

Abstract
Epithelial ovarian cancer (EOC) is the deadliest gynecological cancer. High-grade serous carcinoma (HGSC) is the most frequently diagnosed and lethal histosubtype of EOC. A significant proportion of HGSC patients relapse with chemoresistant disease. Therefore, there is an urgent need for novel therapeutic strategies for HGSC. Metabolic reprogramming is a hallmark of cancer cells, and targeting metabolism for cancer therapy may be beneficial. Here, we found that in comparison to normal fallopian tube epithelial cells, HGSC cells preferentially utilize glucose in the TCA cycle and not for aerobic glycolysis. This correlated with universally increased TCA cycle enzyme expression in HGSC cells under adherent conditions. HGSC disseminates as tumor cell spheroids within the peritoneal cavity. We found that wildtype isocitrate dehydrogenase I (IDH1) is the only TCA cycle enzyme upregulated in both adherent and spheroid conditions and is associated with reduced progression-free survival. IDH1 protein expression is also increased in primary HGSC patient tumors. Pharmacological inhibition or knockdown of IDH1 decreased proliferation of multiple HGSC cell lines by inducing senescence. Mechanistically, suppression of IDH1 increased the repressive histone mark H3K9me2 at multiple E2F target gene loci, which led to decreased expression of these genes. Altogether, these data suggest that increased IDH1 activity is an important metabolic adaptation in HGSC and that targeting wildtype IDH1 in HGSC alters the repressive histone epigenetic landscape to induce senescence.
Introduction
Among all gynecological cancers, epithelial ovarian cancer (EOC) is the most lethal due to dissemination into the peritoneal cavity and omental seeding in late stage disease (1). High-grade serous carcinoma (HGSC) is the most common histosubtype of EOC and has a poor prognosis (2). The five-year survival rate for HGSC patients is approximately 47% due to limited screening options and late stage diagnosis. This percentage decreases to 20–35% in women diagnosed with stage III and IV HGSC. Due to their characteristic TP53 mutations, many HGSC patients initially respond to standard-of-care platinum and taxol chemotherapies; however, a significant portion relapse with chemoresistant disease (3). Poly(ADP-ribose) polymerase (PARP) inhibitors were recently approved as a maintenance therapy for ovarian cancer (4). Although these inhibitors show promise, especially for homologous recombination-deficient ovarian cancer, some patients do not respond and resistance to these drugs has recently become evident. Therefore, there is an urgent need for novel therapeutic strategies for HGSC patients.
Recent evidence suggests that therapy-induced senescence leads to a better 5-year survival rate for HGSC patients (5). Cellular senescence is a state of stable cell cycle arrest that is induced by multiple stimuli, including shortened telomeres, oncogene activation, DNA damage, and certain therapeutics (6). Senescent cells are characterized by many hallmarks such as increased β-galactosidase activity (termed senescence-associated β-galactosidase or SA-β-gal), increased repressive histone marks, such as repressive di- and tri-methylation marks of histone 3 lysine 9 (H3K9me2/3) at E2F target genes (termed senescence-associated heterochromatin foci, SAHF), decreased incorporation of the thymine analog bromodeoxyuridine (BrdU), and decreased proliferation (7–9). Due to the decrease in proliferation, therapy-induced senescence is considered a desirable therapeutic outcome (5,10).
Tumor cells are characterized by metabolic reprogramming in order to maintain uncontrolled cell proliferation and growth (11). Tumors specifically reprogram metabolic pathways to allow for the increased need for biomass, such as nucleotides, lipids, and other macromolecules (12,13). This metabolic reprogramming is increasingly thought to modulate epigenetic marks, including histone and DNA methylation and histone acetylation (14). Altered metabolism in cancer cells provides a unique opportunity to exploit these changes as a targeted therapy (11). Cancer cells undergo anaerobic glycolysis and generate lactate, even in the presence of oxygen, termed the Warburg Effect (12). Although the Warburg effect is a common feature of many cancers, TCA cycle metabolism remains critical for both ATP production and macromolecule synthesis. Indeed, recent evidence suggests that multiple cancer types have increased TCA cycle metabolism (15), although less is known about TCA cycle activity in HGSC. Therefore, targeting the TCA cycle in cancer may serve as a novel therapeutic strategy.
Isocitrate dehydrogenase I (IDH1) catalyzes the oxidative decarboxylation of isocitrate to alpha-ketoglutarate (αKG) in a reversible reaction and produces NADPH. IDH1 is well-known for its mutation and the resulting production of the oncometabolite D-2-hydroxyglutarate (D-2HG, aka (R)-2HG) in secondary glioblastoma, acute myeloid leukemia (AML), and other cancers (16,17). However, recent work demonstrates that overexpression of wildtype IDH1 also promotes cancer progression in primary glioblastoma (18). Previous work identified a role for IDH1 in redox homeostasis and lipid metabolism (18,19). Additionally, IDH1 regulates histone marks through production of αKG, which is a co-substrate for the Jumonji C (JmjC) histone demethylases (20,21). Increased repressive histone methylation is a hallmark of senescence, and JmjC histone demethylases have been implicated in altering the epigenome of senescent cells (21). Therefore, depleting αKG pools through suppression of IDH1 may increase repressive histone methylation to induce senescence. While mutant IDH1 is well-characterized in several cancers (16,17), the role of wildtype IDH1 in metabolism and epigenetics has never been investigated in the context of HGSC.
In this study, we determined that HGSC cells preferentially utilize glucose in the TCA cycle. While most TCA cycle enzymes were upregulated in adherent HGSC cells compared to the cell-of-origin fallopian tube (22), only wildtype IDH1 expression was both increased in HGSC cell lines under spheroid conditions and associated with decreased progression-free survival. Functionally, knockdown of IDH1 induced senescence by increasing repressive histone methylation at multiple E2F target gene loci. Together, these data indicate that targeting wildtype IDH1 in HGSC represents a novel strategy for this patient population by inducing senescence through epigenetic silencing.
Materials and Methods
Cells and Culture Conditions
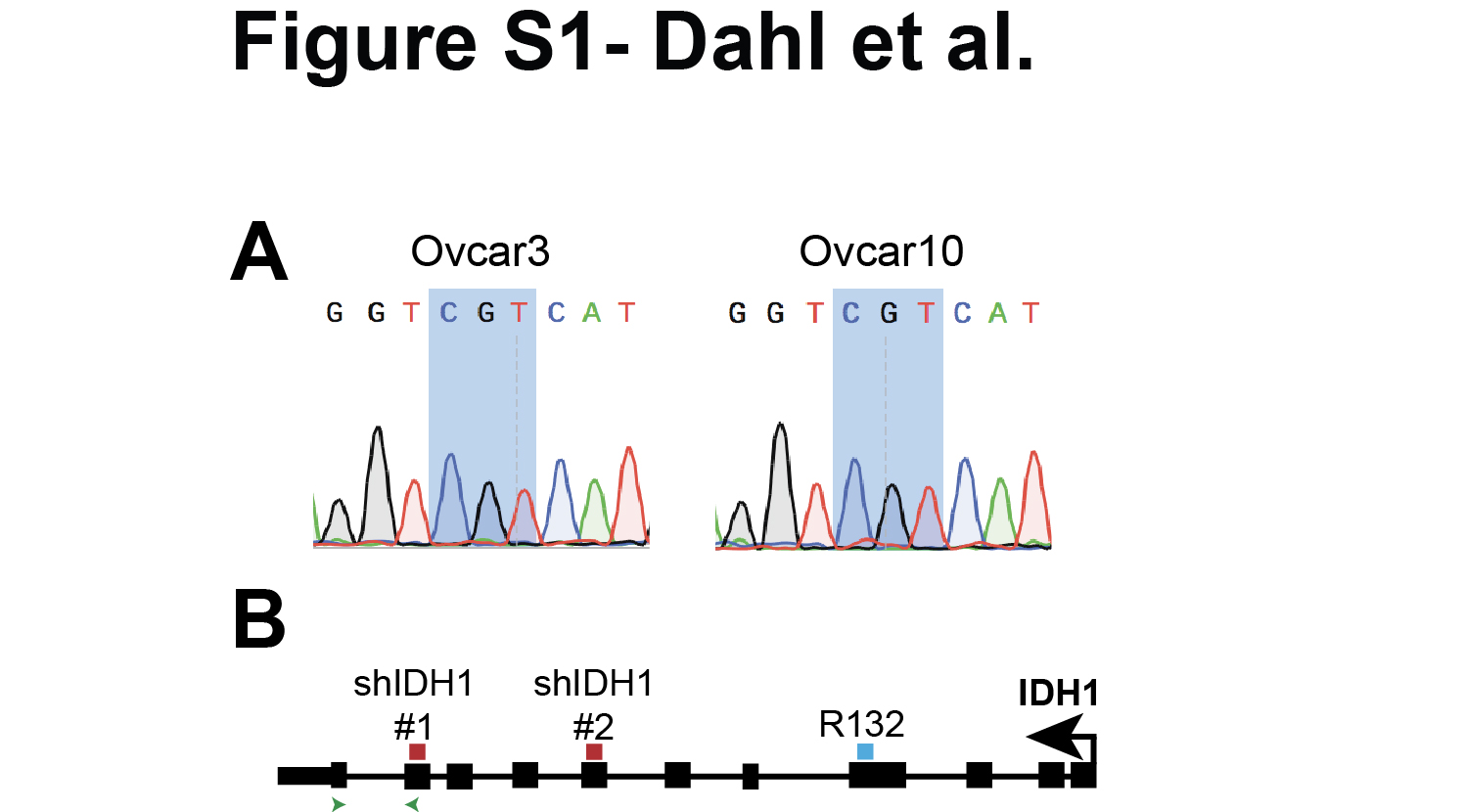
The following human HGSC cells were used: Ovcar3 (ATCC, obtained July 2017) and Ovcar10 (kind gift from Dr. Rugang Zhang, The Wistar Institute, obtained November 2016). Both Ovcar3 and Ovcar10 cells are wildtype for IDH1 (Fig. S1A). Ovcar3 and Ovcar10 cells lines were cultured in RPMI-1640 medium (Gibco) supplemented with 10% FBS. HGSC cell lines were thawed, and experiments were performed within 30 passages. The FT282 fallopian tube cell line was a kind gift from Dr. Ronny Drapkin (University of Pennsylvania, obtained July 2017). PFTE4, FT4-Tag, FT6-Tag, and FT33-Tag fallopian tube cell lines were a kind gift from Dr. Anna Loshkin (University of Pittsburgh, obtained November 2017). Fallopian tube cells were cultured in DMEM:F12 supplemented with 2% FBS and 1% penicillin/streptomycin under 2% oxygen conditions. Fallopian tubes cells were thawed, and experiments were performed within 4 passages. HEK-293FT cells were used for lentiviral packaging and were cultured in DMEM medium (Corning) supplemented with 10% FBS according to ATCC. For spheroid cultures, HGSC Ovcar3 and Ovcar10 cells lines were cultured in ultra-low attachment (ULA) plates (Corning) for 4 days. Cells were treated with 15μM GSK864 (Sigma Aldrich) or 1mM αKG (Sigma Aldrich) where indicated. All cell lines were cultured in Mycozap (Lonza) and were routinely tested every 3 months for mycoplasma as described (23). Cell lines were authenticated using STR profiling in November 2018 (Genetica).
