

Abstract
Ischemia-reperfusion (IR) injury is a process defined by the temporary loss of blood flow and tissue perfusion followed later by restoration of the same. Brief periods of IR can be tolerated with little permanent deficit, but sensitivity varies for different target cells and tissues. IR injuries have multiple causes including peripheral vascular disease and surgical interventions that disrupt soft tissue and organ perfusion as occurs in general and reconstructive surgery. IR injury is especially prominent in organ transplantation where substantial effort has been focused on protecting the transplanted organ from the consequences of IR. A number of factors mediate IR injury including the production of reactive oxygen species and inflammatory cell infiltration and activation. In the kidney, IR injury is a major cause of acute injury and secondary loss of renal function. Transplant-initiated renal IR is also a stimulus for innate and adaptive immune-mediated transplant dysfunction. The cell surface molecule CD47 negatively modulates cell and tissue responses to stress through limitation of specific homeostatic pathways and initiation of cell death pathways. Herein, a summary of the maladaptive activities of renal CD47 will be considered as well as the possible therapeutic benefit of interfering with CD47 to limit renal IR.
Keywords: thrombospondin-1, CD47, SIRP-α, ischemia reperfusion injury, kidney, renal
Ischemia reperfusion injury (IRI)
The cessation of blood flow and secondary restoration of flow is the mechanical basis for ischemia reperfusion. This process can manifest in a wide range of disease processes and medical interventions. Medical diseases and conditions associated with IRI include myocardial infarction, stroke, emergent resuscitation from shock and neurologic conditions that impede motor function, that latter resulting in pressure wounds. Medical interventions associated with IRI include procedures to restore blood flow such a thrombectomy and vascular bypass, elective microsurgical transplantation of tissues for complex wound reconstruction, emergent replantation of amputated tissues and acral parts (digits, etc.), surgical treatment of compartment syndrome and solid vital organ transplantation. Unique pre- and post-natal circumstances contribute to and are associated with pediatric IRI. Nonetheless, all IRI situations share common aspects. First, cells are deprived of adequate oxygen to as a result of cessation of blood flow. Lack of sufficient oxygen delivery results in an inability of cells to maintain sufficient energy sources and oxygen required for efficient synthesis of adenosine triphosphate (ATP). Ischemic cells, if possible, will revert to anaerobic metabolic pathways to meet energy needs. However, many critical processes are dysregulated in such situations including membrane channel function and cytoplasmic electrolyte balance, especially calcium (Ca2+) handling. The integrity of cell mitochondria cannot be sustained under these conditions for any length and will contribute to cell death. Such a sequence of events is common to all cell types in a given tissue including vascular cells, bloodborne circulating cells and parenchymal cells, although the degree of sensitivity to ischemia and oxygen deprivation varies greatly. For example, neural cells have no ability to tolerate even the briefest of ischemic intervals before undergoing cell death. In contrast, some cells such as chondrocytes are naturally less sensitive to periods of ischemia. In the vasculature the normal function of the endothelial cell lining is degraded by ischemia, and the ability to retain fluid within the vascular compartment is compromised. The movement of fluid from the vascular to parenchymal space can elevate pressure in the region and compress the microvasculature.
The second major event in IR injury occurs when blood flow is restored to the ischemic area. While it might be expected that restoration of flow would have beneficial effects, reperfusion also triggers several pathways that result in permanent tissue damage, loss of function, and necrosis. Reperfusion sets in motion inflammatory pathways that are detrimental to blood flow, and cell function and survival. In the vasculature damage to endothelial cells leads to activation of cell adhesion receptors that allow platelets and circulating inflammatory cells to adhere to the vessel wall, and this in turn stimulates thrombosis and decreases capillary blood flow. In some cases, blood flow, while being restored in the macrovasculature, does not re-establish in the microcirculation. At the cellular level, reperfusion results in production of pathologic levels of superoxide anion (O2-) and other ROS, that deplete the vasodilator nitric oxide (NO), adversely modify protein function, and promote vasoconstriction. O2- reacts rapidly with NO with a second order rate constant of 6.7 × 109 M−1/s−1 to yield peroxynitrite (ONOO-) [1], effectively removing NO from the immediate environment and thus eliminating the beneficial effects of NO in the vasculature including its vasodilation and anti-inflammatory activities. Peroxinitrite, in turn, is rapidly converted to the nitrosating species N2O3, which reacts with glutathione and thiols on critical proteins and impairs their functions [1]. Altered redox signaling will be seen later to be of importance in relation to how CD47 operates to promote renal IRI.
Age-related sensitivity to IRI
Animal studies provide evidence for age-mediated IRI sensitivity. In piglets, organ perfusion was assessed in response to hypothermia (ischemic phase) and rewarming (reperfusion phase). Among preterm, newborn and 2-week-old piglets cardiac output dropped and remained below baseline after reperfusion. Interestingly, loss of intestinal blood flow in newborn and 2-week-old animals also remained below baseline post-reperfusion, suggesting resistance to this form of IRI injury in premature but not older animals [2]. Hearts from premature guinea pigs produced less O2- post-IRI compared to hearts from 7-week-old animals [3]. Similarly, rabbits showed an age-related loss of recuperation post-cardiac IRI [4]. In another study, young 4-month-old rats were found to activate a protective gene profile post-cardiac IRI, whereas 25-month-old rats were found to upregulate genes associated with cell death and hypertrophy following cardiac IRI [5]. One-year-old dogs had attenuation of cardiac IRI compared to 10-year-old animals [6]. Young 8–12-week-old mice tolerated hepatic IRI better than 12–13-month-old animals with less liver injury and decreased levels of the injury markers alanine aminotransferase, heat shock protein 70 [7], and tumor necrosis factor-α in the blood [8]. Hind limb IRI in young 6-month-old rats was associated with better functional outcomes at 2 weeks post-reperfusion and elevated levels of insulin like growth factor-1 (IGF-1) compared to 24–27-month-old rats [9]. Likewise, 3-month-old mice showed faster and more complete hind limb ischemia-driven reperfusion compared to 31-month old animals, although decreased vasculature density (rarefaction) compounded interpretation of these findings [10]. Interestingly, ROS species that contribute to IRI-mediated organ damage are also markers of aging [11].
In contrast to the preceding data, 15–21-day-old infant rabbits experienced greater lung injury post-IRI compared to 5–6-month-old adult animals [12], while neonatal rabbit hearts showed more post-IRI arrhythmias compared to adult hearts [13], suggesting sensitivity to IRI is species-, age- and organ-specific. At the cellular level, increased ROS and cytokine production may contribute to age-related IRI susceptibility [14]. Taking these observations further, perfusion with neonatal rodent mesenchymal stem cells (MSCs) improved myocardial function after ex vivo IRI compared to adult MSCs [15], suggesting stem cell reparative capacity under IRI-stress is age biased.
Human data on non-renal IRI in neonates, children and adults are limited. In liver transplantation, retrospective analysis of organ performance from young (≤13 years old) versus older (≥18 years old) donors revealed comparable levels of post-transplant complications [16], suggesting that organs procured from younger donors were able to tolerate transplant-related IRI as well as more mature organs. In cardiac surgery-related IRI, circulatory bypass is a recognized source of injury to visceral organs. This assumes importance in pediatric patients experiencing surgery for congenital heart malformations [17]. Neonates appear to be especially sensitive to this form of IRI [18]. Conversely, ‘young’ myocardial infarction (MI) patients (<50 years) maintained MI-stress related levels of IGF-1 to a greater degree than older (>50 years) MI patients [19], suggesting age-related differences in the reparative response of the human heart to IRI. In a novel study, young (18–30 years) and old (60–80 years) human subjects were exposed to three episodes of upper extremity IRI as a preconditioning stress. Plasma samples were then harvested and used to perfuse isolated rat hearts prior to IRI. Infarct size was reduced in those hearts that were pre-treated with plasma from young human subjects [20], suggesting that youth-derived resistance to IRI is transferrable.
Renal IRI
Reports from experimental models of renal IRI confirm the clinical experience, namely an acute period of ischemia reperfusion followed by the development of long term progressive organ damage to more or lesser degrees. In the clinical scenario of organ transplantation, the first event is loss of blood flow to the organ as the renal arteries and veins are ligated. This interval of warm ischemia is then followed by a period of cold ischemia of varying length wherein the organ is perfused with preservation fluid and transported to the recipient.
The renal vasculature
The kidney is one of the most well vascularized organs on the body. It has two separate circulatory patterns, one that provides blood to the medulla (some 80% of the total flow) and the other that perfuses the renal cortex (about 20% of the total flow) [21]. Under certain limited conditions in which renal blood flow decreases, such as changes in blood pressure, renal cells can modify their oxygen consumption to limit injury [22]. Loss of adequate renal blood flow can be elective, as with renal transplantation, or non-elective, as in hypotension of several etiologies. Lack of adequate renal oxygen delivery, a usual result of loss of blood flow, can also occur under conditions of severe anemia or hemodilution, as with intravenous fluid administration concurrent with loss of red cell mass through bleeding.
Non-transplant induced renal IRI
As previously noted, certain emergent situations can foster renal IRI including hypotension and shock. These, in turn, may arise from critical blood loss as in trauma, from cardiovascular complications leading to decreased cardiac output, from overwhelming infection (septic shock) or in rare instances thrombosis or dissection of the main renal artery. Under any of these conditions there may be both normo-thermic or sub-thermic periods of decreased blood flow (the latter being often clinically associated with global hypotension) followed by periods of restoration of blood flow. The hematologic disorder sickle cell disease, while well known to induce vascular occlusion in the lung and bone, has recently be found to be associated with infarction of the renal microcirculation and thus IRI [23].
Transplant-induced renal IRI
Ischemia reperfusion related to organ transplant is segregated into three primary phases - warm ischemia, cold ischemia and reperfusion. However, it should be noted that these delineations overlap. Technical methods to minimize ischemia and lower the metabolic activity of the transplant have been somewhat standardized [24], although research continues to investigate optimization of transplant protocols. The end results of renal transplantation can vary in terms of kidney function. In the most severe of instances the transplant fails to re-perfuse (so-called no reflow events) and is rendered non-functional during the transplant procedure. This is a rare occurrence in the modern era of renal transplantation. Less dramatic outcomes include delay in renal function, acute rejection of the graft and long-term graft inflammation, fibrosis and functional decline.
Pediatric renal IRI
Renal maturation occurs both before and after birth as demonstrated by the finding that in newborns and premature infants the renal tubules are not fully developed in comparison with tubules in older infants and children. This suggests that age is an important factor in susceptibility to renal IRI. At the same time, reparative mechanisms are believed to be more prominent in young subjects. Experimental data in animals supports the notion of youth-based insensitivity to renal IRI. Young 6–7-week-old juvenile rats tolerated unilateral nephrectomy and 60 minutes of arterial occlusion to the remaining kidney better that 60–65-week-old animals [25]. This age-associated sensitivity to renal IRI may be in part secondary to a loss of renal reparative capacity. Young rats were found to have greater numbers of proliferating tubular epithelial cells following renal IRI compared to old animals [26]. Aged 27-month-old rats challenged with renal IRI displayed increased rates of tubular cell apoptosis concurrent with upregulation of mitochondrial-driven apoptotic genes compared to young animals [27]. Not unexpectedly, young 8–10-week-old mice fared better after bilateral renal IRI compared to aged 46–49-week-old animals [28]. In contrast, pre-term piglets challenged with hemorrhagic hypovolemia displayed decreased renal blood flow compared to the heart and brain [29], suggesting that blood flow under these conditions excludes the neonatal kidney in favor of other visceral organs. More recent work emphasizes the importance of the renal microvasculature in this process and has defined several novel molecular pathways that may contribute to pediatric renal IRI [30].
