

Abstract
The nervous and immune systems both serve as essential assessors and regulators of physiological function. Recently, there has been a great interest in how the nervous and immune systems interact to modulate both physiological and pathological states. In particular, the autonomic nervous system has a direct line of communication with immune cells anatomically, and moreover, immune cells possess receptors for autonomic neurotransmitters. This circumstantial evidence is suggestive of a functional interplay between the two systems, and extensive research over the past few decades has demonstrated neurotransmitters such as the catecholamines (i.e. dopamine, norepinephrine, and epinephrine) and acetylcholine have potent immunomodulating properties. Furthermore, immune cells, particularly T-lymphocytes, have now been found to express the cellular machinery for both the synthesis and degradation of neurotransmitters, which suggests the ability for both autocrine and paracrine signaling from these cells independent of the nervous system. The details underlying the functional interplay of this complex network of neuroimmune communication is still unclear, but this crosstalk is suggestive of significant implications on the pathogenesis of a number of autonomic-dysregulated and inflammation-mediated diseases. In particular, it is widely accepted that numerous forms of cardiovascular diseases possess imbalanced autonomic tone as well as altered T-lymphocyte function, but a paucity of literature exists discussing the direct role of neurotransmitters in shaping the inflammatory microenvironment during the progression or therapeutic management of these diseases. This review seeks to provide a fundamental framework for this autonomic neuroimmune interaction within T-lymphocytes, as well as the implications this may have in cardiovascular diseases.
Keywords: Catecholamines, Acetylcholine, Neurotransmitters, T-lymphocytes, Hypertension, Anti-inflammatory reflex
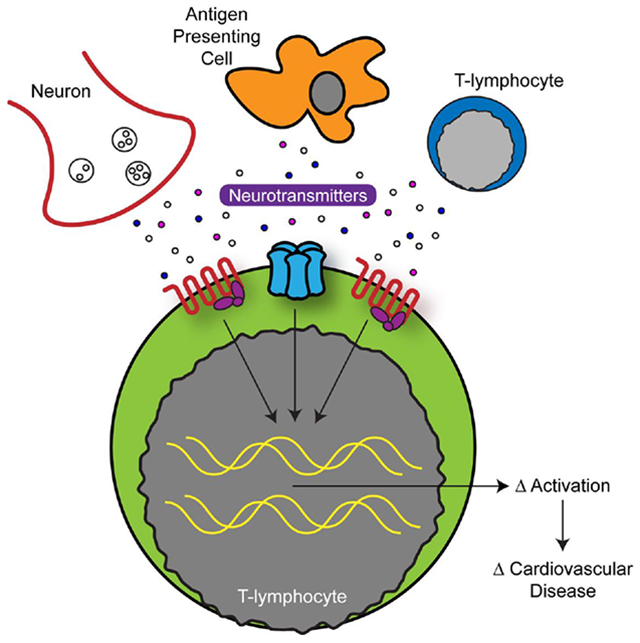
Graphical Abstract
1. Introduction
“The night before Easter Sunday of [1920] I awoke, turned on the light and jotted down a few notes on a tiny slip of thin paper. Then I fell asleep again. It occurred to me at 6:00 o’clock in the morning that during the night I had written down something important, but I was unable to decipher the scrawl. The next night, at 3:00 o’clock, the idea returned. It was the design of an experiment to determine whether or not the hypothesis of chemical transmission that I had uttered 17 years ago was correct. I got up immediately, went to the laboratory, and performed a simple experiment on a frog heart…”
From the autobiography of Nobel Laureate Otto Loewi [1], this passage details the initial experiment that opened the door for our understanding of chemical signal transmission between neurons. This “simple experiment” involved two frog hearts beating in perfusate—one vagally denervated, and one with the vagus nerve yet intact. Dr. Loewi stimulated the intact vagus nerve and found a decrease in the heart rate; a well-known phenomenon even in 1920. The addition of the perfusate from the vagally-intact heart to the second denervated heart still showed a decrease in heart rate, as if its vagus nerve had been stimulated. He called this unknown, humoral factor “Vagustoff”. The discovery earned Otto Loewi and Henry Dale the Nobel Prize in Medicine in 1936, and introduced the world of neuroscience to acetylcholine (Ach). This insight also served as fodder for a greater debate raging within the field of neuroscience. Dubbed “the war of sparks and soups”, neuroscientists familiar with denervation studies argued for purely electrical transmission while early pharmacologists believed there to be a chemical messenger [2]. The integral link between ligand binding and electrical potential changes in post-synaptic neurons was yet to be elucidated.
It is now evident that neurons communicate through both chemical and electrical signal transmission. Experiments on giant squid axons earned Hodgkin and Huxley the Nobel Prize in 1963 while answering many questions about the nature of electrical synapses [3]. Yet, the breadth and depth of variation in the chemical messengers is something that, almost a century later, continues to elude the full understanding of physiologists. One area in particular that remains elusive is the role of neurotransmitters within the immune system. These mobile defensive cells share uncanny similarities to their neuronal counterparts in that they possess the ability to both produce and respond to neurotransmitters. Experimental evidence has suggested their responsiveness to neurotransmitters may play a potential role in various inflammatory diseases, such as cardiovascular disease, but the mechanisms of neurotransmission in immune cells remains unclear. Herein, we discuss the known effects of autonomic regulation on a specific class of immune cell (i.e. T-lymphocytes), and further examine the interplay of these cells in various forms of cardiovascular disease.
2. Neurotransmitters: The Basics
2.1. What defines a neurotransmitter?
The term neurotransmitter generally describes a heterogeneous, yet essential, signaling molecule. While there does not exist a single, explicitly stated definition, the traditional one is predicated on the neurotransmitter meeting a few criteria; it must be a molecule or peptide, synthesized in neurons, that is released at the synaptic cleft to bind a respective receptor [4]. Recent discoveries across a number of fields have begun to redefine this relatively limited definition of neurotransmitters. Steroid molecules have been shown to have neuromodulating properties [5], while small ubiquitous molecules like ATP can function as effector molecules when co-released with neurotransmitters [6]. In cancer biology, neurotransmitters released from non-neuronal cancers are being investigated for their ability to dramatically alter the tumor microenvironment [7]. Even pathways of “neurotransmitters” that are synthesized centrally, yet act distant sites (like vasopressin and oxytocin), do not fit neatly into traditional definitions of neurotransmitters.
While our understanding of neurotransmitter physiology may be incomplete, the clinical usage of neurotransmitter modulation serves as the foundation of pharmacotherapy in a broad array of pathologies, ranging from cardiovascular disease to psychiatric disorders. The diverse range of their pharmacological targets, and thus effects (e.g. elements that make neurotransmission modulating therapies most useful) also is their downfall. The dose-limiting side effects of these drugs continue to be a major barrier to effective treatment of many disease states. It is well known that the majority of anti-depressive, anti-psychotic, and anti-epileptic drugs have off-target effects, with long-term sequelae being discovered constantly.
2.2. Synthesis of neurotransmitters
2.2.1. Catecholamine synthesis
While the discovery of epinephrine is decidedly much less elegant than that of Ach, it is an equally rewarding look into the past. Oliver and Schafer injected adrenal extracts into a variety of animals and found changes in heart and respiration rates; all changes now classically associated with sympathetic activation [8]. They found that this phenomena was unique to injection of the medullary portion of the adrenal gland, not when extract from the cortex was injected [8]. The molecule in this extract, later termed and trademarked adrenaline/epinephrine, is one of several biogenic amines.
Biogenic amines (also termed monoamines) are a crucial class of neurotransmitters that can be further subdivided into 3 groups: catecholamines (epinephrine, norepinephrine, and dopamine), indolamines (serotonin and melatonin), and histamine, but this review will focus specifically on catecholamines. The catecholamines are a class of neurotransmitters based on their namesake in that they all contain a hydroxylated phenol ring known as a catechol moiety. This electron rich structure has been shown to participate in redox chemistry, with both enzymatic and non-enzymatic reactions forming cytotoxic free radicals that have been proposed to contribute to catecholamine-induced neurotoxicity [9–12]. This is in addition to evidence of catecholamine-receptor binding-induced changes in the cellular redox environment [13–15], which is further discussed in section 3.2.1. All catecholamines are synthesized from the non-essential amino acid L-tyrosine, which enters the cytosol of a catecholaminergic neuron through Na+-dependent transporters. The first step in this pathway is the hydroxylation of tyrosine to dihydroxyphenylalanine (L-DOPA) by tyrosine hydroxylase (Figure 1). This reaction requires molecular oxygen, iron, and the reduced co-factor tetrahydrobiopterin (BH4). This is the rate-limiting enzyme for catecholamine synthesis that is subject to negative feedback by its downstream products, and has complex regulation which has been reviewed elsewhere [16].
Figure 1: Catecholamine synthetic pathway.

The catecholamines (dopamine, norepinephrine, and epinephrine) all originate from the amino acid tyrosine. Classically, dopamine β-hydroxylase is membrane-bound, which is indicated with a circle.
The next step in the catecholamine synthetic pathway is mediated by DOPA decarboxylase, which results in the generation of dopamine (Figure 1). Dopamine’s central production in the substantia nigra of the basal ganglia in the brain plays an essential role in the neuro-circuitry involved in voluntary movement, and dysfunctional production of this neurotransmitter in this nuclei leads to Parkinson’s disease [17]. Modulation of dopamine levels (with precursors able to cross the blood brain barrier) are the mainstay of pharmacotherapy in Parkinson’s disease [17]. The role of dopamine in Parkinson’s provides insight into the complex chemistry of dopamine synthesis and metabolism, where dopamine is both central to treatment of the disease and implicated in its pathophysiology [18]. Dopamine is also applied clinically in the management of shock because of its selective ability to dilate the renal vasculature, while providing both ionotropic and chronotropic support through an indirect noradrenergic mechanism [19].
Dopamine is then transported into monoamine storage vesicles by the vesicular monoamine transporter (VMAT), where the second carbon of the ethylamine side chain on dopamine is hydroxylated by the vesicular-membrane bound dopamine β-hydroxylase (DβH; Figure 1), forming norepinephrine (NE) within the vesicle [20]. NE is the major neurotransmitter used at the sympathetic post-ganglionic synapse. Action potentials mediate the influx of calcium (Ca2+), which cause vesicular translocation and expulsion of vesicular NE into the synapse [21], where it perpetuates signal transmission via binding to one of the respective adrenergic receptors, discussed in 2.3.1.
The final step in catecholamine synthesis is the formation of epinephrine, and mainly occurs in the adrenal medulla (Figure 1). Sympathetic preganglionic neurons terminate at neuroendocrine chromaffin cells in the adrenal medulla that contain the cytosolic enzyme phenylethanolamine-N-methyltransferase (PNMT) that methylates the amine group on NE to produce epinephrine [22]. These neuroendocrine cells bear resemblance to sympathetic post-ganglionic neurons, only different in that they release their vesicular contents into the systemic circulation by way of a rich vascular network. Within the cortex of the adrenal gland, glucocorticoids released from the fasiculata layer increase the synthesis of epinephrine by increasing the half-life of PNMT mRNA and protein [23]. Both epinephrine and NE are stored in chromaffin cell vesicles and released into the circulation, although the ratio of release into the bloodstream in humans and rats is approximately 4:1, respectively [24].
2.2.2. Cholinergic synthesis
The molecule Ach was synthesized in a German lab by Adolf von Baeyer in 1867, decades before its physiological role was ever identified [25]. After its identification by Otto Loewi in 1921, Ach was characterized as a natural product after extraction from spleens of horses and oxen by Dale and Dudley in 1929 [26]. Ach is now known to be a crucial neurotransmitter, serving as the major chemical signal in the neuromuscular junction, autonomic ganglia, parasympathetic nervous system, as well as the cholinergic anti-inflammatory reflex during sepsis.
Choline is an essential nutrient that circulates in human plasma. At cholinergic nerve terminals, choline is transported from the extracellular space by a Na+-dependent choline transporter (ChT) that serves as the rate-limiting step in the synthesis, with the majority of choline utilized in Ach synthesis derived from recycled choline from Ach metabolism at the synaptic cleft [27]. The mitochondrial-derived and Krebs cycle substrate acetyl coenzyme A (CoA) serves as a substrate for the acetylation of choline in the cytoplasm by choline acetyltransferase (CHAT) to form Ach. Ach is then transported into synaptic vesicles by vesicular acetylcholine transporter (VAChT), which functions as an antiporter to couple the translocation of Ach into the vesicle for the efflux of protons [27]. Ach is released from the synaptic vesicle to bind its respective receptor subtypes, which will be discussed in detail in sections 2.3.3 and 2.3.4.
2.3. Neurotransmitter receptors
2.3.1. Adrenergic receptors
The adrenergic receptors (ARs) were initially distinguished in 1948 by Raymond Ahlquist, who divided them into alpha (α) and beta (β) receptors based on the physiological response observed when one of six adrenergic agonists were used [28]. He ranked the effects of agonists on various tissues, and divided them into two groups [i.e. α and β) for convenient notation based on the opposing excitatory and inhibitory effects of each purported [28]. This designation of convenience remains with us today. Catecholamines can bind to ARs of either the α or β isoform, and to date 6 subtypes of α receptors (α1A, α1B, α1D, α2A, α2B, and α2C) and 3 subtypes of β receptors (β1, β2, and β3) have been identified [29]. The location, density, and conformational state of these receptor subtypes depends on a number of physiological inputs.
α1 receptors are linked to Gq heterotrimeric G-proteins, which result in the activation of phospholipase C, leading to inositol triphosphate (IP3) formation from phosphatidylinositol 4,5-bisphosphate (PIP2) causing intracellular Ca2+ release from the endoplasmic reticulum [30]. Diacylglycerol (DAG) is also released, activating protein kinase C (PKC), which may act on a number of different effectors, namely by phosphorylating cellular proteins [30]. α2 post-synaptic receptors are linked to Gi and Go proteins which function to decrease the production of cAMP through inhibition of adenylyl cyclase, resulting in a decreased signaling cascade [31]. α2 receptors are unique in that they have been shown to be present on the pre-synaptic nerve terminal as well [32]. These receptors also activate Gβγ proteins, coupled to inward Potassium (K+) channels that result in the hyperpolarization of cellular membranes, which inhibits voltage-gated Ca2+ channels, thus inhibiting post-synaptic signals [33]. This uniquely opposite effect of the activation of the α2-AR has been utilized pharmacologically by the α2-AR agonist clonidine. An early anti-hypertensive, clonidine is also now used off-label for diseases associated with increased sympathetic outflow and hyperarousal, such as post-traumatic stress disorder and restless legs syndrome [34].
The family of β receptors are all coupled to Gs G-proteins, which associate with adenylyl cyclase to increase cAMP production (for an excellent review of ARs and their ligands, see [35]). This elevation in cAMP activates protein kinase A (PKA), allowing for the phosphorylation of many downstream targets. This canonical pathway is of important note as we discuss AR interactions and non-traditional G-protein coupled receptor signaling in T-lymphocytes, which will be relevant in section 3.2.1.
2.3.2. Dopaminergic receptors
Dopamine, as evident by its place along the catecholamine synthetic pathway, is structurally related to NE and epinephrine. This allows it to interact with α and β receptors at differing affinities, despite having a greater affinity for its respective dopamine receptors (i.e. D1-D5). These receptors are best classified by their intracellular effects into D1-like and D2-like families; coupled to Gα subunits resulting in increased cAMP and coupled to Gi resulting in decreased cAMP synthesis, respectively [36]. However, it has been shown that this exact coupling is not always true, which could explain the differences in cellular effects between cell types expressing the same dopamine receptor subtype [36].
This complexity is further convoluted in that NE has been shown to interact with dopamine receptors, although the effects of this interaction are less clear [37]. Dopamine receptors are involved in a diverse array of physiological functions within a variety of systems. As we will discuss further in section 3.2.2., the existence of dopamine receptors on immune cells is not merely a coincidence. Their responses to dopamine agonism and antagonism are complex and physiologically relevant, and pose significant clinical ramifications during the catecholamine-targeted pharmacotherapy of several conditions.
