

Summary
Despite significant advances in the treatment of myeloid malignancies, many patients become resistant to therapy and ultimately succumb to their disease. Accumulating evidence over the past several years has suggested that the inadequacy of many leukaemia therapies results from their failure to target the leukaemic stem cell (LSC). For this reason, the LSC population currently represents the most critical target in the treatment of myeloid malignancies. However, while LSCs are ideal targets in the treatment of these diseases, they are also the most difficult population to target. This is due to both their heterogeneity within the LSC population, and also their phenotypic similarities with normal haematopoietic stem cells. This review will highlight the current landscape surrounding LSC biology in myeloid malignancies, with a focus on altered energy metabolism, and how that knowledge is being translated into clinical advances for the treatment of chronic and acute myeloid leukaemia and myelodysplastic syndromes.
Keywords: Cancer stem cell (CSC), Leukaemic stem cell (LSC), Chronic myeloid leukaemia (CML), Acute myeloid leukaemia (AML), Myelodysplastic syndrome (MDS)
Introduction
Pluripotent haematopoietic stem cells (HSCs) are capable of either self-renewing and remaining pluripotent, or differentiating into committed lymphoid or myeloid progenitor cells. Leukaemic stem cells (LSCs), on the other hand, also known as leukaemia-initiating cells (LICs) or cancer stem cells (CSCs), originate from the oncogenic transformation of normal haematopoietic stem and progenitor cells (HSPCs), resulting in enhanced self-renewal or proliferation, and a reduced propensity for drug-induced apoptosis (Reya et al., 2001). Normal and malignant blood cell development have served as a paradigm for our current understanding of the properties of normal and malignant stem cells. In either case, HSCs and LSCs reside at the apex of a hierarchy of cells (Bonnet and Dick, 1997), with HSCs giving rise to normal haematopoiesis, and LSCs giving rise to malignant haematopoiesis (Dick, 2008) (Figure 1). A major challenge in cancer treatment, particularly the myeloid malignancies, is the fact that not all cancer cells are susceptible to therapy, and malignant stem cells remaining after therapy can serve as a reservoir for residual disease and relapse. Cytotoxic chemotherapies are often successful at eliminating the bulk of proliferating tumour cells, generally leukaemia progenitor cells, but leave behind aggressive stem cells that continue to propagate disease. Thus, new therapeutic strategies are required for targeting of myeloid LSCs to achieve treatment-free remission in this patient population.
Figure 1. Comparison of normal versus leukaemic haematopoiesis.
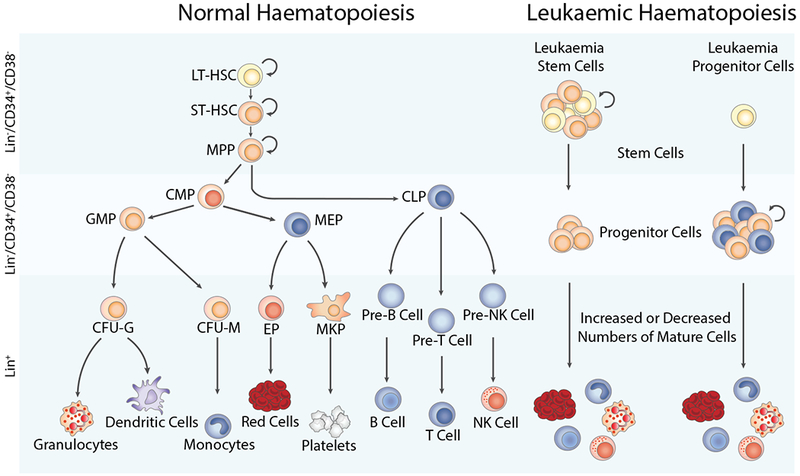
During normal haematopoiesis (left), a small number of haematopoietic stem cells gives rise to a limited number of progenitor cells, which subsequently gives rise to mature members of all blood cell types, including red blood cells, myeloid cell lineages such as granulocytes, dendritic cells, monocytes, or platelets, and lymphoid lineage cells, including NK cells, and T and B lymphocytes. In malignant haematopoiesis (right), leukaemic stem and progenitor cells with enhanced self-renewal or proliferation and reduced apoptosis give rise to abnormal numbers of mature cells in the bone marrow, peripheral blood, and other tissues. CFU-G, colony forming unit-granulocyte; CFU-M, colony forming unit-monocyte; CMP, common myeloid progenitor; EP, erythroid progenitor; GMP, granulocyte-macrophage progenitor; LT-HSC, long term-haematopoietic stem cell; MEP, megakaryocyte-erythroid progenitor; MKP, megakaryocyte progenitor; MPP, multipotent progenitor; NK cell, natural killer cell; ST-HSC, short term-haematopoietic stem cell.
Several signalling pathways have been implicated in the development and maintenance of LSCs, including WNT/β-catenin (Siapati et al., 2011), NF-κB (Kagoya et al., 2014), NOTCH (Reya et al., 2001, Takebe et al., 2011), and Hedgehog (Jagani et al., 2010) (Figure 2). These pathways are often activated in cancer by both genetic and epigenetic mechanisms, and have been linked to cancer initiation, propagation, and even therapy resistance (Eiring et al., 2015a). However, the limitation of these targets is that they are also active during embryonic development and in normal HSCs, complicating any potential treatment approach. Therefore, in order to improve patient outcomes, recent attention has focused on identifying targets that are more highly activated in the LSC population compared to normal HSCs. Several studies have recently demonstrated that LSCs exhibit higher levels of mitochondrial oxidative metabolism compared to normal HSCs, a vulnerability that can be targeted therapeutically in acute myeloid leukaemia (AML), chronic myeloid leukaemia (CML), and possibly myelodysplastic syndrome (MDS). In this review, we highlight recent findings surrounding LSCs in the myeloid malignancies, and how those findings are being translated into clinical advances.
Figure 2. Pathways commonly associated with survival of LSCs from AML, CML, and MDS patients.
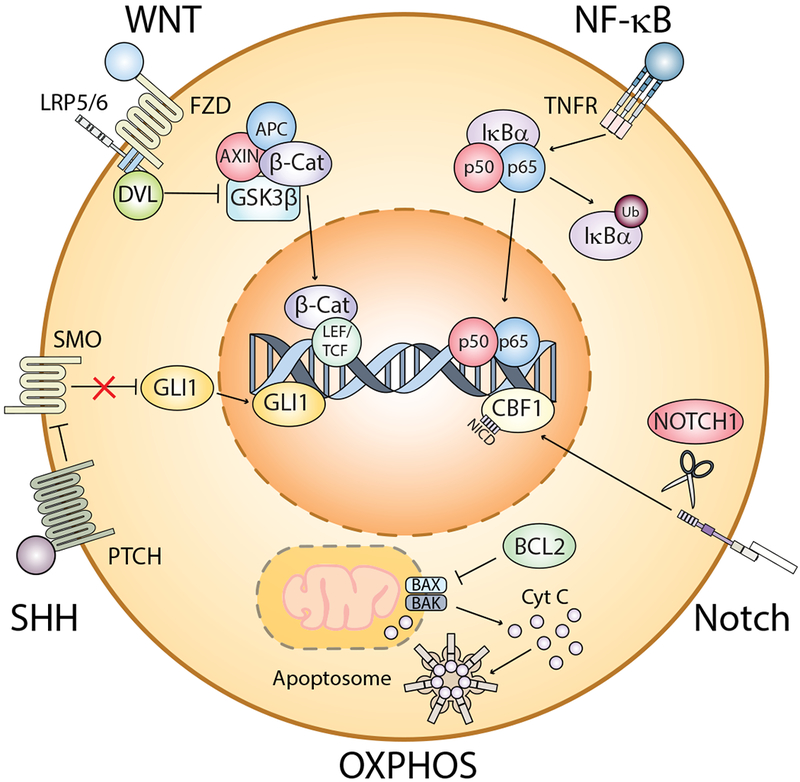
The illustration summarizes some of the major pathways commonly activated in LSCs, including WNT/β-catenin, NF-κB, Sonic hedgehog (SHH), Notch, and oxidative phosphorylation (OXPHOS), which is facilitated by upregulation of the anti-apoptotic protein, BCL-2. All of these pathways can be activated through various mechanisms. To date, the leading commonality among stem cells from each disease is a reliance on OXPHOS. AML, acute myeloid leukaemia; APC, adenomatous polyposis; β-Cat, beta-catenin; BAK, BCL2 homologous antagonist killer; BAX, BCL2-associated X protein; BCL2, B-cell lymphoma 2; CBF1, core binding factor 1;CML, chronic myeloid leukaemia; Cyt C, cytochrome c; DVL, dishevelled; FZD, frizzled; GLI1, glioma-associated oncogene homolog 1; GSK3β, Glycogen Synthase Kinase 3 Beta; IκBα, nuclear factor-kappa B inhibitor alpha; LEF, lymphoid enhancer-binding factor; LRP5/6, low density lipoprotein receptor-related protein 5/6; LSC, leukaemic stem cell;MDS, myelodysplastic syndrome; SMO, smoothened; NF-κB, nuclear factor kappa B; NICD, notch intracellular domain, OXPHOS, oxidative phosphorylation; PTCH, patched; SHH, sonic hedgehog; STCF, T-cell factor; TNFR, tumour necrosis factor receptor; Ub, ubiquitin.
