

Abstract
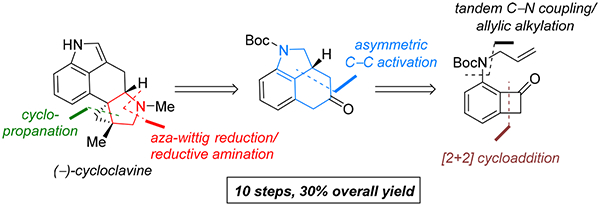
To illustrate the synthetic significance of C–C activation methods, here we describe an efficient strategy for the enantioselective total syntheses of (−)-cycloclavine and (−)-5-epi-cycloclavine, which is enabled by an asymmetric Rh-catalyzed “cut-and-sew” transformation between benzocyclobutenones and olefins. Despite the compact structure of cycloclavine with five-fused rings, the total synthesis was accomplished in 10 steps with a 30% overall yield. Key features of the synthesis include (1) a Pd-catalyzed tandem C–N bond coupling/allylic alkylation sequence to construct the nitrogen-tethered benzocyclobutenone, (2) a highly enantioselective Rh-catalyzed carboacylation of alkenes to forge the indoline-fused tricyclic structure, and (3) a diastereoselective cyclopropanation for preparing the tetrasubstituted cyclopropane ring. Notably, an improved catalytic condition has been developed for the nitrogen-tethered cut-and-sew transformation, which uses a low catalyst loading and allows for a broad substrate scope with high enantioselectivity (94–99% e.e.). The C–C activation-based strategy employed here is anticipated to have further implications for syntheses of other natural products that contain complex fused or bridged rings.
Graphical Abstract
1. INTRODUCTION
Transition metal (TM) catalyzed carbon–carbon bond (C–C) activation has emerged as a rapidly growing field.1 In particular, it allows for new strategic bond disconnection, often leading to more complex structures. However, beyond intriguing trans-formations, synthetic utility of these new C–C activation methods remains underdeveloped. Arguably, one of the most attractive ways to examine the efficacy and scope of new synthetic methods is to use them in the context of complex molecule syntheses. To date, only a handful of examples have been reported on using C–C activation as the key step in total synthesis,2 and even fewer, in the asymmetric synthesis of natural products.2h As an ongoing research interest, our laboratory has been engaged in developing a so-called “cut- and-sew” strategy for constructing bridged and fused rings through intramolecular insertion of an unsaturated unit into the α C–C bond of a cyclic ketone (Figure 1).1q,3 In this article, we hope to describe a complete story on the concise total synthesis of indole alkaloid (–)-cycloclavine and its unnatural analogue enabled by a highly enantioselective cut- and-sew transformation4–6 with nitrogen-tethered benzocyclobutenones and olefins.
Figure 1.

“Cut-and-sew” approach for bridged and fused ring synthesis.
As an indole alkaloid, cycloclavine was first isolated from the seeds of Ipomoea hildebrandtii by Hofmann and co-workers in 19697 and later from Aspergillus japonicas in 1982 (Figure 2).8 It represents the only member in the ergot alkaloid family that contains a cyclopropane ring. During the past decade, ergot alkaloids have attracted significant attention of synthetic chemists due to their striking polycyclic fused scaffolds as well as a broad spectrum of biological activities for potential pharmaceutical and agrichemical applications.9 While the full biological profile of cycloclavine remains to be disclosed, a recent study showed that cycloclavine exhibits promising insecticidal and antiparasitic properties.10
Figure 2.

Structures of representative ergot alkaloids.
Distinct from other members in the ergot alkaloids family, cycloclavine possesses a penta-cyclic core with a unique [3.1.0] structural motif.9k The sterically congested cyclopropane ring along with three contiguous stereogenic centers including two adjacent quaternary carbons presents a considerable challenge for asymmetric total synthesis. The first total synthesis of (±)-cycloclavine was described in 2008 by Száantay.11 In this seminal work, the [3.1.0] fused ring was constructed in the late stage through cyclopropanation of the tetrasubstituted olefin (Scheme 1a), which unfortunately gave only 18% yield (32% based on recovered starting material) likely owing to the steric hindrance of the olefin as well as the presence of nucleophilic indole and pyrrolidine moieties in the substrate. Subsequently, a number of formal syntheses of cycloclavine have been reported utilizing such a late-stage cyclopropanation strategy.12 In contrast, a unique and elegant approach was developed by Wipf in 2011, in which cyclopropane was introduced at the very beginning of the synthesis followed by two intramolecular Diels–Alder cycloaddition to furnish the fused 6–5–6–5 ring systems (Scheme 1b).13 In 2017, the Wipf group reported the first asymmetric total synthesis of (−)-cycloclavine.14 Despite the high novelty and step-efficiency of the synthetic route, the moderate enantioselectivity initially obtained from the asymmetric cyclopropanation step and a low overall yield still leave room for developing an alternative enantioselective synthesis of cycloclavine.
Scheme 1.

Prior Strategies towards Total Synthesis of Cycloclavine
From a retrosynthetic viewpoint (Scheme 2), we envisioned that the pyrrolidine D ring could be installed in the end through an intramolecular reductive amination, and the tetrasubstituted cyclopropane E ring could be constructed via a diastereoselective cyclopropanation between a less bulky 1,1- disubstituted olefin (4) with aryl α-diazoketone 3. The indoline-fused tricycle (A/B/C rings) in intermediate 3 is expected to be synthesized from an enantioselective nitrogen-tethered cut-and-sew reaction via C–C activation of benzocyclobutenone 5, which could be conveniently prepared from the known aryl triflate (6).15
Scheme 2.

