

Abstract
Purpose:
Activating BRAF mutations, most commonly BRAFV600E, are a major oncogenic driver of many cancers. We explored whether BRAFV600E promotes radiation resistance and whether selectively targeting BRAFV600E with a BRAF inhibitor (vemurafenib, BRAFi) sensitizes BRAFV600E thyroid cancer cells to radiotherapy.
Experimental Design:
Immunoblotting, neutral comet, immunocytochemistry, functional reporter and clonogenic assays were used to analyze the outcome and molecular characteristics following radiotherapy with or without BRAFV600E or vemurafenib in thyroid cancer cells.
Results:
BRAFV600E thyroid cancer cell lines were associated with resistance to ionizing radiation (IR), and expression of BRAFV600E into wild type BRAF thyroid cancer cells led to IR resistance. BRAFi inhibited ERK signaling in BRAFV600E mutants but not BRAF wild-type thyroid cancer cell lines. BRAFi selectively radiosensitized and delayed resolution of IR-induced γ-H2AX nuclear foci in BRAFV600E cells. Moreover, BRAFi impaired global DNA repair and altered the resolution of 53BP1 and RAD51 nuclear foci in BRAFV600E cells following IR. BRAFV600E mutants displayed enhanced non-homologous end-joining (NHEJ) repair activity, which was abolished by BRAFi. Intriguingly, BRAFV600E mutation led to upregulation of XLF, a component of NHEJ, which was prevented by BRAFi. Importantly, BRAFi in combination with radiotherapy resulted in marked and sustained tumour regression of BRAFV600E thyroid tumor xenografts.
Conclusions:
BRAFV600E mutation promotes NHEJ activity leading to radioresistance and BRAFi selectively radiosensitizes BRAFV600E thyroid cancer cells through inhibiting NHEJ. Our findings suggest that combining BRAFi and radiation may improve the therapeutic outcome of BRAFV600E mutant thyroid cancer patients.
Keywords: BRAF, vemurafenib, DNA repair, radioresistance, thyroid cancer, non-homologous end-joining
INTRODUCTION
The RAS-RAF-MEK-ERK signaling cascade is the predominant cell growth-promoting signaling pathway deregulated in most human cancers, and thus a major hub for developing molecularly targeted therapies (1–3). The most frequently mutated component of this pathway is RAS, but directly targeting mutant RAS has proved challenging. Thereafter targeting downstream RAF, MEK and ERK is being actively pursued in cancer therapeutics (4,5). RAF kinase family consists of ARAF, BRAF and CRAF members with distinct mutation patterns in different cancer types (6). BRAF mutations are found in about 8% of all human cancers and are commonly present in melanoma (~50%), thyroid cancer (~45%), colorectal cancer (~10%), non-small cell lung carcinoma (~5%) and hairy cell leukemia (~100%) (6–9). The most frequent BRAF mutation is a 1799T>A substitution resulting in constitutively active BRAFV600E mutant, which leads to stimuli independent activation of downstream MEK-ERK signaling (6,9). Thus, specifically targeting the BRAFV600E mutant is a tumor-selective approach and a major goal of cancer therapy. To that end, several BRAFV600E specific small molecule inhibitors have been developed for the treatment of BRAFV600E driving cancers and some have been approved for use in BRAF mutant melanoma, thyroid cancer, and non-small cell lung cancer (6,10). However it has been well documented that BRAFV600E inhibitors are rarely curative, and the response of cancer patients to BRAFV600E inhibitors alone is transient partially due to the paradoxical activation of RAS-ERK signaling through multiple mechanisms (8). The resistance of various cancers to BRAFV600E inhibitors has led to the development of vertical targeting RAS-ERK signaling by simultaneous inhibition of BRAFV600E and MEK kinases. Indeed, combinatory BRAFV600E and MEK inhibitors have shown improved clinical efficacy compared to BRAFV600E inhibitor as monotherapy and has become the standard therapy for BRAFV600E metastatic melanoma (11,12).
Thyroid cancer is the most common cancer of the endocrine system, with an annual incidence of more than 60,000 cases in the USA (13). Thyroid cancers from follicular cells are classified into well differentiated, poorly differentiated, and anaplastic carcinoma. Though well differentiated thyroid cancers such as papillary thyroid cancer (PTC) have a good prognosis, the mortality rates of poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC) are 38-57% and ~100%, respectively (14). Thus, there is an urgent and unmet need to develop novel therapeutic strategies for PDTC and ATC. Recent genomic landscape analyses of thyroid cancers have demonstrated BRAF mutations occur in ~60% of PTC, ~33% of PDTC and ~45% of ATC, and preclinical studies have established that BRAF or RAS mutations are driver mutations in the development of these thyroid cancers (15–18). Of note, BRAF mutations are mutually exclusive with RAS mutations. Interestingly, combined analyses of gene variance, mRNA expression, protein expression and phosphorylation of signaling molecules have unraveled the existence of two distinct PTC groups: BRAFV600E-like and RAS-like. BRAFV600E-like PTCs have over-activation of MEK-ERK signaling companied with high expression of DUSP genes, while RAS-like PTCs have simultaneous activation of PI3K-AKT-mTOR and MEK-ERK pathways (16).
Thus, BRAFV600E inhibitors are potential drugs for the treatment of thyroid cancer patients with BRAFV600E mutation. As proof of principle, it has been shown that BRAFV600E inhibitor vemurafenib (FDA approved for treatment of BRAF mutant melanoma) improves the clinical outcome of thyroid cancer patients with BRAFV600E mutation (3,19–22). The resistance of thyroid cancer cells to vemurafenib has been ascribed to the rapid reactivation of ERK signaling resulting from vemurafenib-induced expression of ERBB3 and its ligand neuregulin-1 (23). Consequently it was found that EGFR kinase inhibitor lapatinib effectively prevents MAPK reactivation and sensitizes BRAFV600E thyroid cancer cells to vemurafenib (24). In addition, it was recently reported that combinatorial administration of BRAFV600E inhibitor dabrafenib and MEK inhibitor trametinib has robust clinical activity in BRAFV600E-mutated ATC patients (3). Thus, rationally designed combinations with targeted drugs, chemotherapy, and radiotherapy may improve the outcome of thyroid cancers with distinct signaling pathways and subvert resistance to drug monotherapy.
DNA damage repair plays an important role in cancer drug resistance (25,26). RAS mutations have been previously shown to promote preclinical radioresistance and targeted inhibition of MEK can effectively radiosensitize RAS mutant tumors (27–31). In our recent pre-clinical study, we found that trametinib attenuates DNA repair pathways in KRAS-mutant pancreatic cancer cells, suggesting that these DNA repair and cell cycle proteins downstream of MEK-ERK are mediating radioresistance in these tumor cells (28). With regard to BRAF inhibition as a radiosensitizing strategy, there is emerging clinical data suggesting that radiosensitization with BRAF inhibitors can be associated with profound increases in treatment efficacy (32).
Local-regional and systemic tumor control in thyroid cancer, especially PDTC and ATC, are very poor due in large part to radiotherapy and chemotherapy resistance. The role of BRAF mutations in radiotherapy resistance is largely unknown. Moreover, whether BRAFV600E inhibitors (BRAFi) sensitize thyroid cancers with a BRAFV600E mutation to radiotherapy remains to be determined. In the present study, we investigated the role of oncogenic BRAF in radioresistance, and tested radiosensitization of thyroid cancer cells to BRAFi in cell culture and mouse xenograft models. We found that BRAFV600E thyroid cancer cells were resistant to radiation, and BRAFV600E mutation resulted in enhanced NHEJ activity leading to radioresistance. In addition, BRAFi selectively radiosensitized BRAFV600E thyroid cancer cells likely through abrogation of heightened DNA double-strand break repair. Our findings suggest that pharmacological BRAFV600E inhibition in combination of radiotherapy may improve the therapeutic outcome of BRAFV600E mutant thyroid cancers.
