

Abstract
Hepatocellular carcinoma (HCC) causes more than half a million annual deaths world-wide. Understanding the mechanisms contributing to HCC development is highly desirable for improved surveillance, diagnosis and treatment. Liver tissue metabolomics has the potential to reflect the physiological changes behind HCC development. Also, it allows identification of biomarker candidates for future evaluation in biofluids and investigation of racial disparities in HCC. Tumor and non-tumor tissues from 40 patients were analyzed by both gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS) platforms to increase the metabolome coverage. The levels of the metabolites extracted from solid liver tissue of the HCC area and adjacent non-HCC area were compared. Among the analytes detected by GC-MS and LC-MS with significant alterations, 18 were selected based on biological relevance and confirmed metabolite identification. These metabolites belong to TCA cycle, glycolysis, purines, and lipid metabolism, and have been previously reported in liver metabolomic studies where high correlation with HCC progression is implied. We demonstrated that metabolites related to HCC pathogenesis can be identified through liver tissue metabolomic analysis. Additionally, this study has enabled us to identify race-specific metabolites associated with HCC.
Keywords: HCC, cirrhosis, alpha-fetoprotein, liver tissues, metabolomics, GC-MS, UPLC-MS
Graphical Abstract
1. Introduction
Hepatocellular carcinoma (HCC) is the third cause of cancer-related death world-wide with mortality rates rapidly increasing over recent decades in most countries.1 The incidence and mortality associated with HCC in the United States have been persistently rising over the past two decades with more than 40,000 new cases of liver cancer estimated to be diagnosed in 2018, 75% of which to be HCC.2 30,200 liver cancer deaths have been estimated to occur in 2018.3,4
Progression of liver fibrosis, primarily caused by chronic viral hepatitis, excessive consumption of alcohol, and non-alcoholic fatty liver disease (NAFLD), results in life-limiting liver cirrhosis and HCC.5 Moreover, HCC disproportionately affects socioeconomically disadvantaged populations.6,7 In a study conducted on data collected from the Surveillance, Epidemiology, and End Results (SEER; https://seer.cancer.gov/) project between 1998 and 2012, among white, black, Asian and native American patients, when controlling for race, differences were detected across groups with specifically, Asian patients less likely to present with cirrhosis and black patients the least likely to present with an elevated AFP at diagnosis.2 Understanding the mechanism of the pathogenesis of HCC with respect to racial disparities would help improve HCC surveillance, diagnosis, and treatment at earlier stages of the disease resulting in lower mortality rates.2,8
The molecular mechanism behind liver carcinogenesis is associated with the regulation of a variety of signal transduction pathways and key molecules in the cells.9 Metabolic alterations play a role in tumor survival and progression, providing support for the vital needs of cancer cells by increasing energy production, macromolecular biosynthesis, and maintenance of redox balance.10 Liver is the key metabolic organ of living systems mediating the expression levels of a large number of metabolites. In view of this, investigation of HCC from a metabolic perspective is highly desired.8
Metabolomics provides insights into the mechanism underlying a pathophysiological condition by dealing with diverse biological properties of small molecules, which are the end products of metabolic pathways.11 Mass spectrometry (MS)-based analysis of metabolites in biospecimens is typically preceded by chromatographic separation to reduce the complexity of the acquired mass spectra. The most common separation methods are gas chromatography (GC) and liquid chromatography (LC).12 LC-MS is usually utilized with a reversed phase column for the analysis of different class of glycerolipids, glycerophospholipids, lysoglycerophospholipids, sphingolipids, carnitines, fatty acyls, amides, etc. GC-MS can be used for the detection of non-polar volatile compounds and medium to polar compounds, such as sugars, carboxylic acids, free fatty acids and other small lipids, after sample derivatization. Because a single analytical technique is not able to measure and identify all metabolites, data from different platforms are combined to increase the metabolomics coverage.12
Metabolomic analysis of tissues allows researchers to study localized responses to stimuli and pathogenesis, gaining biochemical information about the disease mechanisms.13 Compared with biofluids, metabolomic profiling of tissues tend to provide more insight into metabolic changes in HCC development, with better specificity.14 Therefore, systematic metabolic characterization of liver tissues allows us to explore novel diagnostic markers or therapeutic targets for HCC.15 Furthermore, metabolomic analysis conducted by comparing paired tissues from diseased and non-diseased liver regions could identify disease-associated alterations despite any individual differences.16
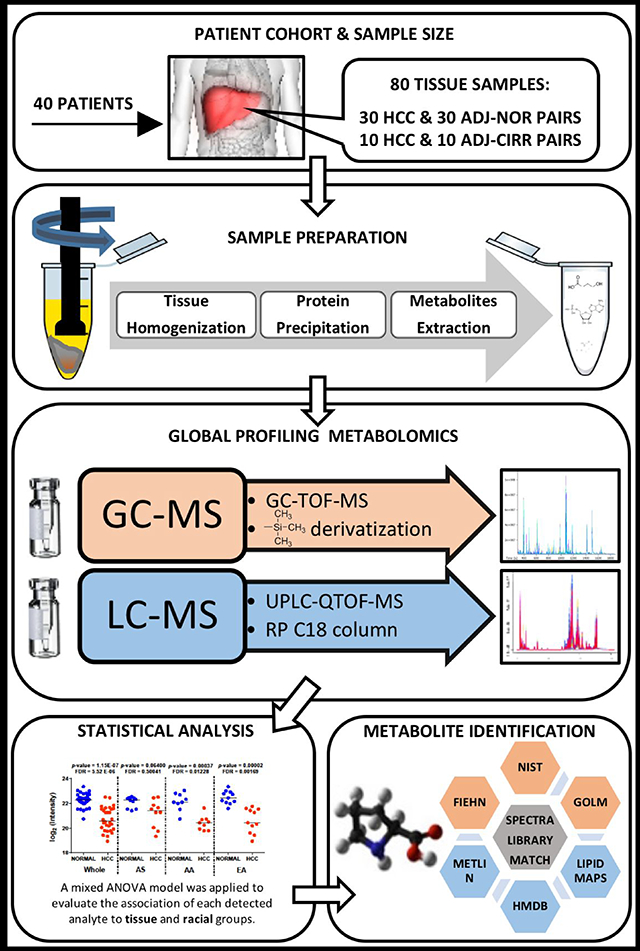
In this paper, we use GC-MS and LC-MS to evaluate changes in metabolite levels in liver tissues from African-American (AA), Asian-American (AS), and European-American (EA) patients that were recruited at MedStar Georgetown University Hospital in Washington, DC. Metabolites extracted from the solid liver tissue corresponding to the HCC area and adjacent non-HCC area are analyzed. The resulting data are analyzed by a mixed model ANOVA to account for the within-subject variability while simultaneously evaluating the effects of tissue status (tumor, normal, or cirrhosis) and racial groups (AA, AS, or EA) on metabolite levels.