2.3.3. Nicotinic Ach receptors
Ach is unique in that it is able to bind both muscarinic and nicotinic receptors, which are metabotropic and ionotropic receptors, respectively. Both of these receptors respond to Ach, thus making them “cholinergic” receptors. To avoid conflation of these two very different receptors, they are referred to by two respective exogenous agonists (nicotine and muscarine), which will be utilized for the remainder of this review. The nicotinic Ach receptor (nAchR) is an ionotropic receptor where ligand binding results in the opening of a water-filled central pore that allows Na+ influx and K+ efflux, down their respective concentration gradients. It is structurally composed of 5 protein subunits, but various combinations of subunits can result in a significant amount of receptor diversity depending on location and cell type [38]. These subunit types can be classified as α, β, γ, δ, and ε based on structure. The always present α-subunit is defined by an extracellular pair of cysteine subunits that are required for ligand binding [39].
2.3.4. Muscarinic receptors
The muscarinic acetylcholine receptor (MR) is another class of receptor for which Ach is a ligand, with subtypes M1-M5. It was originally discovered by Henry Dale, who found its physiological effects on blood pressure to be similar to those seen with the exogenous administration of extracts from the eponymous mushroom, Amantia muscaria [25]. The MR differs from the nAchR in its intracellular effects upon ligand binding, and thus, its functional effects. The M1, M3, and M5 receptors are coupled to a Gq protein that result in the signaling cascade involving IP3 and DAG, similar to that of the α1-AR [40]. The M2 and M4 receptors are coupled to Gi and Go subunits which results in a decrease in intracellular cAMP and downregulation of PKA [40]. These receptors also regulate inward K+ currents that contribute to membrane depolarization and action potential propagation; an effect allowing M2 receptors to negatively regulate chronotropy in the heart [41]. A relative lack of selective ligands for the M5 receptor has limited our ability to selectively study their contributory effects to peripheral physiology, but there is burgeoning interest in its localization and effects in the brain [42].
2.4. Neurotransmitter metabolism and reuptake
2.4.1. Catecholamines
Catecholamines are subject to re-uptake and recycling for future use at the synaptic cleft. The major mechanism of signal termination is this reuptake process, with simple diffusion away from available receptors serving as a more minor mechanism. There are a variety of transporters responsible for transport of endogenous catecholamines, with differential specificities for substrates and varying distribution in tissues. Two transporters dominate neuronal reuptake: the NE transporter (NET) and dopamine transporter (DAT). Each has a greater affinity for their eponymously named catecholamine, but all able to transport dopamine, NE, or epinephrine. NET cotransports an extracellular Na+ as the driving force for transporting catecholamines against their concentration gradient across to the cytoplasmic side [43]. DAT translocates two Na+ and a Cl− in order to energetically favor the transport of catecholamines into nerve terminals [44]. Attenuation of catecholamine reuptake by inhibiting these transporters are the mechanism of drugs such as tricyclic antidepressants and amphetamines [45].
Catecholamine metabolism is a complex process that has been reviewed extensively by Eisenhofer [18]. In brief, monoamine oxidase (MAO) is a flavin-containing enzyme that catalyzes the oxidative deamination of all catecholamines to their respective aldehyde forms. MAO is located on the outer membrane of the mitochondria, and consists of two isoforms: MAO-A and MAO-B [46]. The majority of metabolism of biogenic amines takes place in the cytoplasm (in close proximity to the mitochondria) of sympathetic nerve terminals, secondary to dynamic outward leakage from vesicles. This complex process is balanced with rapid transport by VMAT back into the monoamine vesicles. The majority of circulating NE is transported by the higher affinity VMAT rather than metabolized by MAO [47]. Yet, the inhibition of MAO proved to be a useful therapy in treating depression during the early 1950’s [48]. As we have developed more targeted approaches to modulate monoamine levels, namely through inhibition of reuptake, MAO inhibitors have been mostly abandoned due to extensive side effect profiles.
The deaminated aldehydes generated by MAO are further metabolized in a series of reactions catalyzed by aldehyde reductases, aldehyde dehydrogenases, MAO, and catechol-O-methyltransferase (COMT). This can produce 3-methoxy-4-hydroxyphenylglycol (MHPG), which is subsequently oxidized to produce vanillylmandelic acid (VMA), representing the major-end product of NE, epinephrine, and dopamine metabolism [49]. This MHPG can also be conjugated to sulfate or glucuronide in order to facilitate renal excretion. The more minor pathway, responsible for extraneuronal and adrenally-derived catecholamines, involves the O-methylation by COMT. The subsequent metabolites are then further catabolized by a complex array of the same enzymes, resulting in VMA formation by the liver. VMA and these conjugates of MHPG represent 90% of all catecholamine metabolites in the urine [18].
2.4.2. Ach
Ach metabolism is primarily mediated by two enzymes; acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), also known as pseudocholinesterase. In the synaptic cleft, AChE is responsible for the rapid hydrolysis of Ach into choline and acetate. This choline can then be transported back into the cytosol of the nerve terminal by the aforementioned ChT [27]. In the circulation, BuChE synthesized in the liver is able to hydrolyze Ach, along with many other choline-containing esters ranging from aspirin to cocaine—an area of interest to potentially rescue patients during acute cocaine intoxication [50]. Some individuals are homozygous for an inactivating mutation in BuChE and show complications during surgery due to the inability to hydrolyze succinylcholine, a common surgical neuromuscular-blocking agent. Nerve transmission mediated by Ach is different from that of the catecholamines in that there is no major reuptake process, so metabolism followed by reuptake of the building block choline is the predominant method.
3. Neurotransmitters and T-lymphocytes
3.1. Introduction to T-lymphocytes
T-lymphocytes play a central role in the orchestration of the immune system, as most evidenced by the clinical presentation of deficient disease states such as severe combined immunodeficiency (SCID) and acquired immunodeficiency syndrome (AIDS). This functional role of T-lymphocytes in orchestrating the adaptive immune response makes them an ideal cell type to serve as a conduit for signal generation and reception. As a functional cell designed to coordinate the destruction and clearance of tolerance-failing antigens, T-lymphocyte diversity is most crucially determined by their antigen specificity (or idiotype), as decided during thymic development by their T-lymphocyte Receptor (TCR). T-lymphocytes are able to respond to pre-processed antigens only upon their discrete presentation by antigen-presenting cells (APCs); macrophages, dendritic cells, or B-lymphocytes. These processed antigens are loaded into the major histocompatibility complex (MHC) on these APCs, which together serve as a ligand for the TCR. This resultant interaction is the 1st signal, with a subsequent 2nd signal between the T-lymphocyte and APC being necessary for T-lymphocyte activation and immune system coordination to target the respective antigen. It is an unlikely circumstance that these TCRs do in fact meet their respective epitope; around 1 in 10-100 million, depending on the T-lymphocyte subtype [51,52]. This low likelihood demands that T-lymphocytes are able to proliferate rapidly upon correct antigen presentation, as well as a need for their preservation even after clearance of the antigen.
T-lymphocyte activation is incredibly complex, and has been reviewed elsewhere [53]. In brief, canonical T-lymphocyte activation depends on the TCR-MHC interaction that triggers the activation of phospholipase C-γ (PLC-γ), which generates DAG and IP3, similar to the cascade discussed in sections 2.3.1 and 2.3.4. DAG is responsible for the activation of PKC and the subsequent activation of the mitogen-activated protein kinase/extracellular signal regulated kinase (MAP/ERK) pathway, both of which result in nuclear factor kappa-light-chain enhancer of activated B-cells (NF-κB) activation, resulting in the transcription of genes crucial to activation and proliferation. IP3 allows for the release of Ca2+ from the endoplasmic reticulum (ER). This change in Ca2+ results in numerous events, one of which is the dephosphorylation and thus activation of nuclear factor of activated T-cells (NFAT). NFAT translocates to the nucleus, where it controls the cytokine interleukin 2 (IL-2) transcription, the principle T-lymphocyte growth factor. A milieu of other cytokines (considered the 3rd signal for T-lymphocyte activation) are needed for full T-lymphocyte activation, function, and differentiation, but T-lymphocytes respond very differently to cytokines dependent upon their specific lineage.
T-lymphocytes themselves represent a heterogeneous subtype of lymphocytes, most practically categorized by their display of specific cluster of differentiation (CD) markers. The binary divide of CD4+ and CD8+ T-lymphocytes occurs during their selection in the thymus. CD8+ T-lymphocytes interact with MHC I, which are present on all nucleated cells and platelets. CD8+ T-lymphocytes, also termed cytotoxic T-lymphocytes, respond to cells expressing aberrant intracellular proteins within their MHC I. These could be the result of tumor cells or cells infected with an intracellular pathogen [54]. CD8+ T-lymphocytes release cytotoxic granules that can damage membranes and activate apoptotic pathways, while also secreting cytokines (like IFN-γ) that aid in the mounting of a coordinated immune response to intracellular pathogens or tumor cells [54].
The other division of T-lymphocytes is the CD4+ T-lymphocytes, which communicate with MHC II, found on APCs. CD4+ T-lymphocytes then respond to the presence and/or absence of specific APC-generated cytokine signals that result in further differentiation of CD4+ T-lymphocytes (Figure 2). This too is a highly studied area, and has been reviewed elsewhere [55]. In brief, naïve CD4+ cells can polarize to effector cell subtypes, with the most prominent types known as T-helper type 1 (TH1) cells, T-helper type 2 (TH2) cells, or T-helper type 17 (TH17) cells. IL-12 secretion by dendritic cells (DCs) results in polarization to TH1, which are characterized by IL-2, IFN-γ, and TNF-α secretion [55]. These favor classical cell-mediated immunity, thus making them highly involved in the destruction of intracellular pathogens, tumors, and activation of cytotoxic T-lymphocytes. The absence of IL-12 and presence of IL-4 favors the TH2 subtype, which functions to effectively activate B-lymphocytes and result in isotype switching to immunoglobulin E (IgE) and eosinophil activation [55]. Together, these cells are involved in the effective elimination of helminths and parasites, as well as type 1 hypersensitivity reactions and asthma [55]. TH17 polarization results from DC secretion of IL-23, TGF-β, and IL-6, and these TH17 cells are characterized by their high levels of IL-17 secretion [56]. TH17 cells have been widely implicated in the pathogenesis of autoimmune diseases [56–58]. In addition to polarization to effector subtypes, CD4+ T-lymphocytes can differentiate to regulatory T-lymphocytes (Tregs), which have been antithetically implicated in autoimmune diseases as well [59,60]. These are phenotypically characterized by their expression of CD25 and the transcription factor Foxp3, as well as secretion of IL-10, resulting in the suppression of antigen-specific effector T-lymphocytes [61]. Additional CD4+ subtypes such as TH9, TH22, TFH have also recently been identified, but little is known to date regarding the role of these cells in the adaptive immune response [62–64].
Figure 2: Differentiation of a naïve CD4+ T-lymphocyte.

Naïve CD4+ T-lymphocytes have several different possible differentiation fates that occur upon activation. Classically, cytokines drive this differentiation, however, neurotransmitters also have complex effects affecting these different polarization states.
As our understanding of T-lymphocyte biology continues to grow, it becomes evident that many early investigations erroneously treated T-lymphocytes as a generally homogenous population. Understanding now that numerous subtypes and variations of these cells exist, we will seek to make distinctions in cases wherein specific subsets (CD4+, CD8+, etc) were studied, while also noting cases in which the use of pan T-lymphocytes is relevant. In these cases, we must always be cognizant of the possible type I or II errors due to the dissection of an incredibly complex system using imprecise tools.
3.2. The functional role of neurotransmitter receptors on T-lymphocytes
3.2.1. Adrenergic receptors
Catecholamines may interact with mature T-lymphocytes most readily in three locations in the human body relevant to our discussion: intravascular space, secondary lymphoid organs (e.g. spleen), and the central nervous system. In the plasma, all three catecholamines exist, with NE found at about 20% of the quantity of epinephrine and dopamine [65]. In the intravascular space, T-lymphocytes exist as approximately 70% of peripheral blood mononuclear cells (PBMCs) [66]. In the spleen, sympathetic innervation is regional and specific. Innervation is found in the white pulp that surrounds central arteries, as periarterial lymphoid sheaths (PALS), that are mostly composed of T-lymphocytes [67,68]. Catecholamines may also interact with T-lymphocytes in the central nervous system, as inflammation reduces the fortitude of the blood brain barrier, allowing T-lymphocytes to penetrate and possibly directly interact with catecholamines (a thorough review of this topic can be found here [69]).
In order to best understand the functional effect of catecholamines on T-lymphocytes, we must begin a murky discussion of AR expression. A brief dive into the literature on the expression of ARs on T-lymphocytes will immediately appear contradictory. A number of studies have shown that T-lymphocytes exclusively express β2-AR [70–78]. However, this has been contradicted by other works as well, with reports of expression of β1-ARs, α1-ARs, and α2-ARs on T-lymphocytes. To better dissect this complex discussion, we will review each AR subtype individually.
3.2.1.1. α-Adrenergic receptors
Early studies utilized a agonists and found no changes in T-lymphocyte proliferation [79], which served as proof for lack of α-ARs on T-lymphocytes. Additionally, there was a reported absence of α1-AR mRNA in PBMCs [80], while more recent studies have shown that α1-AR mRNA is present in PBMCs by in-situ hybridization [81]. Heilig et al. found that H3-thymidine incorporation was reduced in a dose-dependent fashion to the α1-AR agonist phenylephrine, with this relationship disappearing with the α-AR antagonist phentolamine [82]. Others have found no expression of α1-ARs on T-lymphocytes until activation with the mitogens (molecules that result in the activation and mitosis of T-lymphocytes, regardless of clonal specificity) phytohemagglutinin (PHA) or lipopolysaccharide (LPS), which both resulted in increased expression of all three α1-AR [83]. In the same study, addition of NE resulted in increased ERK activation, which could be diminished by selective α1-AR antagonism [83]. In another study, PBMCs isolated from patients with juvenile rheumatoid arthritis were treated with phenylephrine, which resulted in cytokine changes such as increased IL-6 compared to healthy control PBMCs [84]. In relation to pan T-lymphocyte proliferation, non-specific activation with the mitogen concanavalin A (ConA) followed by phenylephrine did not show any effect on proliferation, IL-4, or IFN-γ production [85]. In an elegant study by the same group, they found that ConA-activated pan T-lymphocytes, treated with an MAO inhibitor, would polarize to TH2 and produce more IL-4. This effect could be blocked by α1-AR and β2-AR antagonists, but not α2-AR or β1-AR antagonists [86]. This shows a role for endogenous catecholamines binding α1-ARs, but not in all temporal relationships to activation. Our group has additionally observed modulation of the intracellular redox environment with α1 agonism and antagonism, further supporting the presence of this receptor on T-lymphocytes [13]. As discussed previously, α2-AR represents a different mechanism by nature of its downstream intracellular cascade. α2-AR agonism by clonidine of ConA-activated pan T-lymphocytes resulted in inhibited proliferation, as well as decreased IFN-γ and IL-4 production. This effect could be blocked by α2-AR antagonism, and partially attenuated by inhibition of PLC or PKC, indicating a role for this pathway in α2-induced T-lymphocyte inhibition [85]. We have also identified a role for α2-AR in the CD4+ T-lymphocyte redox environment, furthering the complexity of AR signaling in these adaptive immune cells [13].