MATERIALS AND METHODS
Antibodies, chemicals, reagents, cell culture, siRNA transfection
The detailed information of the thyroid cancer cell lines TPC-1, U-Hth-74, SW1736, BCPAP and 8505C is listed in Supplementary Table 1. We also used A375 and HS294T melanoma cells. Cells were maintained at 37°C in 5% CO2 in DMEM media (TPC-1,Hth-74, A375, and HS294T) or RPMI 1640 (SW1736, BCPAP and 8505C) supplemented with 10% fetal bovine serum (Sigma), 1% penicillin/streptomycin and non-essential amino acids (Thermo Fisher Scientific, Waltham, WA). Cells were cultured for no more than 3 months continuously. Vemurafenib (SelleckChem) was dissolved in DMSO and added to appropriate media with a final concentration of no more than 0.1% DMSO. Total ERK-½, phospho-ERK-½ (Thr202/Tyr204), total MEK-½, phospho-MEK-½ (Ser217/221), total BRAF, phospho-BRAF (Ser445), phospho-H2AX, XLF/NHEJ-1, and GAPDH primary antibodies were purchased from Cell Signaling Technology (Danvers, MA). BRAFV600E specific antibody (E19290) was from Spring Bioscience (Pleasanton, CA). Anti-rabbit and anti-mouse immunofluorescent secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE). SMARTpool siGENOME siRNA targeting XLF (NHEJ1) was purchased from Dharmacon Inc. (Lafayette, CO) and 25 nM siRNA was transfected into cells by TransIT-X2 Transfection Reagent according to the manufacturer’s protocol (Mirus Bio, Madison, WI). XLF (NHEJ1) cDNA plasmid was obtained from Genscript (clone OHu23739).
Immunoblotting
Immunoblotting was performed as described previously (33). Briefly, cell lysates were prepared using RIPA buffer (ThermoFisher, Waltham MA) supplemented with 1x protease (Complete, Roche, Indianapolis, IN) and phosphatase inhibitors (PhosSTOP, Roche, Indianapolis, IN, Roche) followed by protein quantification by the Dc protein assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein were loaded and resolved by SDS/PAGE and transferred onto nitrocellulose membranes. Membranes were incubated in 5% BSA in TBS-Tween blocking buffer for 1 hour at room temperature. Primary antibodies were allowed to bind overnight at 4̊C, and used at a dilution of 1:200-1000. After washing in TBS-Tween, the membranes were incubated with immunofluorescent secondary antibodies at a 1:5000 dilution for 1 hour at room temperature. Membranes were washed with TBS-Tween and allowed to air dry prior to imaging via LI-COR Odyssey® CLx Imaging System.
Radiation clonogenic assay
Cells were trypsinized to generate single cell suspensions and seeded onto 60 mm or 100 mm tissue culture plates in triplicate essentially as described (33). Cells were incubated with DMSO or vemurafenib for 3 hours then irradiated with various doses (0-8 Gy). Twenty-four hours after radiation, medium was replaced with fresh medium without DMSO or vemurafenib. Seven to ten days after seeding, colonies were fixed with Methanol/Acetic Acid, and stained with 0.5% crystal violet. The numbers of colonies or colony forming units (CFU) containing at least 50 cells were counted using a dissecting microscope (Leica Microsystems, Inc. Buffalo Grove, IL) and surviving fractions calculated. Experiments were repeated for multiple, independent times.
Cellular proliferation assay
alamarBlue® proliferation assay was performed according to manufacturer’s instructions (Bio-Rad Antibodies, Oxford, UK). Briefly, cells were seeded in 96-well plates in 6 replicates at a density of 1,000-2,000 cells per well in 100 µL medium (Day 0). At various days after plating, alamarBlue® reagent was added and incubated at 37°C for 4 hours, and absorbance was measured at 570 and 600 nm.
Experimental radiation
Irradiation was performed essentially as described previously with 160kV, 25mA at a dose rate of approximately 113cGy/min using a RS-2000 biological irradiator (RadSource, GA) (31).
Production of stable BRAFV600E clones
TPC-1 cells were transfected with empty pBabe-puro plasmid (#1764, Addgene) or pBabe-puro-BRAF-V600E plasmid (#15269, Addgene) using TransIT-2020 Transfection Reagent per manufacturer’s protocol (Mirus Bio, Madison, WI). Cells were seeded in 6-well plates at a density of 2.5×105 cells/ml and cultured overnight. To prepare the TransIT-2020:DNA complexes, 250 μl of OptiMEM I Reduced-Serum Medium, 2.5 μl of plasmid DNA (1µg/μl) and 7.5 µl of TransIT-2020 were mixed in a sterile tube by pipetting. The complex mixtures were allowed to incubate at room temperature for 30 minutes and then added to the cells. After 48 hours of incubation, cells were harvested and cultured overnight. Cells were treated with 0.5 µg/ml of puromycin to select for stable clones. Single stable clone colonies were picked using sterile cloning cylinders and seeded separately in 12-well plates. Each clone was cultured and expanded once confluent. Stable transfection was verified by immunoblotting using a BRAFV600E specific antibody.
Neutral comet assay
Cells were treated with DMSO or vemurafenib for different time points. Neutral comet assay was performed as previously described.(28)
Immunofluorescence for nuclear foci
For γ-H2AX, RAD51, and 53BP1, immunofluorescence for nuclear foci was performed as previously described.(28)
Flow cytometry DNA repair reporter assays
Using TransIT-2020 Transfection Reagent (Mirus), 293T cells were transiently transfected either empty pBabe-puro plasmid or pBabe-puro-BRAF-V600E plasmid. Cells were co-transfected with pDR-GFP plasmid previously characterized (34) or NHEJ-GFP-PEM1 plasmid (generously provided by V. Gorbunova) in order to measure homologous recombination (HR) and non-homologous end-joining (NHEJ) activity respectively. All cells were also transfected with pLVX-mCherry-C1 as a transfection control. Cells were treated with adenovirus expressing the I-SceI restriction enzyme that induces double-strand DNA breaks in the recognition sequence within the reporter construct. Cells were also pretreated with DMSO or vemurafenib using different variations in scheduling as described. Then reporter assays were performed as previously described.(28) Repair efficiency was expressed as the ratio of percent GFP+ cells over percent mCherry+ cells.
In vivo studies
Animal studies were conducted in accordance with an approved protocol adhering to the IACUC policies and procedures at The Ohio State University. Six to eight week-old male athymic nude mice (Taconic Farms Inc., NY) were caged in groups of five or less, and fed a diet of animal chow and water ad libitum. 8505C and BCPAP cells were injected subcutaneously into the flanks of each mouse at 2 × 106 and 2.5 × 106 cells per injection, respectively. Treatment regimens were started once tumors reached ~150mm3 in size, 2-4 weeks post-injection. Vemurafenib powder (Active Biochem) was suspended in 0.9% sodium chloride containing 5% dextrose. Vemurafenib was administered orally using a sterile 18G gavage needle at 50 mg/kg b.i.d. for 5 consecutive days. Using a custom shielding apparatus to block non-targeted areas, 4 Gy of radiation was administered directly to tumors once daily for 5 consecutive days. For combination treatment, mice were treated with radiation 2-3 hours after the daily dose of vemurafenib. To obtain a tumor growth curve, perpendicular diameter measurements of each tumor were measured every 2-5 days from the first day of injection with digital calipers, and volumes were calculated using the formula (L × W × W)/2.
Data analysis
For in vitro experiments, data are presented as the mean ± standard error of the mean (s.e.m.) for clonogenic survival and foci experiments. Statistical comparisons were made between the control and experimental conditions using the two-sided two group t-tests with significance assessed at p-values <0.05.
Correlation between BRAFV600E mutation and radiosensitivity index (RSI): The BRAF mutation status was downloaded from http://www.cbioportal.org/ under the mutation data of well-differentiated thyroid cancer (TCGA, THCA Provisional) named “Tumor samples with mRNA data (RNASeq V2) (n=509). The RSI was calculated based on the following formula using Thyroid cancer TCGA mRNA (RNASeq V2) gene expression data:
RSI=−0.0098009*AR+0.0128283*Jun+0.0254552*STAT1-0.0017589*PRKCB-0.0038171*RELA+0.1070213*ABL1-0.0002509*SUMO1-0.0092431*CDK1-0.0204469*HDAC1-0.0441683*IRF1 (35,36). Note, the RSI value predicts the survival fraction at 2Gy, thus it is inversely correlated with tumor’s radiosensitivity (i.e. lower RSI value with higher radiosensitivity to external radiation). The correlation between BRAFV600E mutation and RSI was tested with two sample t-test. Among TCGA THCA tumor samples, 235 had BRAFV600E mutation and 261 had wild type (WT) BRAF, so data from 496 patients was used to test the correlation between BRAF mutation and RSI.
Correlation between BRAFV600E mutation and XLF expression: Data for BRAF mutations and XLF expression was downloaded from UCSC Xena data portal (https://xena.ucsc.edu/ accessed 1/18/18) for primary tumor samples from TCGA THCA dataset. XLF expression was reported as log2 (normcount+1). Patients with wild-type and BRAFV600E mutations were included (patients with other BRAF mutations were eliminated). The correlation between XLF expression and BRAF mutation was tested with a linear model. Data was available for 288 patients with BRAFV600E mutation and 203 wild type (WT) BRAF tumors for which XLF expression was available.
