

Abstract
Two multiscale algorithms for stochastic simulations of reaction–diffusion processes are analysed. They are applicable to systems which include regions with significantly different concentrations of molecules. In both methods, a domain of interest is divided into two subsets where continuous-time Markov chain models and stochastic partial differential equations (SPDEs) are used, respectively. In the first algorithm, Markov chain (compartment-based) models are coupled with reaction–diffusion SPDEs by considering a pseudo-compartment (also called an overlap or handshaking region) in the SPDE part of the computational domain right next to the interface. In the second algorithm, no overlap region is used. Further extensions of both schemes are presented, including the case of an adaptively chosen boundary between different modelling approaches.
Keywords: Stochastic reaction–diffusion systems, Chemical reaction networks, Markov chain, Gillespie algorithm, Multiscale modelling, Stochastic partial differential equations
Introduction
Stochastic models of well-mixed chemical systems are traditionally formulated in terms of continuous-time Markov chains, which can be simulated using the Gillespie stochastic simulation algorithm (SSA) (Gillespie 1977) or its equivalent formulations (Gibson and Bruck 2000; Cao et al. 2004; Klingbeil et al. 2011). These algorithms provide statistically exact sample paths of stochastic chemical models described by the corresponding chemical master equation (CME). However, they can be computationally expensive for larger chemical systems, because they explicitly simulate each occurrence of each chemical reaction. A number of approaches have been developed in the literature to decrease the computational intensity of SSAs. Taking into account separation of timescales, chemical reaction networks can be simplified by model reduction before they are simulated (Kang 2012; Kang and Kurtz 2013; Kang et al. 2014; Kim et al. 2017; Kang et al. 2019). The idea of model reduction can also be used to develop computational methods which efficiently estimate quantities of interest from stochastic simulations (Cao et al. 2005a, b; Erban et al. 2006; Cotter et al. 2011). Another approach is to describe the molecular populations in terms of their concentrations that change continuously (rather than treating them as discrete random variables). This can be achieved by the chemical Langevin equation, which is a stochastic differential equation (SDE) acting as a bridge between discrete SSAs and deterministic reaction rate equations (Kurtz 1976, 1978; Gillespie 2000). Efficient algorithms which make use of the SDE approximations have been developed for the simulation of chemical systems especially when they include processes occurring on different timescales (Haseltine and Rawlings 2002; Salis and Kaznessis 2005; Griffith et al. 2006; Cotter and Erban 2016). More recently, the SDE approximations have been extensively used to develop hybrid algorithms which use boths SSAs and SDEs for different components of the studied systems (Liu et al. 2012; Ganguly et al. 2015; Duncan et al. 2016; Altintan et al. 2016). The chemical Fokker–Planck equation corresponding to the chemical Langevin equation can also be used to efficiently estimate quantities of interest from stochastic models (Erban et al. 2009; Cotter et al. 2013; Liao et al. 2015; Cucuringu and Erban 2017).
In this paper, we consider spatially distributed (reaction–diffusion) models which can be described in terms of the reaction–diffusion master equation (RDME) (Erban et al. 2007). A spatial domain is discretized into compartments (which are assumed to be well mixed), and diffusion is modelled as a jump process between neighbouring compartments (Erban and Chapman 2009; Kang et al. 2012b; Hu et al. 2014). In the literature, the RDME approach has been adapted to model and simulate spatially distributed systems using uniform meshes (equivalently, subvolumes or compartments) (Stundzia and Lumsden 1996; Elf et al. 2003; Lampoudi et al. 2009), nonuniform meshes (Bernstein 2005) or complex geometries (Isaacson and Peskin 2006). The resulting compartment-based model can be simulated by the Gillespie SSA. Compartment-based reaction–diffusion approaches have been used to model several intracellular processes, including Min oscillations in E. coli (Fange and Elf 2006; Arjunan and Tomita 2010), ribosome biogenesis (Earnest et al. 2015, 2016), actin dynamics in filopodia (Zhuravlev and Papoian 2009; Erban et al. 2014) and pattern formation in morphogen signalling pathways (Kang et al. 2012a). They have also been implemented in a number of software packages including MesoRD (Hattne et al. 2005), URDME (Engblom et al. 2009), STEPS (Wils and De Schutter 2009), SmartCell (Ander et al. 2004), Lattice Microbes (Roberts et al. 2013) and Smoldyn (Robinson et al. 2015). As in the case of the simulation of well-mixed systems, the Langevin approach provides an approximation of the compartment-based model which can reduce the computational intensity of simulations. Spatial Langevin approaches (Kalantzis 2009; Ghosh et al. 2015; Bhattacharjee et al. 2015) and stochastic partial differential equations (SPDEs) (Dogan and Allen 2011; Alexander et al. 2002, 2005; Atzberger 2010; Kim et al. 2017) have been suggested to model stochastic reaction–diffusion systems. A hybrid method has also been introduced using the Langevin approximation for diffusion coupled with the compartment-based model for reactions (Lo et al. 2016).
In the thermodynamic limit (of large populations), compartment-based models lead to reaction–diffusion partial differential equations (PDEs) which are written in terms of spatio-temporal concentrations of chemical species. This property can be exploited to design multiscale (hybrid) algorithms which use the compartment-based Markov chain model in a subset of the simulated system and apply reaction–diffusion PDEs in other parts (Kalantzis 2009; Ferm et al. 2010; Yates and Flegg 2015; Spill et al. 2015; Harrison and Yates 2016). Other hybrid methods have also been developed in the literature including methods which couple more detailed Brownian dynamics (molecular-based) approaches with the compartment-based method (Flegg et al. 2012; Klann et al. 2012; Flegg et al. 2015; Dobramysl et al. 2016) or with reaction–diffusion PDEs (Franz et al. 2013; Schaff et al. 2016; Bakarji and Tartakovsky 2017).
In this paper, we analyse two multiscale algorithms which couple compartment-based models with suitably discretized SPDEs. They can be used when a large number of molecules of some species are located in parts of the computational domain. In the region with a small number of molecules, we use a compartment-based model written as a continuous-time Markov chain. In other regions, we use SPDEs derived from the Markov process. The goal of this multiscale methodology is to get an approximation of the spatio-temporal statistics which we would obtain by running the underlying Markov chain model in the entire computational domain. The paper is organized as follows. In Sect. 2, we present the derivation of the SPDE description from the compartment-based model. In Sect. 3, two multiscale schemes are presented. An illustrative example with a static boundary between the SPDE and Markov chain subdomains is studied in Sect. 4. The algorithm is extended to a time-dependent interface in Sect. 5. In Sect. 6, we discuss an example with multiple species.