As can be seen from this small sample of the literature, T-lymphocyte expression of α-AR is complex with a disparity in findings from the literature. An excellent review by Kavelaars highlights the general thematic immune changes mediated by α1-ARs on immune cells, particularly PBMCs [87]. In summary, in vitro, α1-AR agonism causes increased activation of T-lymphocytes, as evidenced by increased cytokine production and intracellular redox, which can be modulated with α1-AR antagonism. On the other hand, α2-AR agonism results in inhibition of T-lymphocytes through some pathway partially mediated by canonical changes in PLC and PKC. The functional changes represent interesting results in vitro, with little work done to discuss the possibly of these mechanism in vivo, which could be further extended to the discussion of patients taking systemic α-AR modulating drugs.
3.2.1.2. β-Adrenergic receptors
β-AR expression was originally noted in T-lymphocytes indirectly. Application of catecholamines resulted in increased cAMP [88–90], indicative of β-AR expression before any physical receptors were actually detected (reviewed in [91]). We will discuss each β-AR subtype individually, with the focus largely on β2-AR differential expression and function.
In terms of the β1-AR, there is little evidence of its expression, but reports do exist regarding a functional role of this receptor in effector T-lymphocytes. A study by Takayanagi et al. found that NE suppressed IFN-γ and TNF-α production in murine intestinal intraepithelial CD3+ T-lymphocytes through β1-AR, as evidenced by selective pharmacological activation and blockade [92]. Other reports have focused on β1-AR expression in Tregs [93]. Freir et al. noted a greater expression of β1-ARs on Tregs versus CD25- T-lymphocytes, which were both isolated from healthy human patients undergoing an acute physical stressor [94]. β3-AR mRNA has been detected in ConA stimulated pan T-lymphocytes, but without any functional changes upon β3-AR agonism [95]. This could be due to the fact that β3-AR agonism has been shown to generate less cAMP than other β-AR subtypes[96].
In contrast, a breadth of sources demonstrate functional β2-AR expression on T-lymphocytes. For example, naïve CD4+ T-lymphocytes strongly express “high affinity, saturable β2-ARs” (exhaustively reviewed in [97] and [78]), and this expression is tightly regulated as T-lymphocytes differentiate. CD8+ T-lymphocytes express β2-ARs at a higher quantity than CD4+ cells that are differentially regulated in both healthy [98] and RA [99] patients. Work by Sanders and colleagues has shown that upon polarization to the TH1 and TH2 lineages, the expression level of the β2-AR is increased or decreased, respectively [100]. The mechanism of this repression in TH2 cells is driven by methylation of CpG islands in the β2-AR gene promoter, decreased histone 3 (H3) and H4 acetylation, and decreases in histone 3 lysine 4 (H34K) methylation. TH1 cells express opposite responses to upregulate expression of the β2-AR [100]. When naïve CD4+ T-lymphocytes are exposed to NE or selective β2-AR agonists, IL-2 and IFN-γ production upon subsequent activation is reduced [101]. This result is consistent with the traditional view that β2-AR activation results in increased cAMP, resulting in PKA activation and overall suppression of CD4+ T-lymphocytes. This cytokine response varies temporally with the addition of NE; TH1 polarization and activation followed by NE results in increased IFN-γ production from these cells [101 ]. We have recently proposed that this complex regulation allows NE to play both suppressive and activating roles in the T-lymphocyte inflammatory response [102]. This may be due to the intracellular role of cAMP during different activation states, or may even be due to non-canonical β-AR mechanisms [103]. Further investigation into the intracellular signaling pathways is warranted to elucidate these complex interactions.
3.2.2. Dopamine receptors
Dopamine receptors (DRs) of all subtypes (D1-D5) have been described on nearly all T-lymphocyte subtypes [104]. The expression levels of each type of receptor are dynamic and context-dependent, thus, creating a complex interplay between receptor expression and function. Therefore, it is simpler to group these DRs by their intracellular effects, as discussed in section 2.3.2. In theory, D1-like family-mediated increases in cAMP should result in changes in the intracellular signaling cascade such as inhibition of ERK, JNK, and NF-κB activation; all of which would inhibit T-lymphocyte activation, proliferation, and cytokine production (reviewed further in [105]). Conversely to this, D2-like family agonism that functions to decrease adenylyl cyclase activity would result in increased T-lymphocyte activation. Flowever, these global assumptions do not always appear to be true experimentally. In order to further elucidate this mechanistic control, we must look at receptors within each DR grouping more closely.
D1-like receptor agonism with physiological concentrations of dopamine in vitro has been shown to impair the cytotoxicity of CD4+ and CD8+ cells, as well as reduce their proliferation by IL-2 induction [106]. This stimulation of the D1-like family has also been implicated in the polarizing of naïve CD4+ T-lymphocytes to TH2 by a DC-mediated dopamine dose-dependent increase in cAMP levels [107]. In another study by Nakano et al., a small molecule D1-like receptor antagonist resulted in DC-mediated IL-23 production that resulted in TH17 polarization [108]; a result that has been shown both in vitro and in vivo [109]. In addition to this, it has been shown that activation of the D1-like receptors on Tregs results in decreased IL-10 and TGF-β, as well as their decreased proliferation [110]. These cytokines are the primary mechanism by which Tregs are able to inhibit effector T-lymphocyte proliferation [61], thus dopamine appears to inhibit those processes. Dopamine at high concentrations (10-100 μM) can also effectively inhibit already activated effector T-lymphocytes. Ex vivo studies have shown that mitogen-activated CD4+ T-lymphocytes treated with dopamine had reduced the proliferation and synthesis of IFN-γ [111]. Another study by Ghosh et al. showed that 3-5 ng/mL of dopamine added to human pan T-lymphocytes, activated with anti-CD3 antibody, showed significantly reduced proliferation and secretion of IL-2, IFN-γ, and IL-4 [112]. In summary, the activation of D1-like receptors results in decreased functionality of CD4+ T-lymphocytes as well as decreasing the ability of Tregs to suppress effector T-lymphocytes.
D2-like family reception is less consistent than the D1-like family between receptor subtypes. Levite et al. showed that selective agonism of D2R and D3R on CD8+ T-lymphocytes from healthy humans resulted in the induction of adhesion to fibronectin. This high molecular weight glycoprotein, found in the extracellular matrix, functions to promote cellular trafficking and adhesion [113]. In naïve CD8+ T-lymphocytes from both humans and mice, dopamine agonism at D3R-induced adhesion to fibronectin and Intercellular Adhesion Molecule 1 (ICAM-1/CD54) [114], as well increased T-lymphocyte proliferation [115]. In addition to this, agonism at D2R and D3R resulted in increased expression (mRNA and protein) of IL-10 or TNF-α, respectively [116]. This enhanced production of IL-10 would function to inhibit effector T-lymphocytes, while D3R-mediated chemotaxis would promote increased function of these CD8+ cells; a contradictory and complex observation. This overall phenotype promotes cellular immunity, with predominant polarization to TH1 in the CD4+ population [116]. On the other hand, D4R stimulation mediates quiescence by inhibiting the phosphorylation of ERK1/ERK2, thus resulting in the up-regulation of Kruppel-like factor 2 (KLF-2) [117]. This suppressive effect of D4R mimics the D1-like family activity even though these receptors act similarly to the D2-like family.
In total, it should be realized that the role of dopamine in the physiology of T-lymphocytes is decidedly complex. Dopamine can increase the homing and chemotaxis of CD8+ cells, alter the polarization of CD4+ cells, inhibit activated effector T-lymphocytes, and suppress Treg cells. These effects are mediated by the varying dopamine receptor subtypes, which have varying affinities for dopamine itself. This complex interaction is further muddied by the direct interaction of T-lymphocytes with other cell types, like DCs, which also utilize dopamine in their regulation [105].
3.2.3. Ach receptors
It was originally discovered that T-lymphocytes can bind Ach through radiolabeled binding studies of MRs and nAchRs [118,119]. An excellent review by Fujii chronicles the history of both Ach receptors in T-lymphocytes and thymocytes [120]. All five subtypes of MRs have been found on human pan T-lymphocytes at varying densities [121]. The highest expressed is the M3 subtype, followed by the M4 and M5 subtype, with varying expression between individuals of the M1 and M2 receptor [122].
High expression of the M3 receptor (M3R) on T-lymphocytes does not necessarily substantiate claims that it is crucial to T-lymphocyte function. Selective agonism of the M3R in human leukemia T-lymphoblasts induced transient rises in intracellular Ca2+ with a subsequent rise in extracellular Ca2+ oscillations [123]. This M3R-induced Ca2+ current, accompanied by Ca2+ influx through calcium-release activated Ca2+ (CRAC) channels, results in eventual increased c-fos action and IL-2 gene expression. This was demonstrated by CRAC blockade abolishing the increased IL-2 expression seen with muscarinic agonism by the parasympathomimetic oxotremortine [124], while also increasing c-fos mRNA overall DNA synthesis [125]; an effect that could be subsequently abolished by nonselective MR antagonism [123]. This data demonstrates the positive effect of M3R agonism on T-lymphocyte proliferation.
M3R is also crucial in allowing for optimal polarization and respective cytokine production from CD4+ T-lymphocytes when presented with an antigenic challenge expected to elicit a TH1 or TH2 response. Darby et al. found that M3R−/− mice infected with the helminth Nippostrongylus brasiliensis, which is commonly used to elicit a predominantly TH2 immune reaction, had an attenuated immune response when compared to WT controls [126]. This reduced TH2 response previously seen in the M3R−/− group could be recapitulated in the WT group when M3 antagonists were administered. A similar deficiency in adaptive immunity was found in the M3R−/− cohort in response to Salmonella enterica, which elicits primarily a TH1 response. WT mice increased IFN-γ production in response to M3R agonists, while antagonism resulted in an attenuation of IFN-γ production [126]. Taken together, these data show a direct role for M3R in the mounting of an appropriately polarized immune response to helminth or bacterial infection.
M1R and M5R mediate a similar intracellular cascade to M3R, which may partially explain their effects. In an experiment to test the involvement of M1R and M5R specifically, Fujii et al. immunized WT and combined M1R/M5R−/− mice with ovalbumin (OVA) [127]. In the knockout (KO) mice, they found lower levels of the anti-OVA antibodies of the immunoglobulin G1 (IgG1) isotype, but not IgM. In addition, the M1R/M5R−/− group produced less TNF-α and IL-6, while also showing decreased levels of AChE intracellularly [127]. This decreased cytokine production in the KO group could be related to the ability of M1R, M3R, and M5R to increase intracellular Ca2+; a process which is well-known to be essential to effective T-lymphocyte activation. M1R, despite its lower expression, has been implicated in improved IL-2 production and IL-2R expression after TOR activation [125]. In fact, M1R has shown important roles in CD8+ T-lymphocyte differentiation. In M1R−/− mice, naïve CD8+ T-lymphocytes did not effectively differentiate into effector CD8+ T-lymphocytes, resulting in decreased killing of P815 mastocytoma cells (a classical target for cytotoxic T-lymphocytes) [128].
Conversely, M2 and M4 linkage to Go/Gi subunits posits a different functional effect upon their activation, but there is a paucity of studies on their effects on any T-lymphocyte subtypes. There is some discussion of M2R involvement in asthma pathogenesis, with increased expression of M2R and M3R in asthmatic patients [129]. This is complicated by the fact that major basic protein, released by eosinophils and characteristically increased in asthmatics, can also interact with M2R and alter Ach binding [130].
With this, the current literature points toward specifically the Gq linked M1R, M3R, and M5R as having some involvement in the activation and differentiation of both CD4+ and CD8+ T-lymphocytes. These interactions are of great interest for pharmacologists and clinicians alike, with the ubiquitous intentional and off-target effects of cholinomimetics and cholinolytics [131]. The administration of MR agonists might result in increased differentiation and polarization of T-lymphocytes, while antagonists could suppress cholinergic signaling made by and for immune cells.
As discussed in section 2.3.3, Ach can bind not only the MRs on T-lymphocytes, but also nAchRs. This binding of the nAchR mirrors that seen with muscarinic agonism, but differs in mechanism, with nAchR being an ionotropic receptor that allows for direct changes in cation (Na+, Ca2+) flux upon ligand binding (although this fact is convoluted by more recent evidence of nAchR interacting with G-proteins [37, 132–134]). While this simplistic understanding of all nAchRs resulting in a similar effect is enticing, it is clear this is not the case based on structure alone. The heterogeneity of these receptors is evident in T-lymphocytes, where subunits determine the effect. nAchR subunit mRNA has been detected in human PBMCs, as well as human T-lymphocyte leukemic lines, with varied expression between individuals and cell lines [122].
Discussion around the varying detectable subunits has revolved around the expression of α2, α5, α6, α7, α8, α9, and α10 in leukocytes [135, 136], with α2, α5, α9, α10, β1, β2, and β4 nAChR mRNA found in freshly isolated splenic pan T-lymphocytes specifically [137]. Upon ex vivo activation of CD4+ T-lymphocytes by TCR cross-linkage, Qian et al. found the following: α4 and α7 mRNA became discernably expressed; α5, α10, and β4 were upregulated; and α9 and β2 were downregulated [137]. In CD8+ T-lymphocytes, α4 and α7 became expressed upon activation as well, along with dynamic expression changes of other nAchR subunits. For α7 nAchR specifically, mRNA was detected only in activated CD4+ and CD8+ T-lymphocytes, with minimal expression from isolated cells that were not activated.
In recent years, the α7 nAChR has been of great interest to immunologists. In T-lymphocytes, stimulation of the α7 nAchR by nicotine has been shown to reduce indices of CD4+ T-lymphocyte proliferation when exposed to myelin oligodendrocyte glycoprotein (MOG), a commonly used antigen for experimental autoimmune encephalomyelitis (EAE) [138]. In this same study by Nizri et al., nAchR agonism resulted in an overall shift to TH2 differentiation with increased IL-4 production and decreases in TH1 and TH17 cytokines [138]. In α−/−-derived CD4+ T-lymphocytes, nicotine showed no effect on cytokine production, suggesting this variant is the predominant responding isoform to this stimulant. In vivo administration of nicotine attenuated EAE outcomes in WT mice, but α7−/− mice did not respond in a similar fashion, with disease severity reduction being related to impaired antigen presentation [138]. In another study by Qian et al., nicotinergic stimulation of activated CD4+ T-lymphocytes produced upregulation of IFN-γ, while also downregulating IL-10 and IL-17 [137]. This demonstrates the ambiguity in the literature, where nuance such as method of activation can produce seemingly contradictory results. In addition, further differentiation into various phenotypic subtypes can produce differing results. For example, when nAchR is stimulated on Tregs, they have an increased ability to suppress CD4+ T-lymphocytes [139]. Additionally, α7 nAchR has been found to be crucial to the “inflammatory reflex”, which will be discussed in section 4.1.
Overall, the presence of both mAchR and nAchR on T-lymphocytes suggests an important physiological function, but the numerous subtypes of these receptors makes elucidating the functional role of Ach in T-lymphocyte strikingly difficult. Continued research utilizing T-lymphocyte-specific genetic knockouts will hopefully allow researchers to untether these complex interactions in the future.
3.3. T-Lymphocytes synthesize neurotransmitters
Despite their eponymous association with neurons, it has long been known that many cell types, including immune cells, synthesize and respond to neurotransmitters. In 1985, Blalock et al. presented a number of molecular mechanisms by which the neuroendocrine and immune systems integrate to form a more functional unit. It has since been proposed that neurons and immune cells may have a similar evolutionary origin; an idea posited by a few investigators [140,141], but not widely discussed. Aforementioned, T-lymphocytes possess an array of neurotransmitter receptors that allow for critical changes in function, but they may also produce their own neurotransmitters. This ability significantly enhances the complexity of neural-immune crosstalk, and research in this field still is in its infancy.