CSC theory, LSCs, and therapy resistance
The bulk of tumours usually consist of rapidly dividing progenitor cells, as well as fully differentiated, mature cell types. However, similar to the growth and differentiation of normal tissues, the growth of many tumours is fuelled by a small number of dedicated stem cells with the capacity for self-renewal, and only a small, phenotypically distinct population of cancer cells has the ability to proliferate extensively or to form a new tumour. As illustrated in Figure 1 in the case of haematopoiesis, the CSC theory postulates that, like normal tissues, cancers are derived from rare, undifferentiated CSCs at the top of a hierarchy, which are responsible for maintaining the entire population of cells within a given tumour (Reya et al., 2001). While CSCs are not believed to generate all tumour types (Nguyen et al., 2012), they have been described in several different myeloid malignancies, including AML, CML and MDS, as well as solid tumours, such as breast, pancreatic, prostate, colorectal and several different brain cancers. CSCs were first described in AML by Lapidot et al (1994), where they demonstrated that AML LSCs, characterized by the CD34+38− immunophenotype (representing only 0.1-1% of the tumour population), were the only cells capable of generating AML in mice. It has since been demonstrated that the LSC carries the ability to initiate, sustain, and serially propagate leukaemia in vivo, while retaining the capacity to differentiate into more committed progenitor populations that no longer possess leukaemia-initiating activity (Huntly et al., 2004). Due to the ability of LSCs to initiate and sustain leukaemia and to evade therapy-induced apoptosis, LSCs may be critical in disease prognosis, and worth monitoring during therapeutic intervention. Indeed, the frequency of CD34+38− stem cells at diagnosis correlates with response to treatment and survival in AML (Vergez et al., 2011), and the number of residual leukaemia cells remaining after therapy has been correlated with inferior outcomes (Jongen-Lavrencic et al., 2018). Thus, CSCs have been proposed to be good biomarkers to predict treatment failure and relapse, and represent the most critical targets to advance therapeutic options in many different cancers.
Despite the small number of CSCs present in any given tumour, they have been shown to promote cancer initiation, disease progression and therapy resistance in several different tumour types (Lytle et al., 2018). The high rate of tumour relapse following initial therapeutic responses suggests the existence of CSCs that are resistant to conventional therapies. While some cytotoxic cancer therapies have resulted in mutation-based acquired resistance, resulting from clonal evolution during therapy (Branford et al., 2002), other studies have shown that pre-existing resistant clones are present in a tumour that drive disease recurrence following treatment (Lee and Lu, 2014). CSCs are ideal targets for therapy, but they are also challenging cancer targets, due to their heterogeneity within a given tumour (from genetic or epigenetic changes acquired over time), and their similarities with normal stem cells (including properties such as self-renewal, asymmetric division and resistance to chemical insults).
Distinct signalling pathways control stem cell self-renewal in different tissue types, but, in individual tumours, the same pathways are often used by both normal stem cells and CSCs to regulate self-renewal, proliferation and differentiation of the given tissue of origin. Existing therapies largely target the bulk population of tumours, but they are generally ineffective at targeting CSCs. CSC self-renewal and resistance to chemotherapy can be attributed to their reduced proliferation or quiescence, heightened DNA repair and reduced apoptosis, increased clearance of reactive oxygen species (ROS), enhanced drug efflux mechanisms and reduced immune clearance (Lytle et al., 2018). However, perhaps the most compelling phenotype separating CSCs from normal stem cells are aberrations in the dependencies on oxygen and lipid metabolism (reviewed below). These alterations in energy metabolism not only contribute to CSC survival, but also chemotherapy resistance in haematological malignancies (Kuntz et al., 2017, Pollyea et al., 2018, Jones et al., 2018). Just as normal and malignant blood cell development have guided our knowledge of normal and cancer stem cell biology, understanding the role of aberrant energy metabolism in LSCs can guide our understanding of stem cell survival in a variety of malignancies, including solid tumours, with the ultimate goal of identifying novel targets for improved cancer therapy.
Cancer cell metabolism in myeloid LSCs
The production and consumption of energy in living cells are essential for countless numbers of biological processes. Since the middle of the 20th century, cancer cells have traditionally been thought to rely on glycolysis for adenosine triphosphate (ATP) production, as opposed to mitochondrial oxidative metabolism, even in the presence of sufficient oxygen levels. This phenomenon is now referred to as the Warburg effect (Warburg, 1956). This is true in the bulk population of many tumours and also in many different cancer cell lines, because when glucose is highly abundant and rapidly imported into the cell, glycolysis represents a more efficient method of ATP production than aerobic mitochondrial respiration (Vazquez et al., 2010). This is counterintuitive, because it is well established that mitochondrial oxidative phosphorylation is the preferred method of energy production in normal cells, as it is far more metabolically efficient than aerobic glycolysis in terms of ATP generated per molecule of glucose (up to 38 ATP molecules per glucose for oxidative phosphorylation versus 2 for aerobic glycolysis) (Vazquez et al., 2010, Zheng, 2012). However, there is a limited volume of cytosolic space available for components of the ATP-generating machinery and, under conditions of high glucose uptake, the cell is limited in its ability to increase mitochondrial concentration to match the increased respiration capacity (Vazquez et al., 2010). It has been suggested that inefficient ATP production is only a problem when resources are limited, generally not the case for proliferating tumour cells, and that oxidative phosphorylation imposes large requirements for tumour cells in terms of nucleotides, lipids and amino acids required during growth, thus resulting in a preference for glycolysis (Vander Heiden et al., 2009).
The Warburg effect was the first demonstration that malignant tissues exhibit fundamental differences in central metabolic processes when compared to normal tissues. Warburg initially proposed that cancer cells prefer aerobic glycolysis because they have impaired mitochondrial oxidative phosphorylation. However, the function of mitochondrial respiration in cancer cells is indeed intact (Zheng, 2012), and there is increasing evidence that this is the case for LSCs. Recent studies from several different groups have demonstrated that CSCs in particular have a lower glycolytic reserve than more mature cancer cells, and rather depend on oxidative metabolism for their survival. This is true of CSCs from multiple different tumour types, including both solid tumours and haematological malignancies (Lagadinou et al., 2013, Kuntz et al., 2017), a phenomenon now being referred to as the ‘reverse Warburg effect’ (Fu et al., 2017). While the mechanism by which cancer cells regulate the balance between oxidative phosphorylation and glycolysis is not completely understood, it was demonstrated that they undergo waves of gene regulation that suppress and subsequently restore oxidative phosphorylation. This can result from dysregulation of cell signalling pathways, such as the LKB1-AMPK-p53 and PI3K-AKT-mTOR axes (Smolkova et al., 2011), or transcription factors such as c-MYC (Lee et al., 2017). Altogether, these data imply that the Warburg effect is not a universal feature of all cancer cells, particularly in LSC and CSC populations, and therapies targeting oxidative metabolism might be a valuable treatment approach in certain types of cancers.
In fact, there have been several recent reports on an inhibitor of oxidative phosphorylation, IACS-010759, having efficacy in several different cancer models, including lung cancer, melanoma, renal cell carcinoma, and chronic lymphocytic leukaemia (Lissanu Deribe et al., 2018, Sun et al., 2019, Fischer et al., 2019). One particular study reported that IACS-010759 inhibited proliferation and induced apoptosis in models of glioblastoma, neuroblastoma and AML that were shown to be reliant on oxidative phosphorylation (Molina et al., 2018). This was a seminal study demonstrating that pharmacological inhibition of oxidative phosphorylation may be a viable treatment option in several types of cancers. IACS-010759 is a potent, clinical-grade small-molecular inhibitor targeting complex I of the electron transport chain that is currently under clinical development. Their studies in AML demonstrated sensitivity to IACS-010759 in several different AML cell lines and primary AML samples; however, future studies will be required to assess the effects of IACS-010759 on the AML LSC population.
LSCs in acute myeloid leukaemia (AML)
AML is the most common type of acute leukaemia in adults, and although survival has increased steadily over the past several decades, especially among younger patients, AML remains one of the most challenging diseases to cure owing to drug resistance, relapse or complications associated with chemotherapy (e.g. infection). AML is a highly heterogeneous disease, with various cytogenetic rearrangements (e.g. t(8;21) and t(15;17)) and mutations reported (e.g. FLT3-internal tandem duplication [ITD], NPM1, KIT, IDH1, IDH2, CEBPA) (Grimwade et al., 2010). Despite advances in the understanding of AML pathogenesis, the therapeutic backbone has remained largely unchanged for over four decades, and approximately 70% of patients aged 65 years or older will die within 1 year of diagnosis (Derolf et al., 2009, Yang and Wang, 2018). LSCs are considered to play a pivotal role in AML relapse, as these cells have been shown to be resistant to standard chemotherapy regimens. Thus, targeting of the LSC population has the potential for better outcomes or even curative treatment strategies for AML patients (Pollyea and Jordan, 2017). Recent studies have elucidated the role of aberrant energy metabolism in AML LSCs, which is paving the way for new therapeutic strategies to improve outcomes in this high-risk patient population.