From Continuous-Time Markov Jump Processes to Stochastic Partial Differential Equations
We consider a system of N chemically reacting species , ..., , which are diffusing (with diffusion constants , ) in the bounded domain . We use a compartment-based stochastic reaction–diffusion model (Erban et al. 2007), i.e. we divide the domain into K compartments , , and model the diffusion as a jump process between neighbouring compartments. In order to simplify the analysis, we consider that is an elongated pseudo-one-dimensional domain , where , as shown in Fig. 1a. Compartments are rectangular cuboids with the volume where for . Let , , , be the number of molecules of the ith chemical species in the kth compartment at time t. Then, is an N-dimensional column vector with each component representing the number of molecules of the corresponding species in the kth compartment at time t. We define
which is a KN-dimensional column vector and denotes the transpose of a vector. We assume that the chemical system is subject to M chemical reactions with , , being the corresponding N-dimensional stoichiometric vector. Let , , , be a KN-dimensional stoichiometric vector which gives a net molecule change during each occurrence of the jth reaction in the kth compartment. Let (resp. ), , be a KN-dimensional stoichiometric vector which gives a net molecule change during diffusion of the ith species from the kth compartment to the th (resp. th) compartment. Let
be the propensity function of the jth chemical reaction in the kth compartment, i.e. is the probability that the jth reaction occurs in the kth compartment during the time given that the current state at time t is . We denote by , , , a random process which counts the number of times the jth reaction occurs in the kth compartment up to time t. Then,
| 1 |
where are independent unit Poisson processes. We define (resp. ), , (resp. ), random processes counting the numbers of times that one molecule of the ith species in the kth compartment diffuses to the th compartment (resp. to the th compartment) up to time t. Then,
| 2 |
where are independent unit Poisson processes. The governing equation for the state vector is
| 3 |
When the propensities are large (Kurtz 1978), the counting processes in Eqs. (1)–(2) can be approximated as
where and are standard Brownian motions. Using for and changing the index in the last term of Eq. (3), governing Eq. (3) can be approximated by the following SDE (Gillespie 2000; Kurtz 1978)
| 4 |
Since and terms always appear together in Eq. (4), and since the sum of independent normal random variables is normally distributed, Eq. (4) can be rewritten as
| 5 |
where is a standard Brownian motion. Let be the volume of each compartment, and define as a concentration vector for species at time t. Define
Dividing Eq. (5) by , we get
| 6 |
where . The second part of Eq. (6) is consistent with the discretized Langevin scheme for a diffusion equation, as studied in Alexander et al. (2002). We rewrite Eq. (6) using the fact that reaction happens among species in the same compartment and that diffusion occurs between neighbouring compartments. Differentiating Eq. (6), the concentration of the chemical species in the kth compartment satisfies
| 7 |
where are diagonal matrices with on its diagonal for , and is an indicator function. In Eq. (7), is an N-dimensional stoichiometric vector of the jth reaction for , and is a diagonal matrix which has diffusion constants of individual species on its diagonal, i.e.
We approximate white noise processes in Eq. (7) as
where , are spatio-temporal white noise processes (Walsh 1986), i.e.
is normally distributed with zero mean and variance . Matrices are diagonal matrices where diagonal entries are independent spatio-temporal white noise processes. Then, Eq. (7) is a solution of a discretized version of a SPDE in space which can be formally written in the following form
| 8 |
where is a spatio-temporal concentration related to by
The reaction term in Eq. (8) is related to by
Note that Eqs. (6)–(7) are discretized versions of Eq. (8), but the compartment-based model in (3) breaks down as as discussed in Section 2.2 of Engblom et al. (2009). The SPDE in Eq. (8) is consistent to the ones in the previous work (see Equation 1 in Kim et al. 2017 and Equation 3.24 in Dogan and Allen 2011). For more details, see derivations of the SPDE for diffusion in Section 3.1 of Dogan and Allen (2011) and the general version (Equations 8.2.54–8.2.56) in Sections 8.1-8.2 of Gardiner (2004).
Fig. 1.
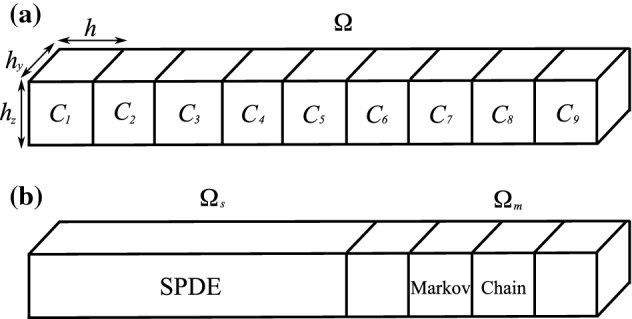
a A schematic illustration of the elongated domain for. b A schematic illustration of the multiscale setup
Multiscale Algorithms Combining Compartment-Based Models with SPDEs
In this section, we present a multiscale approach which uses both SPDEs and Markov chain models. We develop two algorithms, denoted Scheme 1 and Scheme 2 in what follows, which are applied to illustrative examples in Sects. 4, 5 and 6. Considering the same set up as in Sect. 2, we study a system of N chemically reacting species , ..., , which are diffusing (with diffusion constants , ) in an elongated domain , where , given in Fig. 1. The domain is divided into K compartments (rectangular cuboids) with for .
The main goal of this paper is to replace the Markov chain description in a part of the computational domain by the corresponding SPDEs. Let us consider that we use the SPDE in Eq. (8) in the domain where and ; i.e. we consider that the first compartments are described by a suitable discretization of the SPDE in Eq. (8) (see Fig. 1b). We only use the Markov chain model for the remaining compartments, i.e. in subdomain . In this section, we develop an appropriate boundary condition on the interface I between and .
In order to design the numerical scheme, the SPDE in Eq. (8) needs to be appropriately discretized. We denote by the mesh size used in the discretization of the SPDE. There are two important cases: (i) and (ii) . In this section, we focus on case (ii), because we are interested in coupling the SPDE in Eq. (8) with Markov chain models. The case (i) is important when one uses discretized SPDEs to design efficient multiscale schemes, but this introduces additional discretization errors. We will discuss case (i) in Sect. 7. In , each compartment of size h is discretized into grid points () with each grid size equal to . In the remaining part of the computational domain , the compartment-based model is used. The state of the system of the multiscale model is described by vectors , . The vector for represents species ‘numbers’ in the mesh interval in the SPDE region , i.e. it is related to spatio-temporal concentration used in the SPDE description by
The vector for represents species numbers in in the Markov chain region , i.e. it is related to the state vector used in the Markov chain description by
We consider two different schemes to describe transfer of molecules near the interface I coupling discretized SPDEs and the Markov chain model, as shown in Fig. 2. Without loss of generality, both schemes are introduced for diffusion, because the description of reactions does not influence the transfer of molecules across the interface I. In Scheme 1, we assume that there is a virtual compartment, , in , where the molecules are partially treated using a compartment-based approach. Such overlap (handshaking) regions are common in many multiscale methodologies, including coupling molecular dynamics with Brownian dynamics simulations (Erban 2014, 2016), Brownian dynamics with PDEs (Franz et al. 2013), or in atomistic to continuum coupling methods (Miller and Tadmor 2009). We define a state vector
which is a -dimensional column vector. Scheme 1 is described using the following evolution equation for state vector :
| 9 |
where the first sum on the right-hand side represents diffusion in (compare with Eq. (5) replacing h by ). Note that the indicator function is used to make sure the term inside the square root not being negative. Here the symbols describe -dimensional stoichiometric vectors. The second and third sums represent diffusion in the compartment-based region, , where are independent unit Poisson processes (compare with Eq. (2)). The last two sums represent transition of a molecule from to and from to , respectively. A molecule in in the boundary compartment, , jumps to the SPDE domain with a rate . A molecule which jumps is placed to one of the mesh points in the overlap compartment, . To describe this process in Eq. (9), we have defined indicator functions
where are independent random variables uniformly distributed on interval for each t and i. Stoichiometric vectors, for , , give changes due to the diffusion of the ith species between the th SPDE discretization point in and the compartment across the interface I. Transition of a molecule from to is described by the last term of Eq. (9) using time-changed Poisson processes. A molecule, anywhere in the overlap compartment , can be transferred with a rate . The corresponding molecule is then randomly subtracted from one of discretization grid points which are in . Note that the molecular copy number, , in the last term of Eq. (9) can be non-integer value due to the non-integer concentration in . To prevent being negative due to the molecular transfer from to , another indicator function is used in the last term of Eq. (9) to set the propensity as zero if the total molecular copy number in is less than 1.