2. Study cohort and experimental design
Adult patients recruited to participate in this study signed informed consent to a protocol approved by the Georgetown University Institutional Review Board (IRB). Following the participant’s informed consent signature and enrollment, liver tissues were collected at the time of the surgical procedure and stored in liquid nitrogen until the day of metabolite extraction. HCC cases were diagnosed based on diagnostic imaging criteria and/or histology with clinical stages determined on the basis of TNM system. In this study, liver tissues collected from 40 patients with HCC were analyzed by GC-MS and LC-MS. Table 1 presents the characteristics of these patients. From the 40 HCC cases, both tumor and adjacent non-tumor tissues were analyzed comprising 30 HCC tissues (HCC-N) with adjacent normal tissues (ADJ-N), and 10 HCC tissues (HCC-C) with adjacent cirrhotic tissues (ADJ-C). Thus, 80 frozen dissected tissues were homogenized prior to extraction of polar metabolites and secondary lipid enrichment. The polar fraction of the samples, divided into five batches, was analyzed by GC-MS, both fractions were combined for analysis by LC-MS in both positive and negative modes. The experimental design for both GC-MS and LC-MS analyses is illustrated in Supplementary Figures 1 and 2, respectively.
Table 1.
Characteristics of the study cohort.
| AS(N=10) | AA(N=14) | EA(N=16) | ||
|---|---|---|---|---|
| Age | Mean (SD) | 56.8 (11.4) | 59.4 (12.8) | 65.6 (11.9) |
| Gender | Male % | 100 | 78.6 | 62.5 |
| BMI | Mean (SD) | 15.3(12.4) | 31.3(10.9) | 28.4(15.0) |
| HCV Serology | HCV Ab+ % | 30 | 57.1 | 31.2 |
| HCV RNA+ % | 20 | 21.4 | 25 | |
| HBV Serology | anti HBC+ % | 20 | 7.1 | 0 |
| HBs Ag+ % | 20 | 28.6 | 0 | |
| Smoking | Yes % | 80 | 64.3 | 50 |
| No % | 20 | 35.7 | 50 | |
| Alcohol | Yes % | 50 | 42.9 | 43.8 |
| No % | 50 | 57.1 | 56.3 | |
| MELD | Mean (SD) | 8.9 (3.7) | 11.2 (4.7) | 10.8 (4.7) |
| MELD <10 % | 50 | 50 | 50 | |
| AFP | Median (IQR) | 3.6 (0.925) | 11.2 (147.3) | 5.4 (14.025) |
| AST | Median (IQR) | 93 (108) | 132 (140) | 127.5 (171.8) |
| ALT | Median (IQR) | 93 (81) | 141(135) | 101(128.5) |
| Child Pugh score | Mean (SD) | 6.1 (1.7) | 6.2 (1.7) | 6.4 (1.3) |
| Median (IQR) | 5 (1) | 6 (1) | 6 (1.25) | |
| Child Pugh Class | A % | 70 | 78.6 | 62.5 |
| B % | 20 | 7.1 | 37.5 | |
| C % | 0 | 7.1 | 0 | |
| HCC Stage | Stage I % | 50 | 42.9 | 37.5 |
| Stage II % | 30 | 21.4 | 25 |
3. Material and Methods
Chemicals and Reagents
The chemicals and reagents used in this study are presented in Table 2.
Table 2.
Information of the chemicals and reagents used in this study.
| Manufacturer | Chemical & reagents | Product Number |
|---|---|---|
| Millipore Sigma (Burlington, MA) | Alkane standard mixture | 68281 |
| Fatty acid methyl ester standards (FAMEs) | C8 (260673); C9 (245895); C10 (299030); C12 (234591); C14 (P5177); C16 (P5177); C18 (S5376); C20 (10941); C22 (11940); C24 (87115); C26 (H6389); C28 (74701). | |
| Myristic-d27 acid | 366889 | |
| Methoxyamine hydrochloride | 226904 | |
| Pyridine | 360570 | |
| Debrisoquine | D1306 | |
| 4-nitrobenzoic acid | 72910 | |
| TCI chemicals (Portland, OR) | FAME C30 | T0812 |
| Avanti Polar Lipids (Alabaster, AL) | d35-lysophosphocholine | 860398 |
| d7-sphingosine-1-phosphate | 860659 | |
| Thermo Fisher Scientific (Waltham, MA) | MSTFA | TS-48910 |
| Fisher Scientific (Hampton, NH) | HPLC grade water | W6-4 |
| 2-propanol | A-461–212 | |
| Acetonitrile | A955–1 | |
| Formic acid | A117–50 | |
| Ammonium formate | A11550 |
Sample preparation
Figure 1 depicts the sample preparation steps. Briefly, homogenization of 10 mg of liver tissue was performed on ice with 1 mL of pre-chilled methanol:water (1:1) containing five internal standards: 0.001 ppm debrisoquine, 0.004 ppm 4-nitrobenzoic acid, 0.0004 ppb stearoyl (d35)-2-hydroxy-glycerophosphocholine, 0.2 ppb D-erythro-sphingosine (d7)-1-phosphate, and 2 ppm myristic-d27 acid. We centrifuged the homogenized samples at 14,500 g, at 4°C for 15 minutes. The collected supernatant (S1) was split in two aliquots, while the remaining pellet (P1) was kept at −80°C until further extraction. For the extraction of medium to polar compounds, 1:1 volume of pre-chilled acetonitrile was added to the two aliquots, vortex-mixed, kept on ice for 20 minutes and centrifuged again at 14,500 g, at 4°C for 15 minutes. The collected supernatant (S2) was dried-up in a speedvac system operated at room temperature. Pellets (P2 and P3) and supernatants were stored at −80°C. For the extraction of low-polar compounds, P1 was resuspended with 500μ1 of pre-chilled dichloromethane:methanol (3:1) while P2, and P3 with 125 μL of the same mix. Pellets were sonicated on ice for 90sec, combined, and centrifuged at 14,500 g, at 4°C for 20 minutes. Supernatant (S3) was split in two aliquots and a 1:1 volume of pre-chilled acetonitrile was added to both aliquots (GC&LC), vortexed and kept on ice for 20 minutes. Samples were centrifuged at 14,500 g for 15 min at 4°C while pellets were stored at −80 °C for protein quantitation. Finally, supernatants (S4) were concentrated to dryness by speedvac and kept at −80°C until the day of analysis. Blank samples were prepared together with the human samples. They consisted of all reagents and were prepared in the same way as the patients’ samples.