While LSCs exhibit many of the molecular and functional properties of normal HSCs, including self-renewal and a quiescent cell cycle status, there are also important differences that serve as potential targets for therapy (Lagadinou et al., 2013). For instance, several cell surface markers are beginning to show differential expression between LSCs and HSCs (Figure 3), and therapeutic targeting of these markers is emerging in active clinical trials. Cell surface molecules are attractive molecular targets due to the availability of monoclonal antibodies (mAbs) and chimeric antigen receptor T cells (CAR T cells) for therapy. Therapeutic mAbs are broadly characterized into three different classes: i) antibody-drug conjugates that target leukaemia cells directly with cytotoxic compounds, ii) antibodies that target interactions with the bone marrow microenvironment and iii) antibodies that reinforce host immunity. CAR T cells, on the other hand, are T cells engineered to target a specific tumour antigen. However, recent studies have implicated aberrations in energy metabolism as another potential therapeutic liability in stem cells from patients with myeloid malignancies. Over the past several years, Jordan and colleagues have demonstrated that primitive, chemotherapy-resistant AML LSCs are characterized by a low rate of energy metabolism and lower levels of cellular oxidative status, including reactive oxygen species (ROS) (Lagadinou et al., 2013, Jones et al., 2018, Pollyea et al., 2018). This phenotype was associated with increased expression of BCL-2, and could be reversed by BCL-2 inhibition (Lagadinou et al., 2013). ROS-low AML LSCs were shown to be metabolically dormant, have increased levels of glutathioine, and demonstrate a dependency on oxidative respiration rather than glycolysis for energy production, in stark contrast to their normal counterparts or the bulk tumour population (Lagadinou et al., 2013). More recently, the dependence of AML LSCs on oxidative phosphorylation was formally established by assessing the effects of glucose depletion on the LSC population versus the bulk leukaemia population (Jones et al., 2018). In contrast to the previous theory of a dependence on glycolysis, AML LSCs did not respond to glucose depletion, whereas the bulk of AML cells were highly dependent on glucose. In this elegant study, the authors demonstrated that, in the case of newly diagnosed AML, amino acid uptake, catabolism and steady-state levels were increased in LSCs, and that LSCs rely on amino acid metabolism for oxidative phosphorylation and survival.
Figure 3. Energy dependencies and cell surface molecules being targeted in active clinical trials or in preclinical studies.
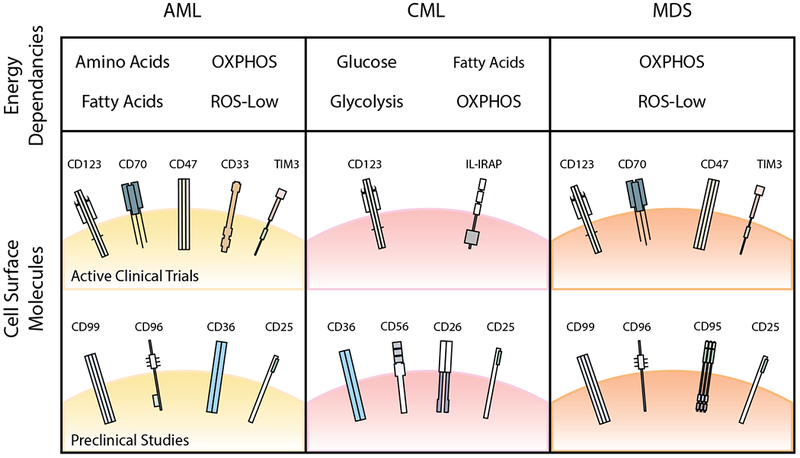
Identifying pathways that are commonly targetable in AML, CML, and MDS will allow for greater clinical utility in the treatment of myeloid malignancies. Here, we summarize some of the energy dependencies and cell surface molecules that are currently being targeted in clinical trials or have been identified to be upregulated in AML, CML, or MDS in preclinical studies. Some of these targets are common across the three disease entities, including oxidative phosphorylation (OXPHOS) and CD123. Commonalities between AML and CML LSCs include fatty acid metabolism and CD36, whereas commonalities between AML and MDS LSCs include CD47, TIM3, CD25, and CD99. AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia; MDS, myelodysplastic syndrome; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species.
In contrast to LSCs from patients with newly diagnosed AML, LSCs from relapsed AML patients were not dependent on amino acid metabolism for their survival, but were rather dependent on increased fatty acid metabolism, suggesting another metabolic vulnerability in patients after relapse (Jones et al., 2018). In fact, evidence suggests there may be fundamental differences between pre-treatment LSCs and cells present after therapy in AML patients. Earlier reports of therapy-resistant AML cells suggested enrichment for LSCs in G0 phase of the cell cycle with an immature cell phenotype ( Saito et al., 2010). However, Farge et al (2017) demonstrated that, in cytarabine-resistant AML cells generated in patient-derived xenografts, the resistant cells present after therapy were not necessarily enriched for LSCs, but were shown to require oxidative metabolism with increased fatty acid oxidation, exhibiting a gene expression signature consistent with oxidative phosphorylation pathways. In contrast to the findings of Jordan and colleagues described above, therapy-resistant LSCs in this model exhibited high levels of ROS (Farge et al., 2017), a difference possibly reflected by the use of in vivo models rather than freshly isolated cells from therapy-resistant AML patients, or the use of phenotypically defined LSCs rather than ROS content.
Despite the differences mentioned above, one phenotype remains consistent across the two groups, and that is a dependency of LSCs and therapy-resistant AML cells on oxidative metabolism (Farge et al., 2017, Jones et al., 2018). This liability is now being translated into clinical advances for high-risk AML patients, with ongoing clinical trials testing the efficacy of the bioavailable BCL-2 inhibitor, venetoclax, in combination with hypomethylating agents, to therapeutically target primitive LSCs both in vitro and in vivo (DiNardo et al., 2018, Pollyea et al., 2018). In these studies, the authors demonstrated that treatment of older AML patients with venetoclax in combination with azacitidine resulted in remissions that were superior to conventional treatment regimens. They demonstrated that LSCs isolated just 6 h after initiation of treatment demonstrated strong downregulation of pathways related to oxidative phosphorylation. Of particular importance, LSCs from patients undergoing combination therapy showed targeted disruption of the tricarboxylic acid (TCA) cycle, with decreased levels of α-ketoglutarate and increased levels of succinate, implicating inhibition of the electron transport chain complex II (Pollyea et al., 2018). They further went on to show in vitro that the combination therapy suppressed oxidative phosphorylation by reducing glutathionylation of succinate dehydrogenase, thereby selectively targeting the AML LSC population. These studies extend recent findings suggesting an important role for mitochondrial respiration in survival of AML LSCs (Skrtic et al., 2011, Lagadinou et al., 2013, Chan et al., 2015), and demonstrate that targeting of these metabolic dependencies in AML might improve patient outcomes. One might argue that venetoclax and other BH3 mimetics work through induction of mitochondrial apoptosis rather than inhibition of oxidative phosphorylation. BH3 mimetics by design inhibit anti-apoptotic BCL-2 family members, leading to BAX and BAK activation (Ni Chonghaile and Letai, 2008). However, in ROS-low AML LSCs, pharmacological inhibition of BCL-2 dramatically reduced oxidative phosphorylation and ATP levels, which was followed by an increase of mitochondrial ROS levels, reduced levels of glutathione, and induction of apoptotic cell death (Lagadinou et al., 2013), thus classifying this compound as an indirect inhibitor of oxidative metabolism that is showing clinical efficacy in several different types of cancers, including AML.
LSCs in chronic myeloid leukaemia (CML)
Perhaps the best understood case of drug-resistant LSCs is in the chronic phase of CML. CML is caused by the Philadelphia (Ph) chromosome, discovered by Nowell and Hungerford in 1960 (Nowell and Hungerford, 1960), which generates a reciprocal translocation known as t(9;22) that gives rise to the BCR-ABL1 fusion oncogenic tyrosine kinase. CML is well-controlled in the clinic by treatment with tyrosine kinase inhibitors (TKIs), such as imatinib, nilotinib and dasatinib (O’Hare et al., 2012, Cortes et al., 2013). BCR-ABL1 targeted therapies are remarkably effective at eliminating most of the Ph-positive cells in chronic phase CML patients, inducing ostensible complete remissions. However, these drugs do not target the CML LSC (Graham et al., 2002, Corbin et al., 2011, Hamilton et al., 2012), and the CML clone can rapidly return after treatment is discontinued, even after many years of therapy. While some authors have reported treatment-free remission following optimal molecular response with first or second generation TKIs (Rousselot et al., 2014, Bocchia et al., 2018), life-long treatment is required to maintain remission in the majority of patients (Chomel et al., 2011, Chu et al., 2011). Long-term TKI treatment comes at a high economic burden and often with significant side effects, as chronic TKI therapy is associated with a reduced quality of life and considerable morbidity, such as cardiovascular adverse events (Minson et al., 2019, Caocci et al., 2019, Jain et al., 2019) and skeletal muscle toxicity (Janssen et al., 2019). Furthermore, patients who have progressed to the blast phase of disease, in which the progenitor population acquires self-renewal properties similar to that of stem cells (see Figure 1), still have a poor prognosis (O’Hare et al., 2012, Cortes et al., 2013). Thus, therapies involving TKIs in combination with drugs that target the LSC population would be a major advancement, leading to eradication of the CML LSC, to cure the majority of patients. Similar to stem cells discovered in AML, LSCs from CML patients have been shown to reside in the CD34+38− population, and are capable of engrafting into immunocompromised mice (Wang et al., 1998). Interestingly, recent evidence suggests that CML LSCs, like AML LSCs, are susceptible to inhibition of oxidative phosphorylation, a vulnerability that could result in improved treatment options for CML patients and eradication of disease (Kuntz et al., 2017).