Metabolomics
Metabolites were quantified by liquid chromatography- high resolution mass spectrometry after extraction of the cells by 80:20 methanol:water at −80 °C, sonication, centrifugation of protein at 17,000 rcf for 10 mins at 4°C, evaporation of the supernatant to dryness under N2 gas and resuspended in 50 μl of 5% 5-sulfosalicylic acid for analysis. For non-acyl-CoA polar metabolites ion pairing reversed phase liquid chromatography- mass spectrometry was conducted by modification of a previously published method on a Ultimate 3000 Binary UHPLC coupled to a Q Exactive HF mass spectrometer (24). Data were processed in Xcalibur (Thermo). Peak areas were normalized to stable isotope labeled internal standards spiked into each sample before extraction as follows; lactate, pyruvate to 13C3-pyruvate, succinate, malate to 13C4-succinate, (D/L)-2-hydroxyglutarate to 13C5-hydroxyglutarate, αKG to 13C5-αKG, fumarate to 13C4-fumarate, citrate to 13C6-citrate, acetyl-CoA, CoA, and succinyl-CoA to 13C2-acetyl-CoA. For malonyl-CoA, reversed phase liquid chromatography- mass spectrometry was conducted by a previously published method on a Ultimate 3000 Binary UHPLC coupled to a Q Exactive Plus mass spectrometer (25). For isotope tracing experiments, analysis was similar except the exact mass corresponding to the detected ion of each 13C isotopologue was integrated in a 5 ppm window in Tracefinder v4.1 (Thermo), and then isotopologue enrichment was calculated by the open, web-based software FluxFix against unlabeled controls (26).
3’mRNA-seq (Lexogen Quantseq)
Total RNA was extracted from cells with Trizol (Life Technologies), DNase treated, and isolated using a RNA Clean an Concentrator kit (Zymo) following the manufacturer’s instructions. The cDNA libraries were prepared using the QuantSeq 3’mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen) supplemented with UMI (unique molecular index) as per the manufacturer’s instructions. Briefly, total RNA was reverse transcribed using oligo (dT) primers. The second cDNA strand was synthesized by random priming, in a manner that DNA polymerase is efficiently stopped when reaching the next hybridized random primer, so only the fragment closed to the 3’ end gets captured for later indexed adapter ligation and PCR amplification. UMIs were incorporated to the first 6 bases of each read, followed by 4 bases of spacer sequences. The processed libraries were assessed for its size distribution and concentration using BioAnalyzer High Sensitivity DNA Kit (Agilent Technologies). Pooled libraries were diluted to 2 nM in EB buffer (Qiagen) and then denatured using the Illumina protocol. The libraries were pooled and diluted to 2 nM using 10 mM Tris-HCl, pH 8.5 and then denatured using the Illumina protocol. The denatured libraries were diluted to 10 pM by pre-chilled hybridization buffer and loaded onto an Illumina MiSeq v3 flow cell for a 150 cycles using a single-read recipe according to the manufacturer’s instructions. De-multiplexed sequencing reads were generated using Illumina BaseSpace.
UMI specific workflows that were developed and distributed by Lexogen were used to extract reads that are free from PCR artifacts (i.e., deduplication). First, the umi2index tool was used to add the 6 nucleotide UMI sequence to the identifier of each read and trims the UMI from the start of each read. This generates a new FASTQ file, which is then processed through trimming and alignment. Second, after the quality and polyA trimming by BBDuk (https://jgi.doe.gov/data-and-tools/bbtools/) and alignment by HISAT2 (version 2.1.0) (27), the mapped reads are collapsed according to the UMI sequence of each read. Reads are collapsed if they have the same mapping coordinates (CIGAR string) and identical UMI sequences. Collapsing reads in this manner removes PCR duplicates. Read counts were calculated using HTSeq (28) by supplementing Ensembl gene annotation (GRCh38.78). EdgeR (29) was used to fit the read counts to the negative binomial model along with generalized linear model (GLM) and differentially expressed genes were determined by the likelihood ratio test method implemented in the edgeR package. Significance was defined to be those with q-value < 0.05 calculated by the Benjamini-Hochberg method to control the false discovery rate (FDR). RNA-seq files are available in the GEO database (GSE128700). The list of differentially expressed genes was analyzed with Ingenuity Pathway Analysis (IPA). Gene Cluster Text files (GTC) and Categorical Class files (CLS) were generated following the Gene Set Enrichment Analysis (GSEA) documentation indications (http://software.broadinstitute.org/gsea/index.jsp). GTC and CLS files were used to run GSEA hallmarks and reactome under default parameters (javaGSEA desktop application).
Patient Samples
Protein lysates of patients’ samples were provided by Dr. Bitler (The University of Colorado, Aurora, CO). The University of Colorado Gynecologic Tumor and Fluid Bank has an Institutional Review Board approved protocol (COMIRB #07–935) in place to collect tissue from gynecologic patients with both malignant and benign disease processes. All participants are counseled regarding the potential uses of their tissue and sign a consent form approved by the Colorado Multiple Institutional Review Board, and studies were conducted in accordance with the US Common Rule. The tissues are processed, aliquoted, and stored at −80 degrees. Benign fallopian tube tissue and primary HGSC tumors were homogenized with a Polytron Homogenizer (Brinkman Instruments) in radioimmunoprecipitation assay (RIPA) buffer (150 mM sodium chloride, Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS [sodium dodecyl sulfate], 50 mM Tris, pH 8.0) supplemented with complete EDTA-free protease inhibitor cocktail (Roche), sodium fluoride (10 mM) and sodium orthovanadate (1 mM).
Plasmids and Antibodies
pLKO.1-shIDH1 plasmids were obtained from Sigma-Aldrich. The TCRN are as follows: shIDH1 #1: TRCN0000027253; shIDH1 #2: TRCN0000027249 (Fig. S1B). To overexpress IDH1, the IDH1 ORF was cloned into the pBABE-puro backbone. The following antibodies were obtained from the indicated suppliers: rabbit anti-IDH1 (Cell Signaling), rabbit anti-Lamin B1 (Abcam), rabbit anti-Cyclin A (Abcam), mouse anti-PCNA (Cell Signaling), mouse anti-MCM3 (Santa Cruz Biotechnologies), mouse anti-Vinculin (Sigma-Aldrich), mouse anti-Beta Actin (Sigma-Aldrich), rat anti-BrdU (Abcam), mouse anti-PML (Santa Cruz Biotechnologies), mouse anti-γH2AX (EMD Millipore), rabbit anti-53BP1 (Bethyl), Fluorescein donkey anti-rat IgG (Jackson ImmunoResearch), Cy™3 donkey anti-mouse (Jackson ImmunoResearch).