Fig 1.

Overview of tissue sample preparation for metabolomics analysis by GC-MS and LC-MS.
Dried supernatant (S2) collected for GC-MS analysis underwent derivatization step. Samples were derivatized prior to injection following oximation and trimethylsilylation (-Si(CH3)3). Briefly, 20 μL of a solution of 20 mg/mL methoxyamine hydrochloride in pyridine, were added to the dried extracts, vortex-mixed and incubated at 30°C for 90 minutes. At room temperature, 80 μL of MSTFA was added, vortex-mixed and incubated at 30°C for 30 minutes. Then, samples were centrifuged at 14,500 g for 15 minutes. Following this, 60 μL of the supernatant was transferred into 250 μL clear glass autosampler vials. Finally, 20 μL of 0.006 μg/μL C18 methyl stearate in hexane were added to the vial prior to injection. For quality assessment, myristic-d27 acid was spiked into the working solution to verify tissue metabolite extraction and derivatization steps. C18 Methyl Stearate was added to monitor each sample injection. QC samples (QCs) were generated by pooling together the supernatant obtained after derivatization of samples of each biological group in each batch separately. A retention index (RI) standard sample was prepared by mixing FAMEs with alkanes. Specifically, all FAMEs were individually suspended in chloroform at concentrations of 0.8 and 0.4 mg/mL for (C8 – C16) and (C18 – C30), respectively, to generate FAME-1 stock solutions. We mixed together 100 μL of each stock and 1.2 mL of chloroform for a final volume of 2.5 mL generating FAME-2 solution. Alkanes, containing all even CnH2n+2 from C10 to C40, were purchased as a mixture at a concentration of 50 mg/L in n-heptane. Alkane mixture were mixed with FAME-2 markers and hexane at a ratio of 1:2:17 and vortex-mixed prior to injection into the GC-MS.
Dried supernatants (S2 & S4) collected from the first and second extraction for LC-MS analysis were reconstituted with 125 μL of methanol:acetonitrile:water (50:25:25) each, and combined for a total volume of 250 μL. d35-lysophosphocholine and d7-sphingosine-1-phosphate were added to evaluate the quality of the metabolite extraction. We used debrisoquine and 4-nitrobenzoic acid for evaluation of the equipment performance. QCs were generated by pooling together the supernatant obtained after resuspension in appropriate solvent of each biological group.
GC-TOF-MS data acquisition
One μL of derivatized metabolites from tissue samples was analyzed by using the Agilent 7890A GC on-line coupled to LECO Pegasus HT GC-TOF-MS system (LECO Corporation). An HP-5 column (Agilent), 30m long, with 0.32mm i.d. and 0.25μm 95% dimethyl 5% diphenyl polysiloxane film was used. GC and MS parameters were set as following: 99.9% Helium with a flow rate of 1.5 mL/min; oven temperature set at 70°C for 4 minutes, then ramped at 10°C/min to 300°C, and remained constant for 4 minutes; transfer line temperature at 275°C; electron impact (EI) ionization at 70V and ion source temperature at 300°C; 300 seconds of solvent delay; full scan (50–600 m/Z) with acquisition rate of 10 scans per second; detector range (1400–1600V) with a voltage offset of 200; 31 minutes of total chromatographic separation. QCs were run before every 7–9 samples for quality assessment. An additional QC sample was run at the beginning of the analysis to equilibrate the chromatographic system. A Retention Index (RI) standard was used for calibration of the RI in each batch by injecting it both at the beginning and the end. To avoid contamination and assure system stability, a routine equipment maintenance was performed before the beginning of each batch, consisting of septum and liner replacement and column cut (2–3 cm).
UPLC-QTOF MS data acquisition
Extracted metabolites from liver tissues were analyzed using an ACQUITY UPLC system on-line coupled to a Synapt G2-Si QTOF-MS (Waters Corporation, Milford, MA, USA) in positive and negative modes. Chromatographic parameters were set as follows: injection volume was 1.5 μL; a reverse-phase ACQUITY UPLC CSH C18 (1.7-μm,100 × 2.1 mm) column (Waters) was employed and thermostated at 55°C; separation of the metabolites was achieved at 0.4 mL/min flow rate with a mobile phase as a gradient composed of acetonitrile:H20 (60:40) containing 0.1% formic acid and 10 mM ammonium formate (A), and isopropanole:acetonitrile (90:10) containing 0.1% formic acid and 10 mM ammonium formate (B); 15 minutes of total chromatographic separation. MS parameters were set as follows: capillary voltage at 1.8 kV and 1.5 kV, and cone voltage at 70 V and 20 V, for positive and negative polarity modes, respectively; desolvation gas flow at 900 Lhr− 1; temperature at 500°C and 550°C for positive and negative polarity mode, respectively; cone gas flow at 100 Lhr− 1; source temperature at 120°C. a LockSpray interface of Leucine enkephalin ([M+H]+ = 556.2766 and [M-H]− = 554.2620) was introduced at a rate of 20 μLmin− 1 to maintain accurate mass. We acquired LC-MS data in centroid full scan mode (50–1200 m/z). By pooling together the supernatants, obtained after resuspension in appropriate solvent, of each biological group, QCs were run before every 16 samples for quality assessment. An additional QC sample was injected at the beginning of the analysis to equilibrate the chromatographic system.