Fig. 2.

Schematic diagrams of a Scheme 1 and b Scheme 2 describing molecular transfer between and . Note that the size of a virtual compartment in is h in panel (a)
Scheme 2 is described in terms of two unknown parameters, denoted and , by the following evolution equation for the state vector :
| 10 |
The first three sums on the right hand side in Eq. (10) are identical to those in Eq. (9). The fourth and fifth sums describe molecular transfer between the last grid point in and the boundary compartment . A molecule in in the boundary compartment, , jumps to the last grid point of the SPDE domain with rate , and the transfer rate in the opposite direction is . Note that in the fifth term of Eq. (10) can be non-integer value due to the non-integer concentration in . To prevent being negative due to the molecular transfer from to , we use an indicator function to set the propensity as zero if the molecular copy number in the last grid point in is less than 1.
To determine parameters and of Scheme 2, we use the discretization of the PDE for diffusion using a finite volume approximation (Bernstein 2005). It gives the jump coefficient of the ith species from the jth compartment to the neighbouring th compartment as , where is the length of the jth compartment and and are the centres of the jth and th compartments, respectively. Considering the size of the domain allowed for molecule transfer across the interface in Scheme 2, we set . We take for the jump coefficient from to and for the jump coefficient from to . Then, we match the jump coefficients to the rate constants for jump across the interface given in Eq. (10) to derive the following formula for the parameters of Scheme 2
The multiscale algorithm for Scheme 1 for the case of diffusion only is given in Table 1. We denote a propensity of diffusion of the ith species in the th compartment in to the left (resp. right) as , for , (resp. , for ) for . This definition also includes the propensity of a diffusive jump of the ith species from the Markov chain domain, given as We denote a propensity of diffusive jump of the ith species from the SPDE domain by
Then, we define total propensity in
| 11 |
Total propensity is used in steps [A] and [B] in the pseudocode in Table 1 to select time when the next event occurs in . The pseudocode denotes the time of the next update in each subdomain as and , and the current time as t. In step [B], we update the compartment-based part of the system. In step [C], we update the SPDE part of the system by
| 12 |
where are independent normally distributed random numbers with zero mean and unit variance.
Table 1.
Pseudocode for the multiscale reaction–diffusion algorithm with Scheme 1 applied to simulation of diffusion
| [A] | Set and . Initialize species numbers, , in and . Then, generate random numbers and uniformly distributed in (0, 1). Set so that , where is defined in Eq. (11). Set the next time when the diffusion occurs in as |
| [B] | If |
| Set | |
| Use to determine which diffusive jump occurs. Each diffusive jump to the left (resp. to the right) has the probability (resp. ) to occur | |
| If the selected diffusive jump only includes internal compartments in , update species numbers in the corresponding compartments | |
| If the diffusion occurs across the interface from to , update the species number in by transferring one molecules from to the corresponding grid point in | |
| If the diffusion occurs across the interface from to , update the species number in by adding one molecule and subtracting one from the corresponding grid point in | |
| Generate random numbers and uniformly distributed in (0, 1). Set so that , where is defined in Eq. (11). Set the next time when the diffusion occurs in as | |
| [C] | If |
| Set | |
| Use Eq. (12) to update the SPDE part of the system from t to | |
| Set the next time of the update of the SPDE part as | |
| [D] | Repeat steps [B]–[C] until the simulation ends |
Application: Static Boundary
In this section, we apply the multiscale approach to examples in which we know a priori the position of the boundary I between and . Generalization to a more complicated case with a moving boundary is presented in Sect. 5.
A Morphogen Gradient Model
We consider a morphogen gradient model in . It consists of one chemical species S, i.e. is a scalar describing the number of molecules of S in . The state of the Markov chain model is described by the K-dimensional column vector . Morphogen is subject to diffusion which is described by Eq. (2). There are also two reactions in the system. Morphogen, S, is produced in the first compartment with rate J, i.e. the propensity is . Morphogen degrades everywhere with rate , i.e. with propensity for . In all stochastic simulations of the morphogen gradient model, we assume that 500 morphogen molecules are initially uniformly distributed in the half of the domain, . The parameters are given in Table 2.
Table 2.
Parameter values in the morphogen gradient model studied in Sect. 4.1
| Notations | Description | Values |
|---|---|---|
| L | Length of the domain | |
| D | Diffusion coefficient | |
| Degradation rate | ||
| J | Production rate | |
| Spatial discretization in | ||
| h | Spatial discretization in | |
| , | Spatial discretization in y and z directions | |
| Time discretization for SPDE |
Denoting c(x, t) the morphogen density at point x and time t, the deterministic model can be written as PDE
where D is the diffusion constant of morphogen S. We apply the multiscale approach using both schemes developed in Sect. 3. Since the morphogen is produced at the left end, the morphogen has a decreasing gradient as it goes towards L. Therefore, we split the spatial domain in half and set the left half as and the right half as , i.e. . The -dimensional state vector of the multiscale model is denoted . Note that the morphogen molecules are only produced in the first discretization mesh point with size in . In Fig. 3, we simulate the morphogen gradient model using Scheme 1 of the multiscale algorithm. We calculate realizations of the sample paths of the stochastic process and present mean and standard deviations of the morphogen numbers in at different times, . Morphogen numbers in grid points of are summed so that they can be compared to the numbers in the underlying Markov chain model. We compare the results with mean and standard deviations of the morphogen numbers which we calculate analytically using matrix analysis for reaction–diffusion Markov chain models (Gadgil et al. 2005; Kang et al. 2012b). In Fig. 3, morphogen numbers in (resp. in ) are expressed as green bars (resp. blue bars). Error bars represent one standard deviations from the mean number of morphogens in each compartment. Mean and standard deviations of the morphogen numbers from the analytic solution are drawn as a red line and blue dotted lines. The results using the multiscale algorithm match perfectly to the ones from the exact solution.
Fig. 3.

Comparison between mean numbers of morphogens and their standard deviations from the mean using the analytic solution (red lines and blue dotted lines) and Scheme 1 (green bars and blue bars for the means in and , respectively, and error bars for the standard deviations) (Color figure online)
In Fig. 4, we present relative errors of the means and standard deviations of the number of molecules between the Markov chain model and multiscale model. The analytic solution is used for the statistics of the Markov chain model, and both schemes are applied numerically for the multiscale approaches. Errors are defined as
| 13 |
where and represent a mean and standard deviation. In Fig. 4a, red and green lines represent and at time using Scheme 1, respectively, and blue and purple lines are for Scheme 2. We observe that the relative errors in Eq. (13) are less than in the entire simulation domain. In Fig. 4b, we compare the maximum absolute values of the relative errors defined in Eq. (13) with and fixed compartment size h where . In both schemes, the relative errors are in a range of less than except for the case when with Scheme 2. The relative errors in the mean and standard deviation become significantly larger when we apply the multiscale algorithm using Scheme 2 with . In this case, the mean in gets larger than the mean in which shows a bias in the method for larger values of (the exact mean number of molecules decreases along the x-axis). We provide an explanation of this phenomenon in the next section.