Lentivirus Infection and Ectopic IDH1 Transfection
Lentivirus was packaged in 293FT cells using the Virapower kit from Invitrogen following the manufacturer’s instructions. Cells infected with viruses encoding the puromycin-resistance gene were selected with 3μg/ml puromycin for 7 days.
IDH1 cDNA was transfected into cells using jetOptimus following manufacturer’s instructions (Polyplus Transfection).
BrdU Labeling and Immunofluorescence
For BrdU, cells on coverslips were incubated with 1μM BrdU for 30min. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and then postfixed with 1% PF + 0.01% Tween-20. Coverslips were DNaseI treated for 10min, and the DNaseI reaction was stopped using 20mM EDTA. Coverslips were then blocked with 3% BSA/PBS and incubated in anti-BrdU primary antibody (1:500) followed by incubation in FITC anti-Rat secondary antibody (1:1000). Finally, coverslips were incubated with 0.15 μg/ml DAPI, mounted, and sealed.
For immunofluorescence, cells on coverslips were fixed, permeabilized, and blocked as above and then incubated with the corresponding primary antibodies followed by incubation in FITC anti-rabbit (1:2000) or Cy3 anti-mouse (1:5000) secondary. Finally, coverslips were counterstained with DAPI, mounted, and sealed. All images were acquired at room temperature using a Nikon Eclipse 90i microscope with a 20x/0.17 objective (Nikon DIC N2 Plan Apo) equipped with a CoolSNAP Photometrics camera.
SA-β-Gal
SA-β-Gal staining was performed as previously described (30). Cells were fixed with 2% formaldehyde/0.2% glutaraldehyde in PBS and incubated at 37°C in staining solution (40 mM Na2HPO4, 150 mM NaCl, 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 1 mg/ml X-gal).
Colony Formation Assay
An equal number of cells was seeded in 6-well plates and cultured for ten to fourteen days. Cells were then fixed with 1% paraformaldehyde/PBS, stained with 0.05% crystal violet, and destained with 10% acetic acid. Absorbance (590nm) was measured using a spectrophotometer (Spectra Max 190).
RT-qPCR
Total RNA was prepared as described above. Relative expression of target genes listed in Table S1 were analyzed using the QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific). Primers were designed using the Integrated DNA Technologies (IDT) tool (https://www.idtdna.com/scitools/Applications/RealTimePCR/). Briefly, 5ng of total RNA was used to One-Step qPCR (Quanta BioSciences) following the manufacturer’s instructions in a final volume of 10μl. Conditions for amplification were: 10 min at 48˚C, 5 min at 95˚C, 40 cycles of 10 s at 95˚C and 7s at the corresponding annealing temperature. Relative quantification was determined normalizing to multiple references genes (B2M, PUM1, PSMC4, and MRPL9) using the delta-delta Ct method. Design of IDH1 primers are shown in Fig. S1B.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as previously described (31) using the ChIP-grade antibody mouse anti-H3K9me2 (Abcam). Cells were fixed in 1% paraformaldehyde for 5 min at room temperature and then quenched with 1 ml of 2.5 M glycine for 5 min at room temperature. After washing, cells were lysed in 1 ml ChIP lysis buffer (50 mM HEPES- KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8.0, 1% Triton X-100, and 0.1% deoxycholate with 0.1 mM PMSF and the EDTA-free Protease Inhibitor Cocktail). Samples were incubated on ice for 10 min and then centrifuged at 3,000 rpm for 3 min at 4°C. The pellet was re- suspended in 500 μl lysis buffer 2 (10 mM Tris, pH 8.0, 200 mM NaCl, 1 mM EDTA, and 0.5 mM EGTA with 0.1 mM PMSF and the EDTA-free Protease Inhibitor Cocktail) and incubated at room temperature for 10 min. Samples were centrifuged at 3,000 rpm for 5 min at 4°C. Next, the pellet was resuspended in 300 μl lysis buffer 3 (10 mM Tris, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% DOC, and 0.5% N-lauroylsarcosine with 0.1 mM PMSF and the EDTA-free Protease Inhibitor Cocktail). Cells were sonicated using a Branson Sonifier 250 for four cycles of 10 seconds on 50 seconds off. Next, 30 μl of 10% Triton X-100 was added to each tube, and then samples were centrifuged at max speed for 15 min at 4°C. 50 μl of the antibody bead conjugate solution was added to the supernatant, and chromatin was immunoprecipitated overnight on a rotator at 4°C. The following washes were performed: ChIP lysis buffer, ChIP lysis buffer + 0.65 M NaCl, wash buffer (10 mM Tris-HCl, pH 8.0, 250 mM LiCl, 0.5% NP-30, 0.5% deoxycholate, and 1 mM EDTA, pH 8.0), and TE (10 mM Tris-HCl, pH 8.0, and 1 mM EDTA, pH 8.0). DNA was eluted with TES (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, pH 8.0, and 1% SDS) for 30 min at 65°C. Reversal of cross-linking was performed by incubating samples overnight at 65°C. Proteins were digested using 1 mg/ml proteinase K and incubating at 37°C for 5 h. Finally, the DNA was purified using the Wizard SV Gel and PCR Clean Up kit (Promega). Immunoprecipitated DNA was analyzed by qPCR using iTaq Universal SYBR® Green Supermix (BioRad). Conditions for amplification were: 5 min at 95°C, 40 cycles of 95°C for 10 sec and 30 sec with 62°C annealing temperature. Enrichment of H3K9me2 was determined by normalizing to a gene desert region (31). Primer sets used for ChIP-qPCR are detailed in Table S1.
Western Blotting
Cells lysates were collected in 1X sample buffer (2% SDS, 10% glycerol, 0.01% bromophenol blue, 62.5mM Tris, pH 6.8, 0.1M DTT), boiled, and sonicated. Protein concentration was determined using the Bradford assay. An equal amount of total protein was resolved using SDS-PAGE gels and transferred to nitrocellulose membranes (Fisher Scientific). Membranes were blocked with 5% nonfat milk followed by overnight incubation in primary antibodies. Membranes were washed, incubated with HRP-conjugated secondary antibodies (Cell Signaling, Danvers, MA), and washed again. Proteins were visualized on film after incubation with chemiluminescent substrate (ThermoFisher, Waltham, MA).
Quantification and Statistical Analysis
GraphPad Prism version 8.0 was used to perform statistical analysis. T-test and One-way ANOVA followed by post hoc Tukey’s HSD tests were applied as appropriate. When indicated, p-values were adjusted according to Benjamini and Hochberg’s false discovery rate (FDR). The significance level was established at p < 0.05. Heatmaps were generated using GraphPad Prism. Kaplan-Meier curves were generated using publicly available ovarian cancer mRNA gene chip data (32). Ovarian cancer patients were filtered by a serous histosubtype and TP53 mutation to signify HGSC.
