (27).Yeo J, Goodman RA, Schirle NT, David SS and Beal PA (2010) RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A 107, 20715–20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Bandaru V, Sunkara S, Wallace SS and Bond JP (2002) A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 1, 517–529. [DOI] [PubMed] [Google Scholar]
- (29).Morland I, Rolseth V, Luna L, Rognes T, Bjoras M and Seeberg E (2002) Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res 30, 4926–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M and Mitra S (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A 99, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shinmura K, Tao H, Goto M, Igarashi H, Taniguchi T, Maekawa M, Takezaki T and Sugimura H (2004) Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis 25, 2311–2317. [DOI] [PubMed] [Google Scholar]
- (32).Boiteux S, Gajewski E, Laval J and Dizdaroglu M (1992) Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization. Biochemistry 31, 106–110. [DOI] [PubMed] [Google Scholar]
- (33).Tchou J, Bodepudi V, Shibutani S, Antoshechkin I, Miller J, Grollman AP and Johnson F (1994) Substrate specificity of Fpg protein. Recognition and cleavage of oxidatively damaged DNA. J Biol Chem 269, 15318–15324. [PubMed] [Google Scholar]
- (34).Karakaya A, Jaruga P, Bohr VA, Grollman AP and Dizdaroglu M (1997) Kinetics of excision of purine lesions from DNA by Escherichia coli Fpg protein. Nucleic Acids Res 25, 474–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Dizdaroglu M, Bauche C, Rodriguez H and Laval J (2000) Novel substrates of Escherichia coli Nth protein and its kinetics for excision of modified bases from DNA damaged by free radicals. Biochemistry 39, 5586–5592. [DOI] [PubMed] [Google Scholar]
- (36).Dizdaroglu M, Laval J and Boiteux S (1993) Substrate specificity of the Escherichia coli endonuclease III: excision of thymine- and cytosine-derived lesions in DNA produced by radiation-generated free radicals. Biochemistry 32, 12105–12111. [DOI] [PubMed] [Google Scholar]
- (37).Prakash A, Carroll BL, Sweasy JB, Wallace SS and Doublie S (2014) Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: activity, structure, and the effect of editing. DNA Repair (Amst) 14, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pallan PS, Lei L, Wang C, Waterman MR, Guengerich FP and Egli M (2015) Research Resource: Correlating Human Cytochrome P450 21A2 Crystal Structure and Phenotypes of Mutations in Congenital Adrenal Hyperplasia. Mol Endocrinol 29, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Guo HH, Choe J and Loeb LA (2004) Protein tolerance to random amino acid change. Proc Natl Acad Sci U S A 101, 9205–9210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Forsbring M, Vik ES, Dalhus B, Karlsen TH, Bergquist A, Schrumpf E, Bjoras M, Boberg KM and Alseth I (2009) Catalytically impaired hMYH and NEIL1 mutant proteins identified in patients with primary sclerosing cholangitis and cholangiocarcinoma. Carcinogenesis 30, 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]