GC-TOF-MS data pre-processing
The raw GC-MS data were pre-processed (outlier screening, peak deconvolution, RI calibration, metabolite identification, and peak alignment) using ChromaTOF software with True Signal Deconvolution package from LECO Corporation (St. Joseph, MI). Supplementary Figure 3 depicts the steps involved. Outlier screening was performed to remove runs that were significantly different from the others based on the number of peaks or the pattern of the chromatogram. Following detection and deconvolution, peaks were aligned on the basis of spectral matching for neighbor peaks across different runs. Data acquired by running the QC samples were used to evaluate the consistency and reproducibility of the peaks across different runs and batches, while the blanks were used to detect possible contamination. We filtered out peaks detected in less than 50% of the QC samples within a group and in less than 75% of the samples within a group. In addition, unrelated peaks, including contamination, column bleeding artifact and internal standards were filtered out along with all unreproducible peaks that showed a relative standard deviation (RSD) greater that 50% in more than one group of QCs. Peaks present in 90% or more of the samples within a biological group were imputed with the value of the median within the group; peaks present between 90 and 75% of the samples were imputed with a value corresponding to half of the minimum one detected in the group. The nearest neighbor averaging method using the R package impute was used for completing the missing data imputation. The ComBat function from the R package sva was applied to correct for batch effect.
UPLC-QTOF data preprocessing
LC-MS data preprocessing involves peak detection, retention time correction, identification of ions that are related to each other (e.g., isotopes, adducts, in-source fragments, etc.), and outlier screening. We used various tools for data pre-processing as illustrated in Supplementary Figure 3. Briefly, the raw UPLC-QTOF-MS data were converted into Network Common Data Format (NetCDF) using MassLynx (Waters). The NetCDF data were then analyzed using the R package XCMS where part of the parameters were optimized using the R package IPO.17 XCMS performed peak detection and peak matching. It also performed automatic missing values imputation by rereading the raw data files and integrating them in the region of the missing peaks. R package CAMERA was used for ion annotation (process of grouping all adducts, cluster ions and charge state entities derived from the same analyte).
Difference Detection
A mixed model ANOVA was applied on peaks detected by GC-MS and LC-MS to evaluate their association with tissue and racial groups. Four tissue groups (HCC-N, ADJ-N, HCC-C, ADJ-C, and three racial groups (AA, AS, EA) are considered in this study. To implement the mixed model ANOVA, we used the lsmeans function from the R package lsmeans.18 The model can be represented by the equation below:
where yijk is the analyte level for subject i from race j and tissue group k, μ is the grand mean, ti is the random effect for patient, i, αj, βk, and (αβ)jk are the main effects and interaction effects for the jth race and kth tissue group respectively. j=1,2,3 indicates race AA, AS, EA respectively; k=1,2,3,4 indicates HCC-C, ADJ-C, HCC-N and ADJ-N, respectively. The mixed model ANOVA was also separately fit using the data from the AA and EA participants only, because no cirrhotic (HCC-C and ADJ-C) samples were available in our AS group. To account for multiple testing, we calculated false discovery rate (FDR) using the Benjamini-Hochberg (BH) method. Fold changes were calculated considering the median of raw intensity values.
Metabolite identification
Identification of analytes detected by GC-TOF-MS was achieved by comparing the experimental spectra with the ones compiled in the NIST 2011 and Fiehn 2013 libraries. In addition, the GOLM database was employed for confirmation. RI calibration was applied to further assist the identification. Putative IDs were assigned to the analytes detected by LC-MS by using MetaboQuest (http://omicscraft.com/tools/) that searches mass values against major compound databases including HMDB, METLIN, KEGG, MMCD and LIPID MAPS. It also calculates putative ID prioritization scores by building a network of putative IDs based on metabolic and biochemical pathways. Furthermore, we acquired MS/MS spectra for a subset of statistically significant analytes. The metabolite extracts for MS/MS analysis were prepared following the same steps described previously. mzXML files were parsed using pyteomics python library. In each mzXML file, expected retention time values from previous experiments were used to access the scans of the targeted analytes. mzXML function in pyteomics library was used to access the m/z and corresponding intensity information within the expected retention time. Top 10 peaks with highest intensities were used for metabolite identification by comparing the observed fragmentation patterns against experimental and in-silico fragments in spectral libraries. We used tools such as MetaboQuest, METLIN, and CEU Mass Mediator to search against libraries and assign spectral matching scores.
4. Results and Discussion
Both GC-MS and LC-MS platforms were used to evaluate the levels of the metabolites extracted from the HCC area and adjacent non-HCC area of liver surgical specimen collected from 40 patients. In each of the five batches for GC-MS and one batch for LC-MS analyses, samples were randomized prior to the metabolite extraction step, GC-MS derivatization step, and MS data generation step. The stability of both platforms was assessed using QC samples analyzed during data acquisition.
As shown in Table 3, 946 peaks were detected by GC-MS, 8197 by LC-POS-MS, and 1799 by LC-NEG-MS. After filtering out some of the peaks, statistical analysis was performed on the remaining ones to identify those with significant changes in HCC-C vs. ADJ-C and HCC-N vs. ADJ-N. Among the analytes with assigned ID, the majority of alterations seemed to occur in the HCC-N vs. ADJ-N comparison with a total of 78 detected by GC-MS and 404 by LC-MS with FDR < 10%. In HCC-C vs. ADJ-C, only 3 analytes by GC-MS and 13 by LC-MS were found significant with FDR < 10%.
Table 3.