Unlike controlled animal-based research, the causes of renal IRI in the neonatal and pediatric population are varied. Lack of clinical correlates in adults also makes assessing the role of age in the susceptibility to renal IRI difficult. Sickle cell disease (SCD) is one cause of pediatric renal IRI. Oxygen-mediated alterations in the shape of sickle red blood cells increases flow resistance in, and thrombosis of, the renal microcirculation with subsequent cortical infarction [31]. Mice expressing human sickle hemoglobin were exceedingly sensitive to renal IRI compared to controls [32]. SCD patients experience focal ischemia in various organs, including the kidney [33], a process that is markedly accentuated during vaso-occlusive crisis [34]. Birth-related asphyxia is another source of renal dysfunction and renal IRI [35, 36]. Infant low birth weight is associated with vascular dysfunction and renal IRI [37]. Also, pediatric renal IRI is a well-recognized complication of cardiac surgery [38, 39]. Organ transplantation is another source of pediatric renal IRI, with 731 pediatric renal transplantations performed in the Unites States in 2016 [40]. Living donor transplantation is associated with less organ dysfunction secondary to decreased ischemic intervals and appears to outweigh the less favorable factor of advanced donor age [41]. Not unexpectantly, organs from older deceased organ donors have increased rates of post-transplant complications and graft failure [42].
Common denominators in renal IRI
Variation in blood flow and metabolic need insure that IRI-related damage is heterogeneous, with certain cell types being more or less sensitive to the process. Analysis has found that epithelial cells lining the proximal tubules of the kidney, especially those in the S3 tubular segment, as well as the ascending loop of Henle, are most sensitive to the oxygen and nutrient deprivation incurred through IR. Given the essential role these cells have in solute and fluid management in the kidney, IRI-induced loss of proximal epithelial cells leads to a decrease in renal filtration and may lead to acute kidney injury.
Damage to other cells types have also been found in post-IR kidneys. The endothelial cell lining of the renal microvasculature is a direct target of IR-related injury [43]. Several pro-blood flow mechanisms supported by normal endothelial cells are lost in renal IR including the production of salutary NO, maintenance of cell-to-cell contacts and vessel barrier function, and suppression of cell adhesion molecules. Concurrently, endothelial cell production of vasoactive agents that cause vessel constriction such as endothelin-1, prostaglandins, and angiotensin and induction of cell surface adhesion receptors such as intracellular adhesion molecule-1 and selectins promote vascular dysfunction.
Matricellular proteins
In complex organisms, cells live within a 3-dimensional scaffold of structural proteins known as the extracellular matrix. The dynamics of cellular interactions with this matrix are fine-tuned by a range of nonstructural molecules that are discharged from cells into the extracellular space. Bornstein and colleagues coined the term matricellular proteins to describe a group of such proteins that modulate interactions between cells and the surrounding extracellular matrix [44]. The defining attributes of matricellular proteins are i) limited constitutive expression but induced expression at specific stages of development and following injury, ii) interactions with cell surface molecules that alter cell responses, iii) binding to the extracellular matrix but not serving as structural proteins in the matrix, and iv) modifying the organization or function of the structural extracellular matrix scaffold either directly or through effects on cells that make and modify the matrix. With some exceptions, studies of mice lacking specific matricellular proteins reveal that they are not generally necessary for life. However, subjecting such transgenic mice to injury and stress has revealed important functions for matricellular proteins in homeostasis.
Thrombospondin-1 (TSP1)
Currently, at least seven families of proteins are classified as matricellular proteins. The first identified and most widely studied family in higher vertebrates is encoded by five thrombospondin (THBS) genes. THBS1 encodes the protein thrombospondin-1 (TSP1), which is secreted by cells as a glycosylated 480 kDa trimer. Each TSP1 monomer has several functional domains, a common feature of the majority of matricellular proteins. The ability of each domain to interact with different cell surface receptors, secreted growth factors, and extracellular matrix proteins results in diverse, and sometimes opposing, actions. Important functions ascribed to TSP1 include effects on inflammation and inflammatory cells, modification of platelets and thrombosis, and suppression of tumor angiogenesis. More recent work has revealed effects of TSP1 in the realm of vascular-mediated events through regulation of endogenous biogases, and in particular NO, H2S, and ROS. These latter properties of TSP1 necessarily require the engagement of its widely expressed cell surface receptor CD47 (Fig. 1). Engaging CD36, an alternative TSP1 receptor, can also mediate NO inhibition, albeit this requires higher concentrations of TSP1 and is only found with co-expression of CD47 [45]. Interestingly, the evolutionary appearance of CD47 occurred close to the appearance of the vascular-based isoform of nitric oxide synthase that produces low levels of NO, namely endothelial nitric oxide synthase (eNOS/NOS3) [46].
Fig. 1. Effects of elevated TSP1 expression induced by ischemia-reperfusion injury on the renal vasculature, renal tubules, and phagocytes.
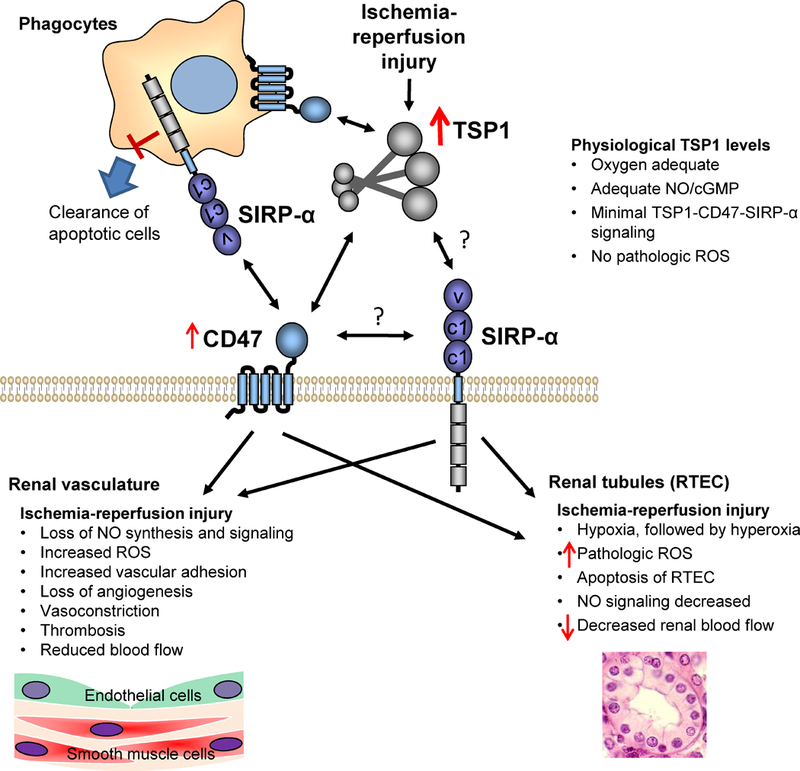
CD47 is a ubiquitously expressed receptor. SIRP-α is highly expressed on phagocytic cells, where it serves as an inhibitory receptor to prevent phagocytosis of healthy cells that express CD47. SIRP-α is also expressed by endothelial cells and RTEC, where it regulates production of ROS by NADPH oxidase. TSP1 binding to CD47 regulates activation of macrophages, NO biosynthesis by endothelial cells, relaxation of vascular smooth muscle by NO, and ROS production and apoptosis in RTEC.
The inhibitory actives of TSP1 occur at multiple levels along the canonical NO pathway, including at the level of eNOS and NO formation [47], at the level of NO activation of its receptor soluble guanylate cyclase (sGC) [48], and at the level of second messenger cyclic guanosine monophosphate (cGMP) signaling [49, 50]. Further, this inhibitory activity is conserved being found in murine, rat, bovine, porcine and human vascular cell types, and in T cells [51] and platelets [49].
Mechanistically, TSP1 exerts its inhibitory activity on canonical NO signaling through more than one signal transduction pathway. In endothelial cells, TSP1 can dysregulate calcium with adverse effects upon eNOS activation and NO production [47]. A second mechanism of inhibition, again at the level of eNOS activation and NO production, occurs via CD47-mediated limitation of vascular endothelial growth factor receptor activation [52]. In human T cells the C-terminal signature domain of TSP1, which interacts with CD47, limits sGC activation, by increasing cytoplasmic Ca2+, possibly through protein phosphorylation [51]. In vascular cells, TSP1 can also act negatively upon NO signaling through indirect pathways via activation of NADPH oxidase and pathologic production of ROS through either CD47 [53] or signal regulatory protein-alpha (SIRP-α) [54]. These effects appear to be mediated directly by TSP1. In human monocytes and murine macrophages TSP1 enhances PMA-mediated O2- production, likely via a CD47-independent manner since recombinant TSP1-derived domains of the N- but not the CD47-interacting C-terminus replicated the effects of the whole protein [55].
CD47 and its interacting partners
CD47, also referred to as integrin associating protein, was initially distinguished by its absence in red blood cells from people with Rhesus (Rh) null hemolytic anemia [56] and association with the integrin αvβ3 in ovarian carcinoma samples [57]. Presently, CD47 is known to be expressed broadly on the cell surface of numerous nontransformed and cancer cells and on extracellular vesicles released by these cells. Both complete and cleaved forms of the molecule can be found on the cell surface, although the signaling implications of the membrane-cytoplasmic only proteolytic fragment of the protein have not been explored. In vitro studies of erythropoiesis have looked at age-dependent CD47 protein expression. CD47 protein was minimally expressed in early stage erythroblasts but over time progressively increased with cell maturation [58]. Similarly, postnatally CD47 expression increased over time in the hippocampal region of rodent brains and in cultured neurons during cell maturation [59]. Besides integrins, CD47 can engage with TSP1 and thrombospondin-2, VEGFR-2, SIRP-α and -γ, ubiquilins, heterotrimeric G proteins, and in lateral complexes with other membrane proteins [60]. The functional implications of most of these interactions have been explored and indicate important roles for CD47 in cell metabolism, self-renewal and inflammatory responses, as well as self-recognition through interaction with the counter-receptor SIRP-α on innate immune cells. In this capacity, inhibitors of the interaction between SIRPα and CD47 have entered clinical trials for cancer treatment [61, 62].
TSP1 in the kidney
Basal TSP1 expression patterns
During murine development days 10 to 13 TSP1 mRNA was demonstrated in primitive kidney cells [63]. As in other tissue types, TSP1 expression levels in the adult kidney are minimal. In a cohort of 11 individuals, without overt chronic diseases or renal pathology, glomerular TSP1 mRNA and protein levels were low [64].
Non-IRI related stress-induced TSP1 expression patterns
Levels of circulating TSP1 can become elevated as a result of platelet activation and in some chronic disease states, which could mediate CD47 signaling in renal endothelial cells. In addition, several pathologic conditions can induce increased local expression of TSP1 in the kidney (Fig. 2). In neonatal kidneys enalapril (an angiotensin-converting enzyme inhibitor) induced tubular injury and decreased glomerular growth and this was associated with upregulation of TSP1 [65]. Renal biopsies of 19 patients with incipient or established diabetic renal disease found increased TSP1 expression mainly in mesangial areas [64]. TSP1 is also increased in anti-Thy1.1 induced glomerulonephritis, aminonucleoside nephrosis, passive Heymann nephritis, and diabetic nephropathy and in some of these was established to correlate with the severity of disease [66, 67]. In mice lacking eNOS, and thus having an inherently low vascular NO environment, renal TSP1 levels were elevated concurrent with mesangiolysis and nodular lesions [68]. Beyond these few reports, renal TSP1 expression patterns have been assessed in chemically-induced Islet β-cell injury rodent models, with later time point analysis being the rule. Thus, TSP1 expression is frequently elevated in renal disease. But in some instances, its pathogenic activity involves CD47-independent functions of TSP1 such as activation of latent TGFβ activation [67, 69].