BCR-ABL1 has been shown to induce aerobic glycolysis (the Warburg effect) in CML cells through activation of the PI3K/AKT pathway (Alvarez-Calderon et al., 2015). Consequently, imatinib-mediated BCR-ABL1 inhibition results in reduced glucose uptake through suppression of glycolysis, increased flux of glucose through the mitochondrial TCA cycle, inhibition of fatty acid synthesis, and restriction of de novo nucleotide production (Gottschalk et al., 2004, Barnes et al., 2005, Kominsky et al., 2009). On the other hand, imatinib resistance in cell line models of CML has been associated with activation of hypoxia-inducible factor 1-α (HIF1α), also resulting in increased glycolysis (Zhao et al., 2010, Kluza et al., 2011). One study used a large-scale loss-of-function RNA interference screen to identify genes whose inhibition synergizes with imatinib in the K562 CML cell line. Interestingly, this screen identified several different enzymes associated with glucose metabolism as synthetically lethal when used in combination with imatinib (Gregory et al., 2010). The authors subsequently demonstrated that an enzyme involved in the pyruvate dehydrogenase (PDH) complex, dihydrolipoamide S-acetyltransferase (DLAT), is a critical mediator of CML cell survival upon TKI-mediated BCR-ABL1 inhibition. These data confirmed that inhibition of glucose utilization is partly responsible for the effects of imatinib treatment, but that mitochondrial pyruvate oxidation can provide protection against TKIs (Alvarez-Calderon et al., 2015). Interestingly, they observed similar results in FMS-like tyrosine kinase 3 receptor (FLT3)-ITD-positive AML cells treated with FLT3 inhibitors, suggesting that this may be a general mechanism of TKI resistance, and not specific for BCR-ABL1 inhibition. Ultimately, inhibition of mitochondrial function has anti-leukaemia effects in combination with TKIs, and TKI treatment creates metabolic perturbations that are potential targets for combination therapies (Alvarez-Calderon et al., 2015).
More recently, Kuntz et al (2017) established the metabolic profile of more primitive CML progenitor cells (CD34+) compared to more differentiated cell populations (CD34−), and discovered that more primitive cells rely on upregulated oxidative metabolism for their survival. Additionally, their work also revealed an increase of oxidative metabolism in CD34+38− CML stem cells compared to the same population from normal individuals. Using liquid chromatography-mass spectrometry, more primitive CML progenitors were shown to have an increase in lipolysis and fatty acid oxidation, with increased levels of glycerol-3-phosphate, carnitine and acylcarnitine, and a decrease of free fatty acids. They further went on to show that CML LSCs were susceptible to combination therapy with TKIs and tigecycline, an antibiotic that inhibits mitochondrial protein translation, both in vitro and in vivo (Kuntz et al., 2017). While further studies will be required to elucidate the mechanism by which CML LSCs become reliant on oxidative mitochondrial metabolism, these results are encouraging for patients who are destined for life-long TKI therapy, and highlight a commonality between LSCs from CML and AML patients.
LSCs in myelodysplastic syndrome (MDS)
Myelodysplastic syndrome (MDS) is a heterogeneous disease encompassing many different conditions categorized by similar clinical characteristics, including peripheral blood cyptopenias involving one or more lineages, bone marrow hypercellularity and dysplastic features of the cytoplasm or nucleus (Greenberg et al., 2011). MDS can arise de novo or as a result of prior chemotherapy or radiotherapy. MDS is primarily a disease of the elderly, with a mean age of 70 years at diagnosis, and can have various clinical presentations, including anaemia, bleeding or infections, in combination with hypercellular or sometimes hypocellular bone marrow. As the disease progresses, haematopoietic cells demonstrate enhanced genomic instability (Kuramoto et al., 2002, Papaemmanuil et al., 2013), leading to additional cytogenetic abnormalities, an increase of blasts in the bone marrow, with transformation to AML occurring in ~30% of patients (Mufti, 2004). MDS patients who have progressed to AML have poor outcomes, with increased rates of chemotherapy resistance and mortality (de Witte et al., 1995, Ruutu et al., 1997). Many genetic mutations have been associated with MDS (Papaemmanuil et al., 2013, Haferlach et al., 2014), with factors involved in RNA splicing identified as the most commonly targeted biological process (Ogawa, 2019). Mutations in RNA splicing or DNA methylation factors were found to be mutated early in disease manifestation, whereas mutations in chromatin remodelling and cell signalling genes occurred at later stages. Due to the role of DNA hypermethylation in MDS, standard therapies currently include hypomethylating agents, such as 5’-azacitidine or decitibine; however, for the majority of patients, this strategy only improves disease symptoms and is not curative (Corey et al., 2007).
MDS is considered a stem cell-derived disease (Stevens et al., 2018), which can affect either a single cell lineage or multiple lineages (Nilsson et al., 2007, Chung et al., 2008). However, research into MDS stem cells is less advanced than AML or CML, partly due to difficulties in establishing mouse models of the disease. The characterization of stem cells requires the use of in vivo repopulation assays, but clonal cells from MDS patients have proven to engraft poorly into immunocompromised mice ( Pang et al., 2013, Rhyasen et al., 2014, Medyouf et al., 2014). In addition to these xenotransplantation models, transgenic mouse models have been developed that display characteristics of MDS. However, the utility of these models is under scrutiny, because these engineered mice develop MDS with features distinct from that of MDS in humans (reviewed in (Zhou et al., 2015)).
Woo et al (2014) were the first to demonstrate unequivocally that MDS is a stem cell-derived disease, establishing that rare lineage-negative CD34+38−90+45RA− MDS cells function as MDS-propagating cells in a xenotransplantation model, and also harbour driver mutations in a subset of the stem cell population. More recently, Stevens et al (2018) added to these findings, showing that upregulation of the interleukin-3 (IL3) receptor alpha chain, CD123, on MDS stem cells designates changes in cellular physiology. Specifically, CD123+ MDS stem cells had elevated levels of protein synthesis and significant changes in cellular energy metabolism that render them susceptible to pharmacological intervention, similar to the groups recent findings in AML (Jones et al., 2018, Pollyea et al., 2018). Targeting protein synthesis and energy metabolism was effective at eliminating MDS stem cells both in vitro and in vivo, yet again revealing similarities between LSCs from multiple different myeloid malignancies.
Opportunities for therapeutic intervention and future directions
It is clear that metabolic dependencies serve as a potent therapeutic target for stem cells from AML, CML and MDS patients, and, as such, could lead to curative treatment strategies. In addition to studies using venetoclax to target BCL-2, other critical metabolic mediators are being targeted, such as isocitrate dehydrogenase (IDH) ½ in IDH-mutated gliomas and AML (Fujii et al., 2016). One target that appears to be active in LSCs from AML, CML and MDS patients is sirtuin-1 (SIRT1). In CML, SIRT1 protein and SIRT1 RNA levels were found to be elevated in CML versus normal stem and progenitor cells, and SIRT1 inhibition in combination with imatinib enhanced elimination of CML LSCs through activation of p53 (also termed TP53) (Li et al., 2012). Additionally, SIRT1 deacetylase activity was shown to promote mutation-based drug resistance in CML (Wang et al., 2013). In AML, SIRT1 was shown to be activated by c-MYC, promoting maintenance and drug resistance in FLT3-ITD-positive AML LSCs (Li et al., 2014). More recently, SIRT1 was shown to also be activated in MDS, resulting in disruption of stem and progenitor cell maintenance by restoring TET2 function (Sun et al., 2018). Nevertheless, SIRT1 also plays a role in normal haematopoiesis, maintaining HSC homeostasis under conditions of oxidative stress or nutrient deprivation (Singh et al., 2013), suggesting that SIRT1 inhibition may have pleiotropic effects. Further investigation will be required to elucidate the effectiveness of SIRT1 inhibition as a LSC-targeted therapy in patients with myeloid malignancies. LSCs not only exhibit selective dependencies on oxidative phosphorylation, but also demonstrate other LSC-specific metabolic properties, including reliance on mitochondrial translation (Skrtic et al., 2011), uncoupling of oxygen consumption from ATP production (Samudio et al., 2009), increased metabolism of branched-chain amino acids (Hattori et al., 2017), and sensitivity to perturbations in the electron transport chain (Chan et al., 2015).
Continuing with the theme of targeting aberrant metabolism to eliminate myeloid LSCs, many groups have turned to fatty acid metabolism. Indeed, CML and AML LSCs have been shown to evade chemotherapy through metabolic adaptation to an adipose tissue niche (Ye et al., 2016), and bone marrow adipocytes have been shown to support HSPCs in vitro (Tabe et al., 2017). Still other groups have focused on targeting of the fatty acid translocase, CD36, which is responsible for transporting free fatty acids into the cell (Landberg et al., 2018, Pascual et al., 2017). CD36 expression is markedly elevated on LSCs versus normal HSCs, resulting in chemotherapy resistance (Ye et al., 2016, Farge et al., 2017, Thomas and Majeti, 2016). It is possible that fatty acid metabolism may be a contributing factor to the increased dependency of myeloid LSCs on oxidative phosphorylation. However, redundancies in fatty acid metabolism and signalling pathways, including a plethora of different fatty acid binding proteins, might limit the utility of targeting this pathway in LSCs. Regardless, these data outline once again the importance of aberrant energy metabolism in LSC survival and therapeutic response.
While there have been many attempts to target the LSC population in pre-clinical models, few approaches have made it to phase I/II clinical trials (Tables I, II), with peer-reviewed publications just starting to emerge (Tables SI,SII). Given the phenotypic similarities between normal HSCs and stem cells from patients with these diseases, targeting of molecules that are upregulated on LSCs but not normal HSCs, such as CD36 discussed above, seems an ideal strategy for use in combination treatment approaches. One such strategy used a monoclonal antibody targeting CD44 in the bone marrow microenvironment (Vey et al., 2016). CD44 is the receptor for hyaluronic acid, for which inhibition prevented engraftment of CML and AML cells in vivo (Jin et al., 2006). Similar pre-clinical studies have been performed targeting other cell surface molecules, including VCAM-1 (Jacamo et al., 2014) and CD33 (Fathi et al., 2018). Indeed, several groups have reasoned that targeting the microenvironment could help eradicate myeloid LSCs, which includes treatment with the CXCR4 antagonist, plerixafor (AMD3100), and strategies targeting the IL3 receptor alpha chain, CD123 (Tables I, SI). Thus, targeting the bone marrow microenvironment appears to be a promising strategy to eradicate AML, CML and MDS LSCs to restore normal haematopoiesis.
Table I.