Results
Wildtype IDH1 is upregulated in high-grade serous ovarian cancer
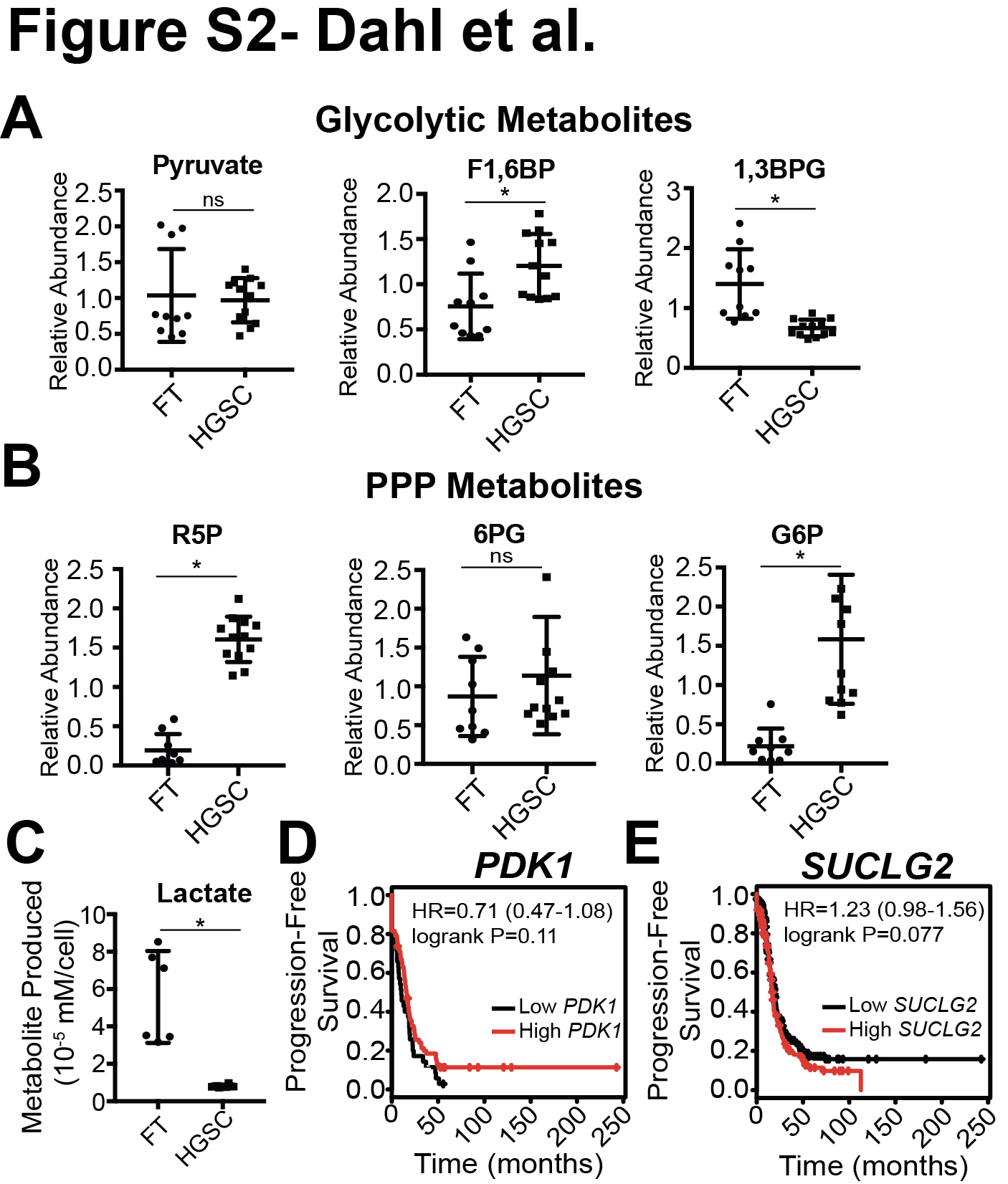
To determine whether changes in metabolism correlate with ovarian cancer disease progression, we performed metabolomics on primary fallopian tube (FT) cells and HGSC cell lines. We observed a significant upregulation in the levels of all TCA cycle metabolites in HGSC cells (Fig. 1A). Metabolites in other pathways such as glycolysis or the pentose phosphate pathway (PPP) were not universally upregulated in HGSC cells versus FT cells (Fig. S2A–B). Additionally, metabolite profiling of cell media showed decreased lactate production in HGSC cells compared to FT cells (Fig. S2C), suggesting that glucose is not being consumed by aerobic glycolysis. To determine whether upregulation of enzyme expression is responsible for the observed increase in TCA cycle metabolism, we performed RT-qPCR analysis of all 27 TCA cycle enzymes in five normal FT cell lines, including two primary and three immortalized lines, and two HGSC cell lines (Fig. 1B). Both HGSC cell lines tested exhibited a significant increase in expression of all TCA cycle enzymes, except IDH2, when compared to FT cells. Together, these data suggest that TCA cycle metabolism is upregulated in HGSC.
Figure 1: TCA cycle metabolism is upregulated in HGSC, and increased expression of IDH1 correlates with decreased progression-free survival of HGSC patients.

(A) LC/MS comparison of 2 FT (FT282 and FT4-Tag) and 2 HGSC (Ovcar3 and Ovcar10) cell lines. Heat map shows z-scores of relative abundance normalized to the mean of each individual TCA cycle metabolite. Significant metabolites are labeled in green. Adjusted p-value (FDR) <0.05.
(B) Differentially expressed transcripts in 5 FT (FT282, FT4-Tag, FT6-Tag, FT33-Tag, and PFTE4) and 2 HGSC (Ovcar3 and Ovcar10) cell lines. Heat map shows z-scores of expression levels relative to B2M of 27 TCA cycle enzymes. Significant transcripts are labeled in green. Adjusted p-value (FDR) <0.05
(C) Differentially expressed transcripts of 27 TCA cycle enzymes. Heat map shows fold change of averaged z-scores of Ovcar3 and Ovcar10 cultured in adherent versus ultra-low attachment conditions. The qPCR for adherent vs. spheroid was performed at the same time with the same conditions. Significantly upregulated transcripts are labeled in green. Adjusted p-value (FDR) <0.001.
(D) Kaplan-Meier curve for progression-free survival of ovarian cancer patients classified according to IDH1 expression level. Ovarian cancer patients were filtered for a serous histosubtype and TP53 mutation. Hazard ratio (HR) and logrank p-values are indicated.
(E) Immunoblot analysis of IDH1 comparing normal human FT and HGSC patient tissue lysates. Vinculin was used as a loading control.
(F) IDH1 protein expression normalized to vinculin and quantified using ImageJ. Data represent mean ± SD. *p<0.03.
(G) IDH1 mRNA expression from a publicly-available dataset comparing fallopian tube and matched HGSC patient samples (GSE10971). *p<0.0006.
HGSC disseminates to the peritoneal cavity in the form of spheroids during late stage disease (1). In order to mimic these conditions in vitro, ultra-low attachment (ULA) plates were used to induce the formation of spheroids. We compared TCA cycle enzyme expression of Ovcar3 and Ovcar10 cells from adherent and spheroid conditions (Fig. 1C). Under these conditions, only three TCA cycle enzymes showed increased expression: PDK1 (pyruvate dehydrogenase kinase 1), SUCLG2 (succinate-CoA ligase GDP-forming beta subunit), and IDH1 (p<0.001). PDK1 is an inhibitor of the TCA cycle (33); however, we found that HGSC cell lines produced a decreased amount of lactate (Fig. S2C) suggesting that PDK1 upregulation may not correlate with enzymatic activity. Further analysis of HGSC patients using mRNA gene chip data (32) demonstrated that only high IDH1 expression was associated with a significant decrease in progression-free survival (Fig. 1D and S2D–E). Consistently, wildtype IDH1 protein is increased in primary HGSC patient tumors when compared to normal FT tissues (Fig. 1E–F). While TCGA datasets lack normal FT samples, we found a significant increase in IDH1 expression using a publicly available dataset of 12 normal fallopian tube and 13 HGSC patient samples (GSE10971; (34)) (Fig. 1G). Data from TCGA demonstrate that no HGSC patients harbor an IDH1 mutation, and we confirmed that Ovcar3 and Ovcar10 HGSC cells express wildtype IDH1 (Fig. S1A). Together, these data demonstrate that wildtype IDH1 is significantly upregulated in both cell lines and patient samples of HGSC compared to FT.