3.3.1. Catecholamines
All three catecholamines have been detected in resting, undifferentiated T-lymphocytes as well as the variety of activated lineages [93, 142, 143]. Gene expression of all enzymes along the catecholamine synthetic pathway have also been reported, including DβH in lymphocytes [144] and PNMT in splenocytes and thymocytes [145, 146]. Tregs constitutively express TH and have been shown to contain relatively large amounts of dopamine when compared to effector T-lymphocytes [93]. The levels of these catecholamines within T-lymphocytes increases during activation, as does content of the rate-limiting TH enzyme, and is intimately linked to the activation of PKC and increases intracellular Ca2+[147]. This upregulation of catecholamine synthesis is mirrored by the reported upregulation of ARs during similar mitogen stimulation or addition of pro-inflammatory cytokines [148,149].
It could be posited that T-lymphocytes could produce catecholamines in a paracrine and autocrine fashion to inhibit growth and proliferation, not unlike reports in macrophages [150]. Studies have shown catecholaminergic-driven inhibition of pan T-lymphocyte function by driving anti-inflammatory cytokine expression as well as changes to the apoptotic factor milieu to potentiate cell death [151], which could be reversed by blocking TH [147]. Upon activation, the increase in TH and other crucial enzymes along the catecholamine synthetic pathway can be thought of as a way for T-lymphocytes to finely titrate the proliferation of specific subpopulations. This type of “catecholamine loop” has come to light in recent literature in macrophage populations during the insidious cytokine storm experienced by some patients receiving immunotherapy [152].
3.3.2. Ach
Ach is a neurotransmitter heavily relied upon to transmit peripheral nervous signals. It is the primary neurotransmitter at the neuromuscular junction, the autonomic pre-ganglion synapse, and the parasympathetic post-ganglionic synapse. At secondary lymphoid organs, there is no evidence of parasympathetic innervation [153]. However, there are significant quantities of Ach in the spleen (as evidenced by the spleen being its first place of its extraction as referenced in 2.2.2 [26]). This raises the question as to the source of this abundant Ach? One potential source of Ach in a secondary lymphoid organ would be from the blood; a topic that was once a matter of debate [154]. However, this is highly unlikely due to the presence of BuChE in plasma, which would lead to its rapid degradation. Interestingly, unlike the catecholamines, Ach is a short-lived molecule, likely due to its labile ester moiety, that can be rapidly hydrolyzed [155]. To this end, the structure of Ach favors its function as an autocrine or paracrine signal produced directly from immune cells, thus, positing immune cells are the source of Ach in lymphoid organs. Indeed, Kawashima et al. discovered that 60% of the total Ach in blood was located in the PBMCs, while there was no Ach detected in the polymorphonuclear leukocyte or red blood cell layer [156]. This has since been validated by the presence of Ach in multiple human T-lymphocyte leukemic cell lines [157, 158] and purified T-lymphocytes—with CD4+ cells showing higher levels than CD8+ cells [159].
If this Ach is produced by T-lymphocytes, by what synthetic mechanism does this occur? This question does not immediately have an obvious answer, since non-neuronal cells also utilize the mitochondrial enzyme carnitine acetyltransferase (CarAT) to synthesize Ach, in addition to the canonical CHAT enzymatic pathway. In a series of experiments on human T-lymphocyte leukemic cell lines (MOLT-3, HSB-2, and CEM), Fujii et al. found that using the CHAT inhibitor, bromoacetylcholine (100 uM), reduced Ach synthesis by 50%, while the application of the CarAT inhibitor bromoacetyl-L-carnitine (100 uM) only reduced the synthesis by 30% [157]. This shows that T-lymphocyte Ach is likely synthesized by CHAT, with a lesser extent of production by CarAT. In experiments of T-lymphocyte activation using PHA on peripheral human mononuclear leukocytes, CHAT mRNA and activity were increased while CarAT remained unchanged [160], thus showing that the T-lymphocyte activation cascade can through some mechanism control CHAT expression and activity [161]. PHA, via the TCR complex, results in increased PLC-mediated production of IP3. In addition to this mechanism, TCR-independent pathways of activation have shown increased CHAT mRNA and activity; such as the calcium ionophore ionomycin, protein kinase C activator phorbol 12-myristate 13-acetate (PMA), and PKA activator dibutyryl cAMP (also called bucladesine) [162, 163].
In summation, T-lymphocytes are able to effectively synthesize both catecholamines and Ach and regulate their production. The source of these neurotransmitters is crucial in situations when there does not appear to be a clear anatomical neuroimmune connection, such as the parasympathetic nervous system communicating with splenic T-lymphocytes.
4. Neurotransmitters & T-Lymphocytes in Cardiovascular Disease
The breadth of literature above paints a complex, yet influential picture of how neurotransmitters of both neuronal and immune origin might result in functional changes. These signals appear to act as a 4th signal of T-lymphocyte activation that further attunes the inflammatory profile of these cells, and may result in significant changes in systemic physiology or even contribute to pathological conditions.
Metabolic syndrome is one of the most important risk factors to the development of cardiovascular disease. This syndrome represents a number of separate but interlinked pathophysiological states (e.g. obesity, insulin resistance, hyperlipidemia, hypertension, and others) that converge to create a phenotype with a high risk for cardiovascular events. Furthermore, autonomic dysregulation as well as chronic low-grade inflammation associated with metabolic syndrome and cardiovascular diseases are both widely accepted [164–167]. This low-grade inflammation in cardiovascular disease begets an etiological question: Is the pro-inflammatory phenotype a response to deranged physiology, or does the pro-inflammation precede the changes in cardiovascular physiology? Moreover, what is the role of interaction between neurotransmitters and T-lymphocytes in this process? While this question is far from fully elucidated in the literature to date, there are valuable studies that have worked to investigate the causality dilemma between inflammation and cardiovascular disease, which is discussed further in sections 4.1 and 4.2.
4.1. The cholinergic anti-inflammatory reflex
A recent body of literature has developed around how NE and Ach are used in CD4+ T-lymphocyte communication with macrophages; this “cholinergic anti-inflammatory pathway” is understood more broadly to be the efferent arm of the “inflammatory reflex” [168–173]. In order to better understand this concept, we must first discuss the autonomic innervation of the spleen.
The discussion of autonomic splenic innervation is a complex one, with the literature asserting that the spleen receives only sympathetic efferent fibers (i.e. no parasympathetic or afferent tracts) in the form of the splenic nerve [174]. The splenic nerve travels in a neurovascular bundle, arising from the celiac ganglion. This ganglion receives sympathetic inputs originating from the rostral ventrolateral medulla (RVLM), after they a synaptic connection in the intermediolateral cell column in the spinal cord [175]. More interestingly, the celiac ganglia has also been reported to receive efferent vagal fibers from the dorsal motor nucleus (DMN) [176, 177]. The splenic nerve itself is exclusively noradrenergic, and thus, has varicosities along its length where synapses en passant release vesicular NE [178]. This NE is released within the spleen, mainly on both B- and T-lymphocytes due to regional distribution, and can reach concentrations in the millimolar range in these local environments [179].
The cholinergic anti-inflammatory pathway is believed to work via neurotransmission between neurons and immune cells, but further propagated by neurotransmission solely between immune cells. As with any neural reflex, afferent information must be transmitted initially. Afferent vagal nerve fibers are activated by various systemic cytokines such as IL-1, TNF, IL-6, IFN-γ, in addition to a host of other inflammatory mediators, reviewed nicely by Goehler and colleagues [180]. After integration in the central nervous system at the suprachiasmatic nucleus, as shown by Guerrero-Vargas et al. [181], The efferent arc sends a neural signal to peripheral organs. While the mechanism of the effect of this efferent signal is agreed upon at the cellular level, the efferent arm itself is wont with controversy [182] which we will discuss later in this section. Reliably, NE is release by the splenic nerve, which then binds β2-AR receptors on CFIAT+ CD4+ T-lymphocytes. This β2-AR stimulation results in the synthesis and secretion of Ach by the aforementioned T-lymphocytes locally within the spleen [183], which then binds α7 nAchRs on macrophages and results in a decrease in serum TNF-α, splenic TNF-α, and macrophage TNF-α release during an LPS challenge [172, 173].
Returning to the discussion of the efferent arm, it has been shown that vagal nerve stimulation results in inhibition of splenic TNF-α production during LPS challenge [169]. This was further demonstrated by the lack of TNF-α suppression in α7 nAchR−/− mice during vagal stimulation [172]. These studies provide evidence for the interplay between the vagus and the spleen, which is purported to occur through the vagal efferent nerves signaling through the celiac ganglia (reviewed further in [184]).
Conversely, strong evidence exists to the contrary that suggests the vagus nerve plays no role in the anti-inflammatory reflex. The aforementioned vagal-splenic sympathetic interaction was not recapitulated by Bratton and colleagues, in which neither an anatomical nor functional connection were found [185]. Martelli and colleagues have shown that this reflexive response to LPS is mediated solely by the sympathetic splanchnic nerve, as only ablation of this nerve (and not bilateral vagotomy) showed a significant decrease in splenic TNF-α levels [186]. This is further validated by recent study from the same group that has shown that bilateral transection of splanchnic sympathetic nerves prevented the anti-inflammatory effects seen with vagal nerve stimulation, providing more evidence that afferent vagal fibers receive information that is integrated centrally, but only sent through an efferent sympathetic arm [187].
Regardless of the mechanism, these autonomic-immune interactions have far-reaching implications in human disease, specifically in cardiovascular diseases like hypertension. TNF-α has been shown to directly increase renal vascular resistance [188], while blockade of TNF-α reduces renal injury and mean arterial pressure [189–191], suggesting a potential role for this reflex in hypertension. Additionally, the hypertension observed in systemic lupus erythematosus (SLE) is thought to be mediated largely by SLE-associated inflammation along with decreased efferent vagal tone (reviewed in [192]). This reduced vagal tone results in a decreased anti-inflammatory reflex and a more pro-inflammatory environment, which could be reversed with central administration of the AchE inhibitor galantamine [193]. The use of this inhibitor, which would cause an accumulation of Ach, resulted in decreased splenic and renal inflammation, helping to explain the functional decrease in pressure seen with galantamine administration.
These experiments address the leading question of the section to demonstrate the importance of neurotransmission in regulating the inflammatory environment, and the impacts it may have on systemic diseases like SLE or hypertension. SLE serves as a well-elucidated example of how pro-inflammatory pathology can potentiate cardiovascular pathology by altering neurotransmission between and through CD4+ T-lymphocytes. Hypertension is another prime example, and recently has been the most widely explored example of T-lymphocytes affecting cardiovascular diseases.
4.2. T-Lymphocytes and hypertension
Enhanced sympathetic outflow is a hallmark of many forms of hypertension [194, 195]. NE activates ARs in the kidney, which results in increased retention of Na+ as well as renin release. This activation of the classical renin-angiotensin-aldosterone system (RAAS) contributes to the maintenance of elevated mean arterial pressure in hypertension (reviewed extensively in [196]). Angiotensin II (Ang II), the main effector peptide of the RAAS system, is a major component and current therapeutic target of hypertension. Intriguingly, Ganta et al. demonstrated that application of Ang II centrally increased splenic pro-inflammatory cytokine production (e.g. IL-1β and IL-6), and was diminished by ablation of the splenic nerve [197]. This phenomenon is suggestive of a neural-immune reflex, but further indicates significant harmony between the cardiovascular, neural, renal, and immune systems.
To this end, we must first discuss that it has already been well established that T-lymphocytes are necessary for the development of hypertension, with investigations starting in the 1960’s [198–201], followed by clinical work with the lymphocyte-depleting drug mycophenolate mofetil resulting in a reversal of hypertension in patients with autoimmune disorders [202]. This has been further validated as a causal link by multiple groups showing that mice lacking lymphocytes are resistant to hypertension of various etiologies with adoptive transfer studies of T-lymphocytes restoring this response [203–205]. This intimate connection is mediated through direct interactions of the nervous and immune components, with local inflammation evident in the kidneys, heart, brain, and vasculature [203,206–208]. It follows that interruption of these sympathetic signals should result in an attenuated inflammatory and hypertensive phenotype.
The central nervous system mediates changes in blood pressure most prominently by altering sympathetic tone and humoral factors [209–211]. The anteroventral third ventricle (AV3V) region includes various circumventricular regions that, when ablated, results in loss of many Ang II-mediated effects, such as vasopressin release and sympathetic tone [212]. Marvar and colleagues found that this ablation of this region in Ang II-treated mice resulted in decreased activation of CD4+ T-lymphocytes and vascular infiltration [213]. This inflammation was found to be mediated by pressure alone, since NE-induced pressure increases bypassed the AV3V lesion and led to inflammation as well as the administration of the anti-hypertensive hydralazine (with no known Ang II effects) resulted in a decrease of the same inflammatory markers.
These investigations provide some insight to the temporal relationship posited in the introduction to this section. It is likely the combined, feed-forward effect of centrally-mediated changes give rise to inflammatory perturbations, which further exacerbate the pathophysiology that then results in frank hypertension. This communication is facilitated at the cellular level by interactions between sympathetic outflow and T-lymphocytes.
One area this neuroimmune interaction may take place is the kidneys, where sympathetic tone and neurohumoral factors can sway the titrated control of intravascular volume and thus, pressure. However, it has become clearer that not all hypertension models are created equal, especially in regards to their effects on the kidneys. Work by Xiao et al. found that renal denervation prevented CD4+ and CD8+ T-lymphocyte infiltration and damage to the kidneys, while also reducing blood pressure in Ang II-treated mice [214]. Additionally, adoptive transfer of splenic DCs from Ang II-treated mice resulted in CD4+ and CD8+ T-lymphocyte activation and renal infiltration, as well as a hypertensive phenotype in untreated mice, an effect that could be ameliorated by bilateral renal denervation [214]. Dexoycorticosterone-acetate salt (DOCA-salt) is another commonly used hypertensive stimulus that has similarly been shown reversible through renal denervation. Interestingly, in this model, targeted chemical or physical afferent renal nerve ablation shows a similar decrease in pressure as compared to complete (i.e. afferent and efferent sympathetic nerves) denervation [215,216]. In addition to the antihypertensive effect of total renal denervation, markers of renal inflammation such as CD4+CD25+ and CD8+ T-lymphocyte infiltration and cytokine content also were reduced [216]. Interestingly, a follow-up study by the same group found that afferent denervation alone did not reduce renal CD3+ T-lymphocyte or macrophage infiltration caused by DOCA-salt treatment [217]. Osborn et al. investigated this work in another model, wherein mice were exposed to either traditional chow or a high-fat diet (45% of caloric content), with the latter group developing the characteristic metabolic syndrome phenotype of increased arterial pressure, hyperglycemia, and increased fat mass. Both groups then underwent complete renal denervation, but only the high fat group displayed a reduction in systemic blood pressure [218]. However, renal denervation did not result in any changes in glucose metabolism or markers of renal inflammation [218], again showing the complexity of hypertension, renal denervation, and renal inflammation dependent on the etiology and possibly extent of the hypertension.
These works provide evidence of a crucial neuroimmune interaction that results in the development of hypertension. They also serve as confirmation that the model system used to produce the hypertension plays a role in its pathophysiological phenotype, with different models responding differently to renal denervation. The ability to treat resistant hypertension with renal nerve ablation is not a new idea clinically, with multiple international clinical trials testing catheter-based ablation methods. Interpretation of these trials is convoluted and nuanced (reviewed exceptionally in [219]), but it stands to reason that modulation of sympathetic signals to an organ heavily involved in the maintenance of hypertension are yet still a potential therapeutic target for hypertension.
In addition to the renal nerves involvement, the splenic nerve provides a potential location for the priming of T-lymphocytes by sympathetic tone (reviewed in [220]). CD4+ and CD8+ T-lymphocytes migrating to the kidneys and vasculature during Ang II hypertension have been shown to originate in the spleen [221]. This was shown elegantly by adoptive transfer of donor mouse splenocytes bearing the leukocyte antigen CD45.1 into a CD45.2 splenectomized recipient mouse [221]. The input these T-lymphocytes receive from the splenic nerve can severely alter their phenotype, as seen from the prior discussions of local NE concentrations in the spleen and its complex effects on T-lymphocytes in vitro. Osborn and colleagues found that celiac ganglionectomized Dahl rats, which upon high salt exposure develop a well-established form of neurogenic hypertension, had decreased arterial pressure when compared to sham controls [222]. This reduction in pressure was shown to have a different mechanism than that of renal denervation, since rats who underwent both ablations had even greater decreases in pressure [222]. This data, in conjunction with evidence of the cholinergic anti-inflammatory reflex in the spleen, represents yet a major anatomical location in which sympathoexcitation can alter T-lymphocytes in a way that can promote proinflammatory states related to hypertension and other cardiovascular diseases.