Retrosynthetic Analysis for the Synthesis of (−)-Cycloclavine: A C–C Activation Strategy
2. RESULTS AND DISCUSSION
2.1. Rh-Catalyzed Enantioselective Cut-and-Sew Re-actions to Construct Fused Indolines.
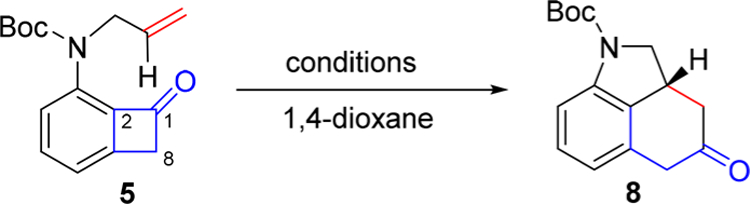
To explore the proposed synthetic strategy, the key cut-and-sew reaction was investigated first. In 2012, we reported a highly enantioselective Rh-catalyzed carboacylation of olefins through cleavage of benzocyclobutenone C–C σ-bonds.5a This reaction is atom-economical and operates under pH- and redox-neutral conditions. While a range of substrates have been demonstrated in this preliminary report, including different substitution patterns on benzocyclobutenones and olefins, the tethering structure has nevertheless been largely restricted to a more flexible oxygen linker.3,5a,f Thus, to enable the synthesis of cycloclavine, the key questions are (1) whether high efficiency and enantioselectivity could be obtained for the cut-and-sew reaction of a more rigid nitrogen-tethered substrate (Scheme 3) and (2) whether a rapid and efficient route could be established for preparing such nitrogen-substituted benzocyclobutenones.
Scheme 3.

Cut-and-Sew Reaction with Nitrogen-Tethered Substrates
In a forward manner, following a reported procedure by Hosoya,15 the ketal-protected OTf-substituted benzocyclo-butenone (6) was prepared in an excellent overall yield from commercially available 2-iodoresorcinol through a sequence of triflation and benzyne-mediated [2 + 2] cycloaddition (Scheme 4). Attempts to directly couple Boc-protected allylamine with triflate 6 were unfruitful likely owing to the steric hindrance of both substrates. However, a Pd-catalyzed C–N bond coupling16 between triflate 6 and tert-butyl carbamate, followed by an allylic alkylation catalyzed by the same Pd species and then acidic workup, provided the nitrogen–tethered benzocyclobutenone (5) in an excellent yield. Hence, this three-step route offers a rapid and high-yielding entry to substrate 5 for the subsequent C–C activation, which otherwise would take around 6 steps using the prior preparation routes.3,17
Scheme 4.

Synthesis of the Nitrogen-Tethered Benzocyclobutenone Substrate
With substrate 5 in hand, the key cut-and-sew reaction was explored (Table 1). Unsurprisingly, under the previous optimal (racemic3 and asymmetric5a) conditions for the ether-linked substrates, the desired tricycle product was only obtained in low yields (entries 1 and 2) and moderate enantioselectivity (entry 2). We hypothesized that the reduced reactivity was likely due to the increased bulkiness and rigidity of the nitrogen linkage, thus use of a less bulky and more electrondeficient Rh catalyst might enhance the binding with the olefin moiety thereby promoting the subsequent 2π insertion. Indeed, the combination of a monodentate phosphine ligand, i.e. PMe2Ph, with π-acidic [Rh(CO)2Cl]2 afforded indoline product 8 in an excellent yield (entry 3). Interestingly, [Rh(CO)2Cl]2 as a precatalyst alone still gave 49% yield (entry 4). However, developing enantioselective transformations based on the [Rh(CO)2Cl]2 system proved to be challenging due to the limitation of using monodentate electron-rich ligands. On the other hand, while a promising level of enantioselectivity could be achieved using chloro-Rh(I)/olefin precatalysts, the yield was difficult to improve (entries 5–7). Finally, we turned our attention to cationic Rh(I) precatalysts, because the olefin coordination would likely be enhanced due to the cationic nature of the metal. To our delight, using Rh(nbd)2BF4 and DTBM-segphos as the metal/ligand combination, 61% yield and 97.5% e.e. were achieved (entry 8). The Rh(cod)2BF4 was found to be more reactive than Rh(nbd)2BF4, and a 95% yield was reached at a lower reaction temperature (90 °C) with the same e.e. (entry 10); in contrast, at 120 °C significant catalyst decomposition was observed leading to a much lower yield (entry 9). Remarkably, with this new catalyst system, the catalyst loading and reaction time could be further reduced. For example, using 3 mol % Rh, tricycle 8 was isolated in 95% yield and 97.5% e.e. in 12 h on a 2.0 mmol scale. Comparing with our first-generation conditions for the ether-linked benzocyclobutenones, the current one shows several advantages: first, the reaction temperature is significantly lower (90 vs 130 °C); second, the loading of the rhodium catalyst could be reduced to 3 mol % (previously, 10 mol %); and finally, the reaction is much faster (12 vs 48 h).
Table 1.
Selected Optimization Study for the Rh-Catalyzed Asymmetric Cut-and-Sew Reactiona
 | ||||
|---|---|---|---|---|
| entry | precatalyst/ligand | temperature (°C) | yieldb | e.e.c |
| 1 | 5 mol % [Rh(cod)Cl]2/10 mol % dppb | 130 | 18%g,h | N/A |
| 2 | 5 mol % [Rh(cod)Cl]2/10 mol % (R)-DTBM-segphos | 130 | 12% (57%) | 75% |
| 3 | 5 mol % [Rh(CO)2Cl]2/10 mol % PMe2Ph | 120 | 86% | N/A |
| 4d | 5 mol % [Rh(CO)2Cl]2 | 120 | 49g | N/A |
| 5 | 5 mol % [Rh(cod)Cl]2/10 mol % (R)-DM-segphos | 130 | 22% (71%) | 26% |
| 6 | 5 mol % [Rh(cod)Cl]2/10 mol % (R)-tol-binap | 130 | 7% (37%) | 72% |
| 7 | 5 mol % [Rh(coe)2Cl]2/10 mol % (R)-DTBM-segphos | 110 | 12% (44%) | 85% |
| 8 | 10 mol % Rh(nbd)2BF4/12 mol % (R)-DTBM-segphos | 100 | 61% | 97.5% |
| 9 | 10 mol % Rh(cod)2BF4/12 mol % (R)-DTBM-segphos | 120 | 22% | 98% |
| 10e | 10 mol % Rh(cod)2BF4/12 mol % (R)-DTBM-segphos | 90 | 95% | 97% |
| 11e | 5 mol % Rh(cod)2BF4/6 mol % (R)-DTBM-segphos | 90 | 96% | 97.5% |
| 12e,f | 3 mol % Rh(cod)2BF4/3.6 mol % (R)-DTBM-segphos | 90 | 95% | 97.5% |
Unless otherwise mentioned, the reaction was run on a 0.1 mmol scale at specified temperature for 24 h.