IDH1 is preferentially utilized through oxidative decarboxylation in HGSC to produce αKG
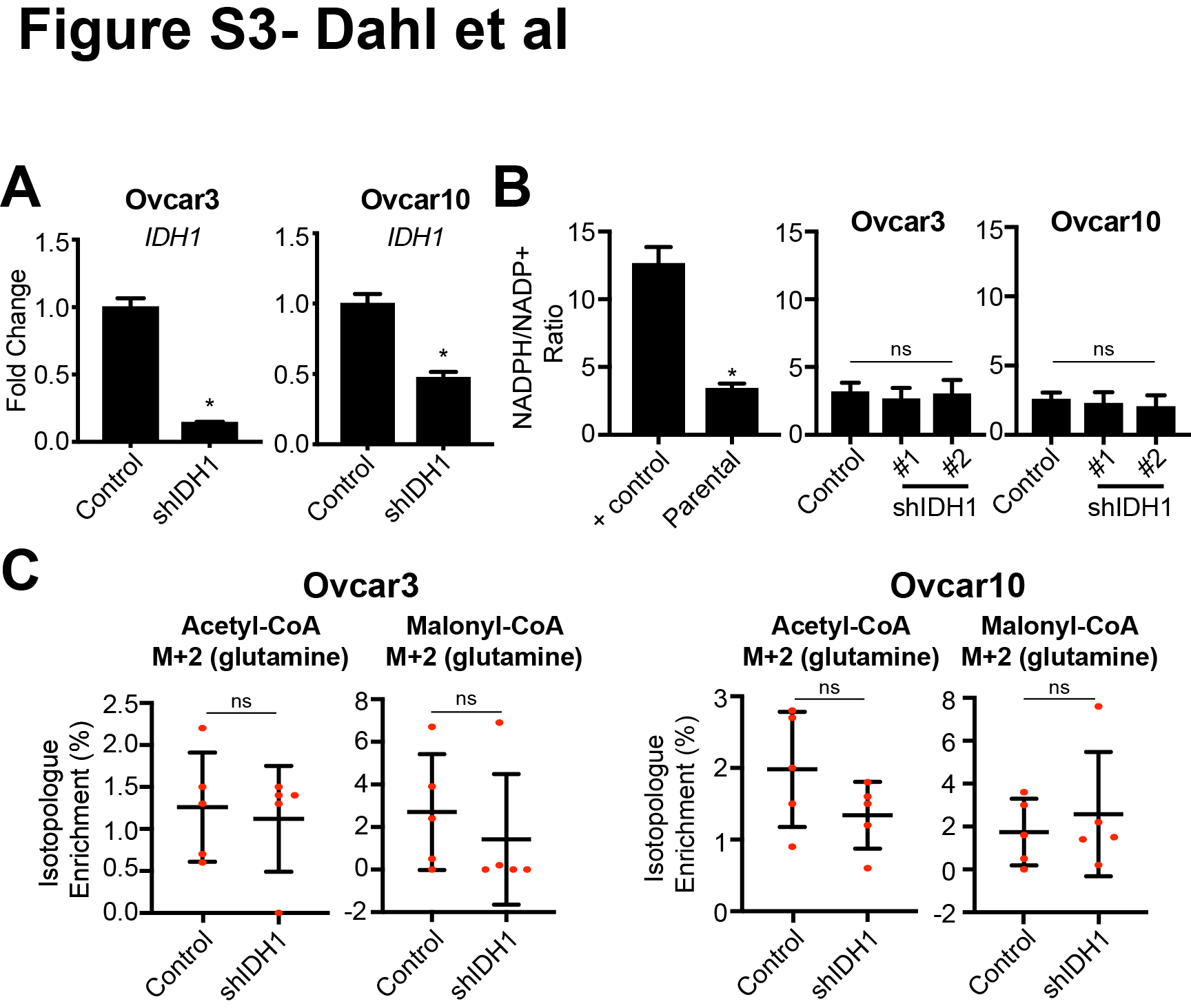
IDH1 catalyzes a reversible reaction to convert isocitrate to αKG (oxidative decarboxylation from glucose) or αKG to isocitrate (reductive carboxylation from glutamine) which is NADP+/NADPH dependent (Fig. 2A). To determine whether HGSC cells utilize IDH1 for oxidative decarboxylation or reductive carboxylation, we knocked down IDH1 (Fig. S3A) and determined αKG and citrate abundance. Knockdown of IDH1 decreased αKG while citrate was increased (Fig. 2B–C). This suggests that IDH1 mainly functions in the oxidative decarboxylation of isocitrate to αKG in HGSC. To further confirm these results, we performed stable isotope labeling using 13C6 glucose or 13C5 glutamine. We observed a decrease in αKG produced through oxidative decarboxylation (M+2) upon IDH1 knockdown (Fig. 2D). Citrate produced through reductive carboxylation (M+5) was undetectable for both control and IDH1 knockdown cells in multiple experiments, suggesting that the reductive carboxylation pathway is not highly active in these cells. Interestingly, knockdown of IDH1 did not alter the NADPH/NADP+ ratio, suggesting that other NADPH pathways are contributing to the total NADPH pool (Fig. S3B). This is consistent with a recent study showing that NADPH is mainly produced by the PPP (35). Lipid metabolism may be influenced by IDH1 expression (18). Production of the lipid precursors acetyl-CoA and malonyl-CoA from glutamine was not changed in either Ovcar3 or Ovcar10 cells (Fig. S3C), suggesting that IDH1 may not play a major role in lipid biosynthesis in HGSC. Together, these data suggest that HGSC cells preferentially utilize glucose for the oxidative decarboxylation reaction of IDH1 to produce αKG.
Figure 2: HGSC cells preferentially utilize glucose for the oxidative decarboxylation reaction of IDH1.

(A) Schematic of glucose and glutamine tracing into the TCA cycle by oxidative decarboxylation (purple) and reductive carboxylation (blue). Glucose tracing into αKG is labeled M+2. Glutamine tracing into citrate through reductive carboxylation is labeled M+5.
(B-D) Ovcar3 and Ovcar10 were infected with lentivirus expressing a short hairpin RNA (shRNA) targeting IDH1 (shIDH1). Empty vector was used as a control. One of two experiments is shown.
(B) Relative abundance of αKG. *p<0.0009.
(C) Relative abundance of citrate *p<0.0006.
(D) Isotopologue enrichment of glucose tracing into αKG (M+2). *p<0.0001.
Knockdown or inhibition of wildtype IDH1 inhibits proliferation of HGSC cells
Next, we sought to determine whether upregulation of IDH1 is critical for HGSC proliferation. Towards this goal, we performed RNA-sequencing analysis of Ovcar3 cells upon IDH1 knockdown (GEO accession no. GSE128700). Indeed, knockdown of IDH1 was associated with a significant decrease in pathways related to the cell cycle and proliferation (Fig. S4A). Importantly, other isocitrate dehydrogenases (IDH2 and IDH3α/β/γ) were not increased upon IDH1 knockdown (Fig. S4B), suggesting there is no compensation from these enzymes. This is also consistent with our metabolomics data (Fig. 2B–D), indicating that IDH1 may be the main isocitrate dehydrogenase enzyme in HGSC cells.
To further confirm that knockdown of IDH1 reduces HGSC cell proliferation, we examined additional proliferation markers upon knockdown of IDH1 in both Ovcar3 and Ovcar10 HGSC cell lines using two independent shRNAs (Fig. 3A and Fig S4C). Knockdown of IDH1 decreased BrdU incorporation, colony formation, and cyclin A expression in both Ovcar3 and Ovcar10 in adherent conditions (Fig. 3B–F and Fig. S4D–G). To limit the potential off-target effects of shRNA knockdown, we overexpressed IDH1 cDNA (Fig. S4H–I). Indeed, overexpression of IDH1 rescued the inhibition of proliferation induced by shIDH1 (Fig. 3G–H). We also assessed proliferation after treatment with an IDH1 inhibitor (IDH1i). There are currently no commercially available inhibitors of wildtype IDH1; therefore, we used a mutant IDH1 inhibitor (IDH1i) that targets the wildtype enzyme at higher concentrations (18,36). Under adherent conditions, inhibition of IDH1 in both Ovcar3 and Ovcar10 cells decreased BrdU incorporation and colony formation (Fig. 3I–L and Fig. S4J–M). Overexpression of IDH1 in IDH1 inhibitor-treated cells (Fig. S4N–O) rescued this phenotype, indicating the observed phenotype is specifically due to inhibition of IDH1 and not off-target effects (Fig. 3M–N). Similar to adherent conditions, proliferation was also decreased by IDH1 suppression in spheroid conditions as indicated by decreased cyclin A, PCNA, and MCM3 (Fig. 3O–P and Fig. S4P). Taken together, we conclude that knockdown or inhibition of wildtype IDH1 suppresses proliferation of HGSC cells.
Figure 3: Knockdown or pharmacological inhibition of IDH1 suppresses proliferation of HGSC cells.

(A-F) Ovcar3 cells were infected with lentivirus expressing two independent short hairpin RNAs (shRNAs) targeting IDH1 (shIDH1). Empty vector was used as a control.
(A) Immunoblot analysis of IDH1. Vinculin was used as a loading control. One of 5 experiments is shown.
(B) BrdU incorporation. One of three experiments is shown.
(C) Quantification of (B). Data represent mean ± SD. *p<0.0001
(D) Colony formation. One of three experiments is shown.
(E) Quantification of (D). Data represent mean ± SD. *p<0.0001
(F) Immunoblot analysis of IDH1 and cyclin A. β-Actin was used as a loading control. One of three experiments is shown.
(G-H) Ovcar3 cells were infected with lentivirus expressing a short hairpin RNA targeting IDH1 (shIDH1) with or without ectopic overexpression of IDH1 cDNA (IDH1 OE). Empty vector was used as a control. Cells were selected with puromycin for 7 days
(G) BrdU incorporation. One of three experiments shown.
(H) Quantification of (G). Data represent mean ± SD. *p<0.005
(I-L) Ovcar3 cells were treated with either DMSO or 15 μM GSK864 (IDH1i) for 7 days.
(I) BrdU incorporation. One of three experiments shown.
(J) Quantification of (I). Data represent mean ± SD. *p<0.0001
(K) Colony formation. One of three experiments is shown.
(L) Quantification of (K). Data represent mean ± SD. *p<0.0001
(M-N) Ovcar3 cells were treated with 15 μM GSK864 (IDH1i) with or without ectopic overexpression of IDH1 cDNA (IDH1 OE). Cells were collected after 7 days.
(M) BrdU incorporation. One of three experiments shown.
(N) Quantification of (M). Data represent mean ± SD. *p<0.02.
(O) Immunoblot of Ovcar3 cells cultured in ULA conditions and treated with 15 μM GSK864 (IDH1i) for cyclin A. β-Actin was used as a loading control. One of three experiments shown.