Fig. 4.

a Errors and given by Eq. (13) are computed at time . b The maximum absolute values of the errors and given by Eq. (13) are computed at time with a static boundary and different values of . The maximum value of the errors is taken over all region, . Red and green lines are relative errors of the means and standard deviations between the analytic solution of the Markov chain model and Scheme 1. Blue and purple lines are relative errors between the Markov chain model and Scheme 2 (Color figure online)
A Diffusion Model with Two Compartments
In Sect. 4.1, we have observed that the error of Scheme 2 increases when we decrease the ratio of the numerical discretization in and the compartment size. When , the mean number of molecules of the morphogen does not have a decreasing gradient across the interface I in Scheme 2. To investigate this numerical error, we consider diffusion in a theoretical two-compartment model. It is similar to the one in Sect. 4.1, but we set so that there is no flux or degradation of the morphogen. We use and () so that and . Then, each region consists of one compartment, , and is an -dimensional vector.
In Fig. 5a, we present simulation results of the two-compartment model using Scheme 1 (red line) and Scheme 2 (green line) with 20, 30, 40, 50 and compare them to the simulation result of the Markov chain model using the Gillespie SSA (purple line). The Markov chain model has numerical grid points where the first ones are with size and the last one with size h. Diffusion of molecules is simulated by jumps from grid points to their nearest neighbours, i.e. the numerical meshes in the Markov chain model are coupled by diffusion in the same way as it is done in Scheme 2. Applying both multiscale algorithms and the Gillespie SSA, we compare the mean morphogen numbers in the second compartment computed from 100 realizations of simulation. Using 50 molecules in total, the exact value of the mean numbers of molecules in , , is 25. Notice that Scheme 1 and the Gillespie SSA with two mesh sizes correctly approximate the means. However, Scheme 2 overestimates the mean morphogen number in as gets large. To understand where the numerical error arises, we also simulate Scheme 2 without the noise term in the SPDEs (marked as a blue line in Fig. 5a), i.e. we remove the term with a square root in Eq. (10).
Fig. 5.

a The mean number of morphogens in at time . Different simulation methods are compared with , 20, 30, 40, 50: the Gillespie SSA with multigrid discretization ( grid points with size and one grid point with size h), Scheme 1, Scheme 2 and “Scheme 2 with no noise” due to diffusion in . b The probability distribution of the normalized morphogen number in with Scheme 2. The probability distributions are computed for , and compared among the cases with , 50 at time , . Initially, 50 molecules are located in in panels (a, b) (Color figure online)
In Fig. 5a, we observe that the mean morphogen number in , , is underestimated as increases when we use Scheme 2 without noise term in the SPDEs. Note that , , always have non-negative integer values due to no noise term in modified Eq. (10). The molecular transfer from to occurs when . However, the frequency of this transfer is not sufficient as gets large, which lowers . On the other hand, with noise terms included in Eqs. (9) and (10), there are more chances that for some due to large fluctuations with a small number of molecules as gets large. Then, it is more frequent that due to the fact that and for some . More frequent molecular transfer from to causes overestimation of the mean morphogen number in in Scheme 2.
In Fig. 5b, we compare distributions of the morphogen numbers when and 50. The distributions are computed from 1000 realizations of simulation when and . Each distribution is computed over all , so that we can display an overall range of the morphogen number in each discretization of . Each is normalized by so that the distributions can be compared for different ’s. The normalized mean morphogen number (density) in decreases significantly in both cases with and 50 as time evolves. On the other hand, the variance of the morphogen density is much greater for than for at due to the lower morphogen number in each discretization of . Therefore, we conclude that the error in Scheme 2 strongly depends on the size of fluctuations close to the interface. On the other hand, the molecular transfer from to is decided by in Scheme 1. This makes Scheme 1 more robust than Scheme 2 for large values of .
Application: Moving Boundary
In some applications (Robinson et al. 2014), it is difficult to decide a position of the interface I a priori. In this section, we extend the presented algorithm to the case when the location of the interface I(t) between and moves in time, based on the number of molecules in each part of the domain. The multiscale approach with the adaptive interface is applied to the example introduced in Sect. 4.
The adaptive algorithm is described in Table 3. Following (Robinson et al. 2014), we introduce two thresholds denoted and (), and one integer parameter . We initialize the position of the interface in step [A’], i.e. we initially model the whole domain using the detailed compartment-based approach. We run the original Scheme 1 until time . We check whether the interface I(t) should be moved in step [C’]. If the number of molecules in the compartment next to the interface in is smaller than , a compartment-based model is used in that region. On the other hand, if the number of molecules in the boundary compartment next to interface I(t) in is larger than threshold , the corresponding compartment is transferred to the SPDE region where the molecules are redistributed uniformly in grid points. Due to the uniform redistribution of the molecules, rapid changing of the interface I(t) introduces more errors. Note that in Scheme 1 with a fixed boundary, one molecule has been chosen randomly from discretizations of in and transferred to in . Similarly, we have taken one molecule from and transferred the molecule to the randomly chosen SPDE mesh point in . However, in the adaptive algorithm, we modify the setting of Scheme 1 so that a molecule is taken uniformly from the entire region of and transferred to , i.e. molecule is subtracted in all SPDE grid points of . Similarly when the molecule is transferred from to , molecule is added in all grid points of . Without this modification of the setting in Scheme 1, the appropriate level of the morphogen gradient is not formed in Fig. 6.
Table 3.
Pseudocode for the adaptive multiscale reaction–diffusion algorithm with Scheme 1 applied to simulation of diffusion
| [A’] | Set , and . Initialize species numbers, , in and , and the boundary location, I(0). Then, generate random numbers and uniformly distributed in (0, 1). Set so that , where is defined in Eq. (11). Set the next time when the diffusion occurs in as |
| [B’] | Repeat steps [B]–[C] of algorithm in Table 1 until time |
| [C’] | Update position of interface I(t), if necessary, by |
| If , set | |
| If and , set | |
| Increase k by 1, i.e. | |
| [D’] | Repeat steps [B’]–[C’] until the simulation ends |
Fig. 6.

Comparison between one realization of the number of morphogens using Scheme 1 with a moving interface, given in Table 3 (green bars and blue bars for the morphogen numbers in and , respectively) and the mean number of molecules given analytically by the compartment-based model (red dots). A blue dotted line represents the location of the interface I(t) (Color figure online)
The adaptive algorithm [A’]–[D’] is applied to the morphogen gradient model introduced in Sect. 4, and the results are presented in Fig. 6. We use , and . Other parameters are given in Table 2. Our initial condition is , for , i.e. the system starts with no molecules and the gradient is formed during the simulation. In Fig. 6, one realization of the algorithm in Table 3 at different times , 2, 10, is presented. The green and blue bars represent the numbers of molecules in the corresponding compartments in and , respectively. The blue dotted line represents interface I(t) between two regions, and the red circles are the mean numbers of molecules obtained from the analytic solution of the stochastic model. Our results show that the boundary between two regions is moving to the right in time as the molecule numbers increase due to the production on the left.