Our group has also examined the role of sympathoexcitation-driven T-lymphocyte activation during hypertension. In a model of increased sympathetic tone through NE-infusion, we found that splenic T-lymphocytes demonstrate an altered phenotype once activated—a result seen in pan T-lymphocytes, as well as CD4+ and CD8+ cells individually [14]. The phenotype of these T-lymphocytes was overall suppression, with decreases in pro-mitotic cyclins and proinflammatory cytokines IFN-γ and TNF-α, which was suggestive of canonical adrenergic stimulation of cAMP leading to diminished activation [14]. However, this inhibition of T-lymphocytes during NE-induced hypertension was at least partially caused by increases in superoxide, as evidenced by antioxidant supplementation modestly restoring the growth and cytokine profile of CD4+ and CD8+ T-lymphocytes. This phenotype of T-lymphocytes during NE-infusion is in contrast to previously discussed findings [213], in which NE infusion resulted in hypertension and activated CD3+ T-lymphocyte infiltration into the vasculature despite AV3V lesions. The differential activation of T-lymphocytes during NE-infusion and hypertension is thus highly dependent on the location of these T-lymphocytes. The dual nature of NE signaling on T-lymphocytes is thus evident, with splenic T-lymphocytes responding to NE in a different manner, likely due to their differing state of activation. This work demonstrates a novel role of redox communication as a downstream mediator in neurotransmission in T-lymphocytes, which is highly relevant to the cardiovascular field where neurotransmission and redox environments are reported to be significantly disrupted.
Another burgeoning area of interest is the role progenitors of T-lymphocytes and other immune cells play in hypertension. The bone marrow serves a primary lymphoid organ, where bone marrow (BM) cells respond to sympathetic input to differentiate and enter the circulation [223,224]. These BM cells express all β-ARs, and their stimulation modulates the release of immune cells into circulation in a diurnal pattern corresponding to increased activity or risk of infection [225,226]. By adoptively transferring β-AR-deficient BM cells into near lethally irradiated mice, Zubcevic and colleagues found that there was an association between the lack of β-ARs on immune cells and the observed decreases in blood pressure [227]. In addition, transcriptomic analysis of these BM cells showed altered expression of transcripts relevant to many immune processes, which pathway analysis revealed were shown to be critical in CD4+ T-lymphocyte activation, proliferation, adhesion, and chemotaxis [227]. This research provides further insight into the etiological question in the introduction to this section by highlighting how adrenergic signaling in immune cells is involved upstream from many of the inflammatory changes seen in hypertension. That is, how early changes in sympathoexcitation could potentiate deleterious proinflammatory changes in T-lymphocytes and other immune cells, as mediated by ARs specifically. Overall, T-lymphocytes have proven to be necessary for the development of hypertension, while also having been shown to be extremely responsive to signals mediated by neurotransmitters. Taken together, these results have important implications for how at various stages of hypertension neuroimmune interactions are crucial, and may elucidate future targets of therapeutic intervention for this prevalent and chronic disease.
5. Conclusion
Neurotransmitters are an essential means of communication within the nervous system, and are only now being understood to be ubiquitous signaling molecules between the nervous and immune systems. T-lymphocytes are able to synthesize and receive neurotransmitters—specifically Ach and the catecholamines—resulting in massive changes in the main outputs of T-lymphocyte function such as proliferation, polarization, cytokine production, and activation. These changes are complex and not as of yet well elucidated, especially in regard to their functional effects in vivo. Published evidence continues to provide mechanistic study of how the master regulators of adaptive immunity, T-lymphocytes, are involved in early pathogenesis and feed-forward maintenance of hypertension. Even further, modulation of both the cholinergic and adrenergic systems are mainstays of clinical pharmacology, with little known about the long-term effects of these therapies [228,229], especially as they relate to cardiovascular disease.
To quote Voltaire, “Doctors are [wo]men who prescribe medicines of which they know little, to cure diseases of which they know less, in human beings of whom they know nothing.” This reminder of our distance from the actual treatment of disease might seem a harsh critique, but a necessary one. Overall, there is a need for continued research on the basics of the neuroimmune interaction between neurotransmitters and T-lymphocytes. By further investigating this intricate biology, we may further validate and explore our ability to utilize therapeutic interventions to treat patients. This basic investigation could provide translational results to improve the lives and outcomes of patients with hypertension and cardiovascular disease.
Figure 3: The dual nature of NE in T-lymphocytes.

On the one side, signals arising centrally in the brain—sent through vagal efferents or splanchnic sympathetic efferents—synapse in the celiac ganglion. The splenic nerve arises here and releases NE locally, wherein it is received by splenic T-lymphocytes, possibly by multiple AR subtypes. In naïve T-lymphocytes, NE reception will result in suppression of cytokine production and increased ROS. Additionally, ChAT+ T-lymphocytes receive NE through the β2-AR, which induces the production of Ach that is received by α7 nAchR-bearing macrophages. This results in a suppression of proinflammatory cytokine release by these splenic macrophages (termed the anti-inflammatory reflex). On the other side, sites of already activated T-lymphocytes and macrophages receiving NE from sympathetic efferents become further activated, contributing to the exacerbation of hypertension.
Acknowledgements
This work was supported by NIH R00HL123471 to AJC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors have declared that no conflict of interest exists.
References
- [1].McCoy AN, ong Tan SY, Otto Loewi (1873-1961): Dreamer and Nobel laureate, Singapore Med. J. (2014). doi: 10.11622/smedj.2014002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Valenstein ES, The discovery of chemical neurotransmitters, Brain Cogn. 49 (2002) 73–95. doi: 10.1006/brcg.2001.1487. [DOI] [PubMed] [Google Scholar]
- [3].Schwiening CJ, A brief historical perspective: Hodgkin and Huxley, J. Physiol 590 (2012) 2571–2575. doi: 10.1113/jphysiol.2012.230458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].editors. Purves D, Augustine GJ, Fitzpatrick D, et al. , What Defines a Neurotransmitter?, in: Neuroscience, Sinauer Associates, 2001. https://www.ncbi.nlm.nih.gov/books/NBK10957/. [Google Scholar]
- [5].Rudolph LM, Cornil CA, Mittelman-Smith MA, Rainville JR, Remage-Healey L, Sinchak K, Micevych PE, Actions of Steroids: New Neurotransmitters, J. Neurosci 36 (2016) 11449–11458. doi: 10.1523/JNEUROSCI.2473-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zimmermann H, Goethe J, ATP and acetylcholine, equal brethren, (2007). doi: 10.1016/j.neuint.2007.09.004. [DOI] [PubMed] [Google Scholar]
- [7].Li ZJ, Cho CH, Neurotransmitters, more than meets the eye-Neurotransmitters and their perspectives in cancer development and therapy ☆, Eur. J. Pharmacol 667 (2011) 17–22. doi: 10.1016/j.ejphar.2011.05.077. [DOI] [PubMed] [Google Scholar]
- [8].Schafer E, Oliver G, The Physiological Effects of Extracts of the Suprarenal Capsules, J. Physiol 18 (1895) 230–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hastings TG, Lewis DA, Zigmond MJ, Reactive dopamine metabolites and neurotoxicity: implications for Parkinson’s disease., Adv. Exp. Med. Biol 387 (1996) 97–106. http://www.ncbi.nlm.nih.gov/pubmed/8794199 (accessed January 14, 2019). [DOI] [PubMed] [Google Scholar]
- [10].Kalyanaraman B, Felix CC, Sealy RC, Peroxidatic oxidation of catecholamines. A kinetic electron spin resonance investigation using the spin stabilization approach., J. Biol. Chem 259 (1984) 7584–9. http://www.ncbi.nlm.nih.gov/pubmed/6330064 (accessed January 14, 2019). [PubMed] [Google Scholar]
- [11].Mason HS, Spencer E, Yamazaki I, Identification by electron spin resonance spectroscopy of the primary product of tyrosinase-catalyzed catechol oxidation, Biochem. Biophys. Res. Commun 4 (1961) 236–238. doi: 10.1016/0006-291X(61)90278-9. [DOI] [PubMed] [Google Scholar]
- [12].YAMAZAKI I, MASON HS, PIETTE L, Identification, by electron paramagnetic resonance spectroscopy, of free radicals generated from substrates by peroxidase., J. Biol. Chem 235 (1960) 2444–9. http://www.ncbi.nlm.nih.gov/pubmed/13846434 (accessed January 14, 2019). [PubMed] [Google Scholar]
- [13].Case AJ, Roessner CT, Tian J, Zimmerman MC, Mitochondrial Superoxide Signaling Contributes to Norepinephrine-Mediated T-Lymphocyte Cytokine Profiles, 2016. doi: 10.1371/journal.pone.0164609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Case AJ, Zimmerman MC, REDOX-REGULATED SUPPRESSION OF SPLENIC T-LYMPHOCYTE ACTIVATION IN A MODEL OF SYMPATHOEXCITATION, Hypertension. 65 (2015) 916–923. doi: 10.1161/HYPERTENSIONAHA.114.05075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Deo SH, Jenkins NT, Padilla J, Parrish AR, Fadel PJ, Norepinephrine increases NADPH oxidase-derived superoxide in human peripheral blood mononuclear cells via -adrenergic receptors, AJP Regul. Integr. Comp. Physiol. 305 (2013) R1124–R1132. doi: 10.1152/ajpregu.00347.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tekin I, Roskoski R, Nurgul C-S, Vrana KE, Complex molecular regulation of tyrosine hydroxylase, (n.d.). doi: 10.1007/s00702-014-1238-7. [DOI] [PubMed] [Google Scholar]
- [17].Medical A, Ontario S, An update on the diagnosis and treatment of Parkinson disease, 188 (2016) 1157–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Eisenhofer G, Kopin IJ, Goldstein DS, Catecholamine Metabolism: A Contemporary View with Implications for Physiology and Medicine, (n.d.). doi: 10.1124/pr.56.3.1. [DOI] [PubMed] [Google Scholar]
- [19].Wakita Y, Inotropic, chronotropic, and arrhythmogenic effects of dopamine on the isolated working heart of rabbit, J Physiol Sci. 57 (2007) 147–157. doi:physiolsci/RP003607 [pii] 10.2170/physiolsci.RP003607. [DOI] [PubMed] [Google Scholar]
- [20].Rush RA, Geffen LB, Dopamine β-Hydroxylase in Health and Disease, CRC Crit. Rev. Clin. Lab. Sci. 12 (1980) 241–277. doi: 10.3109/10408368009108731. [DOI] [PubMed] [Google Scholar]
- [21].editors Purves D, Augustine GJ, Fitzpatrick D, et al. , The Biogenic Amines, in: Neuroscience, 2nd Editio, 2001. https://www.ncbi.nlm.nih.gov/books/NBK11035/. [Google Scholar]
- [22].Wehrwein EA, Orer HS, Barman SM, Overview of the Anatomy, Physiology, and Pharmacology of the Autonomic Nervous System, Compr Physiol. 6 (2016) 1239–1278. doi: 10.1002/cphy.c150037. [DOI] [PubMed] [Google Scholar]
- [23].Ciaranello RD, REGULATION OF PHENYLETHANOLAMINE N-METHYLTRANSFERASE, Biochem. Pharmacol 27 (1978) 1895–1897. [DOI] [PubMed] [Google Scholar]
- [24].Jänig W, Integrative Action of the Autonomic Nervous System: Neurobiology of Homeostasis, Cambridge University Press, 2006. [Google Scholar]
- [25].Burgen ASV, The background of the muscarinic system, Life Sci. 56 (1995) 801–806. doi: 10.1016/0024-3205(95)00013-V. [DOI] [PubMed] [Google Scholar]
- [26].Dale HH, Dudley HW, The Presence of Histamine and Acetyl-Choline in the Spleen of the Ox and the Horse, J. Physiol 68 (1929) 97–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Parsons SM, Prior C, Marshall IG, Acetylcholine transport, storage, and release., Int. Rev. Neurobiol 35 (1993) 279–390. http://www.ncbi.nlm.nih.gov/pubmed/8463062 (accessed January 14, 2019). [DOI] [PubMed] [Google Scholar]
- [28].Ahlquist RP, a Study of the Adrenotropic Receptors, Am. J. Physiol. Content 153 (1948) 586–600. doi: 10.1152/ajplegacy.1948.153.3.586. [DOI] [PubMed] [Google Scholar]
- [29].editors Siegel GJ, Agranoff BW, Albers RW, et al. , α- and β-Adrenergic Receptors, in: Basic Neurochem. Mol. Cell. Med. Asp, 6th ed., Lippincott-Raven, 1999. https://www.ncbi.nlm.nih.gov/books/NBK28138/. [Google Scholar]
- [30].Piascik MT, Perez DM, Alpha1-adrenergic receptors: new insights and directions., J. Pharmacol. Exp. Ther 298 (2001) 403–10. http://www.ncbi.nlm.nih.gov/pubmed/11454900 (accessed January 15, 2019). [PubMed] [Google Scholar]
- [31].Jones SB, Halenda SP, Bylund DB, Alpha 2-adrenergic receptor stimulation of phospholipase A2 and of adenylate cyclase in transfected Chinese hamster ovary cells is mediated by different mechanisms, Mol Pharmacol. 39 (1991) 239–245. [PubMed] [Google Scholar]
- [32].Langer S, Presynaptic Regulation of the Release of Catecholamines, Pharmacol. Rev 32 (1980) 337–362. [PubMed] [Google Scholar]
- [33].Khan Z, Ferguson C, Jones R, Alpha-2 and imidazoline receptor agonists Their pharmacology and therapeutic role, Anaesthesia. 54 (1999) 146–165. [DOI] [PubMed] [Google Scholar]
- [34].Southwick SM, Southwick SM, Bremner JD, Bremner JD, Rasmusson A, Rasmusson A, Morgan CAIII, Morgan CAIII, Arnsten A, Arnsten A, Charney DS, Charney DS, Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder., Biol. Psychiatry. 46 (1999) 1192–1204. papers2://publication/uuid/14E45257-F4BA-4D41-8650-12EB7E9155F9. [DOI] [PubMed] [Google Scholar]
- [35].Tank AW, Wong DL, Peripheral and Central Effects of Circulating Catecholamines, Compr Physiol. 5 (2015) 1–15. doi: 10.1002/cphy.c140007. [DOI] [PubMed] [Google Scholar]