(P) RT-qPCR analysis of IDH1, PCNA, and MCM3 of Ovcar3 shIDH1 cells cultured in ULA conditions. B2M was used as a reference gene. RNA was collected four days after infection. One of three experiments is shown. Data represent mean ± SD. *p<0.02
Knockdown or inhibition of IDH1 induces senescence of HGSC cells
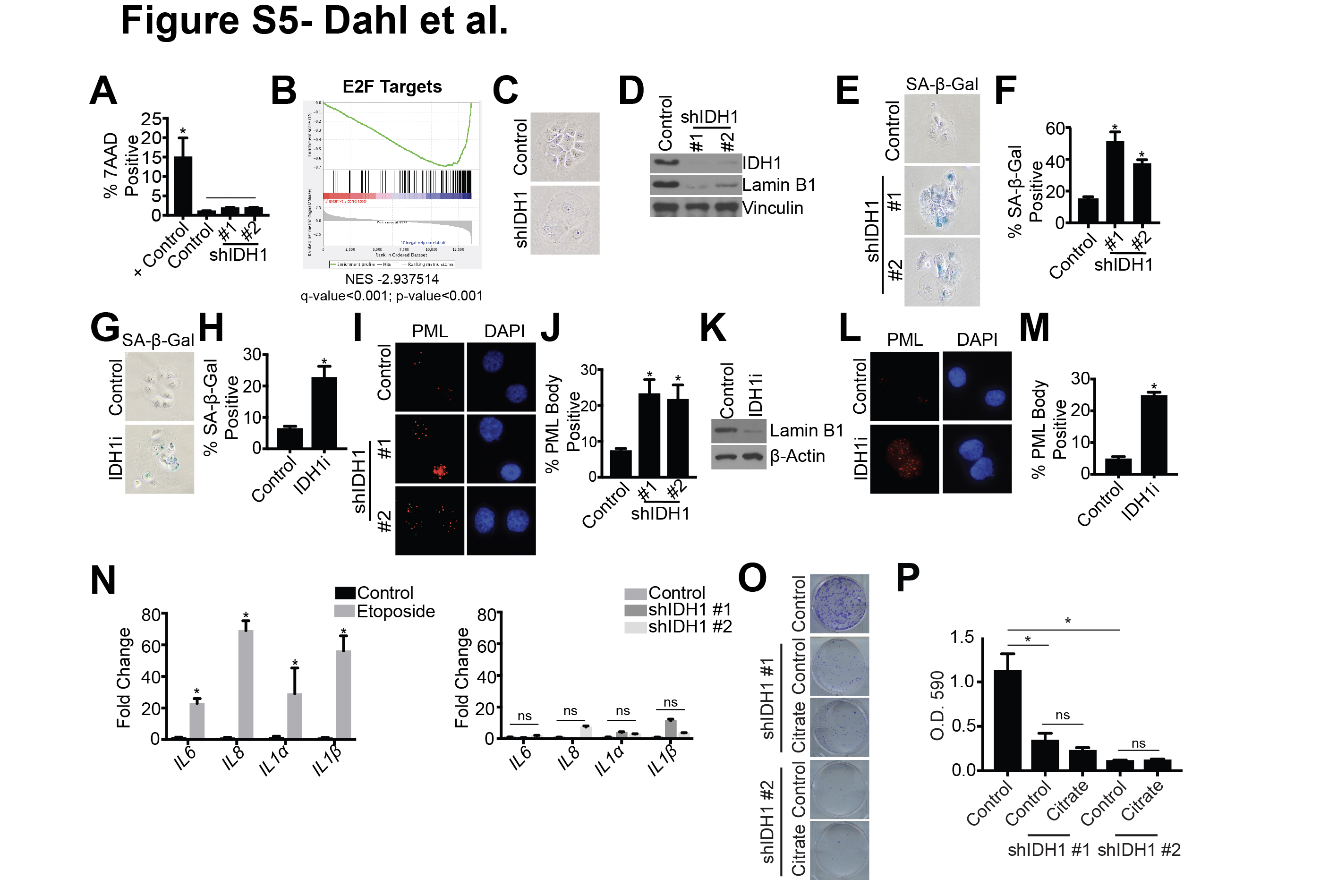
Next, we sought to determine the mechanism by which inhibition or knockdown of IDH1 suppresses HGSC cell proliferation. In adherent conditions, knockdown of IDH1 did not induce cell death (Fig S5A). Interestingly, analysis of our RNA-Seq revealed that the top hit in the GSEA “Hallmark Gene Sets” was E2F target genes (Fig. S5B), which is a characteristic of senescence (37). Indeed, upon suppression of IDH1, cells exhibited a large and flat morphology, which are also hallmarks of senescence (Fig. S5C); therefore, we investigated whether suppression of proliferation was due to senescence induction. Towards this goal, we examined the expression of senescence-associated beta galactosidase (SA-β-Gal) activity. Knockdown or inhibition of IDH1 increased SA-β-Gal activity in both Ovcar3 and Ovcar10 cells (Fig. 4A–E and Fig. S5D–H). Other senescent markers such as decreased lamin B1 and increased PML bodies were also observed when IDH1 was knocked down or inhibited (Fig. 4A, 4F–J and Fig. S5D, S5I–M). Interestingly, the senescence-associated secretory phenotype (SASP) (6) was not robustly increased in IDH1 knockdown cells when compared to etoposide treated cells (Fig. S5N). Overexpression of IDH1 in IDH1 knockdown or IDH1 inhibitor-treated (Fig. S4H–I and S4N–O) cells rescued the senescent phenotype (Fig. 4K–P). Additionally, knockdown or inhibition of IDH1 induced senescence in spheroid conditions as indicated by decreased LMNB1 (Fig. 4Q). Our metabolomics data indicate that knockdown of IDH1 inhibits the oxidative decarboxylation of isocitrate, thereby decreasing αKG levels (Fig. 2B–D). Supplementation of IDH1 knockdown cells with exogenous αKG, but not citrate, partially rescued the senescent phenotype (Fig. 4R–U and Fig. S5O–P), again indicating the important role of IDH1 in oxidative decarboxylation in HGSC cells. These data suggest that inhibition or knockdown of IDH1 induces senescence in both adherent and spheroid conditions and may not lead to the detrimental side effects of senescence that are associated with the SASP (6).
Figure 4: Knockdown or pharmacological inhibition of IDH1 induces senescence in HGSC in both adherent and spheroid conditions.

Ovcar3 cells were infected with lentivirus expressing two independent shRNAs targeting IDH1 or treated with 15μM GSK864 (IDH1i). Empty vector or DMSO was used as a control, respectively.
(A) Immunoblot analysis of IDH1 and lamin B1. β-Actin was used as a loading control. One of three experiments is shown.
(B) SA-β-Gal activity. One of three experiments is shown.
(C) Quantification of (B). Data represent mean ± SD. *p<0.0001
(D) SA-β-Gal activity. One of three experiments is shown.
(E) Quantification of (D). Data represent mean ± SD. *p<0.0001
(F) PML body foci. One of three experiments is shown.
(G) Quantification of (F). Data represent mean ± SD. *p<0.0005
(H) Immunoblot analysis of lamin B1. Vinculin was used as a loading control. One of three experiments is shown.
(I) PML body foci. One of three experiments is shown.
(J) Quantification of (I). Data represent mean ± SD. *p<0.0001
(K-P) Ovcar3 shIDH1 or IDH1i cells were transfected with an IDH1 overexpression vector (IDH1 OE) or control. Cells were collected after 7 days.
(K) SA-β-Gal activity. One of three experiments is shown.
(L) Quantification of (K). Data represent mean ± SD. *p<0.0008.
(M) SA-β-Gal activity. One of three experiments is shown.
(N) Quantification of (M). Data represent mean ± SD. *p<0.0009.
(O) RT-qPCR analysis of LMNB1 in shIDH1 cells. B2M was used as a reference gene. One of three experiments is shown. Data represent mean ± SD. *p<0.02.
(P) RT-qPCR analysis of LMNB1 in IDH1i cells. B2M was used as a reference gene. One of three experiments is shown. Data represent mean ± SD. *p<0.003.
(Q) RT-qPCR analysis of IDH1 and LMNB1 in Ovcar3 shIDH1 cultured in ULA plates. B2M was used as a reference gene. RNA was collected four days after infection. One of three experiments is shown. Data represent mean ± SD. *p<0.0002
(R-U) Ovcar3 cells were infected with lentivirus expressing shRNA targeting IDH1. Empty vector was used as a control. Exogenous αKG (1mM) was added to the indicated cells.
(R) SA-β-Gal activity. One of three experiments is shown.
(S) Quantification of (R). Data represent mean ± SD. *p<0.0001
(T) Colony formation. One of three experiments is shown.
(U) Quantification of (T). Data represent mean ± SD. *p<0.002
Knockdown of IDH1 induces senescence through increased repressive histone methylation at E2F target gene loci
Next, we aimed to determine the mechanism by which targeting IDH1 induces senescence. Inhibition of IDH1 has previously been shown to increase reactive oxygen species (ROS) through decreased NADPH production (38), and ROS are a known inducer of senescence (6). However, we did not observe an increase in ROS upon IDH1 knockdown (Fig. S6A), and treatment with the antioxidant N-acetyl cysteine (NAC) did not rescue senescence due to IDH1 knockdown (Fig. S6B). This is consistent with our results showing no change in the NADPH/NADP+ ratio upon IDH1 knockdown (Fig S3B). Similarly, we did not observe an increase in expression of other NADPH-producing enzymes such as ME3 and G6PD in IDH1 knockdown cells (Fig. S6C). Moreover, we did not observe an increase in DNA damage, another well-known mechanism of therapy-induced senescence (Fig. S6D) (6). These data suggest another mechanism is inducing senescence of HGSC cells upon IDH1 knockdown.