Isolated yield; numbers in parentheses are yields based on recovered starting material (brsm).
Determined by chiral HPLC.
Reaction time was 72 h.
Reaction time was 12 h.
Reaction scale was 2.0 mmol.
NMR yields using 1,1,2,2-tetrachloroethane as an internal standard.
Tetrahydrofuran (THF) was used as solvent.
2.2. Substrate Scope for the Rh-Catalyzed Synthesis of Tricyclic Indolines via C–C Activation.
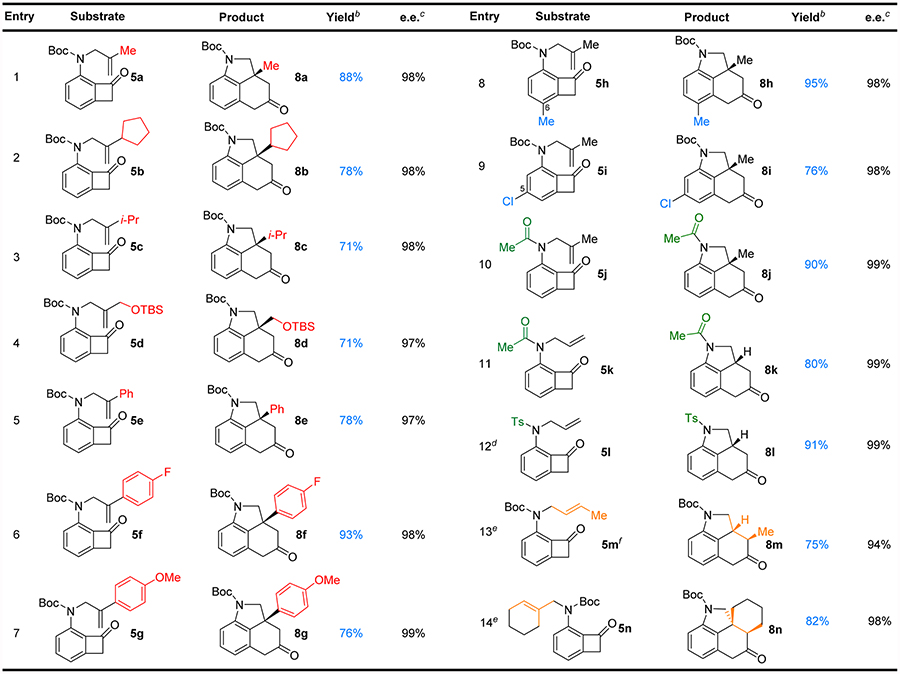
After obtaining the optimal conditions for the Rh-catalyzed carboacylation of olefins with nitrogen-tethered benzocyclobutenones, the substrate scope was further investigated (Table 2). First, alkyl substituents on the olefin with various steric properties all underwent the desired carboacylation reaction giving excellent enantioselectivity (≥97% e.e., entries 1–4). It is not surprising that with increased bulkiness around the olefin, the yield of the reaction slightly decreased from methyl to cyclopentyl to isopropyl, while the enantioselectivity remained the same. Given the pH and redox-neutral reaction conditions, the TBS-protected primary alcohol is well tolerated (entry 4). Both electron-rich and -poor aryl substituents are compatible, giving good yields and excellent enantioselectivity (entries 5–7). In addition, C5 and C6-substituted benzocyclobutenones are competent substrates (entries 8 and 9). Furthermore, besides the Boc moiety, other protecting groups at the nitrogen, such as acyl or tosyl group, are also suitable for this reaction, providing products in 80–91% yields and 99% e.e. (entries 10–12). Gratifyingly, the more challenging substrates that contain 1,2-disubstituted and trisubstituted olefins underwent the desired carboacylation efficiently with excellent e.e.s (entries 13 and 14).
Table 2.
Substrate Scopea
 |
Reaction conditions: [Rh(cod)2]BF4 (5 mol %), (R)-DTBM-segphos (6 mol %) 1,4-dioxane, 90 °C, 12 h.
Isolated yield.
Determined by chiral HPLC.
Run with 10 mol % [Rh(cod)2]BF4 and 12 mol % (R)-DTBM-segphos at 90 °C for 12 h and then 110 °C for 12 h.
Run with 10 mol % [Rh(cod)2]BF4 and 12 mol % (R)-DTBM-segphos.
Substrate 5m was used as an inseparable 4:1 mixture of trans/cis isomers. Control experiment showed that the cis isomer cannot undergo the desired reaction, so the product obtained as a single diastereomer was generated from the trans isomer. The yield was calculated based on the amount of the trans starting material.
In summary, the improved catalytic conditions show a general feature for the nitrogen-tethered substrates, giving high yields and excellent enantioselectivity. The reaction conditions do not contain strong acids/bases or stoichiometric oxidants/reductants, which could be the key for the good functional group tolerance. Thus, this method is expected to be useful for enantioselective synthesis of various indole or indoline-containing complex target molecules.