Ongoing anti-LSC clinical trials targeting cell surface molecules
| LSC Targets | Agent(s) | Disease(s) | Phase | Trial Identifier |
|---|---|---|---|---|
| Cell Surface Molecules | ||||
| CD123 | SL-401 | AML, BPDCN | 1 & 2 | |
| AML | 1 & 2 | |||
| XmAb14045 | AML, CML, BPDCN | 1 | ||
| KHK2823 | AML, MDS | 1 | ||
| IMGN632 | AML, BPDCN, MPN | 1 | ||
| CAR-T Cells | Myeloid malignancies | 1 & 2 | ||
| CAR-T Cells | AML | 1 | ||
| CAR-T Cells | AML | 1 | ||
| CAR-T Cells | AML | 1 | ||
| CD123/CD3 | JNJ-63709178 | AML | 1 | |
| Flotetuzumab (MGD006) | AML, MDS | 1 & 2 | ||
| CML, BPDCN | 2 | |||
| CD123/CLL1 | CAR-T Cells | AML | 2 & 3 | |
| CD33 | IMGN779 | AML | 1 | |
| GEM333 | AML | 1 | ||
| F16IL2, w/BI 836858 | AML | 1 | ||
| Gemtuzumab ozogamicin | AML, MDS | 1 | ||
| Gemtuzumab ozogamicin | AML | 2 | ||
| Lintuzumab (HuM195) | AML | 1 | ||
| CAR-T Cells | AML | 1 | ||
| CAR-T Cells | Myeloid malignancies | 1 & 2 | ||
| CD33/CCL1 | CAR-T Cells | AML, CML, MDS | 1 | |
| CD33/CD3 | AMV564 | AML | 1 | |
| JNJ-67571244 | AML, MDS | 1 | ||
| AMV564 | MDS | 1 | ||
| CD33/38/56/123/117/133/34/Mucl | CAR-T Cells | AML | N/A | |
| CD33/38/123/56/Mucl/CLL1 | CAR-T Cells | AML | 1 & 2 | |
| CD47 | Hu5F9-G4 | AML, MDS | 1 | |
| AML, MDS | 1 | |||
| TTI-621 | AML, MDS, MPN | 1 | ||
| CD70/CD27 | ARGX-110 | AML, MDS | 1 & 2 | |
| c-KIT | Midostaurin (PKC412) | AML | 2 | |
| AML | 2 | |||
| c-KIT, SRC family | Ponatinib | AML | 1 & 2 | |
| CXCR4/CXCL12 | CX-01 | AML, MDS | 1 | |
| TIM3 | MBG453 | AML, MDS | 1 | |
AML, acute myeloid leukaemia; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CAR-T cells, chimeric antigen receptor-T cells; CCL1, chemokine ligand 1; CD, cluster of differentiation; CML, chronic myeloid leukaemia; CXCR4/CXCL12, chemokine receptor type 4 / chemokine ligand 12; c-KIT, KIT proto-oncogene receptor tyrosine kinase; LSC, leukaemic stem cell; MDS, myelodysplastic syndrome; MPN, myeloproliferative neoplasms; SRC, proto-oncogene tyrosine-protein kinase SRC.
Table II.
Ongoing anti-LSC clinical trials targeting cell signaling and metabolism
| LSC Targets | Agent(s) | Disease(s) | Phase | Trial Identifier |
|---|---|---|---|---|
| Cell Signalling Molecules | ||||
| IL-1RAP | CAR-T cells | CML | N/A | |
| NF-κB | Bortezomib | AML | 2 | |
| AML | 1 | |||
| mTOR | Dactolisib (BEZ235) | AML, CML | 1 | |
| Sirolimus | AML | 1 | ||
| Sonic hedgehog | Glasdegib (PF-04449913) | AML | 2 | |
| MDS, CMML | 2 | |||
| AML, MDS | 1 | |||
| AML, MDS | 2 | |||
| AML | 3 | |||
| Sonidegib (LDE225) | AML, CMML, MDS | 1 | ||
| Vismodegib | AML | 2 | ||
| Cellular Metabolism | ||||
| BCL-2 | Venetoclax (ABT-199) | AML | 1 & 2 | |
| AML | 1 & 2 | |||
| AML | 1 & 2 | |||
| AML | 1 & 2 | |||
| S65487 | AML | 1 | ||
| BCL-2/IDH1 | Venetoclax (ABT-199) and Ivosidenib (AG120) | AML | 1 & 2 | |
| IDH1 | Ivosidenib (AG120) | AML | 1 | |
| AML | 1 | |||
| AML | Approved | |||
| AML | 3 | |||
| AML | 1 & 2 | |||
| IDH305 | AML, MDS | 1 | ||
| FT-2102 | AML, MDS | 1 & 2 | ||
| BAY-1436032 | AML | 1 | ||
| IDH2 | Enasidenib (AG221) | AML | 1 | |
| AML | 3 | |||
| AML, CMML | 1 | |||
| AML | 1 & 2 | |||
| Complex I (ETC) | IACS-010759 | AML | 1 | |
| Biomarker-based (The Beat AML Trial) | Various compounds | AML | 1 & 2 | |
AML, acute myeloid leukaemia; BCL-2, B-cell lymphoma 2; CAR-T cells, chimeric antigen receptor T cells; CML, chronic myeloid leukaemia; CMML, chronic myelomonocytic leukaemia; ETC, Electron Transport Chain; IDH, isocitrate dehydrogenase; IL-1RAP, interleukin 1 receptor accessory protein; LSC, leukaemic stem cell; MDS, myelodysplastic syndrome; mTOR, mechanistic target of rapamycin; N/A, not available; NF-κB, nuclear factor kappa B.
In addition to targeting the microenvironment, molecules targeting cell surface receptors and intracellular signalling pathways have also gained attention. Microenvironment interactions work together with a network of cytokine receptors, cytokines and chemokines to promote survival and self-renewal of normal and leukaemic stem cells. Cell surface receptor targets that were shown to be upregulated in LSCs versus normal HSCs include CD123, as mentioned above, as well as molecules such as the IL2 receptor, CD25, and CD33, CD47 and CD70 (Figure 3). Studies targeting intracellular signalling pathways include clinical trials with molecules targeting the PI3K/AKT pathway, the Sonic hedgehog pathway, the ubiquitin proteasome system, and histone methyltransferases (Table II, Table S2). Our group previously demonstrated that combined inhibition of BCR-ABL1 and STAT3 induces synthetic lethality in TKI-resistant CML stem and progenitor cells (Eiring et al., 2015b). Canonical STAT3 function involves phosphorylation-dependent dimerization followed by nuclear translocation to activate transcription. However, TKI resistance is not associated with a STAT3 transcriptional signature (Eiring et al., unpublished observations) suggesting an alternative, non-canonical function for STAT3 in drug resistance of CML. Interestingly, STAT3 has been linked to mitochondrial respiration (Lee et al., 2018), and was shown to activate CD36 expression in lymphoid leukaemias (Rozovski et al., 2018). Thus, it is tempting to speculate whether STAT3 plays a role in aberrant energy metabolism in CML LSCs, TKI-resistant CML progenitor cells, and possibly stem cells from other cancers. STAT3-mediated metabolic regulation might tie into bone marrow microenvironment-mediated support of LSC quiescence and survival, but further studies will be required to fully elucidate the role of STAT3 in LSC survival and self-renewal.
It is clear that a better understanding of the unique metabolic dependencies of CSCs and LSCs will be critical to achieving better outcomes for patients with different types of malignancies. However, despite our increased understanding of the survival and self-renewal of LSCs, experimental studies have yet to be translated into improved long-term survival outcomes for leukaemia patients. Therefore, additional studies of normal and leukaemic stem cell biology will be required to instruct new treatment approaches. While translating new therapeutic approaches into the clinic will probably be accompanied by many challenges, more effective targeting of LSCs has the potential to improve outcomes for patients with myeloid malignancies and, indeed other types of cancers.
Supplementary Material
Acknowledgements
The authors would like to thank Idaly M. Olivas and Joshua J. Lara for editorial assistance. This work was supported in part by the Department of Molecular and Translational Medicine at Texas Tech University Health Sciences Center El Paso, and by the National Cancer Institute of the National Institutes of Health under award number 1K22CA216008-01 (AME). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. AME would also like to acknowledge support from the European Hematology Association (EHA) and the American Society of Hematology (ASH) through mentorship from the 2018 EHA-ASH Translational Research Training in Hematology program.