We observed a significant decrease in E2F target genes (Fig. S5B). E2F target genes are downregulated by increased repressive histone H3 lysine 9 di-methylation (H3K9me2) in senescent cells (7,8,39). Increased histone methylation may be due to decreased histone demethylase activity. The JmjC family of histone demethylases require αKG as a co-substrate (20). As we observed a decrease in αKG levels upon IDH1 knockdown (Fig. 2B), we hypothesized that this would increase repressive histone methylation of E2F target genes to induce senescence. Chromatin-immunoprecipitation (ChIP) assays using an antibody specific for H3K9me2 demonstrated increased H3K9me2 occupancy at well-established E2F target genes PCNA and MCM3 in both Ovcar3 and Ovcar10 cells with IDH1 knockdown compared to controls (Fig. 5A). Consistently, PCNA and MCM3 mRNA and protein expression was decreased in these cells (Fig. 5B–C). Addition of exogenous αKG rescued H3K9me2 occupancy at these loci (Fig. S6E), indicating that decreased αKG upon IDH1 depletion was responsible for histone methylation changes at PCNA and MCM3. To expand upon these findings, we used our RNA-Seq analysis to find additional E2F targets known to play a role in proliferation and senescence that may be regulated by H3K9me2 upon IDH1 knockdown. From that analysis, we identified 6 additional gene loci that show increased H3K9me2 occupancy, which correlated with decreased expression in our RNA-Seq analysis (CCNA2, MKI67, MCM2, CDC45, STMN1, CDC25A) (Fig. S6F–G). Together, these data suggest that knockdown of IDH1 increases histone methylation at E2F target gene loci to decrease their expression and induce senescence.
Figure 5: Knockdown of IDH1 increases repressive H3K9me2 at E2F gene target loci.

Ovcar3 and Ovcar10 cells were infected with two independent hairpins targeting IDH1. Empty vector was used as a control.
(A) H3K9me2 ChIP was performed, and H3K9me2 binding to PCNA and MCM3 was determined by qPCR and is normalized to a gene desert region control. One of two experiments is shown. Data represent mean ± SD. *p<0.02
(B) RT-qPCR analysis PCNA and MCM3 expression. B2M was used as a reference gene. One of three experiments is shown. Data represent mean ± SD. *p<0.008
(C) Immunoblot analysis of PCNA and MCM3. Vinculin was used as a loading control. One of two experiments is shown.
Discussion
In this study, we found that the TCA cycle enzyme IDH1 is significantly increased in HGSC cells compared to FT cells. Patient HGSC samples also indicated a significant increase in IDH1 when compared to normal FT, which correlated with worse progression-free survival. Inhibition or knockdown of wildtype IDH1 suppressed HGSC cell proliferation and induced senescence, a stable cell cycle arrest. Mechanistically, targeting IDH1 induced senescence by increasing repressive H3K9me2 at the loci of E2F gene targets. Taken together, these data indicate that high expression of IDH1 is a poor prognostic factor for HGSC and inhibiting its activity represents a novel strategy for HGSC progression and dissemination.
IDH1 is well-known for its mutation in secondary glioblastoma and AML, which produces (R)-2HG, an oncometabolite (16,17). Indeed, we observed an increase in 2HG in HGSC (Fig. 1A); however, this methodology cannot distinguish between (R) and (S)-2HG. While both Ovcar3 and Ovcar10 cells have wildtype IDH1 (Fig. S1A), we cannot definitively rule out the possibility of (S)-2HG function in these cells. It is known that IDH1 functions in a reversible reaction between isocitrate and αKG (40). Although other studies have shown IDH1 undergoes the reductive carboxylation reaction (38), we found that IDH1 in HGSC cells is preferentially utilized in the forward oxidative decarboxylation reaction under adherent conditions (Fig. 2). The oxidative reaction produces NADPH, and previous studies have shown that IDH1 upregulation protects cells from oxidative stress (38,41). Interestingly, we did not observe a change in NADPH/NADP+ ratio, which is consistent with a recent study demonstrating that PPP activity is necessary and sufficient for NADPH pools and not IDH1 (35). Additionally, previous studies have also identified a role for IDH1 in lipid metabolism (18,19). Our results indicate that precursors to lipid metabolism are unaltered by IDH1 knockdown, suggesting that this is a context- and cell type-dependent effect. Since we observed an increase in acetyl-CoA in HGSC cells compared to FT cells, future studies will need to be performed using lipidomics approaches to determine the exact pathways leading to lipid synthesis in HGSC cells.
Senescence is characterized by marked chromatin remodeling and increased repressive histone methylation, collectively termed the senescence-associated heterochromatin foci (SAHF) (7,8). Specifically, di- and tri- methylation of histone H3 lysine 9 at E2F target gene loci is implicated in senescence (7). We found that knockdown of IDH1 induced H3K9 methylation at multiple E2F target gene loci (Fig. 5A and Fig. S6F), leading to decreased expression of those genes (Fig. 5B and Fig. S6G). Previous publications found that mutant IDH1, through the oncometabolite (R)-2HG, alters histone methylation at DNA damage response and differentiation gene loci through competitive inhibition of histone demethylases that require αKG for their activity (42,43). Indeed, supplementation of shIDH1 cells with αKG rescued the repressive histone phenotype (Fig. S6E), suggesting that αKG loss is affecting histone demethylase activity. Multiple histone demethylases have been connected to senescence induction, although very little is known about the role of αKG in senescence (21). Our data suggest that IDH1 and αKG levels maintain H3K9 in a demethylated state that is critical for HGSC cells to proliferate, and inhibition of αKG production increases repressive histone marks to induce senescence. Future work is needed to determine the specific histone H3K9 demethylase responsible for the senescent phenotype upon IDH1 knockdown.
Approximately 70% of HGSC receiving platinum-based standard-of-care therapy will relapse with chemoresistant disease (3). Currently, there are few therapies available for these patients after relapse, ultimately leading to patient mortality. Aberrant metabolism is a global characteristic of cancer cells (11), and altered metabolism of cancer cells affects the response to many chemotherapies (44). Therefore, targeting metabolism may serve as a novel therapeutic in many cancer types. Previous studies have implicated altered metabolic pathways in ovarian cancer, and multiple studies have targeted glycolytic enzymes for ovarian cancer treatment (45–47). However, our study demonstrates that FT cells also undergo aerobic glycolysis (Fig. S2A&C), suggesting that targeting this pathway may result in toxicity to normal tissue. We found that TCA cycle metabolites and enzymes are universally and significantly upregulated in HGSC cells compared to FT, which suggests that targeting this pathway may lead to less toxicity. Interestingly, PDK1 was also upregulated in HGSC cells, especially under spheroid conditions (Fig. 1B–C). PDK1 inhibits the TCA cycle (33); however, we did not observe an increase in lactate production in HGSC when compared to FT, suggesting no increase in PDK1 activity (Fig. S2C). We also observed changes in other pathway metabolites, such as glycolysis and the PPP (Fig. S2A–B). Future work will determine whether these pathways are critical for HGSC proliferation and survival. Notably, we identified wildtype IDH1 as a potential target for HGSC. Recently, the IDH1 inhibitor Tibsovo (ivosidenib, AG-120) was approved for relapsed AML with mutant IDH1. This inhibitor is also effective against wildtype IDH1, but not wildtype IDH2 (48). Our data and others suggest that targeting wildtype IDH1 is a rational therapy for multiple cancers (18).
Late stage HGSC primarily disseminates to the peritoneal cavity in the form of spheroids (1). These cells then seed on organs within the peritoneal cavity, which in part leads to the morbidity and mortality of late-stage ovarian cancer (1). We found that inhibition and shRNA-mediated knockdown of IDH1 induced senescence in both adherent and spheroid conditions (Fig. 4 and Fig. S5). This observation suggests that targeting IDH1 may be a therapeutic strategy for HGSCs at various stages including early and late stage diagnoses. Moreover, HGSCs are universally characterized by p53 mutations and ~20% have decreased RB1 (encoding for retinoblastoma protein, RB) (49). While these two pathways are implicated in the senescence-associated cell cycle arrest (6), our data suggest that targeting IDH1 downregulates proliferation promoting genes through epigenetic modifications independent of p53 and RB status.
Senescence is considered a tumor suppressive mechanism, and senescence induction is considered a beneficial therapeutic outcome (5,10). However, senescence may also promote cancer and chemoresistance through the senescence-associated secretory phenotype (SASP), which increases inflammation and alters the surrounding microenvironment milieu (6). We found that SASP gene expression was not increased upon IDH1 knockdown (Fig. S5N), suggesting that targeting IDH1 may lead to a sustained cell cycle arrest without the harmful side effects of the SASP. Our results, in addition to others, suggest that senescence induction in the cell-type specific context of ovarian cancer may overall be tumor-suppressing (5,50). Altogether, we propose that targeting IDH1 may act as a novel pro-senescent therapy for HGSC patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications.