2.3. Rh-Catalyzed Diastereoselective Cyclopropanation.
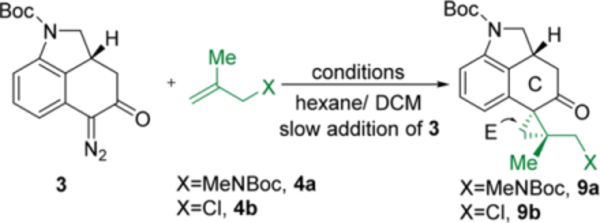
With a reliable route to access key intermediate 8, we continued to explore the forward synthesis of (−)-cycloclavine. First, the diazo-transfer reaction proceeded smoothly to afford compound 3 in 92% yield (Scheme 5). Clearly, the next formidable challenge is to construct the tetra-substituted cyclopropane E ring in an efficient and diastereoselective fashion. While cyclopropanation with donor–acceptor carbenoids, pioneered by the Davies group,18 has been extensively developed, the use of ketone-derived diazo compounds is less common compared to the widely used ester-derived ones.19 In particular, the α-diazoketone-based cyclopropanation with 1,1-disubstituted olefins for constructing tetrasubstituted cyclopropanes diastereoselectively remains elusive.
Scheme 5.

Synthesis of Compound 3 as the Substrate for Cyclopropanation
One potential side reaction with α-diazoketones could be the Wolff rearrangement,20 which would possibly lead to ring contraction. However, given the low temperature operated by the dirhodium-catalyzed cyclopropanation reaction, the Wolff rearrangement could likely be avoided. Thus, the reactions with racemic α-diazoketone 3 were tested initially under various dinuclear Rh(II)-catalyzed cyclopropanation conditions (entries 1–4, Table 3). Davies’ DOSP catalyst21 was found to give higher reactivity than other commercially available Rh(II) catalysts. While the use of protected allyl amine 4a could indeed provide the desired cyclopropane product 9a, the efficiency was not satisfying, likely owing to the bulkiness or chelating effect of the carbamate moiety. Hence, less sterically hindered 2-methylallyl chloride (4b) was then employed as the olefin substrate. To our delight, 4b exhibited significantly enhanced reactivity and could even yield the desired cyclopropane product 9b at −60 °C using Rh2(R-DOSP)4 as the catalyst (entry 5). After a further survey of the reaction temperature and solvent (entries 6–8), −40 °C and hexane/toluene as a mixed solvent proved to be more efficient; cyclopropane 9b could be isolated in 78–80% yield, and ~6:1 d.r. favoring the desired diastereomer with either enantiopure or racemic catalyst (entries 7 and 8). It is noteworthy that both diastereomers are easily separable, and the relative stereo-chemistry of the major diastereomer (9b) was confirmed by X-ray crystallography (Figure 3).
Table 3.
Selected Condition Optimization for the Cyclopropanation Stepa
 | |||||
|---|---|---|---|---|---|
| entry | X | catalyst (l mol %) | T[°C] | Yieldb | d.r.b |
| 1 | MeNBoc | Rh2(OAc)4 | 40 | <10% | N/D |
| 2 | MeNBoc | Rh2(esp)2 | r.t. | trace | N/D |
| 3 | MeNBoc | Rh2(R-DOSP)4 | r.t. | 25% | N/D |
| 4 | MeNBoc | Rh2(R-DOSP)4 | −40 | trace | N/D |
| 5 | Cl | Rh2(R-DOSP)4 | −60 | 27% | 5:1 |
| 6 | Cl | Rh2(R-DOSP)4 | −40 | 45% | 6:1 |
| 7c | Cl | Rh2(R-DOSP)4 | −40 | 80% | 5.9:1 |
| 8c,d | Cl | Rh2(R/S-DOSP)4 | −40 | 78% | 5.8:1 |
| 9c,e,f | Cl | Rh2(R-DOSP)4 | −40 | 85% | 5.8:1 |
| 10c,e | Cl | Rh2(S-DOSP)4 | −40 | 52% | 4.3:1 |
| 11c,e | Cl | Rh2(esp)2 | −40 | 67% | 6.3:1 |
Unless otherwise mentioned, the reaction was run on a 0.1 mmol scale at specified temperature for 6 h using racemic compound 3; a solution of compound 3 was added to the stirring solution of catalyst and 4a or 4b with a speed of 2 mL/h.
Yield and d.r. were determined based on isolated compounds.
Using hexane/toluene (10:1) as a mixed solvent.
0.5 mol % of Rh2(R-DOSP)4 and 0.5 mol % of Rh2(S-DOSP)4 were mixed to provide the racemic catalyst.
Starting material 3 was prepared from compound 8 with 97.5% e.e..
Reaction was run on a 0.7 mmol scale.
Figure 3.

X-ray structures of compound 9b (racemic).
The optimal reaction conditions have also been applied to compound 3 with 97.5% e.e. using 1 mol % Rh2(R-DOSP)4 as the catalyst (entry 9). On a larger scale the desired product 9b was still isolated in 85% yield with satisfactory diastereoselectivity. Interestingly, use of the other enantiomer of the Rh catalyst, i.e. Rh2(S-DOSP)4, gave diminished diastereoselectivity, suggesting a mismatched situation (entry 10). Given that both enantiomers of the Rh catalysts gave the same major diastereomer of the product, this indicates that the diastereoselectivity was majorly controlled by the substrate. Finally, as a control experiment, the achiral Du Bois’ Rh2(esp)222 catalyst was also found to be effective under the optimized conditions (entry 11), though the yield was lower than the DOSP one.