Fig. 2.
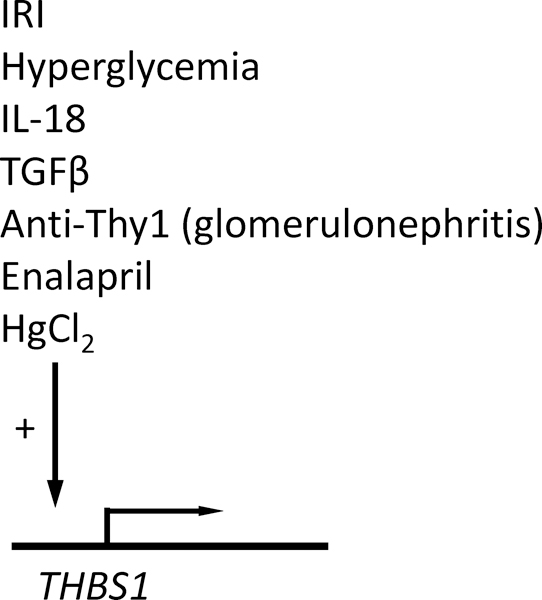
Inducers of TSP1 expression in kidney.
CD47 in the kidney
Basal CD47 expression patterns
Information remains modest in regard to the normal and pathologic roles of CD47 in kidney cells or in the kidney. In the normal human kidney glomeruli, epithelial cells and mesangial cells express CD47 [70]. Cultured human renal tubular epithelial cells (RTEC) also displayed immuno-fluorescent CD47 [71]. Similarly, the human kidney proximal tubular cell line (HK-2 cells) express CD47 [72]. In the healthy rat kidney, CD47 was found via immunofluorescence and immuno-gold techniques to localize along the plasma membrane of mesangial calls, but to not be expressed in podocytes and endothelial cells [73]. In Madin-Darby canine kidney epithelial cells, CD47 co-localized with E-cadherin at points of cell-cell adhesion. Over-expression of CD47 in these cells resulted in disruption of cell-cell adhesions, increased cell spreading and migration [74]. Clinical data from the kidneys of middle-age and older subjects (46–78 years old) demonstrated CD47 immuno-staining in the renal tubules, with markedly less expression in the glomeruli. In these same studies, renal fibroblasts also expressed CD47 in some, but not, every instance (https://www.proteinatlas.org/ENSG00000196776-CD47/tissue/kidney). CD47 mRNA expression was found in all 32 renal samples studied from individuals over the age range 20–69 years.
Non-IRI related stress-induced CD47 expression patterns
CD47 levels are modified in the kidney under certain stress-related conditions. Interestingly, CD47 expression levels in human mesangial cells was reported to decrease in acute post-infectious and membrano-proliferative glomerulonephritis, as well as diabetic nephropathy [70]. Treatment with hypoxia (30 min, FiO2 1%) followed by 24 hours at room air increased TSP1 protein levels in cultured human RTEC, with no change in CD47 protein or TSP1 and CD47 mRNA [71]. Treatment of HK-2 cells with LPS increased CD47 protein expression, while LPS-mediated polymorphonuclear leukocyte migration across a monolayer of HK-2 cells was limited by a putative CD47-blocking antibody [72]. CD47 protein was upregulated in rat kidneys following intra-peritoneal administration of ferric nitrilotriacetate, while CD47 mRNA remained upregulated in tubular epithelial cells one year after treatment [75]. Administration of Thy1.1 antibody to rats, initially decreased CD47 expression in association with mesangial cell death, but CD47 expression was subsequently found increased with cell proliferation and repair to levels above those pre-injury [73]. Glomerular epithelial and endothelial cells did not upregulate CD47 in this rodent nephritic model.
SIRP-α in the kidney
Basal SIRP-α expression patterns
Even less is known about the role of SIRP-α in kidney cells and in the whole organ. Cultured human RTEC express SIRP-α protein [54]. In rodent kidneys, immunofluorescence of tissue sections localized SIRP-α primarily to the glomeruli. Co-localization studies further narrowed expression to podocytes while excluding mesangial and endothelial cells [73]. Gold immune electron microscopy found enrichment in SIRP-α at podocyte slit diaphragms. SIRP-α expression has been reported in kidneys from children (aged 7 and 16 years). In these instances, immunohistochemical expression was modest in the glomeruli and absent in the tubules. However, one individual’s kidneys were found to show robust immuno-reactive protein expression in both the tubules and glomeruli (https://www.proteinatlas.org/ENSG00000198053-SIRPA/tissue/kidney#imid_839000). In adult kidneys, SIRP-α expression varied being undetectable in some cases and highly expressed in both glomeruli and tubules in others. Such a range in expression might be secondary to associated metabolic and renal diseases, although these details were not available. SIRP-α mRNA was found in every renal sample obtained from 32 individuals, none below the age of 20.
Non-IRI related stress-induced SIRP-α expression patterns
Unlike CD47, SIRP-α expression did not increase in rats following induction of nephritis with Thy1.1 antibody [73]. Interestingly, the phosphorylated (active) form of SIRP-α was expressed in normal podocytes and decreased significantly post-injury. It is speculated that post-injury activity between mesangial CD47 and podocyte slit diaphragm SIRP-α modulates downstream activity of cytoplasmic SHP-2 (a SIRP-α signal transducer) [73]. In contrast, treating human RTEC with exogenous TSP1 increased expression of phosphorylated SIRP-α as well as the alternative SIRP-α signal transducer SHP-1 [54], whereas a SIRP-α antibody blocked TSP1-mediated increases in phosphorylation of both proteins. These findings are relevant given that renal biopsies from patients with nephritis have demonstrated increased glomerular TSP1 expression [76]. The potential role TSP1 plays in mesangial CD47-podcyte SIRP-α cross-talk remains to be tested.
TSP1, CD47 and SIRP-α in non-renal IRI
Initial evidence that TSP1 might play a role in ischemia reperfusion injury (IRI) came from studies in newborn mice exposed to high oxygen concentrations (FiO2 75%) for 5 days post-birth followed by room air for another 8 days. This protocol has the effect of inducing IRI to the retina. Retinal TSP1 mRNA levels were increased peaking by day 15 post-birth [77]. Interestingly, in situ hybridization localized TSP1 mRNA to the vasculature, especially those retinal vessels suffering the greatest injury. TSP1 protein expression paralleled mRNA changes, while in control mice retinal TSP1 was undetectable via immunofluorescence. Providing genetic priority to this signal, mice lacking TSP1 were resistant to hyperoxia/normoxia-induced retinal IRI injury compared to control TSP1 positive mice [78]. Peptides derived from the central stalk region of TSP1 inhibited the reactive angiogenesis in the newborn mouse model, suggesting involvement of TSP1 effector pathways other than CD47 [79].
Stroke is a disease process that leads to IRI of the brain. This can be modeled through temporary occlusion of one of the primary blood vessels to the brain. In rats subjected to temporary middle cerebral artery occlusion followed by 14 days of reperfusion TSP1 expression was increased in leptomeningeal endothelial cells at 24 hours post-reperfusion and in endothelial, glial, neuronal, and macrophage cells at 72 hours post-reperfusion [80]. TSP1 was induced in canine and murine hearts after IRI and localized to the extracellular matrix, microvascular endothelium, and a subset of mononuclear cells at the infarct border zone 5 at 28 days post-reperfusion [81]. Results in mice lacking TSP1 suggested that the protein limited extension of the tissue injury in these circumstances, although as cardiac hemodynamic function was not determined, the potential clinical impact of these findings was not clear. Similarly, elevation of TSP1 mRNA in the infarct border zone was noted in a rat heart IRI model [82]. In a murine model of liver IRI, wild type mice (that express TSP1) demonstrated significantly more liver damage and inflammation post-reperfusion compared to mice lacking TSP1 [83]. In a model of large vessel IRI in pigs (30 minutes of aortic cross-clamping followed by 5 hours of reperfusion) luminal endothelial cell TSP1 mRNA was significantly increased [84]. Interpretation of these data are complicated by the role that laminar versus turbulent blood flow can play upon endothelial TSP1 expression [85]. Also, if not captured via proximal aortic cross-clamping, segmental vasa vasorum blood flow could provide some degree of perfusion to the vessel wall. Further studies in rats found that intravenous administration of TSP1 (60 μg/kg body weight) approximately 1 hour prior to IRI (20 minutes of femoral artery occlusion followed by 40 minutes of reperfusion) limited hind limb reperfusion [53].
To a greater or lesser extent all surgical interventions disrupt tissue perfusion and, depending upon the redundancy of tissue collateral blood flow, inflict varying amounts of reperfusion injury [86]. Induction of and/or the presence of TSP1 has been reported to be associated with decreased soft tissue survival to surgically created soft tissue flaps in mice, and this was correlated with decreased tissue cyclic guanidine monophosphate (cGMP), an essential downstream signaling agent of nitric oxide (NO) [87].
TSP1 and CD47 expression often run parallel under conditions of injury, and thus the roles of these two proteins in IRI may be closely linked. Some therapeutic strategies may arise from this linkage. TSP1 has protective roles in certain situations mediated via non-CD47 interactions, and targeting CD47 does not interfere with these. Second, TSP1 is present pre-formed in platelets and provides a continued stimulus of post-injury TSP1-CD47 signaling. In rats, treatment with a CD47 blocking antibody, to limit TSP1-CD47 signaling, resulted in improved soft tissue survival, elevated tissue cGMP, less tissue lipid peroxidation and diminished tissue elastase (a marker of neutrophil activation) after IRI compared to animals treated with an IgG control antibody [88]. Taking a different approach, suppressing CD47 with RNAi attenuated heart IRI injury (30 minutes of occlusion of the left anterior descending coronary artery followed by 3 hours of reperfusion) in rats [89]. In a porcine model of surgically-induced soft tissue IRI, flaps treated with either a TSP1 blocking antibody or a gene silencing CD47 morpholino oligonucleotide displayed enhanced tissue survival [90].
Whole organ transplantation unavoidably induces IRI. Here too, a role for TSP1, signaling through CD47, has been elucidated. Rats undergoing orthotopic syngeneic cold liver transplantation (18 hours at 4° C, with Wisconsin solution flush) were treated with either a CD47 blocking antibody to the transplant vasculature only (1 μg/kg liver weight) or the same to the transplant plus systemic administration of the CD47 antibody to the recipient. Those animals receiving systemic and organ-targeted CD47 antibody therapy showed greater survival 48 hours post-transplantation compared to controls [91]. Of note, hepatic TSP1 and CD47 protein levels were increased in control organs following transplantation, while organs subjected to just cold storage were also found to express more CD47.