RESULTS
BRAFV600E mutation leads to resistance of thyroid cancer cells to ionizing radiation
To assess whether BRAFV600E mutation is correlated with radioresistance of thyroid cancer cells, the cells of BRAF wild type (BRAFWT: U-Hth-74, TPC-1) or V600E mutation (BRAFV600E: 8505C, BCPAP, SW-1736) thyroid cancer cell lines were treated with increasing doses of ionizing radiation (IR) followed by colony formation (clonogenic) assay. Compared to BRAFWT cell lines, there was a relatively higher surviving fraction of thyroid cancer cells with BRAFV600E in response to IR (Fig. 1A). To confirm this observation, stable TPC-1 cells expressing BRAFV600E (V600E) or empty vector control (EMP) were established (western blot inset, Fig. 1B), and subjected to radiation clonogenic assay. We found that expression of BRAFV600E in BRAF wild type TPC-1 stable cells led to marked radioresistance (Fig. 1B), without significant changes in proliferation (Supplementary Figs. 1–2). Finally, we evaluated whether BRAFV600E thyroid tumors in the TCGA database are predicted to be radioresistant using a validated RNA-based molecular signature predicting relative radiosensitivity across tumor cells, the radiosensitivity index (RSI)(35,36), where higher RSI is associated with radioresistance. In support of our preclinical data, BRAFV600E thyroid cancers are generally predicted to be more radioresistant than BRAF wild type thyroid cancer (Fig. 1C–D). The mean calculated RSI for tumors with BRAFV600E mutation (n=235) is 0.7216 +/− 0.1324, while the mean calculated RSI for tumors with WT BRAF (n=261) is 0.6945 +/− 0.1264 with (p=0.0203). Furthermore, we examined radiation sensitivity data from a collection of over 500 cell lines from Yard et al. (37), and pooled radiation sensitivity data from the top three BRAFV600E expressing tumor types that contained more than one BRAF mutant cell line (thyroid, melanoma, and large bowel/colorectal). We found that BRAFV600E mutant cell lines were associated with radioresistance compared to cells lacking BRAFV600E (Supplementary Figure 3). Taken together, these results support that BRAFV600E mutation contributes to radioresistance of thyroid cancer cells (and potentially other tumor types).
Figure 1. Thyroid cancer cells harboring BRAFV600E are resistant to ionizing radiation therapy.
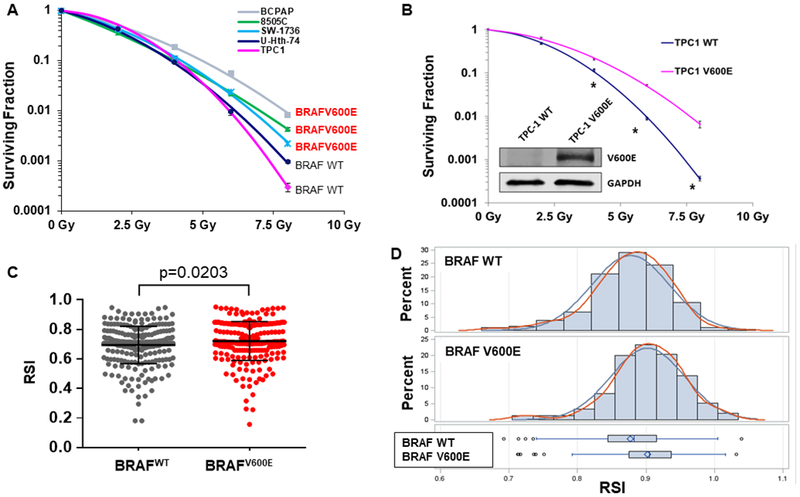
(A) Radiation clonogenic assays from a panel of thyroid cancer cell lines with BRAF wild type (U-Hth-74, TPC1) or V600E mutation (8505C, BCPAP, SW-1736). (B) Exponentially growing stable TPC-1 cells expressing BRAF V600E (V600E) or empty vector control (EMP) were immunoblotted for BRAFV600E (with V600E specific antibody) and β-ACTIN. Radiation clonogenic assays of TPC-1 V600E and TPC-1 control vector (EMP) cells. Each dose was prepared in triplicate per experiment, and no less than 2 experiments were performed per cell line. *p<0.05. (C-D) Plot (C) or distribution bar graph (D) of BRAFV600E status with radiosensitivity index (RSI) using TCGA thyroid cancer data, showing that BRAFV600E tumors are relatively radioresistant with a higher mean RSI value (p=0.0203). Standard error of the mean (s.e.m.) shown for 1C.
BRAFV600E inhibition selectively radiosensitizes BRAFV600E thyroid cancer cells
Vemurafenib is a potent BRAFV600E mutant kinase specific inhibitor (BRAFi) and inhibits ERK signaling in BRAFV600E mutant cells with minimal effect in BRAFWT cells (10). Consistently, vemurafenib reduced pMEK and pERK½ in 8505C and BCPAP cells with BRAFV600E mutation in a concentration-dependent manner after 3 hours of treatment (Fig. 2A). In contrast, treatment of BRAFWT TPC-1 and U-Hth-74 cells with vemurafenib did not alter the phosphorylation of MEK (pMEK) and ERK½ (pERK½) except at perhaps higher doses of BRAFi (e.g. mild decrease at 500 nM), consistent with sustained MEK and ERK activation. We postulated that vemurafinib would sensitize BRAFV600E thyroid cancer cells to radiotherapy by reversing BRAFV600E –induced radioresistance. To test this hypothesis, BRAFWT U-Hth-74 and TPC-1 cells, and BRAFV600E 8505C and BCPAP cells were pretreated with 100 nM BRAFi for 3 hrs and then exposed to different doses of IR, followed by radiation clonogenic assays. We found that BRAFi treatment significantly sensitized BRAFV600E 8505C and BCPAP cells, but not BRAFWT U-Hth-74 and TPC-1 cells to IR (Fig. 2B, Supplementary Fig. 4). Differences in survival were unlikely related to toxicity of vemurafenib, as plating efficiency/colony formation in the absence of radiation was virtually unaffected by vemurafenib (Supplementary Fig. 5). We extended our analysis to other BRAFV600E mutant cell lines, A375 and HS294T melanoma cells, and likewise showed that BRAFi caused marked radiosensitization (Supplementary Fig. 6). Our results showed that inhibition of BRAFV600E mutant kinase with vemurafenib reversed the radioresistance of thyroid cancer (and melanoma) cells harboring a BRAFV600E mutation.
Figure 2. BRAFV600E inhibition selectively radiosensitizes thyroid cancer cells harboring a BRAFV600E mutation.
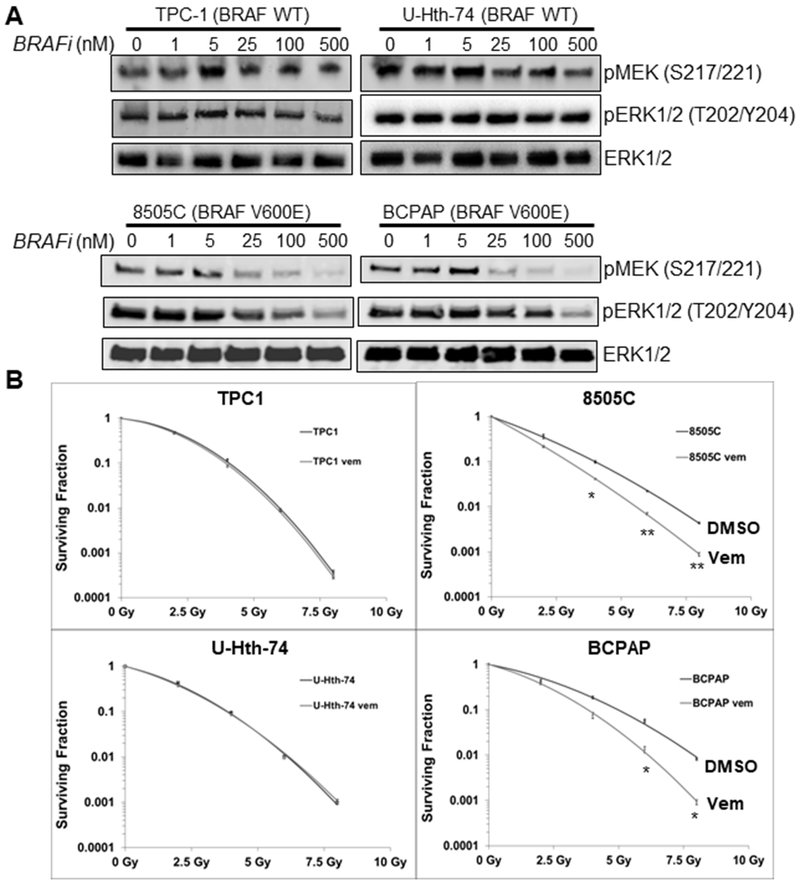
(A) Cells were treated with 0 (0.1% DMSO), 1, 5, 25, 100 and 500 nM vemurafenib (BRAFi) for 3 hrs and total proteins were extracted for immunoblotting of pMEK½, pERK½ and total ERK½. (B) Cells were cultured in media containing 100 nM vemurafenib (Vem) 3 hrs prior to irradiation with 0 (no IR), 2, 4, 6 and 8 Gy doses, followed by radiation clonogenic survival assays. Each dose was prepared in triplicate per experiment, and no less than 2 experiments were performed per cell line. s.e.m. shown. *p<0.05, **p<0.01.