A plausible model has been proposed to explain the observed diastereoselectivity (Figure 4). According to Davies’ model on the cyclopropanation with donor–acceptor carbenoids,23 the aryl group is generally considered as a larger group and the acceptor part is considered as a smaller group. Thus, we rationalized that during the transition state for the cyclopropanation, first, the olefin would approach to the less bulky convex face of the tricycle; second, the smaller methyl substituent of the olefin would prefer to stay at the more sterically hindered side (the aryl side) of the carbenoid and the bulkier chloromethyl group of the olefin would favor the less sterically hindered side (the ketone side). The minor diastereomer likely comes from the other olefin orientation relative to the carbenoid due to the moderate steric difference between methyl and chloromethyl groups.
Figure 4.
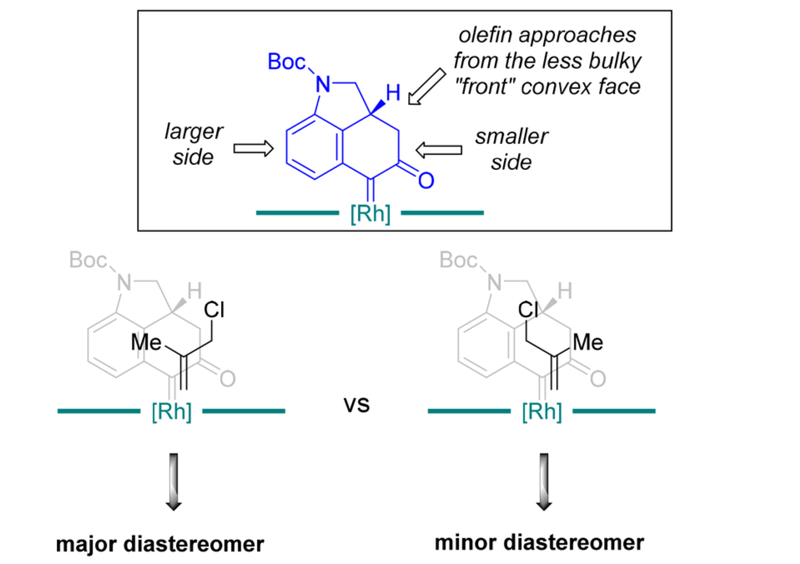
Stereochemical model for the cyclopropanation step.
2.4. End Game.
The stage is now set for closing of the last D ring. An SN2 reaction between alkyl chloride 9b and sodium azide went smoothly to deliver azido compound 10 in an excellent yield (Scheme 6). The subsequent one-pot aza-Wittig, imine reduction and reductive amination sequence24 furnished the pyrrolidine ring and N-methyl substitution in a high overall efficiency and complete diastereoselectivity. However, the C5 stereocenter was found to be opposite from the one of natural cycloclavine (1). Nevertheless, removal of the Boc protecting group with TFA, followed by a seleniummediated dehydrogenation,25 accomplished the synthesis of (−)-5-epi-cycloclavine (12), which spectroscopically matched the one reported by Wipf and co-workers.13 It is noteworthy that indole was added as a scavenger to avoid oxidative decomposition of product 12 formed.25c
Scheme 6.
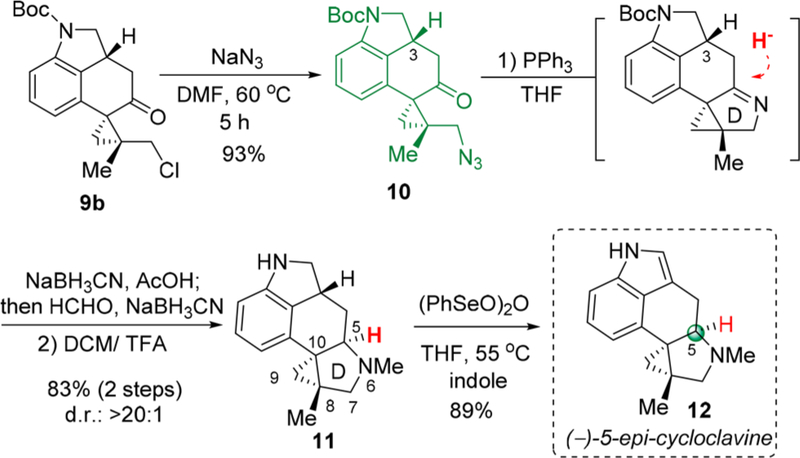
Synthesis of (−)-5-epi-Cycloclavine
We postulated that the undesired diastereoselectivity during the imine reduction step is likely influenced by the remote C3 stereocenter. As depicted from the X-ray crystal structure (Figure 3) of 9b, the cyclohexanone C ring is highly twisted, which forces the C8 quaternary center pointing to the same face as the C3 hydrogen. Thus, we hypothesized that removing the C3 stereocenter might significantly alter the conformation of the imine intermediate, thereby providing a different stereochemical outcome for the reduction. To test this hypothesis, the indoline moiety in azide 10 was converted to a flat indole structural motif through Boc-deprotection and then dehydrogenation (Scheme 7). Using indole 2 as the substrate, to our delight, the same aza-Wittig reduction/reductive amination sequence indeed provided (−)-cycloclavine in 78% yield as a single diastereomer, which is spectroscopically identical to the natural sample but with opposite specific rotation sign.7,14
Scheme 7.