- [36].Neve KA, Seamans JK, Trantham-Davidson H, Dopamine receptor signaling., J. Recept. Signal Transduct. Res. 24 (2004) 165–205. http://www.ncbi.nlm.nih.gov/pubmed/15521361 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [37].Lei S, Cross interaction of dopaminergic and adrenergic systems in neural modulation, Int. J. Physiol. Pathophysiol. Pharmacol 6 (2014) 137–142. [PMC free article] [PubMed] [Google Scholar]
- [38].Albuquerque Edson X., Pereira Edna F.R., Alkondon Manickavasagom, Rogers Scott W., Mammalian nicotinic acetylcholine receptors: from structure to function, Physiol. Rev, 89 (2009), pp. 73–120, 10.1152/physrev.00015.2008.Mammalian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].KARLIN A, COX RN, DIPAOLA M, HOLTZMAN E, KAO PN, LOBEL P, WANG L, YODH N, Functional Domains of the Nicotinic Acetylcholine Receptor, Ann. N. Y. Acad. Sci 463 (1986) 53–69. doi: 10.1111/j.1749-6632.1986.tb21503.x. [DOI] [PubMed] [Google Scholar]
- [40].Caulfield M, Birdsall N, International Union of Pharmacology. XVII. Classification of Muscarinic Acetylcholine Receptors, Pharmacol. Rev 48 (1998) 413–463. http://pharmrev.aspetjournals.org/content/50/2/279.long (accessed January 16, 2019). [PubMed] [Google Scholar]
- [41].Migeon JC, Thomas SL, Nathanson NM, Differential coupling of m2 and m4 muscarinic receptors to inhibition of adenylyl cyclase by Giα; and Goα subunits, J. Biol. Chem 270 (1995) 16070–16074. doi: 10.1074/jbc.270.27.16070. [DOI] [PubMed] [Google Scholar]
- [42].Reever CM, Ferrari-DiLeo G, Flynn DD, The M5 (m5) receptor subtype: fact or fiction?, Life Sci. 60 (1997) 1105–12. http://www.ncbi.nlm.nih.gov/pubmed/9121354 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [43].Mandela P, Ordway GA, The norepinephrine transporter and its regulation, J. Neurochem 97 (2006) 310–333. doi: 10.1111/j.1471-4159.2006.03717.x. [DOI] [PubMed] [Google Scholar]
- [44].Vaughan RA, Foster JD, Mechanisms of dopamine transporter regulation in normal and disease states., Trends Pharmacol. Sci. 34 (2013) 489–96. doi: 10.1016/j.tips.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Dale E, Bang-Andersen B, Sánchez C, Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs, Biochem. Pharmacol 95 (2015) 81–97. doi: 10.1016/J.BCP.2015.03.011. [DOI] [PubMed] [Google Scholar]
- [46].Weyler W, Hsu YPP, Breakafield XO, Biochemistry and genetics of monoamine oxidase, Pharmacol. Ther 47 (1990) 391–417. doi: 10.1016/0163-7258(90)90064-9. [DOI] [PubMed] [Google Scholar]
- [47].Eisenhofer G, The role of neuronal and extraneuronal plasma membrane transporters in the inactivation of peripheral catecholamines., Pharmacol. Ther 91 (2001) 35–62. http://www.ncbi.nlm.nih.gov/pubmed/11707293 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [48].Shulman KI, Herrmann N, Walker SE, Current Place of Monoamine Oxidase Inhibitors in the Treatment of Depression, CNS Drugs. 27 (2013) 789–797. doi: 10.1007/s40263-013-0097-3. [DOI] [PubMed] [Google Scholar]
- [49].ARMSTRONG MD, McMILLAN A, SHAW KN, 3-Methoxy-4-hydroxy-D-mandelic acid, a urinary metabolite of norepinephrine., Biochim. Biophys. Acta. 25 (1957) 422–3. http://www.ncbi.nlm.nih.gov/pubmed/13471587 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [50].Mattes CE, Lynch TJ, Singh A, Bradley RM, Kellaris PA, Brady RO, Dretchen KL, Therapeutic Use of Butyrylcholinesterase for Cocaine Intoxication, Toxicol. Appl. Pharmacol 145 (1997) 372–380. doi: 10.1006/taap.1997.8188. [DOI] [PubMed] [Google Scholar]
- [51].Buchholz VR, Schumacher TNM, Busch DH, T Cell Fate at the Single-Cell Level, Annu. Rev. Immunol 34 (2016) 65–92. doi: 10.1146/annurev-immunol-032414-112014. [DOI] [PubMed] [Google Scholar]
- [52].Jenkins MK, Chu HH, McLachlan JB, Moon JJ, On the composition of the preimmune repertoire of T cells specific for Peptide-major histocompatibility complex ligands., Annu. Rev. Immunol 28 (2010) 275–94. doi: 10.1146/annurev-immunol-030409-101253. [DOI] [PubMed] [Google Scholar]
- [53].Smith-Garvin JE, Koretzky GA, Jordan MS, T cell activation., Annu. Rev. Immunol 27 (2009) 591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Zhang N, Bevan MJ, CD8(+) T cells: foot soldiers of the immune system., Immunity. 35 (2011) 161–8. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhu J, Yamane H, Paul WE, Differentiation of effector CD4 T cell populations (*)., Annu. Rev. Immunol 28 (2010) 445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Bedoya SK, Lam B, Lau K, Larkin J, Th17 Cells in Immunity and Autoimmunity, Clin. Dev. Immunol 2013 (2013) 1–16. doi: 10.1155/2013/986789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J, Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders, Molecules. 22 (2017) 134. doi: 10.3390/molecules22010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Shao S, Yu X, Shen L, Autoimmune thyroid diseases and Th17/Treg lymphocytes, Life Sci. 192 (2018) 160–165. doi: 10.1016/j.lfs.2017.11.026. [DOI] [PubMed] [Google Scholar]
- [59].Spence A, Klementowicz JE, Bluestone JA, Tang Q, Targeting Treg signaling for the treatment of autoimmune diseases, Curr. Opin. Immunol 37 (2015) 11–20. doi: 10.1016/j.coi.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chen X, Oppenheim JJ, Th17 cells and Tregs: unlikely allies., J. Leukoc. Biol 95 (2014) 723–731. doi: 10.1189/jlb.1213633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Corthay A, How do regulatory T cells work?, Scand. J. Immunol 70 (2009) 326–36. doi: 10.1111/j.1365-3083.2009.02308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, Qian J, Dong C, Yi Q, Th9 Cells Represent a Unique Subset of CD4+ T Cells Endowed with the Ability to Eradicate Advanced Tumors., Cancer Cell. 33 (2018) 1048–1060.e7. doi: 10.1016/j.ccell.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jia L, Wu C, The Biology and Functions of Th22 Cells, in: Adv. Exp. Med. Biol, 2014: pp. 209–230. doi: 10.1007/978-94-017-9487-9_8. [DOI] [PubMed] [Google Scholar]
- [64].Varricchi G, Harker J, Borriello F, Marone G, Durham SR, Shamji MH, T follicular helper cells in normal immune responses and in allergic disorders, Allergy. 71 (2016) 1086–1094. doi: 10.1111/all.12878. [DOI] [PubMed] [Google Scholar]
- [65].Van Loon GR, Plasma dopamine: regulation and significance., Fed. Proc 42 (1983) 3012–8. http://www.ncbi.nlm.nih.gov/pubmed/6413258 (accessed January 16, 2019). [PubMed] [Google Scholar]
- [66].Autissier P, Soulas C, Burdo TH, Williams KC, Evaluation of a 12-color flow cytometry panel to study lymphocyte, monocyte, and dendritic cell subsets in humans, Cytom. Part A 77 (2010) 410–419. doi: 10.1002/cyto.a.20859. [DOI] [PubMed] [Google Scholar]
- [67].Felten DL, Felten SY, Carlson SL, Olschowka JA,” A N D, Livnat’ S, NORADRENERGIC AND PEPTIDERGIC INNERVATION OF LYMPHOID TISSUE, 1985. http://www.jimmunol.org/ (accessed November 29, 2018). [PubMed]
- [68].Kraal G, Cells in the marginal zone of the spleen., Int. Rev. Cytol 132 (1992) 31–74. http://www.ncbi.nlm.nih.gov/pubmed/1555921 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [69].Varatharaj A, Galea I, The blood-brain barrier in systemic inflammation, Brain. Behav. Immun 60 (2017) 1–12. doi: 10.1016/j.bbi.2016.03.010. [DOI] [PubMed] [Google Scholar]
- [70].Bourne HR, Melmon KL, Adenyl cyclase in human leukocytes: evidence for activation by separate beta adrenergic and prostaglandin receptors., J. Pharmacol. Exp. Ther 178 (1971) 1–7. http://www.ncbi.nlm.nih.gov/pubmed/4325858 (accessed January 16, 2019). [PubMed] [Google Scholar]
- [71].Williams LT, Snyderman R, Lefkowitz RJ, Identification of beta-adrenergic receptors in human lymphocytes by (−) (3H) alprenolol binding., J. Clin. Invest 57 (1976) 149–55. doi: 10.1172/JCI108254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Conolly ME, Greenacre JK, The beta-adrenoceptor of the human lymphocyte and human lung parenchyma., Br. J. Pharmacol 59 (1977) 17–23. http://www.ncbi.nlm.nih.gov/pubmed/13901 (accessed January 16, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pochet R, Delespesse G, Gausset PW, Collet H, Distribution of beta-adrenergic receptors on human lymphocyte subpopulations., Clin. Exp. Immunol 38 (1979) 578–84. http://www.ncbi.nlm.nih.gov/pubmed/43789 (accessed January 16, 2019). [PMC free article] [PubMed] [Google Scholar]
- [74].Loveland BE, Jarrott B, McKenzie IF, The detection of beta-adrenoceptors on murine lymphocytes., Int. J. Immunopharmacol 3 (1981) 45–55. http://www.ncbi.nlm.nih.gov/pubmed/6271692 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [75].Meurs H, van den Bogaard W, Kauffman HF, Bruynzeel PL, Characterization of (−)-[3H]dihydroalprenolol binding to intact and broken cell preparations of human peripheral blood lymphocytes., Eur. J. Pharmacol 85 (1982) 185–94. http://www.ncbi.nlm.nih.gov/pubmed/6295780 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [76].Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE, Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help., J. Immunol 158 (1997) 4200–10. http://www.ncbi.nlm.nih.gov/pubmed/9126981 (accessed January 16, 2019). [PubMed] [Google Scholar]
- [77].Ramer-Quinn DS, Baker RA, Sanders VM, Activated T helper 1 and T helper 2 cells differentially express the beta-2-adrenergic receptor: a mechanism for selective modulation of T helper 1 cell cytokine production., J. Immunol 159 (1997) 4857–67. http://www.ncbi.nlm.nih.gov/pubmed/9366411 (accessed January 16, 2019). [PubMed] [Google Scholar]
- [78].Sanders VM, The Beta2-Adrenergic Receptor on T and B Lymphocytes: Do We Understand It Yet?, Brain Behav Immun. 26 (2012) 195–200. doi: 10.1016/j.bbi.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].J.M. C-M, R.L. C, R.L. P, Inhibition of lymphocyte activation by catecholamines: Evidence for a non-classical mechanism of catecholamine action, Immunology. 85 (1995) 544–549. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emed3&NEWS=N&AN=1995250741. [PMC free article] [PubMed] [Google Scholar]
- [80].Casale TB, Kaliner M, Demonstration that circulating human blood cells have no detectable alpha 1-adrenergic receptors by radioligand binding analysis., J. Allergy Clin. Immunol. 74 (1984) 812–8. http://www.ncbi.nlm.nih.gov/pubmed/6094641 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [81].Tayebati SK, Bronzetti E, Morra Di Cella S, Mulatero P, Ricci A, Rossodivita I, Schena M, Schiavone D, Veglio F, Amenta F, In situ hybridization and immunocytochemistry of alpha1-adrenoceptors in human peripheral blood lymphocytes., J. Auton. Pharmacol 20 (n.d.) 305–12. http://www.ncbi.nlm.nih.gov/pubmed/11350496 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [82].Heilig M, Irwin M, Grewal I, Sercarz E, Sympathetic Regulation of T-Helper Cell Function, Brain. Behav. Immun 7 (1993) 154–163. doi: 10.1006/brbi.1993.1017. [DOI] [PubMed] [Google Scholar]
- [83].Rouppe van der Voort C, Kavelaars A, van de Pol M, Heijnen CJ, Noradrenaline induces phosphorylation of ERK-2 in human peripheral blood mononuclear cells after induction of alpha(1)-adrenergic receptors., J. Neuroimmunol 108 (2000) 82–91. http://www.ncbi.nlm.nih.gov/pubmed/10900341 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [84].Heijnen CJ, Rouppe van der Voort C, Wulffraat N, van der Net J, Kuis W, Kavelaars A, Functional alpha 1-adrenergic receptors on leukocytes of patients with polyarticular juvenile rheumatoid arthritis., J. Neuroimmunol 71 (1996) 223–6. http://www.ncbi.nlm.nih.gov/pubmed/8982123 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [85].Bao J-Y, Huang Y, Wang F, Peng Y-P, Qiu Y-H, Expression of α-AR Subtypes in T Lymphocytes and Role of the α-ARs in Mediating Modulation of T Cell Function, (2007) 344–353. [DOI] [PubMed] [Google Scholar]
- [86].Huang H-W, Fang X-X, Wang X-Q, Peng Y-P, Qiu Y-H, Regulation of Differentiation and Function of Helper T Cells by Lymphocyte-Derived Catecholamines via alpha1- and beta2-Adrenoceptors, Neuroimmunomodulation. 22 (2015) 138–151. doi: 10.1159/000360579. [DOI] [PubMed] [Google Scholar]
- [87].Kavelaars A, Regulated expression of α-1 adrenergic receptors in the immune system, Brain. Behav. Immun 16 (2002) 799–807. doi: 10.1016/S0889-1591(02)00033-8. [DOI] [PubMed] [Google Scholar]
- [88].Bourne HR, Melmon KL, Adenyl cyclase in human leukocytes: evidence for activation by separate beta adrenergic and prostaglandin receptors., J. Pharmacol. Exp. Ther 178 (1971) 1–7. http://www.ncbi.nlm.nih.gov/pubmed/4325858 (accessed January 17, 2019). [PubMed] [Google Scholar]
- [89].Makman MH, Properties of adenylate cyclase of lymphoid cells., Proc. Natl. Acad. Sci. U. S. A 68 (1971) 885–9. http://www.ncbi.nlm.nih.gov/pubmed/5280525 (accessed January 17, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Bach MA, Differences in Cyclic AMP Changes after Stimulation by Prostaglandins and Isoproterenol in Lymphocyte Subpopulations., J. Clin. Invest 55 (1975) 1074–81. doi: 10.1172/JCI108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wolfe BB, Harden TK, Molinoff PB, In vitro study of beta-adrenergic receptors., Annu. Rev. Pharmacol. Toxicol 17 (1977) 575–604. doi: 10.1146/annurev.pa.17.040177.003043. [DOI] [PubMed] [Google Scholar]
- [92].Takayanagi Y, Osawa S, Ikuma M, Takagaki K, Zhang J, Hamaya Y, Yamada T, Sugimoto M, Furuta T, Miyajima H, Sugimoto K, Norepinephrine suppresses IFN-γ and TNF-α production by murine intestinal intraepithelial lymphocytes via the β1 adrenoceptor., J. Neuroimmunol 245 (2012) 66–74. doi: 10.1016/j.jneuroim.2012.02.007. [DOI] [PubMed] [Google Scholar]
- [93].Cosentino M, Fietta AM, Ferrari M, Rasini E, Bombelli R, Carcano E, Saporiti F, Meloni F, Marino F, Lecchini S, Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop., Blood. 109 (2007) 632–42. doi: 10.1182/blood-2006-01-028423. [DOI] [PubMed] [Google Scholar]
- [94].Freier E, Weber CS, Nowottne U, Horn C, Bartels K, Meyer S, Hildebrandt Y, Luetkens T, Cao Y, Pabst C, Muzzulini J, Schnee B, Brunner-Weinzierl MC, Marangolo M, Bokemeyer C, Deter H-C, Atanackovic D, Decrease of CD4+FOXP3+ T regulatory cells in the peripheral blood of human subjects undergoing a mental stressor, Psychoneuroendocrinology. 35 (2010) 663–673. doi: 10.1016/j.psyneuen.2009.10.005. [DOI] [PubMed] [Google Scholar]