BRAFi treatment results in attenuated DNA repair in BRAFV600E mutant thyroid cancer cells in response to ionizing radiation
Ionizing radiation (IR) induces DNA double strand breaks (DSBs), which result in rapid activation of the ATM-CHK2 checkpoint to arrest the cell cycle and promote repair machineries to restore the damaged DNA. If left unrepaired, IR-induced DSBs results in cell death (25,26). Phosphorylation of histone H2AX (γH2AX) is a marker of DNA damage and its dynamics reflects the processes of DNA damage production and repair. To assess the role for BRAFV600E mutation in DNA damage response (DDR) in thyroid cancer cells, 8505C and BCPAP cells were pretreated with 100 nM BRAFi for 3 hrs and then exposed to 4 Gy IR. At different time points after IR, γH2AX were assessed by immunoblotting. In both 8505C and BCPAP cells without BRAFi treatment, there was strong γH2AX induction as expected at 15 min after IR, and γH2AX expression recovered to basal levels between 6-24 hrs after IR, indicative of the repair of IR induced DNA damage. However, under the presence of BRAFi, there was persistence of γH2AX expression even 24 hrs after IR, indicative of persistent presence of DNA damage (Fig. 3A). To confirm this observation, we performed immunofluorescence analysis of γH2AX nuclear foci of these cells after IR in the presence or absence of BRAFi. As expected, γH2AX foci were formed in the nuclei of 8505C and BCPAP cells soon after radiation (15 min). γH2AX signals gradually decreased with time, indicative of recovery from DNA damage, but there were significantly higher levels of γH2AX foci in cells treated with BRAFi than those without BRAFi at 6 and 24 after IR (Fig. 3B). Conversely, changes in γH2AX were absent in BRAFWT cells in similar immunoblotting and immunocytochemistry assays (Supplementary Figure 7).
Figure 3. BRAFV600E inhibition prevents the recovery of IR-induced DNA damage in BRAFV600E mutant thyroid cancer cells.
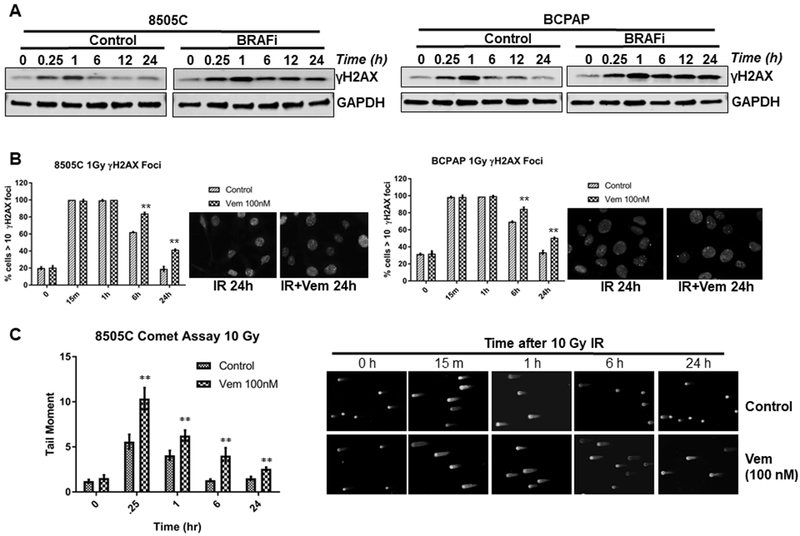
(A) BRAFV600E mutant 8505C and BCPAP cells were cultured in media containing 100 nM vemurafenib (BRAFi) 3 hrs prior to 4Gy dose of irradiation. At the indicated time points in hours following IR, cell lysates were made for immunoblotting of γ-H2AX and GAPDH. The 0 time point indicates no IR, but treated with 100 nM vemurafenib for 3 hrs. (B) BRAFV600E mutant 8505C and BCPAP cells were cultured in media containing 100 nM vemurafenib 3 hrs prior to 1Gy dose of irradiation. At the indicated time points following IR, the cells were prepared for immunofluorescence analysis of γ-H2AX nuclear foci. Mean percentage of cells with greater than 10 γ-H2AX foci and s.e.m. at each time point after IR from two experiments of no less than 100 cells per condition are shown. Representative γ-H2AX foci of the control and vemurafenib treated at 24 h post-IR are shown. (C) DNA damage was quantified as the mean tail-moment taken from no less than 75 cells at different time points relative to 10 Gy in 8505C cells treated with/without 100 nM vemurafenib (Vem). Representative set of photomicrographs of control and vemurafenib treated comets at 24 h post-IR are shown. **p<0.01.
Additionally, comet assay is a sensitive technique for detecting DNA damage at the level of individual cells (38). To test if BRAFi alters the global DNA repair process in irradiated thyroid cancer cells, neutral comet assays were conducted in 8505C cells after 10 Gy IR in the presence or absence of BRAFi. At each time point after IR, there was persistently higher tail moment in the cells treated with BRAFi than those without BRAFi, suggestive of attenuated DNA repair (Fig. 3C). Taken together, these results demonstrate that inhibition of BRAFV600E delays global DNA repair in BRAFV600E mutant thyroid cancer cells.
BRAFi suppresses BRAFV600E-promoted NHEJ repair in thyroid cancer cells
DNA double strand breaks (DSBs) are repaired by multiple DNA repair pathways, including non-homologous end joining (NHEJ) and homologous recombination (HR). Enhanced activity of NHEJ and HR repair pathways play an important role in the resistance of cancer cells to radiotherapy and DNA-damaging based chemotherapy (25,26). Moreover, deregulation of cell growth signaling pathways have been shown to maintain cancer cell survival by promoting DNA damage repair at the cost of increased mutation risk (39–41). DSBs induce a rapid co-localization of p53-binding protein 1 (53BP1) with γH2AX and other DNA repair proteins including RAD51 (a component of HR repair), which can be visualized as discrete nuclear foci (39–41). We determined the effects of BRAFi on radiation-induced 53BP1 and RAD51 foci formation in 8505C and BCPAP cells. Numerous 53BP1 foci were formed in the nuclei of 8505C and BCPAP cells after IR, and these foci were resolved with time as expected; however in the presence of BRAFi, the decrease of 53BP1 foci was significantly impaired during the recovery phase after IR, i.e, at 6 and 24 hrs after treatment (Fig. 4A). Nuclear RAD51 foci increased after IR up to 6-24 hrs after IR as expected. However in the BRAFi treated cells, there was significantly increased number of RAD51 foci particularly at 6 and 24 hrs (Fig. 4B). To more directly determine whether BRAFi induces radiosensitivity of BRAFV600E cells by altering HR and/or NHEJ repair, fluorescent reporter constructs that allow sensitive and quantitative measurement of HR (DR-GFP) or NHEJ repair (Pem1-NHEJ) were transfected in 293T cells. These constructs contain an engineered GFP gene with recognition sites for I-SceI endonuclease for induction of DSBs, which does not express GFP in the absence of repair of these I-SceI-induced DSBs. Proper repair of DSBs in the recognition sites results in GFP expression, and the number of GFP positive cells can be counted by flow cytometry (42). We co-transfected 293T cells with either Pem1-GFP or DR-GFP reporters, with (empty vector) or without BRAFV600E, and tested these cells with or without BRAFi to determine the effects of BRAFV600E inhibition on HR and NHEJ repair capability. Expression of BRAFV600E and subsequent MEK-ERK activation is shown in Supplementary Figure 8. We found that expression of BRAFV600E did not significantly alter HR repair (Fig. 4C, middle panel), and BRAFi only slightly reduced HR repair (but without statistical significance). In the NHEJ repair assay, however, we found that expression of BRAFV600E significantly increased NHEJ mediated-repair, which was significantly attenuated by BRAFi (Fig. 4C, right panel), suggesting that BRAFV600E mutation imparts accelerated DNA repair through an increase in NHEJ repair activity.