References
- Alvarez-Calderon F, Gregory MA, Pham-Danis C, Deryckere D, Stevens BM, Zaberezhnyy V, Hill AA, Gemta L, Kumar A, Kumar V, Wempe MF, Pollyea DA, Jordan CT, Serkova NJ, Graham DK & Degregori J 2015. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clinical Cancer Research, 21, 1360–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes K, Mcintosh E, Whetton AD, Daley GQ, Bentley J & Baldwin SA 2005. Chronic myeloid leukaemia: an investigation into the role of Bcr-Abl-induced abnormalities in glucose transport regulation. Oncogene, 24, 3257–67. [DOI] [PubMed] [Google Scholar]
- Bocchia M, Sicuranza A, Abruzzese E, Iurlo A, Sirianni S, Gozzini A, Galimberti S, Aprile L, Martino B, Pregno P, Sora F, Alunni G, Fava C, Castagnetti F, Puccetti L, Breccia M, Cattaneo D, Defina M, Mulas O, Barate C, Caocci G, Sica S, Gozzetti A, Luciano L, Crugnola M, Annunziata M, Tiribelli M, Pacelli P, Ferrigno I, Usala E, Sgherza N, Rosti G, Bosi A & Raspadori D 2018. Residual peripheral blood CD26+ leukemic stem cells in chronic myeloid leukemia patients during TKI therapy and during treatment-free remission. Frontiers in Oncology, 8, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D & Dick JE 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature Medicine, 3, 730–7. [DOI] [PubMed] [Google Scholar]
- Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, Herrmann R, Lynch KP & Hughes TP 2002. High frequency of point mutations clustered within the adenosine triphosphate-binding region of BCR/ABL in patients with chronic myeloid leukemia or Ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood, 99, 3472–5. [DOI] [PubMed] [Google Scholar]
- Caocci G, Mulas O, Abruzzese E, Luciano L, Iurlo A, Attolico I, Castagnetti F, Galimberti S, Sgherza N, Bonifacio M, Annunziata M, Gozzini A, Orlandi EM, Stagno F, Binotto G, Pregno P, Fozza C, Trawinska MM, De Gregorio F, Cattaneo D, Albano F, Gugliotta G, Barate C, Scaffidi L, Elena C, Pirillo F, Scalzulli E, La Nasa G, Foa R & Breccia M 2019. Arterial occlusive events in chronic myeloid leukemia patients treated with ponatinib in the real-life practice are predicted by the Systematic Coronary Risk Evaluation (SCORE) chart. Hematological Oncology, doi: 10.1002/hon.2606 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong WJ, Zhao F, Medeiros BC, Tyvoll DA & Majeti R 2015. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nature Medicine, 21, 178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomel JC, Bonnet ML, Sorel N, Bertrand A, Meunier MC, Fichelson S, Melkus M, Bennaceur-Griscelli A, Guilhot F & Turhan AG 2011. Leukemic stem cell persistence in chronic myeloid leukemia patients with sustained undetectable molecular residual disease. Blood, 118, 3657–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Mcdonald T, Lin A, Chakraborty S, Huang Q, Snyder DS & Bhatia R 2011. Persistence of leukemia stem cells in chronic myelogenous leukemia patients in prolonged remission with imatinib treatment. Blood, 118, 5565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung YJ, Choi CW, Slape C, Fry T & Aplan PD 2008. Transplantation of a myelodysplastic syndrome by a long-term repopulating hematopoietic cell. Proceedings of the National Academy of Sciences of the United States of America, 105, 14088–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW & Druker BJ 2011. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. Journal of Clinical Investigation, 121, 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC & Schimmer AD 2007. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nature Reviews Cancer, 7, 118–29. [DOI] [PubMed] [Google Scholar]
- Cortes JE, Kim DW, Pinilla-Ibarz J, Le Coutre P, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, Dipersio J, Deangelo DJ, Abruzzese E, Rea D, Baccarani M, Muller MC, Gambacorti-Passerini C, Wong S, Lustgarten S, Rivera VM, Clackson T, Turner CD, Haluska FG, Guilhot F, Deininger MW, Hochhaus A, Hughes T, Goldman JM, Shah NP, Kantarjian H & Investigators P 2013. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. The New England Journal of Medicine, 369, 1783–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Witte T, Suciu S, Peetermans M, Fenaux P, Strijckmans P, Hayat M, Jaksic B, Selleslag D, Zittoun R, Dardenne M & et al. 1995. Intensive chemotherapy for poor prognosis myelodysplasia (MDS) and secondary acute myeloid leukemia (sAML) following MDS of more than 6 months duration. A pilot study by the Leukemia Cooperative Group of the European Organisation for Research and Treatment in Cancer (EORTC-LCG). Leukemia, 9, 1805–11. [PubMed] [Google Scholar]
- Derolf AR, Kristinsson SY, Andersson TM, Landgren O, Dickman PW & Bjorkholm M 2009. Improved patient survival for acute myeloid leukemia: a population-based study of 9729 patients diagnosed in Sweden between 1973 and 2005. Blood, 113, 3666–72. [DOI] [PubMed] [Google Scholar]
- Dick JE 2008. Stem cell concepts renew cancer research. Blood, 112, 4793–807. [DOI] [PubMed] [Google Scholar]
- Dinardo CD, Pratz KW, Letai A, Jonas BA, Wei AH, Thirman M, Arellano M, Frattini MG, Kantarjian H, Popovic R, Chyla B, Xu T, Dunbar M, Agarwal SK, Humerickhouse R, Mabry M, Potluri J, Konopleva M & Pollyea DA 2018. Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. The Lancet Oncology, 19, 216–228. [DOI] [PubMed] [Google Scholar]
- Eiring AM, Khorashad JS, Anderson DJ, Yu F, Redwine HM, Mason CC, Reynolds KR, Clair PM, Gantz KC, Zhang TY, Pomicter AD, Kraft IL, Bowler AD, Johnson K, Partlin MM, O’hare T & Deininger MW 2015a. b-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia, 29, 2328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiring AM, Page BD, Kraft IL, Mason CC, Vellore NA, Resetca D, Zabriskie MS, Zhang TY, Khorashad JS, Engar AJ, Reynolds KR, Anderson DJ, Senina A, Pomicter AD, Arpin CC, Ahmad S, Heaton WL, Tantravahi SK, Todic A, Colaguori R, Moriggl R, Wilson DJ, Baron R, O’hare T, Gunning PT & Deininger MW 2015b. Combined STAT3 and BCR-ABL1 inhibition induces synthetic lethality in therapy-resistant chronic myeloid leukemia. Leukemia, 29, 586–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farge T, Saland E, De Toni F, Aroua N, Hosseini M, Perry R, Bosc C, Sugita M, Stuani L, Fraisse M, Scotland S, Larrue C, Boutzen H, Feliu V, Nicolau-Travers ML, Cassant-Sourdy S, Broin N, David M, Serhan N, Sarry A, Tavitian S, Kaoma T, Vallar L, Iacovoni J, Linares LK, Montersino C, Castellano R, Griessinger E, Collette Y, Duchamp O, Barreira Y, Hirsch P, Palama T, Gales L, Delhommeau F, Garmy-Susini BH, Portais JC, Vergez F, Selak M, Danet-Desnoyers G, Carroll M, Recher C & Sarry JE 2017. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discovery, 7, 716–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fathi AT, Erba HP, Lancet JE, Stein EM, Ravandi F, Faderl S, Walter RB, Advani AS, Deangelo DJ, Kovacsovics TJ, Jillella A, Bixby D, Levy MY, O’meara MM, Ho PA, Voellinger J & Stein AS 2018. A phase 1 trial of vadastuximab talirine combined with hypomethylating agents in patients with CD33-positive AML. Blood, 132, 1125–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer GM, Jalali A, Kircher DA, Lee WC, Mcquade JL, Haydu LE, Joon AY, Reuben A, De Macedo MP, Carapeto FCL, Yang C, Srivastava A, Ambati CR, Sreekumar A, Hudgens CW, Knighton B, Deng W, Ferguson SD, Tawbi HA, Glitza IC, Gershenwald JE, Vashisht Gopal YN, Hwu P, Huse JT, Wargo JA, Futreal PA, Putluri N, Lazar AJ, Deberardinis RJ, Marszalek JR, Zhang J, Holmen SL, Tetzlaff MT & Davies MA 2019. Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discovery, doi: 10.1158/2159-8290.CD-18-1489 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, Li G & Zhou M 2017. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget, 8, 57813–57825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Khawaja MR, Dinardo CD, Atkins JT & Janku F 2016. Targeting isocitrate dehydrogenase (IDH) in cancer. Discovery Medicine, 21, 373–80. [PubMed] [Google Scholar]
- Gottschalk S, Anderson N, Hainz C, Eckhardt SG & Serkova NJ 2004. Imatinib (STI571)-mediated changes in glucose metabolism in human leukemia BCR-ABL-positive cells. Clinical Cancer Research, 10, 6661–8. [DOI] [PubMed] [Google Scholar]
- Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L & Holyoake TL 2002. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood, 99, 319–25. [DOI] [PubMed] [Google Scholar]
- Greenberg PL, Attar E, Bennett JM, Bloomfield CD, De Castro CM, Deeg HJ, Foran JM, Gaensler K, Garcia-Manero G, Gore SD, Head D, Komrokji R, Maness LJ, Millenson M, Nimer SD, O’donnell MR, Schroeder MA, Shami PJ, Stone RM, Thompson JE, Westervelt P & National Comprehensive Cancer N 2011. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes. Journal of the National Comprehensive Cancer Network, 9, 30–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’hare T, Zaberezhnyy V, Williams RT, Druker BJ, Perrotti D & Degregori J 2010. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell, 18, 74–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK & National Cancer Research Institute Adult Leukaemia Working, G. 2010. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood, 116, 354–65. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, Shiraishi Y, Shiozawa Y, Chiba K, Tanaka H, Koeffler HP, Klein HU, Dugas M, Aburatani H, Kohlmann A, Miyano S, Haferlach C, Kern W & Ogawa S 2014. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia, 28, 241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, Nicolini FE, Muller-Tidow C, Bhatia R, Brunton VG, Koschmieder S & Holyoake TL 2012. Chronic myeloid leukemia stem cells are not dependent on Bcr-Abl kinase activity for their survival. Blood, 119, 1501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, Tayyari F, Mcskimming D, Kannan N, Tojo A, Edison AS & Ito T 2017. Cancer progression by reprogrammed BCAA metabolism in myeloid leukaemia. Nature, 545, 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR, Akashi K & Gilliland DG 2004. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell, 6, 587–96. [DOI] [PubMed] [Google Scholar]
- Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, Wang Y, Battula VL, Mak PY, Schallmoser K, Ruvolo P, Schober WD, Shpall EJ, Nguyen MH, Strunk D, Bueso-Ramos CE, Konoplev S, Davis RE, Konopleva M & Andreeff M 2014. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-kappaB mediates chemoresistance. Blood, 123, 2691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagani Z, Mora-Blanco EL, Sansam CG, Mckenna ES, Wilson B, Chen D, Klekota J, Tamayo P, Nguyen PT, Tolstorukov M, Park PJ, Cho YJ, Hsiao K, Buonamici S, Pomeroy SL, Mesirov JP, Ruffner H, Bouwmeester T, Luchansky SJ, Murtie J, Kelleher JF, Warmuth M, Sellers WR, Roberts CW & Dorsch M 2010. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nature Medicine, 16, 1429–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Kantarjian H, Boddu PC, Nogueras-Gonzalez GM, Verstovsek S, Garcia-Manero G, Borthakur G, Sasaki K, Kadia TM, Sam P, Ahaneku H, O’brien S, Estrov Z, Ravandi F, Jabbour E & Cortes JE 2019. Analysis of cardiovascular and arteriothrombotic adverse events in chronic-phase CML patients after frontline TKIs. Blood Advances, 3, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen L, Frambach S, Allard NAE, Hopman MTE, Schirris TJJ, Voermans NC, Rodenburg RJ, Blijlevens NMA & Timmers S 2019. Skeletal muscle toxicity associated with tyrosine kinase inhibitor therapy in patients with chronic myeloid leukemia. Leukemia, doi: 10.1038/s41375-019-0443-7 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F & Dick JE 2006. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature Medicine, 12, 1167–74. [DOI] [PubMed] [Google Scholar]
- Jones CL, Stevens BM, D’alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, Krug A, Reinhold D, Smith C, Degregori J, Pollyea DA & Jordan CT 2018. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell, 34, 724–740 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, Erpelinck-Verschueren CAJ, Gradowska PL, Meijer R, Cloos J, Biemond BJ, Graux C, Van Marwijk Kooy M, Manz MG, Pabst T, Passweg JR, Havelange V, Ossenkoppele GJ, Sanders MA, Schuurhuis GJ, Lowenberg B & Valk PJM 2018. Molecular minimal residual disease in acute myeloid leukemia. The New England Journal of Medicine, 378, 1189–1199. [DOI] [PubMed] [Google Scholar]
- Kagoya Y, Yoshimi A, Kataoka K, Nakagawa M, Kumano K, Arai S, Kobayashi H, Saito T, Iwakura Y & Kurokawa M 2014. Positive feedback between NF-kappaB and TNF-alpha promotes leukemia-initiating cell capacity. Journal of Clinical Investigation, 124, 528–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluza J, Jendoubi M, Ballot C, Dammak A, Jonneaux A, Idziorek T, Joha S, Dauphin V, Malet-Martino M, Balayssac S, Maboudou P, Briand G, Formstecher P, Quesnel B & Marchetti P 2011. Exploiting mitochondrial dysfunction for effective elimination of imatinib-resistant leukemic cells. PLoS One, 6, e21924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominsky DJ, Klawitter J, Brown JL, Boros LG, Melo JV, Eckhardt SG & Serkova NJ 2009. Abnormalities in glucose uptake and metabolism in imatinib-resistant human BCR-ABL-positive cells. Clinical Cancer Research, 15, 3442–50. [DOI] [PubMed] [Google Scholar]
- Kuepper MK, Butow M, Herrmann O, Ziemons J, Chatain N, Maurer A, Kirschner M, Maie T, Costa IG, Eschweiler J, Koschmieder S, Brummendorf TH, Muller-Newen G & Schemionek M 2019. Stem cell persistence in CML is mediated by extrinsically activated JAK1-STAT3 signaling. Leukemia, doi: 10.1038/s41375-019-0427-7 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, Helgason GV & Gottlieb E 2017. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nature Medicine, 23, 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramoto K, Ban S, Oda K, Tanaka H, Kimura A & Suzuki G 2002. Chromosomal instability and radiosensitivity in myelodysplastic syndrome cells. Leukemia, 16, 2253–8. [DOI] [PubMed] [Google Scholar]
- Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, Ashton JM, Pei S, Grose V, O’dwyer KM, Liesveld JL, Brookes PS, Becker MW & Jordan CT 2013. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell, 12, 329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landberg N, Von Palffy S, Askmyr M, Lilljebjorn H, Sanden C, Rissler M, Mustjoki S, Hjorth-Hansen H, Richter J, Agerstam H, Jaras M & Fioretos T 2018. CD36 defines primitive chronic myeloid leukemia cells less responsive to imatinib but vulnerable to antibody-based therapeutic targeting. Haematologica, 103, 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA & Dick JE 1994. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature, 367, 645–8. [DOI] [PubMed] [Google Scholar]
- Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, Nixon MJ, Estrada MV, Sanchez V, Sanders ME, Lee T, Gomez H, Lluch A, Perez-Fidalgo JA, Wolf MM, Andrejeva G, Rathmell JC, Fesik SW & Arteaga CL 2017. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metabolism, 26, 633–647 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS & Lu B 2014. Targeting PINK1 and MQC in brain tumors. Oncotarget, 5, 2864–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Hirpara JL, Eu JQ, Sethi G, Wang L, Goh BC & Wong AL 2018. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biology, doi: 10.1016/j.redox.2018.101073 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang L, Li L, Wang Z, Ho Y, Mcdonald T, Holyoake TL, Chen W & Bhatia R 2012. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell, 21, 266–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Osdal T, Ho Y, Chun S, Mcdonald T, Agarwal P, Lin A, Chu S, Qi J, Li L, Hsieh YT, Dos Santos C, Yuan H, Ha TQ, Popa M, Hovland R, Bruserud O, Gjertsen BT, Kuo YH, Chen W, Lain S, Mccormack E & Bhatia R 2014. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell, 15, 431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissanu Deribe Y, Sun Y, Terranova C, Khan F, Martinez-Ledesma J, Gay J, Gao G, Mullinax RA, Khor T, Feng N, Lin YH, Wu CC, Reyes C, Peng Q, Robinson F, Inoue A, Kochat V, Liu CG, Asara JM, Moran C, Muller F, Wang J, Fang B, Papadimitrakopoulou V, Wistuba Ii, Rai K, Marszalek J & Futreal PA 2018. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nature Medicine, 24, 1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle NK, Barber AG & Reya T 2018. Stem cell fate in cancer growth, progression and therapy resistance. Nature Reviews Cancer, 18, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medyouf H, Mossner M, Jann JC, Nolte F, Raffel S, Herrmann C, Lier A, Eisen C, Nowak V, Zens B, Mudder K, Klein C, Oblander J, Fey S, Vogler J, Fabarius A, Riedl E, Roehl H, Kohlmann A, Staller M, Haferlach C, Muller N, John T, Platzbecker U, Metzgeroth G, Hofmann WK, Trumpp A & Nowak D 2014. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell, 14, 824–37. [DOI] [PubMed] [Google Scholar]
- Minson AG, Cummins K, Fox L, Costello B, Yeung D, Cleary R, Forsyth C, Tatarczuch M, Burbury K, Motorna O, Shortt J, Fleming S, Mcquillan A, Schwarer A, Harrup R, Holmes A, Ratnasingam S, Chan KL, Hsu WH, Ashraf A, Putt F & Grigg A 2019. The natural history of vascular and other complications in patients treated with nilotinib for chronic myeloid leukemia. Blood Advances, 3, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, Mcafoos T, Morlacchi P, Ackroyd J, Agip AA, Al-Atrash G, Asara J, Bardenhagen J, Carrillo CC, Carroll C, Chang E, Ciurea S, Cross JB, Czako B, Deem A, Daver N, De Groot JF, Dong JW, Feng N, Gao G, Gay J, Do MG, Greer J, Giuliani V, Han J, Han L, Henry VK, Hirst J, Huang S, Jiang Y, Kang Z, Khor T, Konoplev S, Lin YH, Liu G, Lodi A, Lofton T, Ma H, Mahendra M, Matre P, Mullinax R, Peoples M, Petrocchi A, Rodriguez-Canale J, Serreli R, Shi T, Smith M, Tabe Y, Theroff J, Tiziani S, Xu Q, Zhang Q, Muller F, Depinho RA, Toniatti C, Draetta GF, Heffernan TP, Konopleva M, Jones P, Di Francesco ME & Marszalek JR 2018. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nature Medicine, 24, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Mufti GJ 2004. Pathobiology, classification, and diagnosis of myelodysplastic syndrome. Best Practice & Research: Clinical Haematology, 17, 543–57. [DOI] [PubMed] [Google Scholar]
- Ni Chonghaile T & Letai A 2008. Mimicking the BH3 domain to kill cancer cells. Oncogene, 27, Suppl 1, S149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson L, Eden P, Olsson E, Mansson R, Astrand-Grundstrom I, Strombeck B, Theilgaard-Monch K, Anderson K, Hast R, Hellstrom-Lindberg E, Samuelsson J, Bergh G, Nerlov C, Johansson B, Sigvardsson M, Borg A & Jacobsen SE 2007. The molecular signature of MDS stem cells supports a stem-cell origin of 5q myelodysplastic syndromes. Blood, 110, 3005–14. [DOI] [PubMed] [Google Scholar]
- Nowell PC & Hungerford D 1960. A minute chromosome in human chronic granulocytic leukemia. Science, 132, 1497. [DOI] [PubMed] [Google Scholar]
- O’Hare T, Zabriskie MS, Eiring AM & Deininger MW 2012. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nature Reviews Cancer, 12, 513–26. [DOI] [PubMed] [Google Scholar]