Summary of detected and statistically significant analytes for each platform utilized in this study.
| No FEATURES after | GC-MS | LC-POS-MS | LC-NEG-MS | |||
| peak detection and alignment | 946 | 8197 | 1799 | |||
| data filtering | 728 | 2286 | 593 | |||
| Putative ID assigned by MS-based search | 250 | 1122 | 429 | |||
| Significant features | HCC-C vs. ADJ-C | HCC-N vs. ADJ-N | HCC-C vs. ADJ-C | HCC-N vs. ADJ-N | HCC-C vs. ADJ-C | HCC-N vs. ADJ-N |
| p< 0.05; FDR <10% | 3 | 78 | 8 | 265 | 5 | 139 |
Supplementary Tables 1 (ST1) and 2 (ST2) present the complete list of the statistically significant analytes with assigned putative identifications in HCC-C vs. ADJ-C and HCC-N vs. ADJ-N comparisons, respectively. The tables present FDR obtained by univariate statistical analysis, and fold changes for the entire cohort (WP) as well as stratified by racial groups (AS, AA, EA). While the first putative ID (with score ≥ 750), returned after spectral matching with Fiehn 2013 library, is presented for those detected by GC-MS, multiple putative IDs obtained from the MS-based search results (with 10 ppm tolerance) by MetaboQuest are listed for analytes detected by LC-POS-MS and LC-NEG-MS.
Among the analytes with statistically significant alterations in HCC-N vs. ADJ-N, 18 were selected for further investigation based on their biological relevance and verification of their putative identification. These include 8 metabolites detected by GC-MS, 7 by LC-MS in the positive mode, and 6 by LC-MS in the negative mode (3 metabolites were detected by both GC-MS and LC-MS). Table 4 presents these metabolites and their statistical levels considering all racial groups together (Whole population) as well as the AS, AA, and EA groups. Based on the FDR <10% cutoff, hypoxanthine, adenosine monophosphate, PC (34:2), and glycerylphosphorylethanolamine showed statistically significant alteration only in the whole population, while the rest of the metabolites showed statistically significance in one or two of the racial groups in addition to the whole population. Supplementary Table 3 presents the information (EI and/or ESI fragments) we used to confirm the identities of the metabolites reported in Table 4. Some of these metabolites were further confirmed by comparing their retention time values and MS/MS fragmentation pattern with those from reference compounds that we run side by side under the same LC-MS/MS conditions.
Table 4.
Significant metabolites identified by GC-MS and LC-MS in HCC-N vs. ADJ-N liver tissues.
| Metabolite | Mode | Mmi (Da) | RT (min.) | HCC-N vs. ADJ-N | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Whole population | AS | AA | EA | ||||||||||||
| p-value | FDR† | FC | p-value | FDR† | FC | p-value | FDR† | FC | p-value | FDR† | FC | ||||
| fumaric acid | EI | 116.01 | 11.2 | 2.72 E-09 | 2.88 E-07 | ↓−3.8 | 0.00211 | 0.11830 | ↓−2.6 | 0.00004 | 0.00217 | ↓−6.7 | 0.00001 | 0.00122 | ↓−4.3 |
| malic acid | EI | 134.02 | 13.1 | 1.15 E-07 | 5.52 E-06 | ↓−3.2 | 0.06400 | 0.50041 | ↓−1.8 | 0.00037 | 0.01228 | ↓−3.1 | 0.00002 | 0.00169 | ↓−4.0 |
| succinic acid | EI | 118.02 | 10.7 | 1.92 E-10 | 2.80 E-08 | ↓−2.9 | 0.00483 | 0.16747 | ↓−2.4 | 2.37E-06 | 0.00025 | ↓−3.6 | 0.00022 | 0.00954 | ↓−3.3 |
| glucose | EI | 180.06 | 18.1 | 2.30 E-06 | 0.00006 | ↓−2.8 | 0.03068 | 0.39253 | ↓−2.5 | 0.00114 | 0.02523 | ↓−4.2 | 0.00375 | 0.06199 | ↓−2.2 |
| lactic acid | EI | 90.03 | 6.6 | 0.00039 | 0.00409 | ↑+1.3 | 0.00071 | 0.05139 | ↑+1.4 | 0.41617 | 0.99999 | ↑+1.4 | 0.21269 | 0.78597 | ↑+1.2 |
| hypoxanthine | EI | 136.03 | 16.7 | 0.00497 | 0.02847 | ↑+1.6 | 0.04964 | 0.48374 | ↑+2.6 | 0.19531 | 0.78501 | ↑+1.8 | 0.44452 | 0.99999 | ↑+1.2 |
| ESI− | 0.6 | 0.01060 | 0.04336 | ↑+1.9 | 0.01038 | 0.14321 | ↑+3.3 | 0.15320 | 0.54342 | ↑+1.9 | 0.99807 | 0.99999 | ↑+1.3 | ||
| xanthosine | EI | 284.07 | 24.4 | 1.95 E-06 | 0.00006 | ↓−2.9 | 0.30885 | 0.99999 | ↓−1.7 | 0.00406 | 0.07082 | ↓−2.8 | 0.00002 | 0.00156 | ↓−3.9 |
| ESI− | 0.6 | 0.00003 | 0.00030 | ↓−4.1 | 0.32362 | 0.99999 | ↓−3.1 | 0.00360 | 0.05202 | ↓−4.4 | 0.00237 | 0.05550 | ↓−4.0 | ||
| adenosine monophosphate | EI | 347.06 | 27.1 | 0.00008 | 0.00115 | ↓−1.8 | 0.06100 | 0.49899 | ↓−1.8 | 0.01610 | 0.18028 | ↓−2.1 | 0.02881 | 0.23563 | ↓−1.6 |
| ESI− | 0.6 | 0.00100 | 0.00589 | ↓−2.6 | 0.05909 | 0.38933 | ↓−2.5 | 0.07575 | 0.36520 | ↓−3.5 | 0.17217 | 0.60745 | ↓−1.8 | ||
| propionyl carnitine | ESI+ | 217.13 | 0.6 | 1.75 E-06 | 0.00004 | ↓−5.3 | 0.00034 | 0.01806 | ↓−6.5 | 0.00154 | 0.03235 | ↓−4.5 | 0.07789 | 0.54289 | ↓−5.0 |
| linoleic acid | ESI− | 280.23 | 8.8 | 0.00003 | 0.00031 | ↓−2.0 | 0.02878 | 0.24500 | ↓−1.4 | 0.00326 | 0.05029 | ↓−3.9 | 0.07603 | 0.34681 | ↓−1.5 |
| PC(34:2) | ESI+ | 495.33 | 8.8 | 0.00067 | 0.00530 | ↓−2.1 | 0.14583 | 0.86099 | ↓−1.4 | 0.01737 | 0.17488 | ↓−2.5 | 0.15721 | 0.83788 | ↓−2.0 |