In Fig. 7a and b, we simulate the adaptive algorithm with fixed thresholds for a range of values of , 10, , , , which are the numbers of time steps to check the criterion to move the interface I(t) in step [C’]. Two sets of fixed thresholds are chosen, in (a) and in (b). In Fig. 7c and d, we simulate the adaptive algorithm with fixed numbers of time steps, , for different values of and , which are the threshold values to check before we move the interface I(t) in step [C’]. We use the following pairs of the values for the thresholds: , (10, 40), (15, 25), (20, 20). Two fixed numbers of time steps are used, in (c) and in (d). As shown in Fig. 7b and c, we observe that the maximum absolute values of the relative errors increase as the number of time steps, , or the size of the threshold window, , gets smaller. This is because the small size of the number of time steps (or the threshold window) makes the interface location change frequently, which causes additional errors. On the other hand, Fig. 7a and d do not show similar pattern since large size of the threshold window () and the number of time steps () prevent frequent movement of the interface location. Overall, Scheme 2 has slightly smaller errors than Scheme 1. In Fig. 7, the maximum absolute values of the relative errors are calculated using realizations of simulation using Scheme 1 or 2 for each value of and for each set of values of and using the analytic solution of the Markov chain model.
Fig. 7.

The maximum absolute values of the errors defined by Eq. (13) at time using the multiscale algorithms, with a moving boundary. Different values of , 10, , , , are used with fixed thresholds a and b. Different threshold values , (10, 40), (15, 25), (20, 20) are used with, c and d. Red and green lines are the maximal relative errors of the means and standard deviations between the analytic solution of the Markov chain model and Scheme 1, given by Eq. (13). Blue and Purple lines are maximal relative errors between the Markov chain model and Scheme 2, given by Eq. (13) (Color figure online)
Applications: Multiple Species
In this section, we illustrate the applicability of the presented multiscale approaches to chemical systems with multiple species. Since different chemical species can have very different molecular distributions in the computational domain, the partition of the computational domain into subdomains and can be species dependent. We use the pom1p gradient model from Saunders et al. (2012) to illustrate a multiscale approach, where each species has a different partition into and depending on its molecular distribution. The model consists of two species, slow-diffusing pom1p clusters, denoted , and fast-diffusing pom1p particles, denoted . We use pseudo 1-dimensional domain as in Fig. 1, where , which is divided into compartments, , . Both and are produced in the whole computational domain with space-dependent rates given by Saunders et al. (2012), i.e. with propensities
where , , and , and are constants given in Table 4. In addition to production, species and are subject to the following reactions which take place in the whole domain
with the corresponding propensities given by
where and , and are constants given in Table 4.
Table 4.
Parameter values in the two-state model for pom1p gradient
| Notations | Description | Values |
|---|---|---|
| L | Length of the Domain | |
| Left boundary of | ||
| Right boundary of | ||
| Diffusion coefficient of | ||
| Diffusion coefficient of | ||
| Production parameter of | ||
| Production parameter of | ||
| Fragmentation rate of | ||
| Aggregation rate | ||
| Disassociation rate | ||
| Parameter of production | 0.1089 | |
| Spatial discretization in | ||
| h | Compartment size in | |
| Time discretization for SPDE |
In Fig. 8, we present an illustrative simulation of pom1p gradient model. We plot spatial distributions of and at times and . We observe that the spatial distribution of contains a region with high abundance of molecules in the centre of the computational domain. The chemical species has low copy numbers in the entire domain. Therefore, we introduce the SPDE region in the middle of the domain by (note that we fix in this example)
where the coarse graining is only applicable to in . In particular, we have introduced two interfaces, and between and . Diffusion of chemical species is simulated using the algorithm in Table 1. Similarly, production of is implemented using the SPDE and Markov chain model in and , respectively, as it is done in Eq. (9). The chemical species is simulated by the Markov chain model in the entire domain, because the average number of molecules of is relatively low. In particular, diffusion, production and degradation of are implemented as in the underlying Markov chain model. The only complications are reactions
| 14 |
because they include both species and , which are in described by different modelling approaches. We treat these reactions as time-changed Poisson processes in both subdomains and . Discretizing each compartment, , , into grid points, the state of variable is described by vector, where (resp. ) are the numbers of molecules of in the left (resp. right) part of . The values of SPDE description in compartment , , are given by , . The state of variable is described by vector, where is the number of molecules of in compartment , . The propensity of the first reaction in (14) of the multiscale model is given by
| 15 |
The propensity of the second reaction in (14) of the multiscale model is given by
| 16 |
We simulate reactions in (14) as time-changed Poisson processes with propensities in Eqs. (15)–(16). If the first of these reactions occurs in , , we subtract from each , , and we add one to . If the second reaction in (14) occurs in , , we add to each , , and we subtract one from . Note that the conversion of in , , is applied equally to the entire grid points of rather than to one randomly chosen grid point in as it is done (consistently with Eq. (9)) for diffusion across the interfaces.
Fig. 8.

Mean numbers of the molecules of slow-diffusing pom1p clusters, and fast-diffusing pom1p particles, and their standard deviations from the means at , computed by averaging over realizations of simulation using the SSA and the multiscale algorithm with Scheme 1 (Color figure online)
In Fig. 8, green bars and blue bars represent the mean numbers of molecules of the pom1p clusters and particles in and using the multiscale algorithm with Scheme 1. Error bars represent one standard deviation from the mean in the multiscale approach. Red lines and blue dotted lines are the mean numbers and their standard deviations from the means computed by the Gillespie SSA simulating the compartment-based approach in the entire domain. Both statistics using the compartment-based approach and the multiscale algorithm are computed by averaging over realizations of the simulations for each case.
Discussion
A Markov chain model (compartment-based model) has been widely used to describe the discrete nature of the molecular copy numbers and inherent stochasticity in reaction–diffusion systems (Erban and Chapman 2019), but it can be computationally intensive. A possible approach to increase efficiency of simulations is to approximate a part of the model by some coarse-grained methods. In this paper, we have introduced two multiscale algorithms coupling the SPDEs and the Markov chain model, which provide good approximations to the solutions obtained by the Markov chain model applied in the entire spatial domain. Two coupling methods of the Markov chain model and the SPDEs across the interface have been studied. In this section, we compare the presented approach with methods in the literature.
Several Langevin formulations have been introduced to model fluctuating hydrodynamics for chemically reactive species (Bhattacharjee et al. 2015) and stochastic reaction–diffusion systems (Kalantzis 2009; Ghosh et al. 2015). In particular, the spatial chemical Langevin equation was applied to the Gray–Scott model, and its pattern formation was compared to the ones obtained by the reaction–diffusion master equation and PDEs (Ghosh et al. 2015). The spatial chemical Langevin equation consists of a system of stochastic differential equations, and it corresponds to Eq. (4) in Sect. 2. On the other hand, several approaches using SPDEs (Atzberger 2010; Dogan and Allen 2011; Alexander et al. 2002, 2005; Kim et al. 2017) have been introduced to model stochastic reaction–diffusion systems. In Atzberger (2010), the SPDE was derived for reaction–diffusion systems, and discretization of PDEs and stochastic fields was discussed. Unlike Eq. (5), the stochastic fields in the discretized SPDEs account for fluctuations due to diffusion but not for reaction. In Kim et al. (2017), the SPDE for reaction–diffusion systems was derived which is consistent with Eq. (8). In their formulation, diffusion was implemented by the SPDE while the reaction was simulated using the exact or modified SSA.