The first evidence that CD47 plays a role in IRI arose from studies in mice challenged with liver ischemia reperfusion. Age and sex matched C57BL6 and Thbs1- and Cd47-null mice were exposed to 45 minutes of occlusion of the portal triad through application of a non-crushing microvascular clamp [83]. This was then followed by 6 hours of reperfusion. Interesting, reperfusion liver blood flow, as quantified by real-time laser Doppler, was significantly greater in Thbs1- and Cd47-null mice compared to wild type. This was associated with lower levels of serum alanine aminotransferase (sALT) and aspartate aminotransferase (sAST), decreased hepatic leukocyte infiltration, and less hepatocyte apoptosis in null versus wild type mice. In other experiments, wild type mice received either a CD47 blocking antibody or an IgG2a control antibody via intraperitoneal injection 90 minutes prior to ischemia. Animals pre-treated with a CD47 blocking antibody prior to liver IR enjoyed increased blood flow post-reperfusion, lower levels of sAST and sALT and less leukocyte infiltration compared to mice pre-treated with an IgG2a control antibody. Consistent with TSP1 being a stress response protein, hepatic expression of TSP1 was lower in livers from mice treated with the CD47 blocking antibody and elevated in livers from animals treated with the control antibody.
Several mechanisms could contribute to these findings. First, TSP1 [48], via interaction with CD47 [45] limits the salutary effects of vascular NO (Fig. 1). Loss of vascular-based NO signaling negatively impacts IRI injury [92], whereas upregulation of eNOS is associated with protection from visceral organ IRI [93]. Alternatively, CD47 impacts central hemodynamics [94], and organ perfusion may be enhanced under basal and injury-mediated conditions in the absence or attenuation of CD47 signaling. At a minimum, laser Doppler data suggest enhanced tissue reperfusion in the absence of CD47 signaling. Other factors that impede reperfusion might also be at play in the setting of low or absent CD47 signaling, including suppression of inflammation, decreased platelet activation [49], reduced endothelium adhesion protein expression and monocyte attachment [95] and less ROS production. Also, TSP1 has anti-angiogenic activity suggesting that reperfusion benefits in the absence of CD47 in visceral organs could be secondary to a baseline gain in vascularity. As of yet, a direct role for SIRP-α in non-renal IRI has not been described. However, SIRP-α may play an indirect role in this process.
As described above, a number of studies from multiple groups of investigators have found enhanced blood flow in mice lacking TSP1 or CD47 immediately following femoral artery ligation-induced ischemia or surgically-induced soft-tissue IRI [96], and in rats, less blood flow with reperfusion after intravenous TSP1 administration. Taken together, these results define a role for TSP1 and CD47 in promoting non-renal IRI. Yet, they also raise interesting questions in relation to the current understanding of IRI. If reperfusion is established sooner and putatively to a greater extent in the absence or diminution of TSP1-CD47 signaling, one could hypothesize that the amount of IRI damage should be greater lacking TSP1 or CD47. And yet, this has not been born out in the data. A more precise analysis of the relationship between the degree and length of the ischemic interval and the relation this has to the initiation and degree of reperfusion may help to clarify this, both generally, and in relation to the TSP1-CD47 signaling axis.
TSP1, CD47 and SIRP-α in acute non-IRI renal injury
Acute non-IRI renal injury, and associated renal failure, can arise from several etiologies. These can include processes such as system-wide or organ-specific trauma, trauma-related and non-traumatic sources of hemodynamic instability, auto-immune conditions such as lupus erythematosus and scleroderma, renal toxins and intravenous radio-contrast agents and post-kidney urinary obstruction. In situations that lower the systemic blood pressure or that negatively impact renal artery or vein blood flow, some degree of IRI is inherent. That TSP1 is involved in any of these situations has not been systematically assessed. Still, there are some data establishing a possible role. In rats administered mercuric chloride (HgCl2, 1 ml/kg body weight subcutaneously) proximal tubule epithelial cell necrosis was found within 5 days and overlapped with a spike in renal cortex TSP1 mRNA levels [97]. Further, mice lacking TSP1 were protected from the renal toxic effects, and especially podocyte loss, of adriamycin. Wild type mice experienced rapid induction renal TSP1 protein post-injury along with podocyte death, elevated serum creatinine and proteinuria [98]. In another study, rats underwent ligation of a single ureter and were given a TSP1-blocking peptide daily thereafter. Renal injury within the first and second week post-ureteral obstruction was significantly less in the peptide-treated cohort and this was associated with amelioration of injury-associated increases in renal TSP1 protein expression [99]. Likewise, in mice subject to unilateral ureteral obstruction, treatment with short hairpin RNA vector that targeted TSP1 reduced early markers of renal injury [100]. Acute renal injury is known to complicate transplantation of other organs, especially the liver. The reasons for this are possibly many and an area of ongoing investigation. In a study of renal injury following orthotopic liver transplantation, renal TSP1 mRNA was acutely increased and associated with acute tubular necrosis [101]. In this study the authors occluded the inferior vena cava for 25 minutes, although the location of the point of occlusion in relation to the renal vein was not described. Nonetheless, some change in renal blood flow may have occurred, but this was not specifically monitored.
In vitro, porcine kidney PK15 cells co-cultured with human inflammatory cells experienced increased cell injury when inflammatory cell SIRP-α phosphorylation was increased [102]. Antigen presentation provides for immune regulation and a means to respond to the stress induced by host-pathogen (host non-host) interactions. A recent study identified a role for SIRP-α in monocyte activation in response to foreign protein. Foreign cells carrying a polymorphism of SIRP-α induced recipient inflammation [103]. While the results of this study did not directly assess renal injury, there are implications for transplant-related rejection separate from any effects mediated by IRI.
TSP1, CD47 and SIRP-α in renal IRI
Non-transplant-mediated renal IRI
Non-transplant renal IRI is experimentally induced via temporary occlusion of the renal vascular pedicle followed by various intervals of reperfusion. Rat and murine kidneys were exposed to 45 and 30 minutes of ischemia respectively and variable periods of reperfusion from 3 to 48 hours. Analysis of tissue samples via cDNA microarray found THBS1 to be among the most upregulated genes, and TSP1 protein levels were similarly elevated [104]. Northern hybridization found TSP1 mRNA levels increased at 3 and 12 hours but decreased to levels in control sham injured kidneys by 24 and 48 hours. Renal cortical mRNA levels of the TSP1 receptor CD36 followed an expression pattern similar to TSP1. Distribution of TSP1 in IRI injured kidney tissue sections localized the protein exclusively to damaged tubules. Finally, mice lacking TSP1 were resistant to IRI-mediated alterations in renal micro-architecture and function compared to controls. The finding of induction of CD36 in the setting of renal IRI is relevant for several reasons. First, TSP1 can block some salutatory effects via CD36 such as NO signaling [105], although a role for this through CD47-mediated cross talk is possible [45]. Additionally, glycerol-mediated acute renal injury was found to dysregulate cholesterol transporters, of which CD36 is a member [106], while a TSP1-based peptide was reported to impede the fatty acid translocase activity of CD36 [107]. It is unknown under conditions of renal IRI if global absence of CD36 would confer protection. But rodent data from soft tissue injury models suggest this would not be the case [108, 109]. Thus, it is possible that the non-transplant mediated renal IRI protection enjoyed by Thbs1-null mice was mediated through the loss of TSP1-CD47 signaling. While speculative, this hypothesis is further supported by data that suggests CD47 is the dominant, high affinity receptor for TSP1 [110].
In another experiment, rats underwent bilateral renal pedicle occlusion and were treated just before reperfusion with IL-18BP to block the inflammatory cytokine IL-18. Within a window of 6 to 72 hours of reperfusion, renal TSP1 expression was decreased compared to controls [111]. IL-18 is known to increase TSP1 mRNA and protein in cultured cells [112], indicating some protective effects from treatment with IL-18BP may be through targeting TSP1.
Experiments in mutant mice lacking CD47 demonstrated a role for TSP1-CD47 signaling in non-transplant-mediated IRI. Cd47-null mice enjoyed complete survival from bilateral renal IRI compared to control (Cd47+/+ mice) that all expired by 50 hours post-reperfusion concurrent with tubular cell death and cast formation [113]. Mutant mice may compensate through changes not simply related to loss of a certain gene. To assess the role of CD47 in control mice, animals were pre-treated with a CD47 blocking or IgG control anybody 90 minutes before non-transplant bilateral renal IRI and tissue injury determined 24 hours after reperfusion. Those animals given the CD47 blocking antibody had minimal renal histologic changes compared to control antibody-treated mice. Short-term hypoxia and re-oxygenation (to mimic organ IRI) upregulated CD47 and TSP1 protein in cultured human RTEC [71]. Both TSP1 and CD47 protein and mRNA were increased in the kidneys of control mice following 22 minutes of unilateral ischemia and 24 hours of reperfusion compared to sham operated mice. Interestingly, in kidney samples from Cd47-null mice renal TSP1 protein was minimally induced post-reperfusion, suggesting a link between ligand and receptor expression levels. Laser Doppler analysis of kidneys in CD47 expressing and null mice confirmed equal degrees of ischemia and comparable reperfusion within 30 minutes. However, by 24 post-reperfusion null animals had greater renal blood flow [71]. Unlike previously noted findings of increased vascularity in skeletal muscle samples from Thbs1-null mice, these real-time data speak to equivalent vascularity and blood flow in kidneys from TSP1-CD47 replete and deficient mice. Kidneys from post-IRI null mice also had marked decreases in inflammatory cell invasion, tubular cell death, inflammatory cytokine transcript levels, and markers of redox stress compared to CD47 expressing mice. Of some importance, wild type mice transplanted with wild type (Cd47+/+) bone marrow experienced more renal damage following renal IRI compared to Cd47-null mice transplanted with Cd47+/+ bone marrow. These data provide evidence for a major role for parenchymal, as opposed to circulating, TSP1-CD47 signaling in non-transplant renal IRI.
Renal recovery following both transplant and non-transplant-induced renal IRI is limited by the rate of tubular epithelial cell self-renewal/proliferation. Interestingly, several key transcription factors know to govern cell self-renewal (Oct¾, Sox2, Klf4 and cMyc) have been found constitutively upregulated in Cd47-null mice and cells [114]. These advantages in self-renewal transcription factor expression were found to extend to kidneys from null mice prior to and after non-transplant-induced renal IRI, and this was associated with enhanced tubular cell proliferation in vitro as well as in vivo [115]. Conversely, tubular epithelial cells from mice and humans were sensitive to TSP1-mediated suppression of self-renewing transcription factors, while cMyc protein stability was greater in Cd47-null epithelial cells. Together, these findings reveal another important mechanism through which TSP1-via CD47 acts to promote renal IRI.
As pointed out, SIRP-α is an important non-TSP1 ligand for CD47. Further, SIRP-α has been linked to ROS production [116]. TSP1, via CD47, stimulates pathologic ROS production in several cell types and tissues [53, 117], while ROS is crucial to IRI-related tissue injury. As all three molecules could co-exist in the tissue microenvironment under stress, SIRP-α is probably mediating IRI-related responses. In a murine IRI model, SIRP-α and its cytoplasmic effector SHP1 were upregulated in the kidney [54]. In vascular cells, exogenous TSP1, via SIRP-α, targeted NADPH oxidase 1 and stimulated increased O2- production, although some degree of CD47 signaling may also be acting under these conditions. In human RTEC, TSP1, via SIRP-α, also stimulated increased O2- production. Finally, administration of a SIRP-α blocking antibody prior to renal IRI mitigated renal damage. While preliminary, these data support a role for TSP1-SIRP-α signaling in non-transplant-mediated renal IRI, separate from the role SIRP-α has in regulating macrophage activation and phagocytic activity (Fig. 1). To more fully define this role, it will be important to consider the impact of renal IRI on mice lacking SIRP-α and how blocking of TSP1 and CD47 under these conditions alters outcomes.