- [95].Borger P, Hoekstra Y, Esselink MT, Postma DS, Zaagsma J, Vellenga E, Kauffman HF, β -Adrenoceptor-mediated Inhibition of IFN-γ , IL-3, and GM-CSF mRNA Accumulation in Activated Human T Lymphocytes Is Solely Mediated by the β2 - Adrenoceptor Subtype, Am. J. Respir. Cell Mol. Biol. 19 (1998) 400–407. doi: 10.1165/ajrcmb.19.3.2765. [DOI] [PubMed] [Google Scholar]
- [96].Hollenga C, Brouwer F, Zaagsma J, Differences in functional cyclic AMP compartments mediating lepolysis by isoprenaline and BRL 37344 in four adipocyte types, Eur. J. Pharmacol 200 (1991) 325–330. doi: 10.1016/0014-2999(91)90590-M. [DOI] [PubMed] [Google Scholar]
- [97].Kohm AP, Sanders VM, Norepinephrine and 2-Adrenergic Receptor Stimulation Regulate CD4 T and B Lymphocyte Function in Vitro and in Vivo, 2001. http://pharmrev.aspetjournals.org (accessed December 1, 2018). [PubMed] [Google Scholar]
- [98].Wahle M, Stachetzki U, Krause A, Pierer M, Häntzschel H, Baerwald CGO, Regulation of beta2-adrenergic receptors on CD4 and CD8 positive lymphocytes by cytokines in vitro, Cytokine. 16 (2001) 205–209. doi: 10.1006/cyto.2001.0965. [DOI] [PubMed] [Google Scholar]
- [99].Baerwald CG, Laufenberg M, Specht T, von Wichert P, Burmester GR, Krause A, Impaired sympathetic influence on the immune response in patients with rheumatoid arthritis due to lymphocyte subset-specific modulation of beta 2-adrenergic receptors., Br. J. Rheumatol 36 (1997) 1262–9. http://www.ncbi.nlm.nih.gov/pubmed/9448586 (accessed April 17, 2019). [DOI] [PubMed] [Google Scholar]
- [100].McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM, Epigenetic regulation of beta2-adrenergic receptor expression in TH1 and TH2 cells, Brain. Behav. Immun 25 (2011) 408–415. doi: 10.1016/j.bbi.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kin NW, It takes nerve to tell T and B cells what to do, J. Leukoc. Biol. 79 (2006) 1093–1104. doi: 10.1189/jlb.1105625. [DOI] [PubMed] [Google Scholar]
- [102].Case AJ, Zimmerman MC, Sympathetic-mediated activation versus suppression of the immune system: Consequences for hypertension, J. Physiol 594 (2016) 527–536. doi: 10.1113/JP271516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Lorton D, Bellinger DL, Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells, Int. J. Mol. Sci. 16 (2015) 5635–5665. doi: 10.3390/ijms16035635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Levite M, Dopamine and T cells: Dopamine receptors and potent effects on T cells, dopamine production in T cells, and abnormalities in the dopaminergic system in T cells in autoimmune, neurological and psychiatric diseases, Acta Physiol. 216 (2016) 42–89. doi: 10.1111/apha.12476. [DOI] [PubMed] [Google Scholar]
- [105].Pacheco R, Prado CE, Barrientos MJ, Bernales S, Role of dopamine in the physiology of T-cells and dendritic cells, J. Neuroimmunol 216 (2009) 8–19. doi: 10.1016/j.jneuroim.2009.07.018. [DOI] [PubMed] [Google Scholar]
- [106].Saha B, Mondal AC, Majumder J, Basu S, Dasgupta PS, Physiological Concentrations of Dopamine Inhibit the Proliferation and Cytotoxicity of Human CD4+ and CD8+ T Cells in vitro: A Receptor-Mediated Mechanism, Neuroimmunomodulation. 9 (2001) 23–33. doi: 10.1159/000049004. [DOI] [PubMed] [Google Scholar]
- [107].Nakano K, Higashi T, Takagi R, Hashimoto K, Tanaka Y, Matsushita S, Dopamine released by dendritic cells polarizes Th2 differentiation, Int. Immunol 21 (2009) 645–654. doi: 10.1093/intimm/dxp033. [DOI] [PubMed] [Google Scholar]
- [108].Nakano K, Higashi T, Hashimoto K, Takagi R, Tanaka Y, Matsushita S, Antagonizing dopamine D1-like receptor inhibits Th17 cell differentiation: Preventive and therapeutic effects on experimental autoimmune encephalomyelitis, Biochem. Biophys. Res. Commun 373 (2008) 286–291. doi: 10.1016/j.bbrc.2008.06.012. [DOI] [PubMed] [Google Scholar]
- [109].Prado C, Contreras F, Gonzalez H, Diaz P, Elgueta D, Barrientos M, Herrada AA, Lladser A, Bernales S, Pacheco R, Stimulation of Dopamine Receptor D5 Expressed on Dendritic Cells Potentiates Th17-Mediated Immunity, J. Immunol 188 (2012) 3062–3070. doi: 10.4049/jimmunol.1103096. [DOI] [PubMed] [Google Scholar]
- [110].Cosentino M, Fietta AM, Ferrari M, Rasini E, Bombelli R, Saporiti F, Meloni F, Marino F, Lecchini S, Carcano E, autocrine / paracrine inhibitory functional loop Human CD4 ϩ CD25 ϩ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine / paracrine inhibitory functional loop, October 109 (2010) 632–642. doi: 10.1182/blood-2006-01-028423. [DOI] [PubMed] [Google Scholar]
- [111].Bergquist J, Tarkowski A, Ekman R, Ewing A, Discovery of endogenous catecholamines in lymphocytes and evidence for catecholamine regulation of lymphocyte function via an autocrine loop., Proc. Natl. Acad. Sci 91 (1994) 12912–12916. doi: 10.1073/pnas.91.26.12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Ghosh MC, Mondal AC, Basu S, Banerjee S, Majumder J, Bhattacharya D, Dasgupta PS, Dopamine inhibits cytokine release and expression of tyrosine kinases, Lck and Fyn in activated T cells., Int. Immunopharmacol 3 (2003) 1019–26. doi: 10.1016/S1567-5769(03)00100-0. [DOI] [PubMed] [Google Scholar]
- [113].Levite M, Chowers Y, Ganor Y, Besser M, Hershkovits R, Cahalon L, Dopamine interacts directly with its D3 and D2 receptors on normal human T cells, and activates beta1 integrin function., Eur. J. Immunol 31 (2001) 3504–12. doi:10.1002/1521-4141(200112)31:12<3504::AID-IMMU3504gt;3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- [114].Watanabe Y, Nakayama T, Nagakubo D, Hieshima K, Jin Z, Katou F, Hashimoto K, Yoshie O, Dopamine Selectively Induces Migration and Homing of Naive CD8+ T Cells via Dopamine Receptor D3, J. Immunol 176 (2006) 848–856. doi: 10.4049/jimmunol.176.2.848. [DOI] [PubMed] [Google Scholar]
- [115].Davis LS, Oppenheimer-Marks N, Bednarczyk JL, McIntyre BW, Lipsky PE, Fibronectin promotes proliferation of naive and memory T cells by signaling through both the VLA-4 and VLA-5 integrin molecules., J. Immunol 145 (1990) 785–93. doi: 10.4049/jimmunol.1103487. [DOI] [PubMed] [Google Scholar]
- [116].Besser MJ, Ganor Y, Levite M, Dopamine by itself activates either D2, D3 or D1/D5 dopaminergic receptors in normal human T-cells and triggers the selective secretion of either IL-10, TNFα or both, J. Neuroimmunol 169 (2005) 161–171. doi: 10.1016/j.jneuroim.2005.07.013. [DOI] [PubMed] [Google Scholar]
- [117].Sarkar C, Das S, Chakroborty D, Chowdhury UR, Basu B, Dasgupta PS, Basu S, Cutting Edge: Stimulation of Dopamine D4 Receptors Induce T Cell Quiescence by Up-Regulating Kruppel-Like Factor-2 Expression through Inhibition of ERK1/ERK2 Phosphorylation, J. Immunol 177 (2006) 7525–7529. doi: 10.4049/jimmunol.177.11.7525. [DOI] [PubMed] [Google Scholar]
- [118].Kawashima K, Fujii T, Moriwaki Y, Misawa H, Horiguchi K, Non-neuronal cholinergic system in regulation of immune function with a focus on α7 nAChRs, Int. Immunopharmacol 29 (2015) 127–134. doi: 10.1016/j.intimp.2015.04.015. [DOI] [PubMed] [Google Scholar]
- [119].Kawashima K, Expression of non-neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function, Front. Biosci 9 (2004) 2063. doi: 10.2741/1390. [DOI] [PubMed] [Google Scholar]
- [120].Kawashima K, Fujii T, The lymphocytic cholinergic system and its contribution to the regulation of immune activity, Life Sci. 74 (2003) 675–696. doi: 10.1016/j.lfs.2003.09.037. [DOI] [PubMed] [Google Scholar]
- [121].Fujii T, Takada-Takatori Y, Kawashima K, Basic and clinical aspects of nonneuronal acetylcholine: expression of an independent, non-neuronal cholinergic system in lymphocytes and its clinical significance in immunotherapy., J. Pharmacol. Sci 106 (2008) 186–92. http://www.ncbi.nlm.nih.gov/pubmed/18285654 (accessed January 18, 2019). [DOI] [PubMed] [Google Scholar]
- [122].Sato KZ, Fujii T, Watanabe Y, Yamada S, Ando T, Kazuko F, Kawashima K, Diversity of mRNA expression for muscarinic acetylcholine receptor subtypes and neuronal nicotinic acetylcholine receptor subunits in human mononuclear leukocytes and leukemic cell lines, Neurosci. Lett 266 (1999) 17–20. doi: 10.1007/s10021-011-9426-x. [DOI] [PubMed] [Google Scholar]
- [123].Fujii T, Kawashima K, Ca2+ oscillation and c-fos gene expression induced via muscarinic acetylcholine receptor in human T- and B-cell lines, Naunyn. Schmiedebergs. Arch. Pharmacol 362 (2000) 14–21. [DOI] [PubMed] [Google Scholar]
- [124].Mashimo M, Yurie Y, Kawashima K, Fujii T, CRAC channels are required for [Ca2 +]ioscillations and c-fos gene expression after muscarinic acetylcholine receptor activation in leukemic T cells, Life Sci. 161 (2016) 45–50. doi: 10.1016/j.lfs.2016.07.014. [DOI] [PubMed] [Google Scholar]
- [125].Nomura J, Hosoi T, Okuma Y, Nomura Y, The presence and functions of muscarinic receptors in human T cells: The involvement in IL-2 and IL-2 receptor system, Life Sci. 72 (2003) 2121–2126. doi: 10.1016/S0024-3205(03)00071-7. [DOI] [PubMed] [Google Scholar]
- [126].Darby M, Schnoeller C, Vira A, Culley F, Bobat S, Logan E, Kirstein F, Wess J, Cunningham AF, Brombacher F, Selkirk ME, Horsnell WGC, The M3 Muscarinic Receptor Is Required for Optimal Adaptive Immunity to Helminth and Bacterial Infection, PLoS Pathog. 11 (2015) 1–15. doi: 10.1371/journal.ppat.1004636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Fujii YX, Tashiro A, Arimoto K, Fujigaya H, Moriwaki Y, Misawa H, Fujii T, Matsui M, Kasahara T, Kawashima K, Diminished antigen-specific IgG1 and interleukin-6 production and acetylcholinesterase expression in combined M1 and M5 muscarinic acetylcholine receptor knockout mice, J. Neuroimmunol 188 (2007) 80–85. doi: 10.1016/j.jneuroim.2007.05.017. [DOI] [PubMed] [Google Scholar]
- [128].Zimring JC, Kapp LM, Yamada M, Wess J, Kapp JA, Regulation of CD8+ cytolytic T lymphocyte differentiation by a cholinergic pathway, J. Neuroimmunol 164 (2005) 66–75. doi: 10.1016/j.jneuroim.2005.03.018. [DOI] [PubMed] [Google Scholar]
- [129].Ricci A, Mariotta S, Amenta F, Tayebati SK, Terzano C, Changes in muscarinic cholinergic receptor expression in human peripheral blood lymphocytes in allergic rhinitis patients, Pulm. Pharmacol. Ther 21 (2008) 79–87. doi: 10.1016/J.PUPT.2006.12.006. [DOI] [PubMed] [Google Scholar]
- [130].Jacoby DB, Gleich GJ, Fryer AD, Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor., J. Clin. Invest 91 (1993) 1314–8. doi: 10.1172/JCI116331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].van Zwieten PA, Doods HN, Muscarinic receptors and drugs in cardiovascular medicine., Cardiovasc. Drugs Ther. 9 (1995) 159–67. http://www.ncbi.nlm.nih.gov/pubmed/7786837 (accessed January 19, 2019). [DOI] [PubMed] [Google Scholar]
- [132].Paulo JA, Brucker WJ, Hawrot E, Proteomic Analysis of an α7 Nicotinic Acetylcholine Receptor Interactome, J. Proteome Res. 8 (2009) 1849–1858. doi: 10.1021/pr800731z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].King JR, Nordman JC, Bridges SP, Lin M-K, Kabbani N, Identification and Characterization of a G Protein-binding Cluster in α7 Nicotinic Acetylcholine Receptors., J. Biol. Chem 290 (2015) 20060–70. doi: 10.1074/jbc.M115.647040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Hawrot E, Connecting the dots between G proteins, G protein coupled receptors, and neuronal nicotinic acetylcholine receptors, BioEssays. 35 (2013) 1022–1022. doi: 10.1002/bies.201300148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Grando SA, Kawashima K, Kirkpatrick CJ, Kummer W, Wessler I, Recent progress in revealing the biological and medical significance of the non-neuronal cholinergic system, Int. Immunopharmacol 29 (2015) 1–7. doi: 10.1016/j.intimp.2015.08.023. [DOI] [PubMed] [Google Scholar]
- [136].Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, Kawashima K, Expression and function of the cholinergic system in immune cells, Front. Immunol 8 (2017) 1–18. doi: 10.3389/fimmu.2017.01085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Qian J, Galitovskiy V, Chernyavsky AI, Marchenko S, Grando SA, Plasticity of the murine spleen T-cell cholinergic receptors and their role in in vitro differentiation of nave CD4 T cells toward the Th1, Th2 and Th17 lineages, Genes Immun. 12 (2011) 222–230. doi: 10.1038/gene.2010.72. [DOI] [PubMed] [Google Scholar]
- [138].Nizri E, Irony-Tur-Sinai M, Lory O, Orr-Urtreger A, Lavi E, Brenner T, Activation of the Cholinergic Anti-Inflammatory System by Nicotine Attenuates Neuroinflammation via Suppression of Th1 and Th17 Responses, J. Immunol 183 (2009) 6681–6688. doi: 10.4049/jimmunol.0902212. [DOI] [PubMed] [Google Scholar]
- [139].Wang D. -w., Zhou R. -b., Yao Y. -m., Zhu X. -m., Yin Y. -m., Zhao G. -j., Dong N, Sheng Z. -y., Stimulation of 7 Nicotinic Acetylcholine Receptor by Nicotine Increases Suppressive Capacity of Naturally Occurring CD4+CD25+ Regulatory T Cells in Mice In Vitro, J. Pharmacol. Exp. Ther 335 (2010) 553–561. doi: 10.1124/jpet.110.169961. [DOI] [PubMed] [Google Scholar]
- [140].Kioussis D, Pachnis V, Essay Immune and Nervous Systems: More Than Just a Superficial Similarity?, Immunity. 31 (n.d.) 705–710. doi: 10.1016/j.immuni.2009.09.009. [DOI] [PubMed] [Google Scholar]
- [141].Ottaviani E, Malagoli D, Franceschi C, Common evolutionary origin of the immune and neuroendocrine systems: from morphological and functional evidence to in silico approaches, Trends Immunol. 28 (2007) 497–502. doi: 10.1016/j.it.2007.08.007. [DOI] [PubMed] [Google Scholar]
- [142].Marino F, Cosentino M, Adrenergic modulation of immune cells: An update, Amino Acids. 45 (2013) 55–71. doi: 10.1007/s00726-011-1186-6. [DOI] [PubMed] [Google Scholar]
- [143].Josefsson E, Bergquist J, Ekman R, Tarkowski A, Catecholamines are synthesized by mouse lymphocytes and regulate function of these cells by induction of apoptosis., Immunology. 88 (1996) 140–6. http://www.ncbi.nlm.nih.gov/pubmed/8707341 (accessed January 19, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Giubilei F, Calderaro C, Antonini G, Sepe-Monti M, Tisei P, Brunetti E, Marchione F, Caronti B, Pontieri FE, Increased lymphocyte dopamine betahydroxylase immunoreactivity in Alzheimer’s disease: compensatory response to cholinergic deficit?, Dement. Geriatr. Cogn. Disord 18 (2004) 338–41. doi: 10.1159/000080128. [DOI] [PubMed] [Google Scholar]