| PC(38:3) | ESI+ | 811.60 | 10.3 | 0.00311 | 0.01969 | ↑+3.7 | 0.28471 | 0.99999 | ↑+5.5 | 0.00134 | 0.02888 | ↑+5.9 | 0.95680 | 0.99999 | ↑+1.4 |
| PC(36:1) | ESI+ | 787.60 | 10.4 | 2.39 E-06 | 0.00005 | ↑+1.9 | 0.00694 | 0.10500 | ↑+1.7 | 0.00011 | 0.00699 | ↑+2.2 | 0.13370 | 0.75426 | ↑+1.8 |
| PC(38:2) | ESI+ | 813.62 | 10.5 | 0.00092 | 0.00698 | ↑+1.6 | 0.00483 | 0.08333 | ↑+3.0 | 0.03028 | 0.26524 | ↑+1.8 | 0.89013 | 0.99999 | ↑+1.5 |
| glycerol-3-phosphate | ESI− | 172.01 | 0.6 | 3.05 E-06 | 0.00006 | ↓−3.9 | 0.10341 | 0.61322 | ↓−1.7 | 0.00083 | 0.02246 | ↓−6.5 | 0.00173 | 0.04690 | ↓−4.9 |
| glycerylphosphoryl ethanolamine | ESI− | 215.05 | 0.6 | 0.00023 | 0.00165 | ↓−2.9 | 0.11243 | 0.64653 | ↓−1.9 | 0.02312 | 0.18281 | ↓−4.5 | 0.04236 | 0.26761 | ↓−2.9 |
| glycerophospho choline | ESI+ | 257.10 | 0.7 | 5.86 E-06 | 0.00011 | ↓−3.7 | 0.02022 | 0.22227 | ↓−3.6 | 0.00233 | 0.04207 | ↓−5.6 | 0.00957 | 0.17291 | ↓−3.5 |
| coenzyme Q10 | ESI+ | 862.68 | 12.4 | 0.00015 | 0.00153 | ↓−2.8 | 0.53056 | 0.99999 | ↓−1.6 | 0.00267 | 0.04506 | ↓−4.2 | 0.01850 | 0.24819 | ↓−2.9 |
FDRs <10% are marked in bold
The selected metabolites belong to tricarboxylic acid (TCA) cycle, glycolysis pathway, purine metabolism, and energy metabolism. Specifically, succinic acid, fumaric acid, and malic acid, are metabolites that belong to TCA cycle. In this study they were found significantly downregulated in HCC-N vs. ADJ-N, consistently with other liver tissue metabolomic studies where HCC and non-tumor tissues were compared,14,15,19,20 in whole population, AA, and EA groups. The TCA cycle allows the release of stored energy into adenosine triphosphate (ATP) and CO2, through the oxidation of acetyl-CoA derived from carbohydrates, fats, and proteins, regulating the oxidative metabolism in the cells. The decrease in the level of TCA intermediate metabolites together with the decrement of glucose, and the increment of lactic acid, as also shown in our study, suggests the rapid consumption of glucose during glycolysis having a low level of aerobic oxidation via the TCA cycle itself.14
Hypoxanthine, xanthosine, and adenosine monophosphate (AMP) belong to purine metabolism. In this study they were detected by both GC-MS and LC-MS showing the same trend between HCC-N and ADJ-N tissues across all racial groups. While hypoxanthine and AMP show their statistically significance only in the whole group, xanthosine shows it in the AA, and EA group in addition to the whole population. Previous metabolomic studies on liver tissue have reported similar alterations between HCC and non-HCC tissues.15,20 Purines, which are the basic components of nucleotides, provide the necessary energy and cofactors for cell survival and proliferation. Therefore, impaired purine metabolism is related to progression of cancer.21 A new viewpoint on how a cell regulates purine need has been reported underlying how purine metabolism may contribute into cancer progression.22
Also reported in HCC tumors is the significant changes of the acylcarnitines class. Consistent with previous studies,14,23 we found that short-chain acylcarnitines such as propionylcarnitine was significantly downregulated in the AS, AA in addition to the whole population. These metabolites are primarily involved in lipid metabolism in cells by transporting fatty acids inside the mitochondria where they are subjected to oxidization and conversion into energy through the TCA cycle. Their downregulation together with the downregulation of TCA intermediates suggests a reduced consumption of carboxylic acids in the mitochondria.15
Coenzyme Q10, found in this study significantly decreased in HCC-N v ADJ-N, particularly among the AA, is an essential compound for the transport of electrons in the mitochondrial respiratory chain and for antioxidant defense. Its downregulation has been observed in many solid tumors and recently it has been investigated in a study where HCC tumors were compared to paired non-tumor tissues. A correlation between the decreased expression of coenzyme Q10 and prenyl diphosphate synthase subunit 2 (PDSS2), a key factor in its synthesis, was also observed and in vitro and in vivo investigated implying that PDSS2 deficiency, due to mutation in its gene, could induce hepatocarcinogenesis. 24
Fatty acids are the metabolites required for both structural and functional purposes needed for membrane synthesis. We found linoleic acid significantly altered in the whole population, and AA groups. Its downregulation is consistent with previous liver metabolomics studies.15,25 Specifically, Beyoğlu et al. found that among the fatty acids, linoleic acid was the only one statistically significantly decreased in HCC tissues suggesting that HCC may engage in enhanced arachidonic acid synthesis, because linoleic acid is the starting point for de novo synthesis of arachidonic acid.