In Yates and Flegg (2015), two hybrid algorithms are suggested for coupling a compartment-based model and a PDE model when the size of the PDE discretization is less than or equal to the compartment size. Both algorithms extend the PDE approach to the systems with low copy numbers of molecules in a part of the computational domain. The first algorithm considers the PDE solution as the probability density to find a particle within the region and is applied to both cases of low and high copy numbers of molecules in the PDE region. The second algorithm is a simplified and more efficient version of the first one when the PDE region involves the high copy number of molecules. Like in this paper, both algorithms implement a pseudo-compartment with size h in the PDE region where h represents the compartment size. The second algorithm in Yates and Flegg (2015) is similar to Scheme 1 if a discretized version of SPDEs replaces the PDEs. However, the interface between the two modelling regimes is assumed to be fixed in Yates and Flegg (2015). In Harrison and Yates (2016), a hybrid algorithm is introduced coupling a compartment-based model and PDEs where the size of the PDE discretization is much finer than the compartment size. In the model, an overlap region is defined with two interfaces (corresponding to the pseudo-compartment in Scheme 1) where both modelling regimes are valid, and both cases with fixed and adaptive interfaces are considered. Unlike our pseudo-compartment in Scheme 1, the overlap region can contain multiple compartments if needed. On one interface between the compartment-based model and the overlap region, the population of the PDE solution on the interface is matched to the average of the population in the neighbouring compartments. On the other interface between the PDE region and the overlap region, flux on the interface was matched. The hybrid algorithm in Harrison and Yates (2016) approximates the mean population numbers in the compartment-based model if it was possible to apply it over the entire spatial region. The use of the overlap region allows matching the variance between two models in the compartment-based region when the fixed interface is used. On the other hand, the goal of Scheme 1 and Scheme 2 is to approximate the compartment-based model by employing the discretized version of SPDEs in the region with high molecules. Therefore, we can match both the mean and variance of the population numbers computed by our multiscale algorithms to the results obtained by the compartment-based model in the whole spatial domain. This is done for both cases with a fixed or adaptive boundary. Unlike the previous approaches in Yates and Flegg (2015); Harrison and Yates (2016), the presented multiscale algorithms can apply to systems with multiple species as it is shown in Sect. 6 where each species has a different partition of the spatial domain into subdomains where different models are used, depending on the spatial distribution of molecules of each species. In Spill et al. (2015), a hybrid algorithm is presented using a compartment-based model and PDEs, where the size of the compartment and numerical discretization for the PDE model is equal.
In this paper, we have discussed the case when the mesh size of the numerical discretization of the SPDEs is smaller (or equal) than the compartment size in the Markov chain model (). This case is useful when we add inherent stochasticity in the PDE model where a fine spatial resolution of the PDE solution is required to describe the solution of the SPDE. This case was also discussed in other hybrid algorithms coupling the compartment-based model and the macroscopic PDEs (Yates and Flegg 2015; Harrison and Yates 2016). The other case, , discussed, for example, in the hybrid algorithm coupling a random walk on a lattice and the PDE model (Flekkoy et al. 2001), is helpful when the PDE or SPDE model is used as a coarse-grained approximation of the compartment-based model. Such approximation can be used in the region where spatial concentration gradients are not large, so they do not require a fine resolution in space. Although we have focused on the case , the presented approach can be extended to as well. In fact, if , both Scheme 1 and Scheme 2 will be the same. If , we may be able to consider an overlap region (like a pseudo-compartment) in the compartment-based region to extend Scheme 1. The presented SPDE-based approach provides a bridge between the stochastic approach (using the Markov chain compartment-based model) and the deterministic approach (using the macroscopic PDEs) by incorporating a discretized version of SPDEs. The SPDEs can be utilized to build other hybrid models, for example, by coupling them with macroscopic PDEs. Then, some approaches used in the hybrid algorithms coupling the compartment-based model with the PDEs (Yates and Flegg 2015; Harrison and Yates 2016; Spill et al. 2015; Smith and Yates 2018) will naturally apply to the case with the SPDEs.
Acknowledgements
RE would like to thank the Royal Society for a University Research Fellowship and the Leverhulme Trust for a Philip Leverhulme Prize. This prize money was used to support research visits of HWK to Oxford. The research leading to these results has received funding from the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ ERC Grant Agreement No. 239870. This research has been supported in part by the Mathematical Biosciences Institute and the National Science Foundation under Grant DMS-1440386 and DMS-1620403 (HWK), and by the University of Maryland Baltimore County under Grant UMBC KAN3STRT (HWK). The authors would like to thank the Isaac Newton Institute for Mathematical Sciences, Cambridge, for support and hospitality during the programme “Stochastic Dynamical Systems in Biology: Numerical Methods and Applications”, where work on this paper was undertaken. This work was supported by EPSRC Grant No. EP/K032208/1. This work was supported by a grant from the Simons Foundation.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hye-Won Kang, Email: hwkang@umbc.edu.
Radek Erban, Email: erban@maths.ox.ac.uk.
References
- Alexander F, Garcia A, Tartakovsky D. Algorithm refinement for stochastic partial differential equations: I. Linear diffusion. J Comput Phys. 2002;182(1):47–66. [Google Scholar]
- Alexander F, Garcia A, Tartakovsky D. Algorithm refinement for stochastic partial differential equations: II. Correlated systems. J Comput Phys. 2005;207(2):769–787. [Google Scholar]
- Altintan D, Ganguly A, Koeppl H. Efficient simulation of multiscale reaction networks: a multilevel partitioning approach. Am. Control Conf. 2016;2016:6073–6078. [Google Scholar]
- Ander M, Beltrao P, Di Ventura B, Ferkinghoff-Borg J, Foglierini M, Kaplan A, Lemerle C, Tomás-Oliveira I, Serrano L. SmartCell, a framework to simulate cellular processes that combines stochastic approximation with diffusion and localisation: analysis of simple networks. Syst Biol. 2004;1(1):129–138. doi: 10.1049/sb:20045017. [DOI] [PubMed] [Google Scholar]
- Arjunan S, Tomita M. A new multicompartmental reaction–diffusion modeling method links transient membrane attachment of E. coli MinE to E-ring formation. Syst Synth Biol. 2010;4(1):35–53. doi: 10.1007/s11693-009-9047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzberger P. Spatially adaptive stochastic numerical methods for intrinsic fluctuations in reaction–diffusion systems. J Comput Phys. 2010;229(9):3474–3501. [Google Scholar]