Transplant-mediated renal IRI
In a porcine model designed to mimic the clinical scenario, renal transplants were performed with organs harvested from heart-beating and non-beating donors. Renal cortex sampling was done at several time points throughout the pre- and post-transplant period including following induction of anesthesia, after warm and cold ischemia, after one hour of reperfusion and five days after transplant. Cortical TSP1 mRNA levels were found to be increased in samples from non-beating donor organs at day 5 only. All other time points sampled, and all samples from organs from heart-beating donors, showed no change in cortical TSP1 mRNA compared to samples obtained at the time of anesthetic induction [118]. While in the present study, renal TSP1 levels did change, these results are interesting as they differ from data obtained in other animal models of non-transplant-mediated renal IRI were changes in TSP1 were uniformly upregulated. Still, variation could be expected given differences in species, model design, etc.
In a related study, again in pigs, the effects on renal function, tubular architecture and tissue levels of TSP1 following one week of reperfusion were assessed. Some organs underwent 24 hours of cold ischemia while others were subjected to 24 hours of cold ischemia and 1 hour of warm ischemia prior to transplant. As expected, renal dysfunction was greatest in organs exposed to both cold and warm ischemia and TSP1 expression was found to be increased concurrent with decreased vascular endothelial growth factor [119].
The role of TSP1 in human transplant-mediated IRI has not been explored to any substantial degree. This is surprising given the number of key redox pathways TSP1 dysregulates [50, 113]. In renal transplant recipients the presence of organ dysfunction was associated with increased urinary TSP1 mRNA [120]. However, information as to the time of urine sampling in relation to transplantation for each patient was not provided. Further, urine sampling was not repeated in individual patients. Nonetheless, this study raises the interesting idea that urinary TSP1 levels may presage renal damage, and thus, have a role as a marker of organ injury. TSP1 expression was found increased in biopsy samples from 16 patients with organ nephropathy compared to levels in renal biopsies from 6 non-transplant individuals with normal renal function [121]. Together, these data suggest that post-transplant renal graft dysfunction is associated with persistent TSP1 expression. These findings however, may not reflect acute changes in the context of renal transplantation and indicate an opportunity for further study.
CD47 has been interrogated in several models of transplant-mediated renal IRI. In mice, the pre-transplant administration of a CD47 blocking antibody (0.8 μg/g body weight anti-mouse CD47 clone 301 via intraperitoneal injection) mitigated renal injury after syngeneic renal transplantation with bilateral nephrectomy compared to vehicle (saline) treated animals [115]. This was associated with enhanced renal expression of several self-renewal transcription factors and decreased cytokine mRNA levels in CD47 antibody-treated mice. Animals given a lower dose of CD47 antibody did not enjoy renal protection following transplantation indicating that the antibody effect was dose-dependent.
In a more clinically relevant model, rats underwent bilateral nephrectomy followed by syngeneic renal transplant. On harvest, kidneys were flushed with Wisconsin solution containing a CD47 blocking (clone OX101) or an IgG control antibody and then stored at 4° C for 6 hours prior to transplant [122]. Analysis of renal tissue sections revealed that the CD47 blocking antibody bound exclusively to the vascular endothelial compartments of the renal tubules and glomeruli. Eighty percent of animals receiving a kidney pre-treated with the CD47 antibody survived while all animals receiving a kidney treated with an IgG antibody died by the fifth post-transplant day. Renal function of CD47 antibody-treated kidneys, while modestly altered at 48 hours post-transplant had returned to normal by the seventh day. Laser Doppler analysis of renal blood flow found minimal to no blood flow in a number of IgG antibody-treated organs at 24 hours post-transplant. This raises the question of possible surgical factors, such as acute thrombosis at the vascular anastomosis, intervening adversely in this cohort. In a subsequent study in rats, donor kidneys, either syngeneic or allogeneic, were treated with a CD47 blocking antibody (clone OX101) or an IgG control antibody flush followed by 1 hour of warm ischemia. Recipients of organs flushed with the CD47 antibody did markedly better than animals given control antibody perfused transplants [123]. Further, syngeneic organs performed better than autologous. Lastly, in a porcine model of organ donation after cardiac death, transplanted kidneys flushed with a CD47 blocking antibody fared better than those receiving preservation fluid only [123].
Summary and perspectives
Preclinical studies have shown renal cellular and organ expression changes in TSP1, CD47 and SIRP-α to a range of stresses (Table 1). Further, deletion of Thbs1 and Cd47 or signal blockade strategies in wild type animals indicate a role for TSP1-CD47, and TSP1-SIRPα, signaling in promoting renal IRI, both non- and transplant-related. While most of these reports assessed immediate post-injury time points, there is a body of work that has shown TSP1, through its ability to enhance subacute and chronic fibrosis [124, 125] and to modify immunity, may play a role in other aspects of renal IRI-initiated organ damage. In this latter regard, liver transplant patients were found to have TSP1 in circulating immune complexes specifically in those individuals experiencing acute cellular rejection [126]. Review of published data indicates that TSP1 and CD47 function to promote or limit immune responses in a context-dependent manner.
Table 1.
Renal Cell and Organ Stress- and Disease-related TSP1, CD47 and SIRP-α Expression
| Stress/Disease | Expression (protein, mRNA or immunoreactive) | ||
|---|---|---|---|
| TSP1 | CD47 | SIRP-α | |
| Stress: | |||
| Hypoxia | Increased | Increased | NK |
| TSP1 | NK | NK | Increased activity |
| Enalaparil | Increased | NK | NK |
| Ferric nitrilotriacetate | NK | Increased | NK |
| Mercuric chloride | Increased | NK | NK |
| LPS | NK | Increased | NK |
| Thy1.1 Ab | Increased | Increased | Decreased activity |
| Immune-associated | Increased in multiple diseases | NK | NK |
| Infection | NK | Decreased | NK |
| Metabolic (Diabetes) | Increased | Decreased | NK |
| eNOS deletion (low vascular NO) | Increased | NK | NK |
| Ureteral ligation | Increased | NK | NK |
| Non-renal transplant-related IRI: | |||
| Orthotopic liver transplant | Increased | NK | NK |
| Increased | Increased | Increased | |
| Renal vascular occlusion | |||
| Transplant-related IRI | Increased | NK | NK |
NK – Not Known
The role of CD47 has not been assessed in human renal transplant-mediated IRI, and the role of SIRP-α has not been determined in either animal or human transplant-mediated renal IRI. Based on existent data, it is reasonable to expect that TSP1-CD47-SIRP-α targeting therapies will prove effective at mitigating transplant and non-transplant IRI in multiple visceral organs, including the kidney. In vitro animal and human cell studies and numerous in vivo studies in rodents and higher mammals support expanded preclinical and clinical research in this direction. The potential importance of such investigations is emphasized by recent reports of the development of humanized CD47 antibodies [127] and SIRP-α-derived molecules [62] for the treatment of cancer.
Acknowledgments
Funding: This study was funded in part by the Intramural Research Program of the NIH, NCI, CCR
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Thomas DD, Heinecke JL, Ridnour LA, Cheng RY, Kesarwala AH, Switzer CH, McVicar DW, Roberts DD, Glynn S, Fukuto JM, Wink DA, Miranda KM (2015) Signaling and stress: The redox landscape in NOS2 biology. Free Radic Biol Med 87:204–225. 10.1016/j.freeradbiomed.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powell RW, Dyess DL, Collins JN, Roberts WS, Tacchi EJ, Swafford AN Jr., Ferrara JJ, Ardell JL (1999) Regional blood flow response to hypothermia in premature, newborn, and neonatal piglets. J Pediatr Surg 34:193–198. [DOI] [PubMed] [Google Scholar]
- 3.Southworth R, Shattock MJ, Hearse DJ, Kelly FJ (1998) Developmental differences in superoxide production in isolated guinea-pig hearts during reperfusion. J Mol Cell Cardiol 30:1391–1399. 10.1006/jmcc.1998.0707. [DOI] [PubMed] [Google Scholar]
- 4.Ataka K, Chen D, Levitsky S, Jimenez E, Feinberg H (1992) Effect of aging on intracellular Ca2+, pHi, and contractility during ischemia and reperfusion. Circulation 86:II371–376. [PubMed] [Google Scholar]
- 5.Simkhovich BZ, Marjoram P, Poizat C, Kedes L, Kloner RA (2003) Age-related changes of cardiac gene expression following myocardial ischemia/reperfusion. Arch Biochem Biophys 420:268–278. [DOI] [PubMed] [Google Scholar]
- 6.Jugdutt BI, Jelani A, Palaniyappan A, Idikio H, Uweira RE, Menon V, Jugdutt CE (2010) Aging-related early changes in markers of ventricular and matrix remodeling after reperfused ST-segment elevation myocardial infarction in the canine model: effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation 122:341–351. 10.1161/CIRCULATIONAHA.110.948190. [DOI] [PubMed] [Google Scholar]
- 7.Okaya T, Blanchard J, Schuster R, Kuboki S, Husted T, Caldwell CC, Zingarelli B, Wong H, Solomkin JS, Lentsch AB (2005) Age-dependent responses to hepatic ischemia/reperfusion injury. Shock 24:421–427. [DOI] [PubMed] [Google Scholar]
- 8.Selzner M, Selzner N, Chen L, Borozan I, Sun J, Xue-Zhong M, Zhang J, McGilvray ID (2009) Exaggerated up-regulation of tumor necrosis factor alpha-dependent apoptosis in the older mouse liver following reperfusion injury: targeting liver protective strategies to patient age. Liver Transpl 15:1594–1604. 10.1002/lt.21864. [DOI] [PubMed] [Google Scholar]