In a previous untargeted metabolomics study conducted on liver tissues, lysophosphorylcholine (LPC) levels were observed to have decreased in cancer tissues while phosphorylcholine (PC) levels were increased.15 Comparable results were found in this study with significant downregulation of PC (34:2) and upregulation of PC (36:1), PC (38:2), and PC (38:3). The majority of the metabolomic studies involving both liver tissues and sera have reported imbalance of these metabolites suggesting their participation in different biological processes that promote HCC development.26
Metabolites belonging to the glycerolipid metabolism (glycerol 3-phosphate, glycerylphosphorylethanolamine, glycerophosphocholine) displayed statistically significant changes in tumor vs. non-tumor tissues in this study, as previously reported,27 with glycerol 3-phosphate significantly altered in both AA and EA, and glycerophosphocholine in AA only. Dysregulation of glycerolipid metabolism may contribute to HCC by affecting the insulin uptake and glucose consumption, leading to a reinforced capacity for the cell to proliferate under conditions that would normally be considered unviable.28
Overall, as shown in Table 4, among the 18 metabolites selected in this study, the majority (thirteen out of 18) showed statistically significant changes (FDR < 10%) in AA, six of which belonging to lipid metabolism. All seven metabolites that were found statistically significant in EA are a subset of the fourteen significant metabolites selected for AA. Among the three metabolites found significant for AS, only one overlaps with those selected for AA, the other two are unique to AS. Figure 2 depicts the dot plots of three metabolites that most exhibited statistically significant changes in AA, coenzyme Q10, linoleic acid, and PC (38:3). The dot plots for the remaining fifteen metabolites selected in this study are shown in Supplementary Figures 4–6. Confirmation of the observed changes in metabolites level by targeted quantitation is a pre-requisite for subsequent biological interpretation and for their investigation in peripheral blood aimed at identifying accurate noninvasive HCC biomarkers.
Fig 2.

Metabolites with statistically significant change (FDR < 10%) in HCC-N vs. ADJ-N for AA.
In summary, the results obtained in this study demonstrate that metabolomic analysis of liver tissues not only offers holistic information on dynamic metabolic responses to the HCC mechanism, but also allows the investigation of racial disparity by identifying metabolites associated to HCC in a race-specific manner.
5. Conclusion
Metabolic reprogramming is crucial for carcinogenesis; therefore it is generally known that metabolic profile variations are present in HCC patients at the time of their diagnosis.8 In this study, we report a metabolomic approach to find HCC-associated metabolites by comparing paired liver tissues obtained as surgical specimen from the participants in this study. The levels of metabolites in tissues were assessed by both GC-MS and LC-MS platforms. We selected 18 metabolites that showed statistically significant change in their levels between HCC and non-HCC liver tissues. These metabolites belong to tricarboxylic acid (TCA) cycle, glycolysis pathway, purine metabolism, and energy metabolism that are specifically involved in HCC formation. In addition, we investigated the metabolites that showed significant association to HCC in a race-specific manner. In view of these, we believe the metabolites identified in this study may have the potential not only to elucidate HCC pathophysiology but also to help investigate racial disparities in HCC. Future work will focus on further investigating the observed changes in metabolites level by targeted quantitation in liver tissues and peripheral blood.
Supplementary Material
S1 Fig. Experimental design for GC-MS analysis.
S2 Fig. Experimental design for LC-MS analysis.
S3 Fig. Summary of the software tools used for GC-MS and LC-MS data pre-processing.
S4 Fig. Dot plots of selected metabolites detected by GC-MS.
S5 Fig. Dot plots of selected metabolites detected by LC-POS-MS.
S6 Fig. Dot plots of selected metabolites detected by LC-NEG-MS.
S1 Table Statistically significant analytes (FDR <10%) with assigned putative identification for the HCC-C v ADJ-C comparison.
S2 Table Statistically significant analytes (FDR <10%) with assigned putative identification for the HCC-N v ADJ-N comparison.
S3 Table EI and/or ESI fragments used to confirm the metabolites identification of analytes detected by GC-MS and LC-MS.
Acknowledgments
This work was supported by NIH/NCI U01CA185188 and NIH/NIGMS R01GM123766 grants, awarded to HWR. Tissue samples were provided by the Histopathology and Tissue Shared Resource (HTSR) and LC-MS/MS data were generated by Proteomics and Metabolomics Shared Resource (PMSR).
Abbreviations
- ADJ-C
Adjacent cirrhotic
- ADJ-N
Adjacent normal
- AA
African-American
- AFP
Alphafetoprotein
- ALT
Alanine aminotransferase
- AS
Asian-American
- AST
Aspartate aminotransferase
- EA
European-American
- FAME
Fatty acid methyl esters
- GC
Gas chromatography
- HBV
Hepatitis B virus
- HCV
Hepatitis C virus
- HCC
Hepatocellular carcinoma
- HCC-C
Hepatocellular carcinoma paired to adjacent cirrhotic tissue
- HCC-N
Hepatocellular carcinoma paired to adjacent normal tissue
- LC
Liquid chromatography
- MS
Mass spectrometry
- QTOF
Quad-Time-of-flight
- TOF
Time-of-flight
- TCA
Tricarboxylic acid cycle
- TNM
Tumor, Node, Metastasis
- UPLC
Ultra performance liquid chromatography
Footnotes
Metabolomic Data
Metabolomic data have been submitted to the Metabolomics Workbench (http://www.metabolomicsworkbench.org).