- Bakarji J, Tartakovsky D. On the use of reverse Brownian motion to accelerate hybrid simulations. J Comput Phys. 2017;334:68–80. [Google Scholar]
- Bernstein D. Simulating mesoscopic reaction–diffusion systems using the Gillespie algorithm. Phys Rev E. 2005;71(4):041103. doi: 10.1103/PhysRevE.71.041103. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Balakrishnan K, Garcia A, Bell J, Donev A. Fluctuating hydrodynamics of multi-species reactive mixtures. J Chem Phys. 2015;142(22):224107. doi: 10.1063/1.4922308. [DOI] [PubMed] [Google Scholar]
- Cao Y, Gillespie D, Petzold L (2005a) Multiscale stochastic simulation algorithm with stochastic partial equilibrium assumption for chemically reacting systems. J Comput Phys 206:395–411
- Cao Y, Gillespie D, Petzold L (2005b) The slow-scale stochastic simulation algorithm. J Chem Phys 122(1):14116 [DOI] [PubMed]
- Cao Y, Li H, Petzold L. Efficient formulation of the stochastic simulation algorithm for chemically reacting systems. J Chem Phys. 2004;121(9):4059–4067. doi: 10.1063/1.1778376. [DOI] [PubMed] [Google Scholar]
- Cotter S, Zygalakis K, Kevrekidis I, Erban R. A constrained approach to multiscale stochastic simulation of chemically reacting systems. J Chem Phys. 2011;135:094102. doi: 10.1063/1.3624333. [DOI] [PubMed] [Google Scholar]
- Cotter S, Vejchodsky T, Erban R. Adaptive finite element method assisted by stochastic simulation of chemical systems. SIAM J Sci Comput. 2013;35(1):B107–B131. [Google Scholar]
- Cotter S, Erban R. Error analysis of diffusion approximation methods for multiscale systems in reaction kinetics. SIAM J Sci Comput. 2016;38(1):B144–B163. [Google Scholar]
- Cucuringu M, Erban R. ADM-CLE approach for detecting slow variables in continuous time Markov chains and dynamic data. SIAM J Sci Comput. 2017;39(1):B76–B101. [Google Scholar]
- Dobramysl U, Rudiger S, Erban R. Particle-based multiscale modeling of calcium puff dynamics. Multiscale Modell Simul. 2016;14(3):997–1016. [Google Scholar]
- Dogan E, Allen E. Derivation of stochastic partial differential equations for reaction–diffusion processes. Stoch Anal Appl. 2011;29(3):424–443. [Google Scholar]
- Duncan A, Erban R, Zygalakis K. Hybrid framework for the simulation of stochastic chemical kinetics. J Comput Phys. 2016;326:398–419. [Google Scholar]
- Earnest T, Lai J, Chen K, Hallock M, Williamson J, Luthey-Schulten Z. Toward a whole-cell model of ribosome biogenesis: kinetic modeling of SSU assembly. Biophys J. 2015;109(6):1117–1135. doi: 10.1016/j.bpj.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earnest T, Cole J, Peterson J, Hallock M, Kuhlman T, Luthey-Schulten Z. Ribosome biogenesis in replicating cells: integration of experiment and theory. Biopolymers. 2016;105:735–751. doi: 10.1002/bip.22892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elf J, Doncic A, Ehrenberg M. Mesoscopic reaction–diffusion in intracellular signaling. Fluct Noise Biol Biophys Biomed Syst. 2003;5110:114–125. [Google Scholar]
- Engblom S, Ferm L, Hellander A, Lötstedt P. Simulation of stochastic reaction–diffusion processes on unstructured meshes. SIAM J Sci Comput. 2009;31:1774–1797. [Google Scholar]
- Erban R, Chapman SJ. Stochastic modelling of reaction–diffusion processes: algorithms for bimolecular reactions. Phys Biol. 2009;6(4):046001. doi: 10.1088/1478-3975/6/4/046001. [DOI] [PubMed] [Google Scholar]
- Erban R, Chapman SJ, Maini P (2007) A practical guide to stochastic simulations of reaction–diffusion processes, 35 p. arXiv:0704.1908 [DOI] [PubMed]
- Erban R, Kevrekidis I, Adalsteinsson D, Elston T. Gene regulatory networks: a coarse-grained, equation-free approach to multiscale computation. J Chem Phys. 2006;124(8):084106. doi: 10.1063/1.2149854. [DOI] [PubMed] [Google Scholar]
- Erban R, Chapman SJ, Kevrekidis I, Vejchodsky T. Analysis of a stochastic chemical system close to a SNIPER bifurcation of its mean-field model. SIAM J Appl Math. 2009;70(3):984–1016. [Google Scholar]
- Erban R. From molecular dynamics to Brownian dynamics. Proc R Soc A. 2014;470:20140036. doi: 10.1098/rspa.2014.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erban R. Coupling all-atom molecular dynamics simulations of ions in water with Brownian dynamics. Proc R Soc A. 2016;472:20150556. doi: 10.1098/rspa.2015.0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erban R, Chapman SJ (2019) Stochastic modelling of reaction-diffusion processes. Cambridge texts in applied mathematics. Cambridge University Press
- Erban R, Flegg M, Papoian G. Multiscale stochastic reaction–diffusion modeling: application to actin dynamics in filopodia. Bul Math Biol. 2014;76(4):799–818. doi: 10.1007/s11538-013-9844-3. [DOI] [PubMed] [Google Scholar]
- Fange D, Elf J. Noise-induced Min phenotypes in E. coli. PLoS Comput Biol. 2006;2(6):637–648. doi: 10.1371/journal.pcbi.0020080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferm L, Hellander A, Lötstedt P. An adaptive algorithm for simulation of stochastic reaction–diffusion processes. J Comput Phys. 2010;229:343–360. [Google Scholar]
- Flegg M, Chapman J, Erban R. The two-regime method for optimizing stochastic reaction–diffusion simulations. J R Soc Interface. 2012;9(70):859–868. doi: 10.1098/rsif.2011.0574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flegg M, Hellander S, Erban R. Convergence of methods for coupling of microscopic and mesoscopic reaction–diffusion simulations. J Comput Phys. 2015;289:1–17. doi: 10.1016/j.jcp.2015.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flekkoy E, Feder J, Wagner G. Coupling particles and fields in a diffusive hybrid model. Phys Rev E. 2001;64:066302. doi: 10.1103/PhysRevE.64.066302. [DOI] [PubMed] [Google Scholar]
- Franz B, Flegg M, Chapman SJ, Erban R. Multiscale reaction–diffusion algorithms: PDE-assisted Brownian dynamics. SIAM J Appl Math. 2013;73(3):1224–1247. [Google Scholar]
- Gadgil C, Lee C, Othmer H. A stochastic analysis of first-order reaction networks. Bull Math Biol. 2005;67:901–946. doi: 10.1016/j.bulm.2004.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly A, Altintan D, Koeppl H. Jump-diffusion approximation of stochastic reaction dynamics: error bounds and algorithms. Multiscale Model Simul. 2015;13(4):1390–1419. [Google Scholar]
- Gardiner CW. Handbook of Stochastic Methods. 3. Berlin: Springer; 2004. [Google Scholar]
- Ghosh A, Leier A, Marquez-Lago T. The spatial chemical Langevin equation and reaction diffusion master equations: moments and qualitative solutions. Theor Biol Med Model. 2015;12(1):5. doi: 10.1186/s12976-015-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson M, Bruck J. Efficient exact stochastic simulation of chemical systems with many species and many channels. J Phys Chem A. 2000;104:1876–1889. [Google Scholar]
- Gillespie D. Exact stochastic simulation of coupled chemical reactions. J Phys Chem. 1977;81(25):2340–2361. [Google Scholar]
- Gillespie D. The chemical Langevin equation. J Chem Phys. 2000;113(1):297–306. [Google Scholar]
- Griffith M, Courtney T, Peccoud J, Sanders W. Dynamic partitioning for hybrid simulation of the bistable HIV-1 transactivation network. Bioinformatics. 2006;22(22):2782–2789. doi: 10.1093/bioinformatics/btl465. [DOI] [PubMed] [Google Scholar]