- Ogawa S 2019. Genetics of MDS. Blood, 133, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang WW, Pluvinage JV, Price EA, Sridhar K, Arber DA, Greenberg PL, Schrier SL, Park CY & Weissman IL 2013. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proceedings of the National Academy of Sciences of the United States of America, 110, 3011–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, Raine K, Hinton J, Mudie LJ, Mclaren S, Hardy C, Latimer C, Della Porta MG, O’meara S, Ambaglio I, Galli A, Butler AP, Walldin G, Teague JW, Quek L, Sternberg A, Gambacorti-Passerini C, Cross NC, Green AR, Boultwood J, Vyas P, Hellstrom-Lindberg E, Bowen D, Cazzola M, Stratton MR, Campbell PJ & Chronic Myeloid Disorders Working Group Of The International Cancer Genome, C. 2013. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood, 122, 3616–27; quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, Bescos C, Di Croce L & Benitah SA 2017. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature, 541, 41–45. [DOI] [PubMed] [Google Scholar]
- Pollyea DA & Jordan CT 2017. Therapeutic targeting of acute myeloid leukemia stem cells. Blood, 129, 1627–1635. [DOI] [PubMed] [Google Scholar]
- Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D’alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, Hesselberth JR, Abbott D, Schatz D, Gutman JA, Purev E, Smith C & Jordan CT 2018. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nature Medicine, 24, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Morrison SJ, Clarke MF & Weissman IL 2001. Stem cells, cancer, and cancer stem cells. Nature, 414, 105–11. [DOI] [PubMed] [Google Scholar]
- Rhyasen GW, Wunderlich M, Tohyama K, Garcia-Manero G, Mulloy JC & Starczynowski DT 2014. An MDS xenograft model utilizing a patient-derived cell line. Leukemia, 28, 1142–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousselot P, Charbonnier A, Cony-Makhoul P, Agape P, Nicolini FE, Varet B, Gardembas M, Etienne G, Rea D, Roy L, Escoffre-Barbe M, Guerci-Bresler A, Tulliez M, Prost S, Spentchian M, Cayuela JM, Reiffers J, Chomel JC, Turhan A, Guilhot J, Guilhot F & Mahon FX 2014. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. Journal of Clinical Oncology, 32, 424–30. [DOI] [PubMed] [Google Scholar]
- Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, Burger J, Thompson P, Jain N, Wierda W, Keating MJ & Estrov Z 2018. STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget, 9, 21268–21280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruutu T, Hanninen A, Jarventie G, Koistinen P, Koivunen E, Katka K, Nousiainen T, Oksanen K, Pelliniemi TT, Remes K, Timonen T, Volin L & Elonen E 1997. Intensive chemotherapy of poor prognosis myelodysplastic syndromes (MDS) and acute myeloid leukemia following MDS with idarubicin and cytarabine. Leukemia Research, 21, 133–8. [DOI] [PubMed] [Google Scholar]
- Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, Taniguchi S, Shultz LD & Ishikawa F 2010. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nature Biotechnology, 28, 275–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samudio I, Fiegl M & Andreeff M 2009. Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Research, 69, 2163–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siapati EK, Papadaki M, Kozaou Z, Rouka E, Michali E, Savvidou I, Gogos D, Kyriakou D, Anagnostopoulos NI & Vassilopoulos G 2011. Proliferation and bone marrow engraftment of AML blasts is dependent on b-catenin signalling. British Journal of Haematology, 152, 164–74. [DOI] [PubMed] [Google Scholar]
- Singh SK, Williams CA, Klarmann K, Burkett SS, Keller JR & Oberdoerffer P 2013. Sirt1 ablation promotes stress-induced loss of epigenetic and genomic hematopoietic stem and progenitor cell maintenance. The Journal of Experimental Medicine, 210, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai CK, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge AC, Datti A, Ketela T, Moffat J, Robinson BH, Cameron JH, Wrana J, Eaves CJ, Minden MD, Wang JC, Dick JE, Humphries K, Nislow C, Giaever G & Schimmer AD 2011. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell, 20, 674–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolkova K, Plecita-Hlavata L, Bellance N, Benard G, Rossignol R & Jezek P 2011. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. The International Journal of Biochemistry & Cell Biology, 43, 950–68. [DOI] [PubMed] [Google Scholar]
- Stevens BM, Khan N, D’alessandro A, Nemkov T, Winters A, Jones CL, Zhang W, Pollyea DA & Jordan CT 2018. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nature Communications, 9, 3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, He X, Zhu Y, Ding Z, Dong H, Feng Y, Du J, Wang H, Wu X, Zhang L, Yu X, Lin A, Mcdonald T, Zhao D, Wu H, Hua WK, Zhang B, Feng L, TOHYAMA K, BHATIA R, Oberdoerffer P, Chung YJ, Aplan PD, Boultwood J, Pellagatti A, Khaled S, Kortylewski M, Pichiorri F, Kuo YH, Carlesso N, Marcucci G, Jin H & Li L 2018. SIRT1 activation disrupts maintenance of myelodysplastic syndrome stem and progenitor cells by restoring TET2 function. Cell Stem Cell, 23, 355–369.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Bandi M, Lofton T, Smith M, Bristow CA, Carugo A, Rogers N, Leonard P, Chang Q, Mullinax R, Han J, Shi X, Seth S, Meyers BA, Miller M, Miao L, Ma X, Feng N, Giuliani V, Geck Do M, Czako B, Palmer WS, Mseeh F, Asara JM, Jiang Y, Morlacchi P, Zhao S, Peoples M, Tieu TN, Warmoes MO, Lorenzi PL, Muller FL, Depinho RA, Draetta GF, Toniatti C, Jones P, Heffernan TP & Marszalek JR 2019. Functional genomics reveals synthetic lethality between phosphogluconate dehydrogenase and oxidative phosphorylation. Cell Reports, 26, 469–482 e5. [DOI] [PubMed] [Google Scholar]
- Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, Mogushi K, Shikami M, Ruvolo V, Ishizawa J, Hail N Jr., Kazuno S, Igarashi M, Matsushita H, Yamanaka Y, Arai H, Nagaoka I, Miida T, Hayashizaki Y, Konopleva M & Andreeff M 2017. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Research, 77, 1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe N, Harris PJ, Warren RQ & Ivy SP 2011. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature Reviews Clinical Oncology, 8, 97–106. [DOI] [PubMed] [Google Scholar]
- Thomas D & Majeti R 2016. Burning fat fuels leukemic stem cell heterogeneity. Cell Stem Cell, 19, 1–2. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC & Thompson CB 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 324, 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Liu J, Zhou Y & Oltvai ZN 2010. Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Systems Biology, 4, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergez F, Green AS, Tamburini J, Sarry JE, Gaillard B, Cornillet-Lefebvre P, Pannetier M, Neyret A, Chapuis N, Ifrah N, Dreyfus F, Manenti S, Demur C, Delabesse E, Lacombe C, Mayeux P, Bouscary D, Recher C & Bardet V 2011. High levels of CD34+CD38low/-CD123+ blasts are predictive of an adverse outcome in acute myeloid leukemia: a Groupe Ouest-Est des Leucemies Aigues et Maladies du Sang (GOELAMS) study. Haematologica, 96, 1792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vey N, Delaunay J, Martinelli G, Fiedler W, Raffoux E, Prebet T, Gomez-Roca C, Papayannidis C, Kebenko M, Paschka P, Christen R, Guarin E, Broske AM, Baehner M, Brewster M, Walz AC, Michielin F, Runza V, Meresse V & Recher C 2016. Phase I clinical study of RG7356, an anti-CD44 humanized antibody, in patients with acute myeloid leukemia. Oncotarget, 7, 32532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Lapidot T, Cashman JD, Doedens M, Addy L, Sutherland DR, Nayar R, Laraya P, Minden M, Keating A, Eaves AC, Eaves CJ & Dick JE 1998. High level engraftment of NOD/SCID mice by primitive normal and leukemic hematopoietic cells from patients with chronic myeloid leukemia in chronic phase. Blood, 91, 2406–14. [PubMed] [Google Scholar]
- Wang Z, Yuan H, Roth M, Stark JM, Bhatia R & Chen WY 2013. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene, 32, 589–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O 1956. On the origin of cancer cells. Science, 123, 309–14. [DOI] [PubMed] [Google Scholar]
- Woll PS, Kjallquist U, Chowdhury O, Doolittle H, Wedge DC, Thongjuea S, Erlandsson R, Ngara M, Anderson K, Deng Q, Mead AJ, Stenson L, Giustacchini A, Duarte S, Giannoulatou E, Taylor S, Karimi M, Scharenberg C, Mortera-Blanco T, Macaulay IC, Clark SA, Dybedal I, Josefsen D, Fenaux P, Hokland P, Holm MS, Cazzola M, Malcovati L, Tauro S, Bowen D, Boultwood J, Pellagatti A, Pimanda JE, Unnikrishnan A, Vyas P, Gohring G, Schlegelberger B, Tobiasson M, Kvalheim G, Constantinescu SN, Nerlov C, Nilsson L, Campbell PJ, Sandberg R, Papaemmanuil E, Hellstrom-Lindberg E, Linnarsson S & Jacobsen SE 2014. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer Cell, 25, 794–808. [DOI] [PubMed] [Google Scholar]
- Yang X & Wang J 2018. Precision therapy for acute myeloid leukemia. Journal of Hematology & Oncology, 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM, Klemm DJ, Woolthuis CM, Stranahan AW, Park CY & Jordan CT 2016. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell, 19, 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, Sanchez PV, Lum JJ, Sayed N, Melo JV, Perl AE, Carroll M, Tuttle SW & Thompson CB 2010. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene, 29, 2962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J 2012. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncology Letters, 4, 1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Kinney MC, Scott LM, Zinkel SS & Rebel VI 2015. Revisiting the case for genetically engineered mouse models in human myelodysplastic syndrome research. Blood, 126, 1057–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.