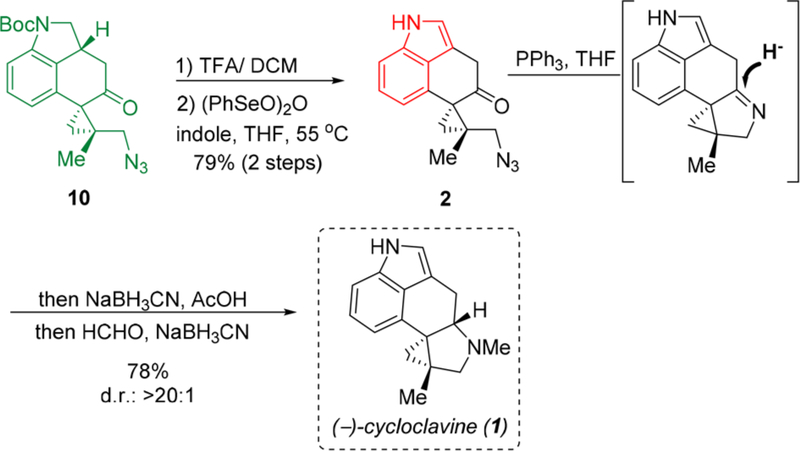
Total Synthesis of (−)-Cycloclavine
3. CONCLUSION
In summary, a concise enantioselective total synthesis of (−)-cycloclavine has been accomplished in 10 steps with 30% overall yield (Scheme 8). The high efficiency of the synthetic strategy is enabled by a number of transition metal-catalyzed transformations. First, a Pd-catalyzed tandem C–N bond coupling/allylic alkylation offers a rapid and high-yielding route to access nitrogen-tethered benzocyclobutenone substrates. Second, an asymmetric Rh-catalyzed cut-and-sew reaction has been employed to build the fused A/B/C core structure in a highly enantioselective manner. Third, a diastereoselective Rh-catalyzed cyclopropanation between α-diazoketone and 1,1-substituted olefin effectively constructs two adjacent quaternary centers, which addresses the “late-stage cyclopropanation challenge” in cycloclavine synthesis. In particular, the C–C activation method has played a central role in this total synthesis, which could have further implications beyond this work. It is anticipated that the simple cut-and-sew methods may inspire future development of new strategic bond disconnections for the syntheses of other natural products and bioactive compounds that contain complex bridged or fused rings.
Scheme 8.
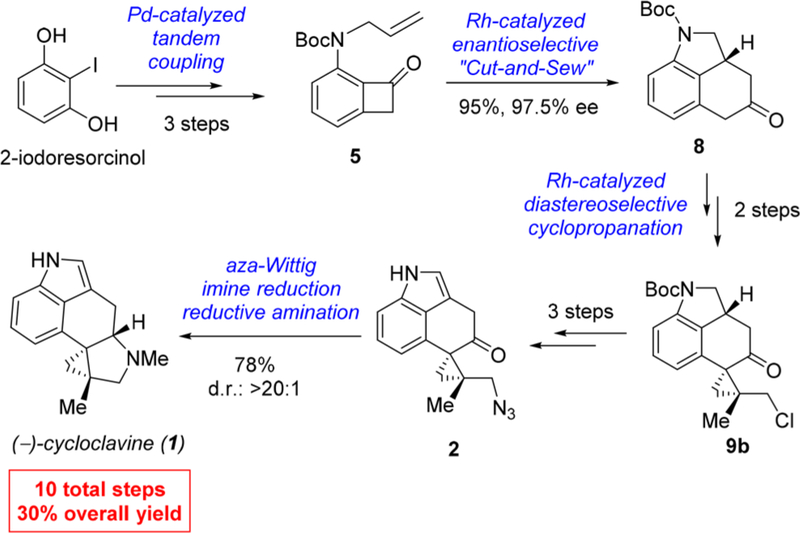
Summary of the Synthetic Route
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge NIGMS (R01GM109054–01) for funding. We thank Mr. Ki-young Yoon for X-ray structures and Dr. Antoni Jurkiewicz for NMR advices. Chiral Technologies is thanked for their generous donation of chiral HPLC columns.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05549.
Experimental procedures; spectral data (PDF)Crystallographic data for 9b (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).For selected reviews, see:; (a) Crabtree RH Chem. Rev 1985, 85, 245. [Google Scholar]; (b) Jones WD Nature 1993, 364, 676. [Google Scholar]; (c) Murakami M; Ito Y Top. Organomet. Chem 1999, 3, 97. [Google Scholar]; (d) Rybtchinski B; Milstein D Angew. Chem., Int. Ed 1999, 38, 870. [DOI] [PubMed] [Google Scholar]; (e) Jun C-H Chem. Soc. Rev 2004, 33, 610. [DOI] [PubMed] [Google Scholar]; (f) Necas D; Kotora M Curr. Org. Chem 2007, 11, 1566. [Google Scholar]; (g) Park YJ; Park J-W; Jun C-H Acc. Chem. Res 2008, 41, 222. [DOI] [PubMed] [Google Scholar]; (h) Murakami M; Matsuda T Chem. Commun 2011, 47, 1100. [DOI] [PubMed] [Google Scholar]; (i) Seiser T; Saget T; Tran DN; Cramer N Angew. Chem., Int. Ed 2011, 50, 7740. [DOI] [PubMed] [Google Scholar]; (j) Korotvicka A; Necas D; Kotora M Curr. Org. Chem 2012, 16, 1170. [Google Scholar]; (k) Chen F; Wang T; Jiao N Chem. Rev 2014, 114, 8613. [DOI] [PubMed] [Google Scholar]; (l) Dermenci A; Coe JW; Dong G Org. Chem. Front 2014, 1, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Dong G C–C bond activation; Springer-Verlag: Berlin, 2014; Vol. 346. [Google Scholar]; (n) Murakami M; Ishida N Fundamental Reactions to Cleave Carbon–Carbon σ-Bonds with Transition Metal Complexes. In Cleavage of Carbon–Carbon Single Bonds by Transition Metals; Wiley-VCH Verlag GmbH & Co. KGaA: 2015; pp 1. [Google Scholar]; (o) Souillart L; Cramer N Chem. Rev 2015, 115, 9410. [DOI] [PubMed] [Google Scholar]; (p) Chen P.-h.; Dong G Chem. - Eur. J 2016, 22, 18290. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Chen P.-h.; Billett BA; Tsukamoto T; Dong G ACS Catal. 2017, 7, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Fumagalli G; Stanton S; Bower JF Chem. Rev 2017, 117, 9404. [DOI] [PubMed] [Google Scholar]; (s) Kim D-S; Park W-J; Jun C-H Chem. Rev 2017, 117, 8977. [DOI] [PubMed] [Google Scholar]