- 9.Hammers DW, Merritt EK, Matheny RW Jr., , Adamo ML, Walters TJ, Estep JS, Farrar RP(2008) Functional deficits and insulin-like growth factor-I gene expression following tourniquet-induced injury of skeletal muscle in young and old rats. J Appl Physiol (1985) 105:1274–1281. 10.1152/japplphysiol.90418.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faber JE, Zhang H, Lassance-Soares RM, Prabhakar P, Najafi AH, Burnett MS, Epstein SE (2011) Aging causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler Thromb Vasc Biol 31:1748–1756. 10.1161/ATVBAHA.111.227314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Labat-Robert J, Robert L (2014) Longevity and aging. Role of free radicals and xanthine oxidase. A review. Pathol Biol (Paris) 62:61–66. 10.1016/j.patbio.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Qiu W, Zheng L, Gu H, Chen D, Chen Y (2008) Comparison between adult and infant lung injury in a rabbit ischemia-reperfusion model. J Thorac Cardiovasc Surg 136:352–359. 10.1016/j.jtcvs.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Chenliu C, Sheng X, Dan P, Qu Y, Claydon VE, Lin E, Hove-Madsen L, Sanatani S, Tibbits GF (2016) Ischemia-reperfusion destabilizes rhythmicity in immature atrioventricular pacemakers: A predisposing factor for postoperative arrhythmias in neonate rabbits. Heart Rhythm 13:2348–2355. 10.1016/j.hrthm.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 14.Calabrese V, Bates TE, Stella AM (2000) NO synthase and NO-dependent signal pathways in brain aging and neurodegenerative disorders: the role of oxidant/antioxidant balance. Neurochem Res 25:1315–1341. [DOI] [PubMed] [Google Scholar]
- 15.Markel TA, Wang Y, Herrmann JL, Crisostomo PR, Wang M, Novotny NM, Herring CM, Tan J, Lahm T, Meldrum DR (2008) VEGF is critical for stem cell-mediated cardioprotection and a crucial paracrine factor for defining the age threshold in adult and neonatal stem cell function. Am J Physiol Heart Circ Physiol 295:H2308–2314. 10.1152/ajpheart.00565.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lan C, Song JL, Yan LN, Yang JY, Wen TF, Li B, Xu MQ (2017) Pediatric Donor to Adult Recipients in Donation After Cardiac Death Liver Transplantation: A Single-Center Experience. Transplant Proc 49:1383–1387. 10.1016/j.transproceed.2017.01.088. [DOI] [PubMed] [Google Scholar]
- 17.Paparella D, Yau TM, Young E (2002) Cardiopulmonary bypass induced inflammation: pathophysiology and treatment. An update. Eur J Cardiothorac Surg 21:232–244. [DOI] [PubMed] [Google Scholar]
- 18.Gill RS, Pelletier JS, LaBossiere J, Bigam DL, Cheung PY (2012) Therapeutic strategies to protect the immature newborn myocardium during resuscitation following asphyxia. Can J Physiol Pharmacol 90:689–695. 10.1139/y2012-041. [DOI] [PubMed] [Google Scholar]
- 19.Reeves I, Abribat T, Laramee P, Jasmin G, Brazeau P (2000) Age-related serum levels of insulin-like growth factor-I, -II and IGF-binding protein-3 following myocardial infarction. Growth Horm IGF Res 10:78–84. 10.1054/ghir.2000.0143. [DOI] [PubMed] [Google Scholar]
- 20.Heinen A, Behmenburg F, Aytulun A, Dierkes M, Zerbin L, Kaisers W, Schaefer M, Meyer-Treschan T, Feit S, Bauer I, Hollmann MW, Huhn R (2018) The release of cardioprotective humoral factors after remote ischemic preconditioning in humans is age- and sex-dependent. J Transl Med 16:112 10.1186/s12967-018-1480-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basile DP, Anderson MD, Sutton TA (2012) Pathophysiology of acute kidney injury. Compr Physiol 2:1303–1353. 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans RG, Ince C, Joles JA, Smith DW, May CN, O’Connor PM, Gardiner BS (2013) Haemodynamic influences on kidney oxygenation: clinical implications of integrative physiology. Clin Exp Pharmacol Physiol 40:106–122. 10.1111/1440-1681.12031. [DOI] [PubMed] [Google Scholar]
- 23.Audard V, Moutereau S, Vandemelebrouck G, Habibi A, Khellaf M, Grimbert P, Levy Y, Loric S, Renaud B, Lang P, Godeau B, Galacteros F, Bartolucci P (2014) First evidence of subclinical renal tubular injury during sickle-cell crisis. Orphanet J Rare Dis 9:67 10.1186/1750-1172-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timsit MO, Tullius SG (2011) Hypothermic kidney preservation: a remembrance of the past in the future? Curr Opin Organ Transplant 16:162–168. 10.1097/MOT.0b013e3283446b07. [DOI] [PubMed] [Google Scholar]
- 25.Kusaka J, Koga H, Hagiwara S, Hasegawa A, Kudo K, Noguchi T (2012) Age-dependent responses to renal ischemia-reperfusion injury. J Surg Res 172:153–158. 10.1016/j.jss.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 26.Miya M, Maeshima A, Mishima K, Sakurai N, Ikeuchi H, Kuroiwa T, Hiromura K, Nojima Y (2012) Age-related decline in label-retaining tubular cells: implication for reduced regenerative capacity after injury in the aging kidney. Am J Physiol Renal Physiol 302:F694–702. 10.1152/ajprenal.00249.2011. [DOI] [PubMed] [Google Scholar]
- 27.Qiao X, Chen X, Wu D, Ding R, Wang J, Hong Q, Shi S, Li J, Xie Y, Lu Y, Wang Z (2005) Mitochondrial pathway is responsible for aging-related increase of tubular cell apoptosis in renal ischemia/reperfusion injury. J Gerontol A Biol Sci Med Sci 60:830–839. [DOI] [PubMed] [Google Scholar]
- 28.Clements ME, Chaber CJ, Ledbetter SR, Zuk A (2013) Increased cellular senescence and vascular rarefaction exacerbate the progression of kidney fibrosis in aged mice following transient ischemic injury. PLoS One 8:e70464. 10.1371/journal.pone.0070464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dyess DL, Powell RW, Roberts WS, Tacchi EJ, Swafford AN Jr., Ferrara JJ, Ardell JL (1995) Regional blood flow redistribution in preterm piglets with hemorrhage and resuscitation. J Surg Res 59:29–34. 10.1006/jsre.1995.1128. [DOI] [PubMed] [Google Scholar]
- 30.Maringer K, Sims-Lucas S (2016) The multifaceted role of the renal microvasculature during acute kidney injury. Pediatr Nephrol 31:1231–1240. 10.1007/s00467-015-3231-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pham PT, Pham PC, Wilkinson AH, Lew SQ (2000) Renal abnormalities in sickle cell disease. Kidney Int 57:1–8. 10.1046/j.1523-1755.2000.00806.x. [DOI] [PubMed] [Google Scholar]
- 32.Nath KA, Grande JP, Croatt AJ, Frank E, Caplice NM, Hebbel RP, Katusic ZS (2005) Transgenic sickle mice are markedly sensitive to renal ischemia-reperfusion injury. Am J Pathol 166:963–972. 10.1016/S0002-9440(10)62318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong WS, Moss AA, Federle MP, Cochran ST, London SS (1984) Renal infarction: CT diagnosis and correlation between CT findings and etiologies. Radiology 150:201–205. 10.1148/radiology.150.1.6689761. [DOI] [PubMed] [Google Scholar]
- 34.Deux JF, Audard V, Brugieres P, Habibi A, Manea EM, Guillaud-Danis C, Godeau B, Galacteros F, Stehle T, Lang P, Grimbert P, Audureau E, Rahmouni A, Bartolucci P (2017) Magnetic Resonance Imaging Assessment of Kidney Oxygenation and Perfusion During Sickle Cell Vaso-occlusive Crises. Am J Kidney Dis 69:51–59. 10.1053/j.ajkd.2016.07.027. [DOI] [PubMed] [Google Scholar]
- 35.Alaro D, Bashir A, Musoke R, Wanaiana L (2014) Prevalence and outcomes of acute kidney injury in term neonates with perinatal asphyxia. Afr Health Sci 14:682–688. 10.4314/ahs.v14i3.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humes HD, Liu S (1994) Cellular and molecular basis of renal repair in acute renal failure. J Lab Clin Med 124:749–754. [PubMed] [Google Scholar]
- 37.Csaicsich D, Russo-Schlaff N, Messerschmidt A, Weninger M, Pollak A, Aufricht C (2008) Renal failure, comorbidity and mortality in preterm infants. Wien Klin Wochenschr 120:153–157. 10.1007/s00508-008-0941-5. [DOI] [PubMed] [Google Scholar]
- 38.Yuan SM (2018) Acute kidney injury after pediatric cardiac surgery. Pediatr Neonatol; 10.1016/j.pedneo.2018.03.007. 10.1016/j.pedneo.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Joffe R, Al Aklabi M, Bhattacharya S, Cave D, Calleja T, Garros D, Majesic N, Ryerson L, Morgan C (2018) Cardiac Surgery-Associated Kidney Injury in Children and Renal Oximetry. Pediatr Crit Care Med 19:839–845. 10.1097/PCC.0000000000001656. [DOI] [PubMed] [Google Scholar]
- 40.Hart A, Smith JM, Skeans MA, Gustafson SK, Wilk AR, Robinson A, Wainright JL, Haynes CR, Snyder JJ, Kasiske BL, Israni AK. Organ Procurement and Transplantation Network (OPTN) and Scientific Registry of Transplant Recipients (SRTR). OPTN/SRTR 2016. In: Department of Health and Human Services HRaSA, editor. Rockville, MD: 2017. p. 1–97. [Google Scholar]
- 41.Dale-Shall AW, Smith JM, McBride MA, Hingorani SR, McDonald RA (2009) The relationship of donor source and age on short- and long-term allograft survival in pediatric renal transplantation. Pediatr Transplant 13:711–718. 10.1111/j.1399-3046.2008.01054.x. [DOI] [PubMed] [Google Scholar]
- 42.Kusaka M, Kubota Y, Sasaki H, Fukami N, Fujita T, Hirose Y, Takahashi H, Kenmochi T, Shiroki R, Hoshinaga K (2016) Combined predictive value of the expanded donor criteria for long-term graft survival of kidneys from donors after cardiac death: A single-center experience over three decades. Int J Urol 23:319–324. 10.1111/iju.13045. [DOI] [PubMed] [Google Scholar]
- 43.Basile DP (2007) The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int 72:151–156. 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 44.Bornstein P (1995) Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 130:503–506. 10.1083/jcb.130.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD (2006) CD47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem 281:26069–26080. 10.1074/jbc.M605040200. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Domenech CM, Munoz-Chapuli R (2010) Molecular evolution of nitric oxide synthases in metazoans. Comp Biochem Physiol Part D Genomics Proteomics 5:295–301. 10.1016/j.cbd.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 47.Bauer EM, Qin Y, Miller TW, Bandle RW, Csanyi G, Pagano PJ, Bauer PM, Schnermann J, Roberts DD, Isenberg JS (2010) Thrombospondin-1 supports blood pressure by limiting eNOS activation and endothelial-dependent vasorelaxation. Cardiovasc Res 88:471–481. https://doi.org/cvq218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isenberg JS, Ridnour LA, Perruccio EM, Espey MG, Wink DA, Roberts DD (2005) Thrombospondin-1 inhibits endothelial cell responses to nitric oxide in a cGMP-dependent manner. Proc Natl Acad Sci U S A 102:13141–13146. 10.1073/pnas.0502977102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Isenberg JS, Romeo MJ, Yu C, Yu CK, Nghiem K, Monsale J, Rick ME, Wink DA, Frazier WA, Roberts DD (2008) Thrombospondin-1 stimulates platelet aggregation by blocking the antithrombotic activity of nitric oxide/cGMP signaling. Blood 111:613–623. 10.1182/blood-2007-06-098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rogers NM, Seeger F, Garcin ED, Roberts DD, Isenberg JS (2014) Regulation of soluble guanylate cyclase by matricellular thrombospondins: implications for blood flow. Front Physiol 5:134 10.3389/fphys.2014.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramanathan S, Mazzalupo S, Boitano S, Montfort WR (2011) Thrombospondin-1 and angiotensin II inhibit soluble guanylyl cyclase through an increase in intracellular calcium concentration. Biochemistry 50:7787–7799. 10.1021/bi201060c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD (2010) Thrombospondin-1 inhibits vascular endothelial growth factor receptor-2 signaling by disrupting its association with CD47. J Biol Chem 285:38923–38932. https://doi.org/M110.172304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Csanyi G, Yao M, Rodriguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, Huang X, Kelley EE, Isenberg JS, Pagano PJ (2012) Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler Thromb Vasc Biol 32:2966–2973. 10.1161/ATVBAHA.112.300031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao M, Rogers NM, Csanyi G, Rodriguez AI, Ross MA, St Croix C, Knupp H, Novelli EM, Thomson AW, Pagano PJ, Isenberg JS (2014) Thrombospondin-1 activation of signal-regulatory protein-alpha stimulates reactive oxygen species production and promotes renal ischemia reperfusion injury. J Am Soc Nephrol 25:1171–1186. 10.1681/ASN.2013040433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin-Manso G, Galli S, Ridnour LA, Tsokos M, Wink DA, Roberts DD (2008) Thrombospondin-1 promotes tumor macrophage recruitment and enhances tumor cell cytotoxicity by differentiated U937 cells. Cancer Res 68:7090–7099. 10.1158/0008-5472.CAN-08-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miller YE, Daniels GL, Jones C, Palmer DK (1987) Identification of a cell-surface antigen produced by a gene on human chromosome 3 (cen-q22) and not expressed by Rhnull cells. Am J Hum Genet 41:1061–1070. [PMC free article] [PubMed] [Google Scholar]