Figure 4. BRAFV600E promotes NHEJ repair, which is attenuated by BRAF inhibition.
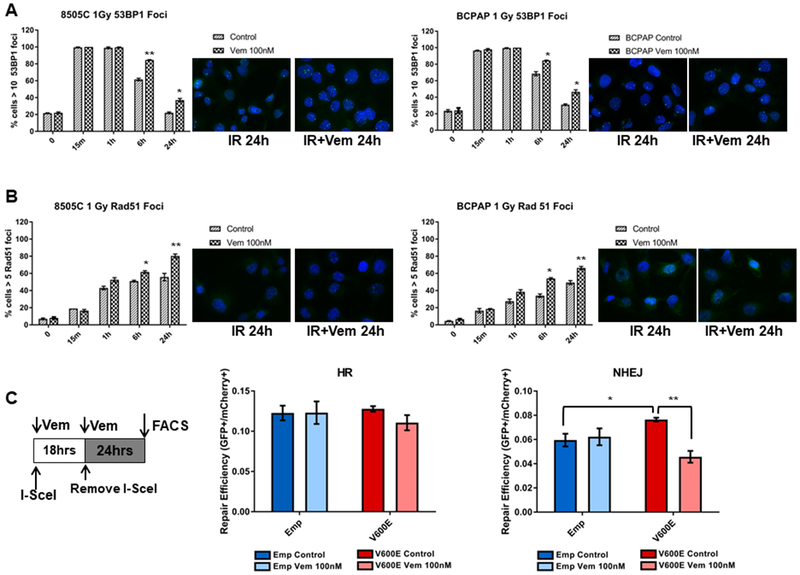
(A-B) BRAFV600E mutant 8505C and BCPAP cells were cultured in media containing 100 nM vemurafenib 3 hrs prior to 1 Gy of IR. At the indicated time points following IR, the cells were prepared for immunofluorescence analysis of 53BP1 (A) and RAD51 (B) foci. Mean percentage of cells with greater than ten 53BP1 foci and greater than five Rad51 foci at each time point after IR are shown (with s.e.m.). Data were calculated from two experiments of no less than 100 cells per condition. Representative set of images of control and vemurafenib treated at 24 h post-IR are shown. (C) Functional DNA repair reporter assays of HR (middle panel) and NHEJ (left panel)-mediated DSB repair efficiency. 293T cells were co-transfected with HR-GFP or NHEJ-GFP reporter plasmids and either empty backbone plasmid (EMP) or BRAFV600E plasmid (V600E), as well as a transfection control plasmid containing mCherry. 24 h after transfection, media (+/− vemurafenib) containing I-SceI-adenovirus was added and allowed to incubate for 18 h. Media was then replaced without adenovirus and allowed to incubate for an additional 24 h before harvesting cells for flow cytometry. Repair efficiency was calculated as the fraction of GFP positive cells divided by the fraction of mCherry positive cells. *p<0.05, **p<0.01.
BRAFV600E mutation leads to upregulation of XLF expression in thyroid cancer cells
To explore the potential mechanisms of the up-regulation of NHEJ by BRAFV600E mutation, we analyzed the thyroid cancer (THCA) TCGA database for the gene expression status of the components of NHEJ and the correlation with BRAFV600E mutation. We found higher levels of XLF RNA expression in the presence of a BRAFV600E mutation (7.28 vs 7.06 log normalized counts, fold-change=1.17, p=2.49e-14) (Fig. 5A), as well as other NHEJ associated genes (Supplementary Table 2). To more directly determine whether BRAFV600E affects XLF expression, we compared XLF protein expression by immunoblotting between BRAFWT (TPC-1 and Hth-74) and several BRAFV600E (8505C, BCPAP, SW1736) thyroid cancer cell lines, and observed an increase of XLF in BRAFV600E thyroid cancer cells at both the protein and mRNA levels (Fig. 5B). In support of our findings, treatment of BRAFV600E thyroid cancer cells with BRAFi led to reduction of XLF mRNA and protein (Fig. 5C). As shown previously, expression of BRAFV600E in BRAFWT TPC-1 cells led to radioresistance in clonogenic assay (Fig. 1C). Ectopic expression of BRAFV600E in these cells resulted in up-regulation of XLF (Fig. 5D). Furthermore, the radioresistance induced by BRAFV600E in TPC-1 cells could be attenuated by BRAFi (Fig. 5D). Consistent with pharmacologic inhibition of BRAFV600E, genetic silencing of XLF by RNA interference (siRNA) effectively radiosensitized TPC-1-BRAFV600E cells with minimal effect on TPC-1 parental cells (Fig. 5E). Importantly, knockdown of XLF by siRNA in BRAFV600E 8505C cells also resulted in radiosensitization (Fig. 5F). Furthermore, we transfected XLF cDNA into BRAF wild-type TPC-1 cells and assessed radiosensitivity with a radiation clonogenic assay. We found that XLF expression was sufficient to confer increased radioresistance (Supplementary Figure 9). These results suggest that up-regulation of XLF may at least in part contribute to the enhancement of NHEJ-mediated repair of DSBs and concomitant radioresistance of BRAFV600E thyroid cancer cells.
Figure 5. BRAFV600E mutation enhances NHEJ by increasing XLF expression in thyroid cancer cells.
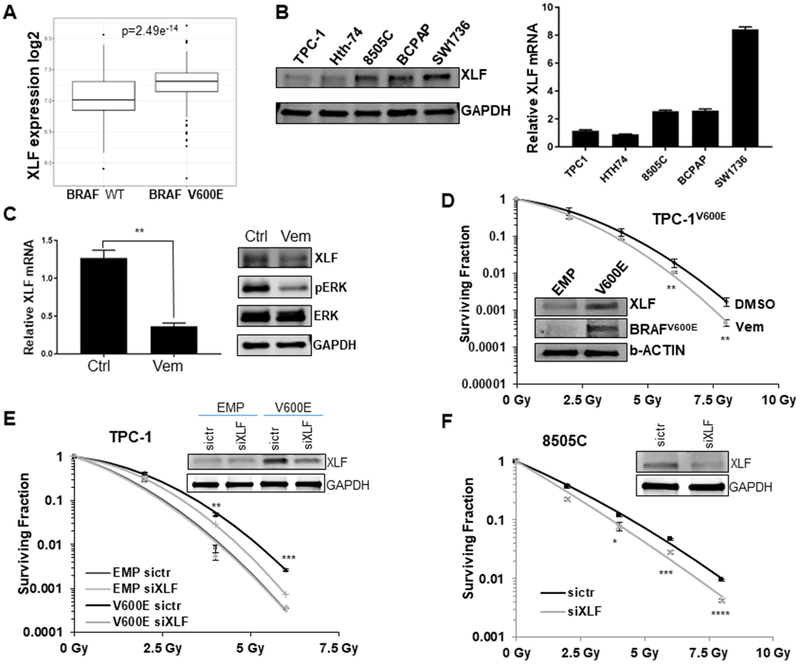
(A) Correlation of XLF expression with BRAFV600E mutation in TCGA thyroid cancer (THCA) data. Box plot showing the relationship between XLF expression and BRAFV600E mutation status. Expression was measured from primary tumor and is reported as log2 of the normalized count plus 1. A linear model shows a significant association (p=2.4e−14) of XLF expression with BRAFV600E mutation status in thyroid cancer. (B) A panel of thyroid cancer cell lines with BRAF wild type (U-Hth-74, TPC1) or V600E mutation (8505C, BCPAP, SW-1736) were analyzed for XLF protein by immunoblotting (left) and XLF mRNA levels by quantitative RT-PCR (right). (C) Exponentially growing BCPAP cells were treated with 100 nM vemurafenib or vehicle control (Ctrl) and analyzed for XLF mRNA levels by quantitative RT-PCR (left) and XLF protein by immunoblotting (right). **p<0.01 Vem vs. Ctrl. (D) TPC-1 cells expressing BRAF V600E (TPC-1 V600E) were cultured in media containing 100 nM vemurafenib (Vem) 3 hrs prior to irradiation with 0 (no IR), 2, 4, 6 and 8 Gy doses, followed by radiation clonogenic survival assays. Inset shows the upregulation of XLF and BRAFV600E in TPC-1 V600E cells by immunoblotting. **p<0.01 Vem vs. DMSO. BRAFV600E specific antibody confirms V600E expression. (E) Stable TPC-1 cells expressing BRAFV600E (TPC-1 V600E) or empty vector control (TPC-1 EMP) were transfected with siRNA of control (sictr) or XLF (siXLF) for 24 hrs. Then the cells were irradiated with 0 (no IR), 2, 4, 6 and 8 Gy doses, followed by radiation clonogenic survival assays. Inset shows the knockdown of XLF in TPC-1 V600E cells by immunoblotting with GAPDH as loading control. **p<0.01, ***p<0.001 V600E sictr vs. V600V siXLF. (F) BRAFV600E mutant 8505C cells were transfected with siRNA of control (sictr) or XLF (siXLF) for 24 hrs. Then the cells were irradiated with 0 (no IR), 2, 4, 6 and 8 Gy doses, followed by radiation clonogenic survival assays. Inset shows the knockdown of XLF in 8505C cells by immunoblotting with GAPDH as loading control. Each dose was prepared in triplicate per experiment, and no less than 2 experiments were performed. *p<0.05, ***p<0.001, ****p<0.0001
BRAFi treatment radiosensitizes BRAFV600E mutant thyroid tumors in vivo
To translate our findings, we tested whether BRAF inhibition could radiosensitize BRAFV600E thyroid cancer cells. When tumors of 8505C and BCPAP cell lines reached about 100-200 mm3, the mice were randomized to be treated with vehicle or BRAFi (vemurafenib delivered by oral gavage, 50 mg/kg × 5 consecutive days), and/or radiation (4 Gy × 5 consecutive days). In the group of mice treated with concurrent BRAFi and radiation, BRAFi was dosed 2-3 hrs before radiation. BRAFi alone led to tumor growth delay, but resulted in rapid re-growth of tumors after discontinuation of BRAFi. Radiation was effective in halting tumor growth for a period of time, but tumors almost universally recurred. However, BRAFi in combination with radiation led to marked and sustained regression of the tumors in multiple BRAFV600E mutant xenograft models (log-rank p<0.0001, Fig. 6A–B), and longer tumor-doubling rates (log-rank p=0.0057, Fig. 6C–D). In terms of tolerance, treatment with BRAF inhibitor did not result in significant weight loss (Supplementary Figure 10). While mice treated with radiation (with or without BRAFi) experienced up to 7-8% weight loss 1 week within the start of radiation, they recovered to their baseline weight by ~2 weeks after therapy was complete, and no significant clinical toxicity was noted.