- (2).For selected reviews, see:; (a) Murakami M; Ishida N Total Syntheses of Natural Products and Biologically Active Compounds by Transition-Metal-Catalyzed C–C Cleavage. In Cleavage of Carbon-Carbon Single Bonds by Transition Metals; Wiley-VCH Verlag GmbH & Co. KGaA: 2015; pp 253. For selected examples, see: [Google Scholar]; (b) South MS; Liebeskind LS J. Am. Chem. Soc 1984, 106, 4181. [Google Scholar]; (c) Wender PA; Fuji M; Husfeld CO; Love JA Org. Lett 1999, 1, 137. [Google Scholar]; (d) Trost BM; Waser J; Meyer AJ Am. Chem. Soc 2007, 129, 14556. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Jiao L; Yuan C; Yu Z-XJ Am. Chem. Soc 2008, 130, 4421. [DOI] [PubMed] [Google Scholar]; (f) Nakao Y; Ebata S; Yada A; Hiyama T; Ikawa M; Ogoshi SJ Am. Chem. Soc 2008, 130, 12874. [DOI] [PubMed] [Google Scholar]; (g) Hirata Y; Yukawa T; Kashihara N; Nakao Y; Hiyama TJ Am. Chem. Soc 2009, 131, 10964. [DOI] [PubMed] [Google Scholar]; (h) Liang Y; Jiang X; Yu Z-X Chem. Commun 2011, 47, 6659. [DOI] [PubMed] [Google Scholar]; (i) Evans PA; Inglesby PA; Kilbride K Org. Lett 2013, 15, 1798. [DOI] [PubMed] [Google Scholar]; (j) Xu T; Dong G Angew. Chem., Int. Ed 2014, 53, 10733. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Sun T; Zhang Y; Qiu B; Wang Y; Qin Y; Dong G; Xu T Angew. Chem., Int. Ed 2018, 57, 2859. [DOI] [PubMed] [Google Scholar]
- (3).Xu T; Dong G Angew. Chem., Int. Ed 2012, 51, 7567. [DOI] [PubMed] [Google Scholar]
- (4).Souillart L; Parker E; Cramer N Asymmetric Transformations via C–C Bond Cleavage In C–C Bond Activation; Dong G, Ed.; Springer: Berlin Heidelberg, 2014; pp 163. [DOI] [PubMed] [Google Scholar]
- (5).For asymmetric C–C activation reactions via oxidative addition, see: [Google Scholar]; (a) Xu T; Ko HM; Savage NA; Dong GJ Am. Chem. Soc 2012, 134, 20005. [DOI] [PubMed] [Google Scholar]; (b) Parker E; Cramer N Organometallics 2014, 33, 780. [Google Scholar]; (c) Souillart L; Cramer N Angew. Chem., Int. Ed 2014, 53, 9640. [DOI] [PubMed] [Google Scholar]; (d) Souillart L; Parker E; Cramer N Angew. Chem., Int. Ed 2014, 53, 3001. [DOI] [PubMed] [Google Scholar]; (e) Zhou X; Dong GJ Am. Chem. Soc 2015, 137, 13715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Deng L; Xu T; Li H; Dong GJ Am. Chem. Soc 2016, 138, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).For asymmetric C–C activation reactions via β-carbon elimination, see: [Google Scholar]; (a) Matsuda T; Shigeno M; Makino M; Murakami M Org. Lett 2006, 8, 3379. [DOI] [PubMed] [Google Scholar]; (b) Seiser T; Cramer N Angew. Chem., Int. Ed 2008, 47, 9294. [DOI] [PubMed] [Google Scholar]; (c) Seiser T; Roth OA; Cramer N Angew. Chem., Int. Ed 2009, 48, 6320. [DOI] [PubMed] [Google Scholar]; (d) Shigeno M; Yamamoto T; Murakami M Chem. - Eur. J 2009, 15, 12929. [DOI] [PubMed] [Google Scholar]; (e) Seiser T; Cramer N Chem. - Eur. J 2010, 16, 3383. [DOI] [PubMed] [Google Scholar]; (f) Seiser T; Cramer N Angew. Chem., Int. Ed 2010, 49, 10163. [DOI] [PubMed] [Google Scholar]; (g) Liu L; Ishida N; Murakami M Angew. Chem., Int. Ed 2012, 51, 2485. [DOI] [PubMed] [Google Scholar]; (h) Zhou X; Dong G Angew. Chem., Int. Ed 2016, 55, 15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stauffacher D; Niklaus P; Tscherter H; Weber HP; Hofmann A Tetrahedron 1969, 25, 5879. [DOI] [PubMed] [Google Scholar]
- (8).Furuta T; Koike M; Abe M Agric. Biol. Chem 1982, 46, 1921. [Google Scholar]
- (9).(a) MacLeod RM; Lehmeyer JE Cancer Res. 1973, 33, 849. [PubMed] [Google Scholar]; (b) Berde B; Stürmer E Introduction to the Pharmacology of Ergot Alkaloids and Related Compounds as a Basis of Their Therapeutic Application In Ergot Alkaloids and Related Compounds; Berde B, Schild HO, Eds.; Springer: Berlin Heidelberg, 1978; pp 1. [Google Scholar]; (c) Horwell DC Tetrahedron 1980, 36, 3123. [Google Scholar]; (d) Lyons PC; Plattner RD; Bacon CW Science 1986, 232, 487. [DOI] [PubMed] [Google Scholar]; (e) de Groot ANJA; van Dongen PWJ; Vree TB; Hekster YA; van Roosmalen J Drugs 1998, 56, 523. [DOI] [PubMed] [Google Scholar]; (f) Somei M; Yokoyama Y; Murakami Y; Ninomiya I; Kiguchi T; Naito T Recent synthetic studies on the ergot alkaloids and related compounds. In The Alkaloids: Chemistry and Biology; Academic Press: 2000; Vol. 54, pp 191. [Google Scholar]; (g) Boichenko LV; Boichenko DM; Vinokurova NG; Reshetilova TA; Arinbasarov MU Microbiology 2001, 70, 