- 57.Mawby WJ, Holmes CH, Anstee DJ, Spring FA, Tanner MJ (1994) Isolation and characterization of CD47 glycoprotein: a multispanning membrane protein which is the same as integrin-associated protein (IAP) and the ovarian tumour marker OA3. Biochem J 304:525–530. 10.1042/bj3040525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N (2009) Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci U S A 106:17413–17418. 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohnishi H, Kaneko Y, Okazawa H, Miyashita M, Sato R, Hayashi A, Tada K, Nagata S, Takahashi M, Matozaki T (2005) Differential localization of Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 and CD47 and its molecular mechanisms in cultured hippocampal neurons. J Neurosci 25:2702–2711. 10.1523/JNEUROSCI.5173-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Soto-Pantoja DR, Kaur S, Roberts DD (2015) CD47 signaling pathways controlling cellular differentiation and responses to stress. Crit Rev Biochem Mol Biol 50:212–230. 10.3109/10409238.2015.1014024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, Willingham S, Howard M, Prohaska S, Volkmer J, Chao M, Weissman IL, Majeti R (2015) Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS One 10:e0137345. 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murata Y, Saito Y, Kotani T, Matozaki T (2018) CD47-signal regulatory protein alpha signaling system and its application to cancer immunotherapy. Cancer Sci 109:2349–2357. 10.1111/cas.13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iruela-Arispe ML, Liska DJ, Sage EH, Bornstein P (1993) Differential expression of thrombospondin 1, 2, and 3 during murine development. Dev Dyn 197:40–56. 10.1002/aja.1001970105. [DOI] [PubMed] [Google Scholar]
- 64.Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, Schaefer R, Mason RM (2005) Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia 48:2650–2660. 10.1007/s00125-005-0006-5. [DOI] [PubMed] [Google Scholar]
- 65.Yoo KH, Yim HE, Bae ES, Hong YS (2018) Capillary rarefaction and altered renal development: the imbalance between pro- and anti-angiogenic factors in response to angiotensin II inhibition in the developing rat kidney. J Mol Histol 49:219–228. 10.1007/s10735-018-9762-7. [DOI] [PubMed] [Google Scholar]
- 66.Hugo C, Shankland SJ, Pichler RH, Couser WG, Johnson RJ (1998) Thrombospondin 1 precedes and predicts the development of tubulointerstitial fibrosis in glomerular disease in the rat. Kidney Int 53:302–311. 10.1046/j.1523-1755.1998.00774.x. [DOI] [PubMed] [Google Scholar]
- 67.Hugo C, Daniel C (2009) Thrombospondin in renal disease. Nephron Exp Nephrol 111:e61–66. 10.1159/000198235. [DOI] [PubMed] [Google Scholar]
- 68.Kosugi T, Heinig M, Nakayama T, Connor T, Yuzawa Y, Li Q, Hauswirth WW, Grant MB, Croker BP, Campbell-Thompson M, Zhang L, Atkinson MA, Segal MS, Nakagawa T (2009) Lowering blood pressure blocks mesangiolysis and mesangial nodules, but not tubulointerstitial injury, in diabetic eNOS knockout mice. Am J Pathol 174:1221–1229. 10.2353/ajpath.2009.080605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Daniel C, Schaub K, Amann K, Lawler J, Hugo C (2007) Thrombospondin-1 is an endogenous activator of TGF-beta in experimental diabetic nephropathy in vivo. Diabetes 56:2982–2989. 10.2337/db07-0551. [DOI] [PubMed] [Google Scholar]
- 70.Hafdi Z, Lesavre P, Nejjari M, Halbwachs-Mecarelli L, Droz D, Noel LH (2000) Distribution of alphavbeta3, alphavbeta5 integrins and the integrin associated protein--IAP (CD47) in human glomerular diseases. Cell Adhes Commun 7:441–451. [DOI] [PubMed] [Google Scholar]
- 71.Rogers NM, Thomson AW, Isenberg JS (2012) Activation of Parenchymal CD47 Promotes Renal Ischemia-Reperfusion Injury. J Am Soc Nephrol 23:1538–1550. 10.1681/ASN.2012020137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bijuklic K, Sturn DH, Jennings P, Kountchev J, Pfaller W, Wiedermann CJ, Patsch JR, Joannidis M (2006) Mechanisms of neutrophil transmigration across renal proximal tubular HK-2 cells. Cell Physiol Biochem 17:233–244. 10.1159/000094128. [DOI] [PubMed] [Google Scholar]
- 73.Kurihara H, Harita Y, Ichimura K, Hattori S, Sakai T (2010) SIRP-alpha-CD47 system functions as an intercellular signal in the renal glomerulus. Am J Physiol Renal Physiol 299:F517–527. 10.1152/ajprenal.00571.2009. [DOI] [PubMed] [Google Scholar]
- 74.Shinohara M, Ohyama N, Murata Y, Okazawa H, Ohnishi H, Ishikawa O, Matozaki T (2006) CD47 regulation of epithelial cell spreading and migration, and its signal transduction. Cancer Sci 97:889–895. 10.1111/j.1349-7006.2006.00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nishiyama Y, Tanaka T, Naitoh H, Mori C, Fukumoto M, Hiai H, Toyokuni S (1997) Overexpression of integrin-associated protein (CD47) in rat kidney treated with a renal carcinogen, ferric nitrilotriacetate. Jpn J Cancer Res 88:120–128. 10.1111/j.1349-7006.1997.tb00356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thompson EM, Hughes J, Van Noorden S, Sharpe J, Savill J (1996) Expression of the multifunctional extracellular matrix protein thrombospondin in crescentic glomerulonephritis. J Pathol 178:89–94. . [DOI] [PubMed] [Google Scholar]
- 77.Suzuma K, Takagi H, Otani A, Oh H, Honda Y (1999) Expression of thrombospondin-1 in ischemia-induced retinal neovascularization. Am J Pathol 154:343–354. 10.1016/S0002-9440(10)65281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang S, Wu Z, Sorenson CM, Lawler J, Sheibani N (2003) Thrombospondin-1-deficient mice exhibit increased vascular density during retinal vascular development and are less sensitive to hyperoxia-mediated vessel obliteration. Dev Dyn 228:630–642. 10.1002/dvdy.10412. [DOI] [PubMed] [Google Scholar]
- 79.Shafiee A, Penn JS, Krutzsch HC, Inman JK, Roberts DD, Blake DA (2000) Inhibition of retinal angiogenesis by peptides derived from thrombospondin-1. Invest Ophthalmol Vis Sci 41:2378–2388. [PubMed] [Google Scholar]
- 80.Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY (2003) Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke 34:177–186. [DOI] [PubMed] [Google Scholar]
- 81.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML (2005) Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 111:2935–2942. 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 82.Sezaki S, Hirohata S, Iwabu A, Nakamura K, Toeda K, Miyoshi T, Yamawaki H, Demircan K, Kusachi S, Shiratori Y, Ninomiya Y (2005) Thrombospondin-1 is induced in rat myocardial infarction and its induction is accelerated by ischemia/reperfusion. Exp Biol Med (Maywood) 230:621–630.. [DOI] [PubMed] [Google Scholar]
- 83.Isenberg JS, Maxhimer JB, Powers P, Tsokos M, Frazier WA, Roberts DD (2008) Treatment of ischemia/reperfusion injury by limiting thrombospondin-1/CD47 signaling. Surgery 144:752–761. 10.1016/j.surg.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Causey MW, Salgar S, Singh N, Martin M, Stallings JD (2012) Valproic acid reversed pathologic endothelial cell gene expression profile associated with ischemia-reperfusion injury in a swine hemorrhagic shock model. J Vasc Surg 55:1096–1103 e1051. 10.1016/j.jvs.2011.08.060. [DOI] [PubMed] [Google Scholar]
- 85.Freyberg MA, Kaiser D, Graf R, Buttenbender J, Friedl P (2001) Proatherogenic flow conditions initiate endothelial apoptosis via thrombospondin-1 and the integrin-associated protein. Biochem Biophys Res Commun 286:141–149. 10.1006/bbrc.2001.5314. [DOI] [PubMed] [Google Scholar]
- 86.Ennis WJ, Koh TJ, Urao N, Jan YK, Sui A, Brown K, Borhani M. Ischemia/reperfusion: A potential cause for tissue necrosis Skin Necrosis;DOI: 10.1007/978-3-7091-1241-0_2: Springer-Verlag; Vienna; 2015. p. 9–17. [DOI] [Google Scholar]
- 87.Isenberg JS, Shiva S, Gladwin M (2009) Thrombospondin-1-CD47 blockade and exogenous nitrite enhance ischemic tissue survival, blood flow and angiogenesis via coupled NO-cGMP pathway activation. Nitric Oxide 21:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maxhimer JB, Shih HB, Isenberg JS, Miller TW, Roberts DD (2009) Thrombospondin-1-CD47 blockade following ischemia reperfusion injury is tissue protective. Plast Reconstr Surg 124:1880–1889. 10.1097/PRS.0b013e3181bceec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang HB, Yang J, Ding JW, Chen LH, Li S, Liu XW, Yang CJ, Fan ZX, Yang J (2016) RNAi-Mediated Down-Regulation of CD47 Protects against Ischemia/Reperfusion-Induced Myocardial Damage via Activation of eNOS in a Rat Model. Cell Physiol Biochem 40:1163–1174. 10.1159/000453170. [DOI] [PubMed] [Google Scholar]