Figure 6. Vemurafenib in combination with radiotherapy results in sustained tumour regression of BRAFV600E thyroid tumor xenografts.
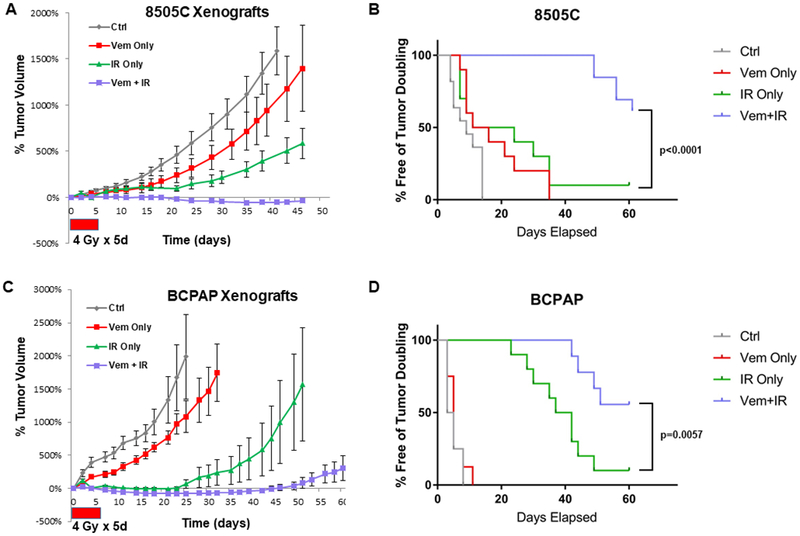
Mice were injected with 2×106 8505C or BCPAP cells in the left flank. Each treatment was started once tumors reached 100-200 mm3. Vemurafenib was administered via oral gavage at 50 mg/kg BID for 5 days (control group received vehicle at same intervals). Irradiation was administered at 4 Gy for 5 days. Tumor growth curves and mice free of tumor doubling in 8505C xenograft tumors (A-B, log-rank p<0.0001 Vem+IR vs. IR Only), or BCPAP xenograft tumors (C-D, log-rank p=0.0057 Vem+IR vs. IR Only) were plotted.
DISCUSSION
In this study, we demonstrate that for the first time that BRAFV600E directly promotes radiation resistance through heightened nuclear DNA double-strand break repair, and that a potent BRAF inhibitor suppresses the resistance of BRAFV600E thyroid cancer cells to radiotherapy. We further showed that BRAFV600E thyroid cancer cells displayed an increase of NHEJ activity with up-regulation of XLF at the protein and RNA level, a key component of NHEJ. Importantly, BRAF inhibition in combination with radiotherapy led to a marked and sustained tumour regression of BRAFV600E thyroid tumor xenografts. Our studies provide the basis for clinical testing of combinatorial radiotherapy (RT) and BRAF inhibitor treatment modalities for thyroid cancers, particularly BRAFV600E mutated ATC.
Another study has demonstrated the efficacy of combining BRAF inhibitors in combination with radiation for BRAF mutant tumors. Dasgupta et al. showed in high grade glioma preclinical models that BRAFi and RT showed greater anti-tumor effects than either treatment alone in BRAFV600E (but not BRAF wild-type) lines.(43) They noted that the combination treatment increased apoptotic cell death, decreased Ki-67 and phospho-MAPK signaling, and increased γH2AX compared to control tumor cells. Our study joins this one in demonstrating the efficacy of combined RT and BRAFi for BRAFV600E tumors.
Among the different subtypes of thyroid cancer, ATC has the worst prognosis with an overall survival at 2 years of 10% and mortality of ~95-100%, making it one of the most lethal solid tumors (44). Recent genomic profiling of ATC has revealed high frequency mutations of the RAS-ERK signaling pathway, which is well-characterized as important for tumor cell proliferation, aggressiveness, and response to therapy (15). Oncogenic mutations in BRAF are the most common amongst the RAS-ERK pathway genes, occurring in ~45% of ATC with the vast majority representing BRAFV600E activating mutations (>95%).(14,15,17,18) BRAF mutations are also commonly found in differentiated thyroid cancers, particularly in 60% of PTC, and have been linked to radioactive iodine resistance (16,45–47). In addition, RAS mutations occur in an additional 20-25% of ATC patients (NRAS 15-20%, HRAS 5%, KRAS 5%), and are mutually exclusive with BRAF mutations (14,15,18). Thus, BRAF represents an attractive target for BRAF mutant thyroid cancers. However, clinical trials have shown that BRAFV600E inhibitors as monotherapy, including BRAFi, are not curative for cancer patients with BRAFV600E, and resistance emerges due to paradoxical activation of RAS-ERK signaling (19–22). Even though vertical targeting of RAS-ERK bypass resistance mechanisms through dual targeting of BRAF and MEK kinases improves the survival of cancer patients with BRAFV600E mutation, the tumors still relapse at least partially due to paradoxical RAS-ERK activation, similar to tumors treated with BRAF inhibitors alone (6). Our preclinical data suggests that BRAFV600E promotes intrinsic radioresistance in thyroid cancer cells and that subversion of oncogene-mediated radioresistance leads to the marked efficacy of the combination of BRAFi and IR in BRAFV600E xenograft models, with profound delays in tumor growth without significant toxicity. Regarding that radioresistance is one of the major mechanisms of the high rate of disease failure of ATC, our findings suggest that targeting BRAFV600E in parallel with radiation is a novel strategy for the management of ATC patients.
RAS mutations have been previously shown to promote preclinical radioresistance in various tumor types, and targeted inhibition of downstream MEK-½ can effectively radiosensitize RAS mutant tumors as shown by our group and others (27–31). Furthermore, RAS mutations have recently been associated with clinical radioresistance in multiple tumor types (48,49). In the present study, we found that BRAFV600E promotes intrinsic radioresistance in thyroid cancer cells through accelerated NHEJ repair activity, which was prevented by BRAFi in both BRAFV600E mutation thyroid cancer cell lines and an isogenic thyroid cancer cell line stably expressing BRAFV600E. Together, it seems that up-regulation of DNA repair capacity by oncogenic proteins such as BRAF and RAS mutations may be a common mechanism by which deregulation of cell growth and survival pathways promotes not only tumorigenesis but therapeutic resistance.