References
- (1).Bertuccio P; Turati F; Carioli G; Rodriguez T; La Vecchia C; Malvezzi M; Negri E Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol 2017, 67, 302–309. [DOI] [PubMed] [Google Scholar]
- (2).Xu L; Kim Y; Spolverato G; Gani F; Pawlik TM Racial disparities in treatment and survival of patients with hepatocellular carcinoma in the United States. Hepatobiliary. Surg. Nutr 2016, 5, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Marcellin P; Kutala BK Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver International 2018, 38, 2–6. [DOI] [PubMed] [Google Scholar]
- (4).American Cancer Society. Cancer Facts & Figures 2018. Atlanta: American Cancer Society; 2018. [Google Scholar]
- (5).Burra P; Zanetto A; Germani G Liver transplantation for alcoholic liver disease and hepatocellular carcinoma. Cancers 2018, 10, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Singal AG; Li X; Tiro J; Kandunoori P; Adams-Huet B; Nehra MS; Yopp A Racial, social, and clinical determinants of hepatocellular carcinoma surveillance. Am. J. Med 2015, 128, 90. e1–90. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stewart SL; Kwong SL; Bowlus CL; Nguyen TT; Maxwell AE; Bastani R; Chak EW; Chen MS Jr Racial/ethnic disparities in hepatocellular carcinoma treatment and survival in California, 1988–2012. World J. Gastroenterol 2016, 22, 8584–8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guo W; Tan HY; Wang N; Wang X; Feng Y Deciphering hepatocellular carcinoma through metabolomics: from biomarker discovery to therapy evaluation. Cancer management and research 2018, 10, 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen C; Wang G Mechanisms of hepatocellular carcinoma and challenges and opportunities for molecular targeted therapy. World J Hepatol 2015, 7, 1964–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).De Matteis S; Ragusa A; Marisi G; De Domenico S; Casadei Gardini A; Bonafè M; Giudetti AM Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxidative medicine and cellular longevity 2018, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Procopet B; Fischer P; Farcau O; Stefanescu H Metabolomics: From liver chiromancy to personalized precision medicine in advanced chronic liver disease. World journal of hepatology 2018, 10, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Armitage EG; Ciborowski M Applications of metabolomics in cancer studies. In Metabolomics: From Fundamentals to Clinical Applications Springer: 2017; pp 209–234. [DOI] [PubMed] [Google Scholar]
- (13).Johnson CH; Ivanisevic J; Siuzdak G Metabolomics: beyond biomarkers and towards mechanisms. Nature reviews Molecular cell biology 2016, 17, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lu Y; Li N; Gao L; Xu YJ; Huang C; Yu K; Ling Q; Cheng Q; Chen S; Zhu M; Fang J; Chen M; Ong CN Acetylcarnitine Is a Candidate Diagnostic and Prognostic Biomarker of Hepatocellular Carcinoma. Cancer Res. 2016, 76, 2912–2920. [DOI] [PubMed] [Google Scholar]
- (15).Huang Q; Tan Y; Yin P; Ye G; Gao P; Lu X; Wang H; Xu G Metabolic characterization of hepatocellular carcinoma using nontargeted tissue metabolomics. Cancer Res. 2013, 73, 4992–5002. [DOI] [PubMed] [Google Scholar]
- (16).Naz S; Moreira dos Santos Délia Chaves; García A; Barbas C Analytical protocols based on LC-MS, GC-MS and CE-MS for nontargeted metabolomics of biological tissues. Bioanalysis 2014, 6, 1657–1677. [DOI] [PubMed] [Google Scholar]
- (17).Libiseller G; Dvorzak M; Kleb U; Gander E; Eisenberg T; Madeo F; Neumann S; Trausinger G; Sinner F; Pieber T IPO: a tool for automated optimization of XCMS parameters. BMC Bioinformatics 2015, 16, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Lenth RV Using lsmeans. J Stat Softw 2017, 69, 1–33. [Google Scholar]
- (19).Budhu A; Roessler S; Zhao X; Yu Z; Forgues M; Ji J; Karoly E; Qin LX; Ye QH; Jia HL; Fan J; Sun HC; Tang ZY; Wang XW Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology 2013, 144, 1066–1075.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Han J; Qin W; Li Z; Xu A; Xing H; Wu H; Zhang H; Li C; Liang L; Quan B Tissue and serum metabolite profiling reveals potential biomarkers of human hepatocellular carcinoma. Clinica Chimica Acta 2019, 488, 68–75. [DOI] [PubMed] [Google Scholar]
- (21).Yin J; Ren W; Huang X; Deng J; Li T; Yin Y Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol 2018, 9, 1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pedley AM; Benkovic SJ A new view into the regulation of purine metabolism: the purinosome. Trends Biochem. Sci 2017, 42, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Warburg O On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [PubMed] [Google Scholar]
- (24).Li Y; Lin S; Li L; Tang Z; Hu Y; Ban X; Zeng T; Zhou Y; Zhu Y; Gao S; Deng W; Zhang X; Xie D; Yuan Y; Huang P; Li J; Cai Z; Guan XY PDSS2 Deficiency Induces Hepatocarcinogenesis by Decreasing Mitochondrial Respiration and Reprogramming Glucose Metabolism. Cancer Res. 2018, 78, 4471–4481. [DOI] [PubMed] [Google Scholar]
- (25).Beyoglu D; Imbeaud S; Maurhofer O; Bioulac-Sage P; Zucman-Rossi J; Dufour JF; Idle JR Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mazzocca A; Dituri F; Lupo L; Quaranta M; Antonaci S; Giannelli G Tumor-secreted lysophostatidic acid accelerates hepatocellular carcinoma progression by promoting differentiation of peritumoral fibroblasts in myofibroblasts. Hepatology 2011, 54, 920–930. [DOI] [PubMed] [Google Scholar]
- (27).Liu SY; Zhang RL; Kang H; Fan ZJ; Du Z Human liver tissue metabolic profiling research on hepatitis B virus-related hepatocellular carcinoma. World J. Gastroenterol 2013, 19, 3423–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Cao H; Gerhold K; Mayers JR; Wiest MM; Watkins SM; Hotamisligil GS Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1 Fig. Experimental design for GC-MS analysis.
S2 Fig. Experimental design for LC-MS analysis.
S3 Fig. Summary of the software tools used for GC-MS and LC-MS data pre-processing.
S4 Fig. Dot plots of selected metabolites detected by GC-MS.
S5 Fig. Dot plots of selected metabolites detected by LC-POS-MS.
S6 Fig. Dot plots of selected metabolites detected by LC-NEG-MS.
S1 Table Statistically significant analytes (FDR <10%) with assigned putative identification for the HCC-C v ADJ-C comparison.
S2 Table Statistically significant analytes (FDR <10%) with assigned putative identification for the HCC-N v ADJ-N comparison.
S3 Table EI and/or ESI fragments used to confirm the metabolites identification of analytes detected by GC-MS and LC-MS.