- 90.Isenberg JS, Romeo MJ, Maxhimer JB, Smedley J, Frazier WA, Roberts DD (2008) Gene silencing of CD47 and antibody ligation of thrombospondin-1 enhance ischemic tissue survival in a porcine model: implications for human disease. Ann Surg 247:860–868. 10.1097/SLA.0b013e31816c4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xiao ZY, Banan B, Jia J, Manning PT, Hiebsch RR, Gunasekaran M, Upadhya GA, Frazier WA, Mohanakumar T, Lin Y, Chapman WC (2015) CD47 blockade reduces ischemia/reperfusion injury and improves survival in a rat liver transplantation model. Liver Transpl 21:468–477. 10.1002/lt.24059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Talukder MA, Yang F, Shimokawa H, Zweier JL (2010) eNOS is required for acute in vivo ischemic preconditioning of the heart: effects of ischemic duration and sex. Am J Physiol Heart Circ Physiol 299:H437–445. 10.1152/ajpheart.00384.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kerkela R, Karsikas S, Szabo Z, Serpi R, Magga J, Gao E, Alitalo K, Anisimov A, Sormunen R, Pietila I, Vainio L, Koch WJ, Kivirikko KI, Myllyharju J, Koivunen P (2013) Activation of hypoxia response in endothelial cells contributes to ischemic cardioprotection. Mol Cell Biol 33:3321–3329. 10.1128/MCB.00432-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Isenberg JS, Qin Y, Maxhimer JB, Sipes JM, Despres D, Schnermann J, Frazier WA, Roberts DD (2009) Thrombospondin-1 and CD47 regulate blood pressure and cardiac responses to vasoactive stress. Matrix Biol 28:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Narizhneva NV, Razorenova OV, Podrez EA, Chen J, Chandrasekharan UM, DiCorleto PE, Plow EF, Topol EJ, Byzova TV (2005) Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium. Faseb J 19:1158–1160. 10.1096/fj.04-3310fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Isenberg JS, Hyodo F, Pappan LK, Abu-Asab M, Tsokos M, Krishna MC, Frazier WA, Roberts DD (2007) Blocking thrombospondin-1/CD47 signaling alleviates deleterious effects of aging on tissue responses to ischemia. Arterioscler Thromb Vasc Biol 27:2582–2588. 10.1161/ATVBAHA.107.155390. [DOI] [PubMed] [Google Scholar]
- 97.Suzuki K, Wang R, Kubota H, Shibuya H, Saegusa J, Sato T (2005) Kinetics of biglycan, decorin and thrombospondin-1 in mercuric chloride-induced renal tubulointerstitial fibrosis. Exp Mol Pathol 79:68–73. 10.1016/j.yexmp.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 98.Maimaitiyiming H, Zhou Q, Wang S (2016) Thrombospondin 1 Deficiency Ameliorates the Development of Adriamycin-Induced Proteinuric Kidney Disease. PLoS One 11:e0156144. 10.1371/journal.pone.0156144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xie XS, Li FY, Liu HC, Deng Y, Li Z, Fan JM (2010) LSKL, a peptide antagonist of thrombospondin-1, attenuates renal interstitial fibrosis in rats with unilateral ureteral obstruction. Arch Pharm Res 33:275–284. 10.1007/s12272-010-0213-6. [DOI] [PubMed] [Google Scholar]
- 100.Sun D, Ma Y, Han H, Yin Z, Liu C, Feng J, Zhou X, Li X, Xiao A, Yu R (2012) Thrombospondin-1 short hairpin RNA suppresses tubulointerstitial fibrosis in the kidney of ureteral obstruction by ameliorating peritubular capillary injury. Kidney Blood Press Res 35:35–47. 10.1159/000330718. [DOI] [PubMed] [Google Scholar]
- 101.Wang J, Du Z, Zhang W, Han B, Peng C, Chen N (2011) Post liver transplantation acute kidney injury in a rat model of syngeneic orthotopic liver transplantation. Lab Invest 91:1158–1169. 10.1038/labinvest.2011.59. [DOI] [PubMed] [Google Scholar]
- 102.Jung SH, Hwang JH, Kim SE, Young Kyu K, Park HC, Lee HT (2017) The potentiating effect of hTFPI in the presence of hCD47 reduces the cytotoxicity of human macrophages. Xenotransplantation 24 10.1111/xen.12301. [DOI] [PubMed] [Google Scholar]
- 103.Dai H, Friday AJ, Abou-Daya KI, Williams AL, Mortin-Toth S, Nicotra ML, Rothstein DM, Shlomchik WD, Matozaki T, Isenberg JS, Oberbarnscheidt MH, Danska JS, Lakkis FG (2017) Donor SIRPalpha polymorphism modulates the innate immune response to allogeneic grafts. Sci Immunol 2: pii: eaam6202. 10.1126/sciimmunol.aam6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thakar CV, Zahedi K, Revelo MP, Wang Z, Burnham CE, Barone S, Bevans S, Lentsch AB, Rabb H, Soleimani M (2005) Identification of thrombospondin 1 (TSP-1) as a novel mediator of cell injury in kidney ischemia. J Clin Invest 115:3451–3459. 10.1172/JCI25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Isenberg JS, Jia Y, Fukuyama J, Switzer CH, Wink DA, Roberts DD (2007) Thrombospondin-1 inhibits nitric oxide signaling via CD36 by inhibiting myristic acid uptake. J Biol Chem 282:15404–15415. 10.1074/jbc.M701638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zager RA, Johnson AC, Hanson SY, Shah VO (2003) Acute tubular injury causes dysregulation of cellular cholesterol transport proteins. Am J Pathol 163:313–320. 10.1016/S0002-9440(10)63655-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Isenberg JS, Yu C, Roberts DD (2008) Differential effects of ABT-510 and a CD36-binding peptide derived from the type 1 repeats of thrombospondin-1 on fatty acid uptake, nitric oxide signaling, and caspase activation in vascular cells. Biochem Pharmacol 75:875–882. 10.1016/j.bcp.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Isenberg JS, Romeo MJ, Abu-Asab M, Tsokos M, Oldenborg A, Pappan L, Wink DA, Frazier WA, Roberts DD (2007) Increasing survival of ischemic tissue by targeting CD47. Circ Res 100:712–720. 10.1161/01.RES.0000259579.35787.4e. [DOI] [PubMed] [Google Scholar]
- 109.Isenberg JS, Pappan LK, Romeo MJ, Abu-Asab M, Tsokos M, Wink DA, Frazier WA, Roberts DD (2008) Blockade of thrombospondin-1-CD47 interactions prevents necrosis of full thickness skin grafts. Ann Surg 247:180–190. 10.1097/SLA.0b013e31815685dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Isenberg JS, Annis DS, Pendrak ML, Ptaszynska M, Frazier WA, Mosher DF, Roberts DD (2009) Differential interactions of thrombospondin-1, −2, and −4 with CD47 and effects on cGMP signaling and ischemic injury responses. J Biol Chem 284:1116–1125. 10.1074/jbc.M804860200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang J, Long Q, Zhang W, Chen N (2012) Protective effects of exogenous interleukin 18-binding protein in a rat model of acute renal ischemia-reperfusion injury. Shock 37:333–340. 10.1097/SHK.0b013e318240bdc8. [DOI] [PubMed] [Google Scholar]
- 112.Kim J, Kim C, Kim TS, Bang SI, Yang Y, Park H, Cho D (2006) IL-18 enhances thrombospondin-1 production in human gastric cancer via JNK pathway. Biochem Biophys Res Commun 344:1284–1289. 10.1016/j.bbrc.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 113.Rogers NM, Yao M, Novelli EM, Thomson AW, Roberts DD, Isenberg JS (2012) Activated CD47 regulates multiple vascular and stress responses: implications for acute kidney injury and its management. Am J Physiol Renal Physiol 303:F1117–1125. 10.1152/ajprenal.00359.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kaur S, Soto-Pantoja DR, Stein EV, Liu C, Elkahloun AG, Pendrak ML, Nicolae A, Singh SP, Nie Z, Levens D, Isenberg JS, Roberts DD (2013) Thrombospondin-1 signaling through CD47 inhibits self-renewal by regulating c-Myc and other stem cell transcription factors. Sci Rep 3:1673 10.1038/srep01673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rogers NM, Zhang ZJ, Wang JJ, Thomson AW, Isenberg JS (2016) CD47 regulates renal tubular epithelial cell self-renewal and proliferation following renal ischemia reperfusion. Kidney Int 90:334–347. 10.1016/j.kint.2016.03.034. [DOI] [PubMed] [Google Scholar]
- 116.van Beek EM, Zarate JA, van Bruggen R, Schornagel K, Tool AT, Matozaki T, Kraal G, Roos D, van den Berg TK (2012) SIRPalpha controls the activity of the phagocyte NADPH oxidase by restricting the expression of gp91(phox). Cell Rep 2:748–755. 10.1016/j.celrep.2012.08.027. [DOI] [PubMed] [Google Scholar]
- 117.Meijles DN, Sahoo S, Al Ghouleh I, Amaral JH, Bienes-Martinez R, Knupp HE, Attaran S, Sembrat JC, Nouraie SM, Rojas MM, Novelli EM, Gladwin MT, Isenberg JS, Cifuentes-Pagano E, Pagano PJ (2017) The matricellular protein TSP1 promotes human and mouse endothelial cell senescence through CD47 and Nox1. Sci Signal 10 10.1126/scisignal.aaj1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lario S, Bescos M, Campos B, Mur C, Luque P, Alvarez R, Campistol JM (2007) Thrombospondin-1 mRNA expression in experimental kidney transplantation with heart-beating and non-heart-beating donors. J Nephrol 20:588–595. [PubMed] [Google Scholar]
- 119.Delpech PO, Thuillier R, Le Pape S, Rossard L, Jayle C, Billault C, Goujon JM, Hauet T (2014) Effects of warm ischaemia combined with cold preservation on the hypoxia-inducible factor 1alpha pathway in an experimental renal autotransplantation model. Br J Surg 101:1739–1750. 10.1002/bjs.9611. [DOI] [PubMed] [Google Scholar]
- 120.Mas VR, Mas LA, Archer KJ, Yanek K, King AL, Gibney EM, Cotterell A, Fisher RA, Posner M, Maluf DG (2007) Evaluation of gene panel mRNAs in urine samples of kidney transplant recipients as a non-invasive tool of graft function. Mol Med 13:315–324. 10.2119/2007-00017.Mas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hotchkiss H, Chu TT, Hancock WW, Schroppel B, Kretzler M, Schmid H, Liu Y, Dikman S, Akalin E (2006) Differential expression of profibrotic and growth factors in chronic allograft nephropathy. Transplantation 81:342–349. 10.1097/01.tp.0000195773.24217.95. [DOI] [PubMed] [Google Scholar]
- 122.Lin Y, Manning PT, Jia J, Gaut JP, Xiao Z, Capoccia BJ, Chen CC, Hiebsch RR, Upadhya G, Mohanakumar T, Frazier WA, Chapman WC (2014) CD47 Blockade Reduces Ischemia-Reperfusion Injury and Improves Outcomes in a Rat Kidney Transplant Model. Transplantation 98:394–401. 10.1097/TP.0000000000000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Xu M, Wang X, Banan B, Chirumbole DL, Garcia-Aroz S, Balakrishnan A, Nayak DK, Zhang Z, Jia J, Upadhya GA, Gaut JP, Hiebsch R, Manning PT, Wu N, Lin Y, Chapman WC (2018) Anti-CD47 monoclonal antibody therapy reduces ischemia-reperfusion injury of renal allografts in a porcine model of donation after cardiac death. Am J Transplant 18:855–867. 10.1111/ajt.14567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hugo C, Kang DH, Johnson RJ (2002) Sustained expression of thrombospondin-1 is associated with the development of glomerular and tubulointerstitial fibrosis in the remnant kidney model. Nephron 90:460–470. 10.1159/000054735. [DOI] [PubMed] [Google Scholar]
- 125.Hohenstein B, Daniel C, Wittmann S, Hugo C (2008) PDE-5 inhibition impedes TSP-1 expression, TGF-beta activation and matrix accumulation in experimental glomerulonephritis. Nephrol Dial Transplant 23:3427–3436. 10.1093/ndt/gfn319. [DOI] [PubMed] [Google Scholar]
- 126.Aibara N, Ohyama K, Hidaka M, Kishikawa N, Miyata Y, Takatsuki M, Eguchi S, Kuroda N (2018) Immune complexome analysis of antigens in circulating immune complexes from patients with acute cellular rejection after living donor liver transplantation. Transpl Immunol 48:60–64. 10.1016/j.trim.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 127.Gholamin S, Mitra SS, Feroze AH, Liu J, Kahn SA, Zhang M, Esparza R, Richard C, Ramaswamy V, Remke M, Volkmer AK, Willingham S, Ponnuswami A, McCarty A, Lovelace P, Storm TA, Schubert S, Hutter G, Narayanan C, Chu P, Raabe EH, Harsh Gt, Taylor MD, Monje M, Cho YJ, Majeti R, Volkmer JP, Fisher PG, Grant G, Steinberg GK, Vogel H, Edwards M, Weissman IL, Cheshier SH (2017) Disrupting the CD47-SIRPalpha anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci Transl Med 9 10.1126/scitranslmed.aaf2968. [DOI] [PubMed] [Google Scholar]