Ionizing radiation causes DSBs, which result in mitotic catastrophe and cell death if left unrepaired (25,26). DSBs are repaired by NHEJ as well as HR. HR repair is restricted mostly to S and G2 phases of the cell cycle, and is a higher fidelity process. NHEJ repair is more error-prone and operates throughout the cell cycle. NHEJ is activated by Ku70/Ku80 sensor proteins, which subsequently activate DNA-PK catalytic subunit to recruit effector proteins to DSB sites, including XLF (NHEJ1) (25,26). XLF, in turn, interacts with XRCC4 and DNA ligase IV and may be involved in the end-bridging or ligation steps of NHEJ. It has been recently established that BRAF and RAS mutations contribute to tumorigenesis and tumor progression of thyroid cancers (14). However, the role of BRAF and RAS mutations in radiotherapy and other genotoxic therapy response is largely unknown, particularly for BRAF mutations. We revealed that BRAFV600E mutation led to radiation resistance through up-regulation of NHEJ, which may in part be mediated by XLF up-regulation, and XLF expression was decreased by a BRAFV600E inhibitor. Thus, the upregulation of XLF-mediated NHEJ repair may be a contributing factor to the radioresistance observed in BRAFV600E thyroid cancer cells, and inhibition of XLF or other NHEJ activity may represent alternative therapeutic strategies. Nevertheless, in the future, it will be important to more thoroughly elucidate the molecular mechanisms of the up-regulation of XLF by BRAFV600E mutation in thyroid cancer as well as whether this is an important mediator of radioresistance in other RAS and BRAF mutated cancers (e.g. melanoma, colorectal cancer, non-small cell lung cancer, pancreatic cancer).
The discovery of oncogene addiction of a subset of cancers to BRAF mutations has triggered the development of a wave of RAF kinase inhibitors for cancer therapeutics. More recently, the rapid appearance of RAF kinase inhibitor resistance has led to the combination of RAF inhibitors with other targeted drugs, and development of third generation RAF kinase inhibitors (6). Multiple mechanisms contribute to the resistance to BRAFV600E inhibitors, including paradoxical activation of RAS-ERK signaling as mentioned above (50–52). Consequently, it has been shown that vertically targeting RAS-ERK signaling by simultaneous inhibition of BRAFV600E and MEK kinases improves clinical efficacy of both thyroid and melanoma with BRAFV600E in comparison to BRAFV600E inhibitors as monotherapy (3,11,12). In addition, activation of PI3K-AKT-mTOR signaling and modulation of the cell apoptosis cascade play important roles in resistance to BRAFV600E inhibitors (50–52). One of the pivotal roles of PI3K-AKT-mTOR signaling is the control of protein translation via regulating eIF4F complex formation. Intriguingly, it was observed that the persistent formation of eIF4F complex contributes to BRAFV600E inhibitors resistance (53). It is possible that the combination of BRAFV600E inhibitors with PI3K-AKT-mTOR signaling inhibitors or apoptosis agonists such as IAP antagonists may reverse the resistance of BRAFV600E driving cancers to BRAFV600E inhibitors, and thereby improve overall clinical outcomes.
In summary, we demonstrate for the first time that BRAFV600E promotes the radioresistance of thyroid cancer cells by enhancing NHEJ-mediated double strand break repair activity, which may be due in part to XLF up-regulation. More importantly, BRAFV600E radioresistance is abrogated by BRAFV600E inhibitor both in cell culture and mouse xenografts. Our findings suggest that upregulation of DNA damage repair is one of the mechanisms by which BRAFV600E exerts therapeutic resistance. Taken together, these results provide strong rationale for clinical testing of radiotherapy and BRAF inhibitors for BRAFV600E mutant thyroid cancers.
Supplementary Material
TRANSLATIONAL RELEVANCE.
We herein evaluated whether BRAFV600E mutation induces radioresistance and whether BRAFV600E selective inhibitor vemurafenib could sensitize cells to radiation using in vitro and in vivo models of thyroid cancer. Our work revealed that BRAFV600E thyroid cancer cell lines are resistant to ionizing radiation, which is accompanied by increased DNA double strand break repair capacity. We uniquely identified that BRAFV600E mutation leads to upregulation of XLF and enhanced activity of non-homologous end-joining repair in thyroid cancer cells following ionizing radiation. Most importantly, we discovered that vemurafenib selectively radiosensitized BRAFV600E mutants but not BRAF wild-type thyroid cancer cells. Our study demonstrates that BRAFV600E mutation promotes DNA damage repair leading to radioresistance, and pharmacological inhibition of BRAFV600E selectively radiosensitizes BRAFV600E thyroid cancer cells. Our findings suggest that combining vemurafenib and radiation may improve the therapeutic outcome of BRAFV600E mutant thyroid cancer patients, providing a novel strategy for managing thyroid cancer patients with BRAFV600E.
ACKNOWLEDGMENTS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the National Institutes of Health.
Funding Support: This work was supported by the following grants: American Cancer Society (RSG-17-221-01-TBG to T.W.), National Institutes of Health P50CA168505 (Thyroid SPORE CDP to T.W.), and National Center for Advancing Translational Sciences (KL2TR001068 to T.W.). Research reported in this article was also supported by The Ohio State University Comprehensive Cancer Center (OSU-CCC) and National Institute of Health (P30 CA016058).
Footnotes
Conflict of Interest: All authors have no conflicts of interest or competing financial interests to disclose.
REFERENCES
- 1.Lavoie H, and Therrien M (2015) Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16, 281–298 [DOI] [PubMed] [Google Scholar]
- 2.Roberts PJ, and Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 [DOI] [PubMed] [Google Scholar]
- 3.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, Mookerjee B, Wang D, Rangwala F, and Keam B (2017) Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600- Mutant Anaplastic Thyroid Cancer. J Clin Oncol, JCO2017736785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines AT, Xu D, and Der CJ (2011) Inhibition of Ras for cancer treatment: the search continues. Future Med Chem 3, 1787–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caunt CJ, Sale MJ, Smith PD, and Cook SJ (2015) MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer 15, 577–592 [DOI] [PubMed] [Google Scholar]
- 6.Karoulia Z, Gavathiotis E, and Poulikakos PI (2017) New perspectives for targeting RAF kinase in human cancer. Nat Rev Cancer 17, 676–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiskus W, and Mitsiades N (2016) B-Raf Inhibition in the Clinic: Present and Future. Annu Rev Med 67, 29–43 [DOI] [PubMed] [Google Scholar]
- 8.Holderfield M, Deuker MM, McCormick F, and McMahon M (2014) Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 14, 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, and Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 10.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, and Bollag G (2008) Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proceedings of the National Academy of Sciences of the United States of America 105, 3041–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, Haanen JB, Hansson J, Utikal J, Ferraresi V, Kovalenko N, Mohr P, Probachai V, Schadendorf D, Nathan P, Robert C, Ribas A, DeMarini DJ, Irani JG, Casey M, Ouellet D, Martin AM, Le N, Patel K, and Flaherty K (2014) Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. The New England journal of medicine 371, 1877–1888 [DOI] [PubMed] [Google Scholar]
- 12.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, Mandala M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, Sovak MA, Chang I, Choong N, Hack SP, McArthur GA, and Ribas A (2014) Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. The New England journal of medicine 371, 1867–1876 [DOI] [PubMed] [Google Scholar]
- 13.Siegel R, Ma J, Zou Z, and Jemal A (2014) Cancer statistics, 2014. CA Cancer J Clin 64, 9–29 [DOI] [PubMed] [Google Scholar]
- 14.Xu B, and Ghossein R (2016) Genomic Landscape of poorly Differentiated and Anaplastic Thyroid Carcinoma. Endocr Pathol 27, 205–212 [DOI] [PubMed] [Google Scholar]
- 15.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, Schultz N, Berger MF, Sander C, Taylor BS, Ghossein R, Ganly I, and Fagin JA (2016) Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 126, 1052–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research, N. (2014) Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeon MJ, Chun SM, Kim D, Kwon H, Jang EK, Kim TY, Kim WB, Shong YK, Jang SJ, Song DE, and Kim WG (2016) Genomic Alterations of Anaplastic Thyroid Carcinoma Detected by Targeted Massive Parallel Sequencing in a BRAF(V600E) Mutation-Prevalent Area. Thyroid 26, 683–690 [DOI] [PubMed] [Google Scholar]