Inhibition of IDH1 may act as a novel therapeutic approach to alter both the metabolism and epigenetics of HGSC as a pro-senescent therapy.
Acknowledgments
This work was partially supported by grants from the National Institutes of Health (F31CA236372 to E.S.D., R00CA194309 to K.M.A., R00CA194318 to B.G.B, R03CA211820 to N.W.S, and the use of core facilities under P30ES013508) the W. W. Charitable Trust (to K.M.A.) and Penn State Cancer Institute Postdoctoral Fellowship (to R.B.).
We would like to thank the members of the Aird Lab for their thoughtful contributions. We would like to thank Dr. Ronny Drapkin (University of Pennsylvania) and Dr. Anna Loshkin (University of Pittsburgh) for the fallopian tube cells. We would like to thank Dr. George-Lucian Moldovan’s lab (Penn State College of Medicine) for the PCNA and MCM3 antibodies.
Abbreviations
- IDH1
isocitrate dehydrogenase 1
- TCA cycle
the citric acid cycle
- HGSC
high grade serous carcinoma
- EOC
epithelial ovarian cancer
- PARP
poly(ADP-ribose) polymerase
- SAHF
senescence-associated heterochromatin foci
- BrdU
bromodeoxyuridine
- αKG
alpha-ketoglutarate
- AML
acute myeloid leukemia
- JmjC
jumonji C
- FT
fallopian tube
- TCGA
The Cancer Genome Atlas
- SASP
senescence-associated secretory phenotype
- PML
promyelocytic leukemia protein
- NAC
n-acetyl cysteine
- PCNA
proliferating cell nuclear antigen
- MCM3
minichromosome maintenance complex 3
Footnotes
Disclosure of Potential Conflicts of Interest: The authors disclose no potential conflicts of interest.
References
- 1.Lengyel E Ovarian cancer development and metastasis. Am J Pathol 2010;177(3):1053–64 doi 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018;68(4):284–96 doi 10.3322/caac.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 2003;3(7):502–16 doi 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 4.D’Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA repair 2018;71:172–6 doi 10.1016/j.dnarep.2018.08.021. [DOI] [PubMed] [Google Scholar]
- 5.Calvo L, Cheng S, Skulimowski M, Clement I, Portelance L, Zhan Y, et al. Cellular senescence is a central response to cytotoxic chemotherapy in high-grade serous ovarian cancer. bioRxiv 2018. [Google Scholar]
- 6.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007;8(9):729–40 doi 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 7.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003;113(6):703–16 doi S009286740300401X [pii]. [DOI] [PubMed] [Google Scholar]
- 8.Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 2007;27(6):2343–58 doi 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence. Trends Cell Biol 2018;28(6):436–53 doi 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 2011;11(7):503–11 doi 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv 2016;2(5):e1600200 doi 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buj R, Aird KM. Deoxyribonucleotide Triphosphate Metabolism in Cancer and Metabolic Disease. Frontiers in endocrinology 2018;9:177 doi 10.3389/fendo.2018.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinnaird A, Zhao S, Wellen KE, Michelakis ED. Metabolic control of epigenetics in cancer. Nat Rev Cancer 2016;16(11):694–707 doi 10.1038/nrc.2016.82. [DOI] [PubMed] [Google Scholar]
- 15.Kim A Mitochondria in Cancer Energy Metabolism: Culprits or Bystanders? Toxicological research 2015;31(4):323–30 doi 10.5487/tr.2015.31.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH½ enzymes and therapy responses. Oncogene 2018;37(15):1949–60 doi 10.1038/s41388-017-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360(8):765–73 doi 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calvert AE, Chalastanis A, Wu Y, Hurley LA, Kouri FM, Bi Y, et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep 2017;19(9):1858–73 doi 10.1016/j.celrep.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badur MG, Muthusamy T, Parker SJ, Ma S, McBrayer SK, Cordes T, et al. Oncogenic R132 IDH1 Mutations Limit NADPH for De Novo Lipogenesis through (D)2-Hydroxyglutarate Production in Fibrosarcoma Cells. Cell Rep 2018;25(6):1680 doi 10.1016/j.celrep.2018.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 2006;7(9):715–27 doi 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- 21.Leon KE, Aird KM. Jumonji C Demethylases in Cellular Senescence. Genes 2019;10(1) doi 10.3390/genes10010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun 2017;8(1):1093 doi 10.1038/s41467-017-00962-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uphoff CC, Drexler HG. Detection of mycoplasma contaminations. Methods Mol Biol 2005;290:13–23. [DOI] [PubMed] [Google Scholar]
- 24.Guo L, Worth AJ, Mesaros C, Snyder NW, Glickson JD, Blair IA. Diisopropylethylamine/hexafluoroisopropanol-mediated ion-pairing ultra-high-performance liquid chromatography/mass spectrometry for phosphate and carboxylate metabolite analysis: utility for studying cellular metabolism. Rapid communications in mass spectrometry : RCM 2016;30(16):1835–45 doi 10.1002/rcm.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frey AJ, Feldman DR, Trefely S, Worth AJ, Basu SS, Snyder NW. LC-quadrupole/Orbitrap high-resolution mass spectrometry enables stable isotope-resolved simultaneous quantification and (1)(3)C-isotopic labeling of acyl-coenzyme A thioesters. Analytical and bioanalytical chemistry 2016;408(13):3651–8 doi 10.1007/s00216-016-9448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trefely S, Ashwell P, Snyder NW. FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC bioinformatics 2016;17(1):485 doi 10.1186/s12859-016-1360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12(4):357–60 doi 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 2015;31(2):166–9 doi 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 2010;26(1):139–40 doi 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995;92(20):9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol 2016;215(3):325–34 doi 10.1083/jcb.201608026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer 2012;19(2):197–208 doi 10.1530/erc-11-0329. [DOI] [PubMed] [Google Scholar]
- 33.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans 2003;31(Pt 6):1143–51 doi 10.1042/. [DOI] [PubMed] [Google Scholar]
- 34.Tone AA, Begley H, Sharma M, Murphy J, Rosen B, Brown TJ, et al. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin Cancer Res 2008;14(13):4067–78 doi 10.1158/1078-0432.Ccr-07-4959. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, et al. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nature Metabolism 2019;1(3):404–15 doi 10.1038/s42255-019-0043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okoye-Okafor UC, Bartholdy B, Cartier J, Gao EN, Pietrak B, Rendina AR, et al. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat Chem Biol 2015;11(11):878–86 doi 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park C, Lee I, Kang WK. E2F-1 is a critical modulator of cellular senescence in human cancer. Int J Mol Med 2006;17(5):715–20. [PubMed] [Google Scholar]
- 38.Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016;532(7598):255–8 doi 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 2012;47(2):203–14 doi 10.1016/j.molcel.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.D AA Jr., Haft DE. An Alternate Pathway of Alpha-Ketoglutarate Catabolism in the Isolated, Perfused Rat Liver. I. Studies with Dl-Glutamate-2- and −5–14c. J Biol Chem 1965;240:613–7. [PubMed] [Google Scholar]
- 41.Itsumi M, Inoue S, Elia AJ, Murakami K, Sasaki M, Lind EF, et al. Idh1 protects murine hepatocytes from endotoxin-induced oxidative stress by regulating the intracellular NADP(+)/NADPH ratio. Cell Death Differ 2015;22(11):1837–45 doi 10.1038/cdd.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue S, Li WY, Tseng A, Beerman I, Elia AJ, Bendall SC, et al. Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 2016;30(2):337–48 doi 10.1016/j.ccell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012;483(7390):474–8 doi 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaal EA, Berkers CR. The Influence of Metabolism on Drug Response in Cancer. Frontiers in oncology 2018;8:500 doi 10.3389/fonc.2018.00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loar P, Wahl H, Kshirsagar M, Gossner G, Griffith K, Liu JR. Inhibition of glycolysis enhances cisplatin-induced apoptosis in ovarian cancer cells. American journal of obstetrics and gynecology 2010;202(4):371.e1–8 doi 10.1016/j.ajog.2009.10.883. [DOI] [PubMed] [Google Scholar]
- 46.Liu Y, Tong L, Luo Y, Li X, Chen G, Wang Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J Cell Biochem 2018;119(7):6162–72 doi 10.1002/jcb.26822. [DOI] [PubMed] [Google Scholar]
- 47.Xintaropoulou C, Ward C, Wise A, Queckborner S, Turnbull A, Michie CO, et al. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer 2018;18(1):636 doi 10.1186/s12885-018-4521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Popovici-Muller J, Lemieux RM, Artin E, Saunders JO, Salituro FG, Travins J, et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS medicinal chemistry letters 2018;9(4):300–5 doi 10.1021/acsmedchemlett.7b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474(7353):609–15 doi 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aird KM, Li H, Xin F, Konstantinopoulos PA, Zhang R. Identification of ribonucleotide reductase M2 as a potential target for pro-senescence therapy in epithelial ovarian cancer. Cell Cycle 2014;13(2):199–207 doi 10.4161/cc.26953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.