- Harrison J, Yates C. A hybrid algorithm for coupling partial differential equation and compartment-based dynamics. J R Soc Interface. 2016;13(122):20160335. doi: 10.1098/rsif.2016.0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haseltine E, Rawlings J. Approximate simulation of coupled fast and slow reactions for stochastic chemical kinetics. J Chem Phys. 2002;117:6959–6969. [Google Scholar]
- Hattne J, Fange D, Elf J. Stochastic reaction–diffusion simulation with MesoRD. Bioinformatics. 2005;21(12):2923–2924. doi: 10.1093/bioinformatics/bti431. [DOI] [PubMed] [Google Scholar]
- Hu J, Kang H-W, Othmer H. Stochastic analysis of reaction–diffusion processes. Bull Math Biol. 2014;76(4):854–894. doi: 10.1007/s11538-013-9849-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson S, Peskin C. Incorporating diffusion in complex geometries into stochastic chemical kinetics simulations. SIAM J Sci Comput. 2006;28(1):47–74. [Google Scholar]
- Kalantzis G. Hybrid stochastic simulations of intracellular reaction–diffusion systems. Comput Biol Chem. 2009;33(3):205–215. doi: 10.1016/j.compbiolchem.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H-W. A multiscale approximation in a heat shock response model of E. coli. BMC Syst Biol. 2012;6(1):143. doi: 10.1186/1752-0509-6-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H-W, KhudaBukhsh W, Koeppl H, Rempala G. Quasi-steady-state approximations derived from the stochastic model of enzyme kinetics. Bull Math Biol. 2019;81(5):1303–1336. doi: 10.1007/s11538-019-00574-4. [DOI] [PubMed] [Google Scholar]
- Kang H-W, Kurtz T. Separation of time-scales and model reduction for stochastic reaction networks. Ann Appl Probab. 2013;23(2):529–583. [Google Scholar]
- Kang H-W, Kurtz T, Popovic L. Central limit theorems and diffusion approximations for multiscale Markov chain models. Ann Appl Probab. 2014;24(2):721–759. [Google Scholar]
- Kang H-W, Zheng L, Othmer H (2012a) The effect of the signalling scheme on the robustness of pattern formation in development. Interface Focus 2(4):465–486 [DOI] [PMC free article] [PubMed]
- Kang H-W, Zheng L, Othmer H (2012b) A new method for choosing the computational cell in stochastic reaction–diffusion systems. J Math Biol 65(6–7):1017–1099 [DOI] [PMC free article] [PubMed]
- Kim C, Nonaka A, Bell J, Garcia A, Donev A. Stochastic simulation of reaction–diffusion systems: a fluctuating-hydrodynamics approach. J Chem Phys. 2017;146(12):124110. doi: 10.1063/1.4978775. [DOI] [PubMed] [Google Scholar]
- Kim J, Rempala G, Kang H-W. Reduction for stochastic biochemical reaction networks with multiscale conservations. Multiscale Model Simul. 2017;15(4):1376–1403. [Google Scholar]
- Klann M, Ganguly A, Koeppl H. Hybrid spatial Gillespie and particle tracking simulation. Bioinformatics. 2012;28(18):i549–i555. doi: 10.1093/bioinformatics/bts384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingbeil G, Erban R, Giles M, Maini P. STOCHSIMGPU: parallel stochastic simulation for the systems biology Toolbox 2 for MATLAB. Bioinformatics. 2011;27(8):1170–1171. doi: 10.1093/bioinformatics/btr068. [DOI] [PubMed] [Google Scholar]
- Kurtz T. Limit theorems and diffusion approximations for density dependent Markov chains, stochastic systems: modeling, identification and optimization. Berlin: Springer; 1976. pp. 67–78. [Google Scholar]
- Kurtz T. Strong approximation theorems for density dependent Markov chains. Stoch Processes Appl. 1978;6(3):223–240. [Google Scholar]
- Lampoudi S, Gillespie D, Petzold L. The multinomial simulation algorithm for discrete stochastic simulation of reaction–diffusion systems. J Chem Phys. 2009;130(9):094104. doi: 10.1063/1.3074302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S, Vejchodsky T, Erban R. Tensor methods for parameter estimation and bifurcation analysis of stochastic reaction networks. J R Soc Interface. 2015;12(108):20150233. doi: 10.1098/rsif.2015.0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Pu Y, Li F, Shaffer C, Hoops S, Tyson J, Cao Y. Hybrid modeling and simulation of stochastic effects on progression through the eukaryotic cell cycle. J Chem Phys. 2012;136(3):034105. doi: 10.1063/1.3677190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W, Zheng L, Nie Q. A hybrid continuous-discrete method for stochastic reaction–diffusion processes. R Soc Open Sci. 2016;3(9):160485. doi: 10.1098/rsos.160485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R, Tadmor E. A unified framework and performance benchmark of fourteen multiscale atomistic/continuum coupling methods. Model Simul Mater Sci Eng. 2009;17:053001. [Google Scholar]
- Roberts E, Stone J, Luthey-Schulten Z. Lattice microbes: high-performance stochastic simulation method for the reaction–diffusion master equation. J Comput Chem. 2013;34(3):245–255. doi: 10.1002/jcc.23130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M, Andrews S, Erban R. Multiscale reaction–diffusion simulations with Smoldyn. Bioinformatics. 2015;31:2406. doi: 10.1093/bioinformatics/btv149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M, Flegg M, Erban R. Adaptive two-regime method: application to front propagation. J Chem Phys. 2014;140(12):124109. doi: 10.1063/1.4868652. [DOI] [PubMed] [Google Scholar]
- Salis H, Kaznessis Y. Accurate hybrid stochastic simulation of a system of coupled chemical or biochemical reactions. J Chem Phys. 2005;122:054103. doi: 10.1063/1.1835951. [DOI] [PubMed] [Google Scholar]
- Saunders T, Pan K, Angel A, Guan Y, Shah J, Howard M, Chang F. Noise reduction in the intracellular pom1p gradient by a dynamic clustering mechanism. Dev Cell. 2012;22(3):558–572. doi: 10.1016/j.devcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaff J, Gao F, Li Y, Novak I, Slepchenko B. Numerical approach to spatial deterministic-stochastic models arising in cell biology. PLOS Comput Biol. 2016;12(12):e1005236. doi: 10.1371/journal.pcbi.1005236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Yates C. Spatially-extended hybrid methods: a review. J R Soc Interface. 2018;15(139):20170931. doi: 10.1098/rsif.2017.0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stundzia A, Lumsden C. Stochastic simulation of coupled reaction–diffusion processes. J Comput Phys. 1996;127(1):196–207. [Google Scholar]
- Spill F, Guerrero P, Alarcon T, Maini P, Byrne H. Hybrid approaches for multiple-species stochastic reaction–diffusion models. J Comput Phys. 2015;299:429–445. doi: 10.1016/j.jcp.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J (1986) An introduction to stochastic partial differential equations, École d’Été de Probabilités de Saint Flour XIV-1984, pp 265–439
- Wils S, De Schutter E. STEPS: modeling and simulating complex reaction–diffusion systems with Python. Front Neuroinformatics. 2009;3(15):1–8. doi: 10.3389/neuro.11.015.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates C, Flegg M. The pseudo-compartment method for coupling partial differential equation and compartment-based models of diffusion. J R Soc Interface. 2015;12(106):20150141. doi: 10.1098/rsif.2015.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravlev P, Papoian G. Molecular noise of capping protein binding induces macroscopic instability in filopodial dynamics. Proc Natl Acad Sci. 2009;106(28):11570–11575. doi: 10.1073/pnas.0812746106. [DOI] [PMC free article] [PubMed] [Google Scholar]