- 18.Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, Rubinstein JC, Choi M, Kiss N, Nelson-Williams C, Mane S, Rimm DL, Prasad ML, Hoog A, Zedenius J, Larsson C, Korah R, Lifton RP, and Carling T (2015) Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Human molecular genetics 24, 2318–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dadu R, Shah K, Busaidy NL, Waguespack SG, Habra MA, Ying AK, Hu MI, Bassett R, Jimenez C, Sherman SI, and Cabanillas ME (2015) Efficacy and tolerability of vemurafenib in patients with BRAF(V600E) -positive papillary thyroid cancer: M.D. Anderson Cancer Center off label experience. The Journal of clinical endocrinology and metabolism 100, E77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, Nolop K, Lee RJ, and Sherman SI (2013) Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid 23, 1277–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, Sherman SI, and Sherman EJ (2016) Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. The lancet oncology 17, 1272–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, Hofheinz RD, Hidalgo M, Chan E, Schuler M, Lasserre SF, Makrutzki M, Sirzen F, Veronese ML, Tabernero J, and Baselga J (2015) Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. The New England journal of medicine 373, 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabanillas ME, Patel A, Danysh BP, Dadu R, Kopetz S, and Falchook G (2015) BRAF inhibitors: experience in thyroid cancer and general review of toxicity. Horm Cancer 6, 21–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, Ryder M, Ghossein RA, Rosen N, and Fagin JA (2013) Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov 3, 520–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown JS, O’Carrigan B, Jackson SP, and Yap TA (2017) Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov 7, 20–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helleday T, Petermann E, Lundin C, Hodgson B, and Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8, 193–204 [DOI] [PubMed] [Google Scholar]
- 27.Chung EJ, Brown AP, Asano H, Mandler M, Burgan WE, Carter D, Camphausen K, and Citrin D (2009) In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase ½ kinase. Clin Cancer Res 15, 3050–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estrada-Bernal A, Chatterjee M, Haque SJ, Yang L, Morgan MA, Kotian S, Morrell D, Chakravarti A, and Williams TM (2015) MEK inhibitor GSK1120212-mediated radiosensitization of pancreatic cancer cells involves inhibition of DNA double-strand break repair pathways. Cell Cycle 14, 3713–3724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna WG, Muschel RJ, Gupta AK, Hahn SM, and Bernhard EJ (2003) The RAS signal transduction pathway and its role in radiation sensitivity. Oncogene 22, 5866–5875 [DOI] [PubMed] [Google Scholar]
- 30.Sklar MD (1988) The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 239, 645–647 [DOI] [PubMed] [Google Scholar]
- 31.Williams TM, Flecha AR, Keller P, Ram A, Karnak D, Galban S, Galban CJ, Ross BD, Lawrence TS, Rehemtulla A, and Sebolt-Leopold J (2012) Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Molecular cancer therapeutics 11, 1193–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seeley AR, De Los Santos JF, and Conry RM (2015) Induction vemurafenib followed by consolidative radiation therapy for surgically incurable melanoma. Melanoma Res 25, 246–251 [DOI] [PubMed] [Google Scholar]
- 33.Chatterjee M, Ben-Josef E, Robb R, Vedaie M, Seum S, Thirumoorthy K, Palanichamy K, Harbrecht M, Chakravarti A, and Williams TM (2017) Caveolae-Mediated Endocytosis Is Critical for Albumin Cellular Uptake and Response to Albumin-Bound Chemotherapy. Cancer Res 77, 5925–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, Arumugarajah S, Hylander-Gans L, Morosini D, Simeone DM, Canman CE, Normolle DP, Zabludoff SD, Maybaum J, and Lawrence TS (2010) Mechanism of radiosensitization by the Chk½ inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer research 70, 4972–4981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott JG, Berglund A, Schell MJ, Mihaylov I, Fulp WJ, Yue B, Welsh E, Caudell JJ, Ahmed K, Strom TS, Mellon E, Venkat P, Johnstone P, Foekens J, Lee J, Moros E, Dalton WS, Eschrich SA, McLeod H, Harrison LB, and Torres-Roca JF (2017) A genome-based model for adjusting radiotherapy dose (GARD): a retrospective, cohort-based study. The lancet oncology 18, 202–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torres-Roca JF, Eschrich S, Zhao H, Bloom G, Sung J, McCarthy S, Cantor AB, Scuto A, Li C, Zhang S, Jove R, and Yeatman T (2005) Prediction of radiation sensitivity using a gene expression classifier. Cancer Res 65, 7169–7176 [DOI] [PubMed] [Google Scholar]
- 37.Yard BD, Adams DJ, Chie EK, Tamayo P, Battaglia JS, Gopal P, Rogacki K, Pearson BE, Phillips J, Raymond DP, Pennell NA, Almeida F, Cheah JH, Clemons PA, Shamji A, Peacock CD, Schreiber SL, Hammerman PS, and Abazeed ME (2016) A genetic basis for the variation in the vulnerability of cancer to DNA damage. Nat Commun 7, 11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh NP, McCoy MT, Tice RR, and Schneider EL (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Experimental cell research 175, 184–191 [DOI] [PubMed] [Google Scholar]
- 39.Shen C, Lancaster CS, Shi B, Guo H, Thimmaiah P, and Bjornsti MA (2007) TOR signaling is a determinant of cell survival in response to DNA damage. Mol Cell Biol 27, 7007–7017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen C, Oswald D, Phelps D, Cam H, Pelloski CE, Pang Q, and Houghton PJ (2013) Regulation of FANCD2 by the mTOR pathway contributes to the resistance of cancer cells to DNA double-strand breaks. Cancer Res 73, 3393–3401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou X, Liu W, Hu X, Dorrance A, Garzon R, Houghton PJ, and Shen C (2017) Regulation of CHK1 by mTOR contributes to the evasion of DNA damage barrier of cancer cells. Sci Rep 7, 1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seluanov A, Mao Z, and Gorbunova V (2010) Analysis of DNA double-strand break (DSB) repair in mammalian cells. Journal of visualized experiments : JoVE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dasgupta T, Olow AK, Yang X, Hashizume R, Nicolaides TP, Tom M, Aoki Y, Berger MS, Weiss WA, Stalpers LJ, Prados M, James CD, Mueller S, and Haas-Kogan DA (2016) Survival advantage combining a BRAF inhibitor and radiation in BRAF V600E-mutant glioma. J Neurooncol 126, 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keutgen XM, Sadowski SM, and Kebebew E (2015) Management of anaplastic thyroid cancer. Gland Surg 4, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL, Bollag G, Kolesnick R, Thin TH, Rosen N, Zanzonico P, Larson SM, Refetoff S, Ghossein R, and Fagin JA (2011) Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest 121, 4700–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franco AT, Malaguarnera R, Refetoff S, Liao XH, Lundsmith E, Kimura S, Pritchard C, Marais R, Davies TF, Weinstein LS, Chen M, Rosen N, Ghossein R, Knauf JA, and Fagin JA (2011) Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proceedings of the National Academy of Sciences of the United States of America 108, 1615–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sabra MM, Dominguez JM, Grewal RK, Larson SM, Ghossein RA, Tuttle RM, and Fagin JA (2013) Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. The Journal of clinical endocrinology and metabolism 98, E829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duldulao MP, Lee W, Nelson RA, Li W, Chen Z, Kim J, and Garcia-Aguilar J (2013) Mutations in specific codons of the KRAS oncogene are associated with variable resistance to neoadjuvant chemoradiation therapy in patients with rectal adenocarcinoma. Annals of surgical oncology 20, 2166–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mak RH, Hermann G, Lewis JH, Aerts HJ, Baldini EH, Chen AB, Colson YL, Hacker FH, Kozono D, Wee JO, Chen YH, Catalano PJ, Wong KK, and Sher DJ (2015) Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clinical lung cancer 16, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lito P, Rosen N, and Solit DB (2013) Tumor adaptation and resistance to RAF inhibitors. Nat Med 19, 1401–1409 [DOI] [PubMed] [Google Scholar]
- 51.Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, Glaspy JA, Sosman JA, van Baren N, Long GV, Ribas A, and Lo RS (2014) Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov 4, 80–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tentori L, Lacal PM, and Graziani G (2013) Challenging resistance mechanisms to therapies for metastatic melanoma. Trends Pharmacol Sci 34, 656–666 [DOI] [PubMed] [Google Scholar]
- 53.Boussemart L, Malka-Mahieu H, Girault I, Allard D, Hemmingsson O, Tomasic G, Thomas M, Basmadjian C, Ribeiro N, Thuaud F, Mateus C, Routier E, Kamsu-Kom N, Agoussi S, Eggermont AM, Desaubry L, Robert C, and Vagner S (2014) eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature 513, 105–109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.