306. [PubMed] [Google Scholar]; (h) Schardl CL; Panaccione DG; Tudzynski P Chapter 2 Ergot Alkaloids–Biology and Molecular Biology. In The Alkaloids: Chemistry and Biology; Cordell GA, Ed.; Academic Press, 2006; Vol. 63, pp 45. [DOI] [PubMed] [Google Scholar]; (i) Schiff PL Am. J. Pharm. Educ 2006, 70, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Liu Q; Jia Y Org. Lett 2011, 13, 4810. [DOI] [PubMed] [Google Scholar]; (k) Wallwey C; Li S-M Nat. Prod. Rep 2011, 28, 496. [DOI] [PubMed] [Google Scholar]; (l) McCabe SR; Wipf P Org. Biomol. Chem 2016, 14, 5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Körber K; Song D; Rheinheimer J; Kaiser F; Dickhaut J; Narine A; Culbertson DL; Thompson S; Rieder J Cycloclavine and derivatives thereof for controlling invertebrate pests.- WO2014096238 A1, 2014. [Google Scholar]
- (11).Incze M; Dörnyei G; Moldvai I; Temesváari-Major E; Egyed O; Száantay C Tetrahedron 2008, 64, 2924. [Google Scholar]
- (12).(a) Jabre ND; Watanabe T; Brewer M Tetrahedron Lett. 2014, 55, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang W; Lu J-T; Zhang H-L; Shi Z-F; Wen J; Cao X-PJ Org. Chem 2014, 79, 122. [DOI] [PubMed] [Google Scholar]; (c) Netz N; Opatz TJ Org. Chem 2016, 81, 1723. [DOI] [PubMed] [Google Scholar]; (d) Chen J-Q; Song L-L; Li F-X; Shi Z-F; Cao X-P Chem. Commun 2017, 53, 12902. [DOI] [PubMed] [Google Scholar]; (e) Chaudhuri S; Ghosh S; Bhunia S; Bisai A Chem. Commun 2018, 54, 940. [DOI] [PubMed] [Google Scholar]
- (13).Petronijevic FR; Wipf PJ Am. Chem. Soc 2011, 133, 7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McCabe SR; Wipf P Angew. Chem., Int. Ed 2017, 56, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yoshida S; Uchida K; Igawa K; Tomooka K; Hosoya T Chem. Commun 2014, 50, 15059. [DOI] [PubMed] [Google Scholar]
- (16).For selected reviews, see:; (a) Hartwig JF Acc. Chem. Res 1998, 31, 852. [Google Scholar]; (b) Wolfe JP; Wagaw S; Marcoux J-F; Buchwald SL Acc. Chem. Res 1998, 31, 805. [Google Scholar]; (c) Yang BH; Buchwald SL J. Organomet. Chem 1999, 576, 125. [Google Scholar]; (d) Yin J; Buchwald SL Org. Lett 2000, 2, 1101. [DOI] [PubMed] [Google Scholar]; (e) Hartwig JF Acc. Chem. Res 2008, 41, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Surry DS; Buchwald SL Angew. Chem., Int. Ed 2008, 47, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen PH; Savage NA; Dong G Tetrahedron 2014, 70, 4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Davies HML; Antoulinakis EG Org. React 2001, 57, 1. [Google Scholar]; (b) Lebel H; Marcoux J-F; Molinaro C; Charette AB Chem. Rev 2003, 103, 977. [DOI] [PubMed] [Google Scholar]; (c) Reissig H-U; Zimmer R Chem. Rev 2003, 103, 1151. [DOI] [PubMed] [Google Scholar]; (d) Bartoli G; Bencivenni G; Dalpozzo R Synthesis 2014, 46, 979. [Google Scholar]
- (19).(a) Denton JR; Davies HM L. Org. Lett 2009, 11, 787. [DOI] [PubMed] [Google Scholar]; (b) Lindsay VNG; Nicolas C; Charette AB J. Am. Chem. Soc 2011, 133, 8972. [DOI] [PubMed] [Google Scholar]
- (20).Kirmse W Eur. J. Org. Chem 2002, 2002, 2193. [Google Scholar]
- (21).Davies HML; Nagashima T; Klino JL Org. Lett 2000, 2, 823. [DOI] [PubMed] [Google Scholar]
- (22).Espino CG; Fiori KW; Kim M; Du Bois JJ Am. Chem. Soc 2004, 126, 15378. [DOI] [PubMed] [Google Scholar]
- (23).Qin C; Boyarskikh V; Hansen JH; Hardcastle KI; Musaev DG; Davies HM L. J. Am. Chem. Soc 2011, 133, 19198. [DOI] [PubMed] [Google Scholar]
- (24).(a) Feng P; Fan Y; Xue F; Liu W; Li S; Shi Y Org. Lett 2011, 13, 5827. [DOI] [PubMed] [Google Scholar]; (b) Hou SH; Tu YQ; Liu L; Zhang FM; Wang SH; Zhang XM Angew. Chem., Int. Ed 2013, 52, 11373. [DOI] [PubMed] [Google Scholar]
- (25).(a) Barton DHR; Lusinchi X; Milliet P Tetrahedron Lett. 1982, 23, 4949. [Google Scholar]; (b) Ninomiya I; Hashimoto C; Kiguchi T; Barton DHR; Lusinchi X; Milliet P Tetrahedron Lett. 1985, 26, 4187. [Google Scholar]; (c) Ninomiya I; Kiguchi T; Hashimoto C; Barton DHR; Lusinchi X; Milliet P Tetrahedron Lett. 1985, 26, 4183. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.