- [145].Andreassi JL, Eggleston WB, Stewart JK, Phenylethanolamine N-methyltransferase mRNA in rat spleen and thymus., Neurosci. Lett 241 (1998) 75–8. http://www.ncbi.nlm.nih.gov/pubmed/9507924 (accessed January 19, 2019). [DOI] [PubMed] [Google Scholar]
- [146].Ziegler MG, Bao X, Kennedy BP, Joyner A, Enns R, Location, development, control, and function of extraadrenal phenylethanolamine N-methyltransferase., Ann. N. Y. Acad. Sci 971 (2002) 76–82. http://www.ncbi.nlm.nih.gov/pubmed/12438093 (accessed January 19, 2019). [DOI] [PubMed] [Google Scholar]
- [147].Cosentino M, Marino F, Bombelli R, Ferrari M, Rasini E, Lecchini S, Frigo G, Stimulation with phytohaemagglutinin induces the synthesis of catecholamines in human peripheral blood mononuclear cells: role of protein kinase C and contribution of intracellular calcium, J Neuroimmunol. 125 (2002) 125–133. doi: 10.1016/S0165-5728(02)00019-X. [DOI] [PubMed] [Google Scholar]
- [148].Zoukos Y, Thomaides T, Chaudhuri KR, Pavitt DV, Cuzner ML, Mathias CJ, Expression of beta-adrenoceptors on circulating mononuclear cells in hypertensives and normotensives before and after reduction of central sympathetic outflow by clonidine., Blood Press. 3 (1994) 172–7. http://www.ncbi.nlm.nih.gov/pubmed/8069405 (accessed January 19, 2019). [DOI] [PubMed] [Google Scholar]
- [149].Roupe van der Voort C, Heijnen CJ, Wulffraat N, Kuis W, Kavelaars A, Stress induces increases in IL-6 production by leucocytes of patients with the chronic inflammatory disease juvenile rheumatoid arthritis: a putative role for alpha(1)-adrenergic receptors., J. Neuroimmunol 110 (2000) 223–9. http://www.ncbi.nlm.nih.gov/pubmed/11024553 (accessed January 16, 2019). [DOI] [PubMed] [Google Scholar]
- [150].Spengler RN, Chensue SW, Giacherio DA, Blenk N, Kunkel SL, Endogenous norepinephrine regulates tumor necrosis factor-alpha production from macrophages in vitro., J. Immunol 152 (1994) 3024–31. http://www.ncbi.nlm.nih.gov/pubmed/8144901 (accessed January 19, 2019). [PubMed] [Google Scholar]
- [151].Jiang JL, Peng YP, Qiu YH, Wang JJ, Effect of endogenous catecholamines on apoptosis of Con A-activated lymphocytes of rats, J. Neuroimmunol 192 (2007) 79–88. doi: 10.1016/j.jneuroim.2007.09.012. [DOI] [PubMed] [Google Scholar]
- [152].Staedtke V, Bai R-Y, Kim K, Darvas M, Davila ML, Riggins GJ, Rothman PB, Papadopoulos N, Kinzler KW, Vogelstein B, Zhou S, Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome, Nature. 564 (2018) 273–277. doi: 10.1038/s41586-018-0774-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Nance DM, Sanders VM, Autonomic innervation and regulation of the immune system (1987-2007), (2007). doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Kawashima K, Fujii T, Extraneuronal cholinergic system in lymphocytes, Pharmacol. Ther 86 (2000) 29–48. doi: 10.1016/S0163-7258(99)00071-6. [DOI] [PubMed] [Google Scholar]
- [155].Franco R, Pacheco R, Lluis C, Ahern GP, O’Connell PJ, The emergence of neurotransmitters as immune modulators, Trends Immunol. 28 (2007) 400–407. doi: 10.1016/j.it.2007.07.005. [DOI] [PubMed] [Google Scholar]
- [156].Kawashima K, Kajiyama K, Fujimoto K, Oohata H, Suzuki T, Presence of acetylcholine in blood and its localization in circulating mononuclear leukocytes of humans, Biog. Amin 9 (1993) 251–258. https://keio.pure.elsevier.com/en/publications/presence-of-acetylcholine-in-bloodand-its-localization-in-circul (accessed January 19, 2019). [Google Scholar]
- [157].Fujii T, Tajima S, Yamada S, Watanabe Y, Sato KZ, Matsui M, Misawa H, Kasahara T, Kawashima K, Constitutive expression of mRNA for the same choline acetyltransferase as that in the nervous system, an acetylcholine-synthesizing enzyme, in human leukemic T-cell lines, Neurosci. Lett 259 (1999) 71–74. doi: 10.1016/S0304-3940(98)00921-5. [DOI] [PubMed] [Google Scholar]
- [158].Fujii T, Tsuchiya T, Yamada S, Fujimoto K, Suzuki T, Kasahara T, Kawashima K, Localization and synthesis of acetylcholine in human leukemic T cell lines, J. Neurosci. Res 44 (1996) 66–72. doi:10.1002/(SICI)1097-4547(19960401)44:1<66::AID-JNR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- [159].Rinner I, Kawashima K, Schauenstein K, Rat lymphocytes produce and secrete acetylcholine in dependence of differentiation and activation., J. Neuroimmunol 81 (1998) 31–7. doi: 10.1016/S0165-5728(97)00155-0. [DOI] [PubMed] [Google Scholar]
- [160].Fujii T, Yamada S, Watanabe Y, Misawa H, Tajima S, Fujimoto K, Kasahara T, Kawashima K, Induction of choline acetyltransferase mRNA in human mononuclear leukocytes stimulated by phytohemagglutinin, a T-cell activator., J. Neuroimmunol 82 (1998) 101–7. doi: 10.1016/S0165-5728(97)00195-1. [DOI] [PubMed] [Google Scholar]
- [161].Fujii T, Mashimo M, Moriwaki Y, Misawa H, Ono S, Horiguchi K, Kawashima K, Physiological functions of the cholinergic system in immune cells, J. Pharmacol. Sci 134 (2017) 1–21. doi: 10.1016/j.jphs.2017.05.002. [DOI] [PubMed] [Google Scholar]
- [162].Fujii T, Watanabe Y, Inoue T, Kawashima K, Upregulation of mRNA encoding the M5muscarinic acetylcholine receptor in human T- and B-lymphocytes during immunological responses, Neurochem. Res 28 (2003) 423–429. doi: 10.1023/A:1022840416292. [DOI] [PubMed] [Google Scholar]
- [163].Nishizuka Y, The role of protein kinase C in cell surface signal transduction and tumour promotion, Nature. 308 (1984) 693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- [164].Paoletti R, Bolego C, Poli A, Cignarella A, Metabolic syndrome, inflammation and atherosclerosis., Vasc. Health Risk Manag. 2 (2006) 145–52. http://www.ncbi.nlm.nih.gov/pubmed/17319458 (accessed January 20, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Monteiro R, Azevedo I, Chronic inflammation in obesity and the metabolic syndrome., Mediators Inflamm. 2010 (2010). doi: 10.1155/2010/289645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Zhu L, Zhao X, Zeng P, Zhu J, Yang S, Liu A, Song Y, Study on autonomic dysfunction and metabolic syndrome in Chinese patients., J. Diabetes Investig. 7 (2016) 901–907. doi: 10.1111/jdi.12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Lumeng CN, Saltiel AR, Inflammatory links between obesity and metabolic disease., J. Clin. Invest 121 (2011) 2111–7. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Tracey KJ, The inflammatory reflex, Nature. 420 (2002) 853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- [169].Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ, Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin, Nature. 405 (2000) 458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- [170].Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey KJ, Pharmacological stimulation of the cholinergic antiinflammatory pathway., J. Exp. Med 195 (2002) 781–8. http://www.ncbi.nlm.nih.gov/pubmed/11901203 (accessed January 20, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, Tracey KJ, Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion, J. Vasc. Surg 36 (2002) 1231–1236. doi: 10.1067/mva.2002.129643. [DOI] [PubMed] [Google Scholar]
- [172].Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ, Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation, Nature. 421 (2002) 384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- [173].Pavlov VA, Tracey KJ, The vagus nerve and the inflammatory reflex—linking immunity and metabolism, Nat Rev Endocrinol. 8 (2012) 743–754. doi: 10.1038/nrendo.2012.189.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Nance DM, Burns J, Innervation of the spleen in the rat: evidence for absence of afferent innervation., Brain. Behav. Immun 3 (1989) 281–90. http://www.ncbi.nlm.nih.gov/pubmed/2611414 (accessed January 20, 2019). [DOI] [PubMed] [Google Scholar]
- [175].Kumagai H, Oshima N, Matsuura T, Iigaya K, Imai M, Onimaru H, Sakata K, Osaka M, Onami T, Takimoto C, Kamayachi T, Itoh H, Saruta T, Importance of rostral ventrolateral medulla neurons in determining efferent sympathetic nerve activity and blood pressure, Hypertens. Res 35 (2012) 132–141. doi: 10.1038/hr.2011.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Berthoud HR, Powley TL, Characterization of vagal innervation to the rat celiac, suprarenal and mesenteric ganglia., J. Auton. Nerv. Syst 42 (1993) 153–69. http://www.ncbi.nlm.nih.gov/pubmed/8450174 (accessed January 20, 2019). [DOI] [PubMed] [Google Scholar]
- [177].Berthoud H-R, Neuhuber WL, Functional and chemical anatomy of the afferent vagal system, Auton. Neurosci 85 (2000) 1–17. doi: 10.1016/S1566-0702(00)00215-0. [DOI] [PubMed] [Google Scholar]
- [178].Smolen AJ, Morphology of Synapses in the Autonomic Nervous System, (1988) 187–204. [DOI] [PubMed] [Google Scholar]
- [179].Shimizu N, Hori T, Nakane H, An Interleukin-1β-Induced Noradrenaline Release in the Spleen Is Mediated by Brain Corticotropin-Releasing Factor: An in Vivo Microdialysis Study in Conscious Rats, Brain. Behav. Immun 8 (1994) 14–23. doi: 10.1006/BRBI.1994.1002. [DOI] [PubMed] [Google Scholar]
- [180].Goehler LE, Gaykema RPA, Hansen MK, Anderson K, Maier SF, Watkins LR, Vagal immune-to-brain communication: A visceral chemosensory pathway, Auton. Neurosci. Basic Clin. 85 (2000) 49–59. doi: 10.1016/S1566-0702(00)00219-8. [DOI] [PubMed] [Google Scholar]
- [181].Guerrero-Vargas NN, Salgado-Delgado R, del M Basualdo C, García J, Guzmán-Ruiz M, Carrero JC, Escobar C, Buijs RM, Reciprocal interaction between the suprachiasmatic nucleus and the immune system tunes down the inflammatory response to lipopolysaccharide, J. Neuroimmunol 273 (2014) 22–30. doi: 10.1016/j.jneuroim.2014.05.012. [DOI] [PubMed] [Google Scholar]
- [182].Martelli D, Farmer DGS, Yao ST, The splanchnic anti-inflammatory pathway: could it be the efferent arm of the inflammatory reflex?, Exp. Physiol 10 (2016) 1245–1252. doi: 10.1113/EP085559. [DOI] [PubMed] [Google Scholar]
- [183].Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ, Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit., Science. 334 (2011) 98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Chavan SS, Pavlov VA, Tracey KJ, Mechanisms and Therapeutic Relevance of Neuro-immune Communication., Immunity. 46 (2017) 927–942. doi: 10.1016/j.immuni.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Bratton BO, Martelli D, Mckinley MJ, Trevaks D, Anderson CR, Mcallen RM, Neural regulation of inflammation: No neural connection from the vagus to splenic sympathetic neurons, Exp. Physiol 97 (2012) 1180–1185. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- [186].Martelli D, Yao ST, Mckinley MJ, Mcallen RM, Reflex control of inflammation by sympathetic nerves, not the vagus, J. Physiol 592 (2014) 1677–1686. doi: 10.1113/jphysiol.2013.268573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Komegae EN, Farmer DGS, Brooks VL, McKinley MJ, McAllen RM, Martelli D, Vagal afferent activation suppresses systemic inflammation via the splanchnic anti-inflammatory pathway, Brain. Behav. Immun 73 (2018) 441–449. doi: 10.1016/j.bbi.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Shahid M, Francis J, Majid DSA, Tumor necrosis factor-α induces renal vasoconstriction as well as natriuresis in mice, Am. J. Physiol. Physiol 295 (2008) F1836–F1844. doi: 10.1152/ajprenal.90297.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM, TNF-α inhibition reduces renal injury in DOCA-salt hypertensive rats, Am. J. Physiol. Integr. Comp. Physiol 294 (2008) R76–R83. doi: 10.1152/ajpregu.00466.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Elmarakby AA, Quigley JE, Pollock DM, Imig JD, Tumor Necrosis Factor α Blockade Increases Renal Cyp2c23 Expression and Slows the Progression of Renal Damage in Salt-Sensitive Hypertension, Hypertension. 47 (2006) 557–562. doi: 10.1161/01.HYP.0000198545.01860.90. [DOI] [PubMed] [Google Scholar]
- [191].Tran LT, MacLeod KM, McNeill JH, Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats, Mol. Cell. Biochem 330 (2009) 219–228. doi: 10.1007/s11010-009-0136-z. [DOI] [PubMed] [Google Scholar]
- [192].Mathis KW, An impaired neuroimmune pathway promotes the development of hypertension in systemic lupus erythematosus, Am. J. Physiol. Integr. Comp. Physiol 309 (2015) R1074–R1077. doi: 10.1152/ajpregu.00143.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Pham GS, Wang LA, Mathis KW, Pharmacological Potentiation of the Efferent Vagus Nerve Attenuates Blood Pressure and Renal Injury in a Murine Model of Systemic Lupus Erythematosus, Am. J. Physiol. Integr. Comp. Physiol (2018) ajpregu.00362.2017. doi: 10.1152/ajpregu.00362.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Julius S, Nesbitt S, Sympathetic overactivity in hypertension. A moving target., Am. J. Hypertens 9 (1996) 113S–120S. http://www.ncbi.nlm.nih.gov/pubmed/8931844 (accessed January 21, 2019). [DOI] [PubMed] [Google Scholar]
- [195].Grassi G, Mark A, Esler M, The Sympathetic Nervous System Alterations in Human Hypertension, Circ. Res 116 (2015) 976–990. doi: 10.1161/CIRCRESAHA.116.303604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Nishi EE, Bergamaschi CT, Campos RR, The crosstalk between the kidney and the central nervous system: the role of renal nerves in blood pressure regulation, Exp. Physiol 100 (2015) 479–484. doi: 10.1113/expphysiol.2014.079889. [DOI] [PubMed] [Google Scholar]
- [197].Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ, Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system, Am. J. Physiol. Circ. Physiol 289 (2005) H1683–H1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- [198].White FN, Grollman A, Autoimmune Factors Associated with Infarction of the Kidney, Nephron. 1 (1964) 93–102. doi: 10.1159/000179322. [DOI] [PubMed] [Google Scholar]
- [199].Okuda T, Grollman A, Passive transfer of autoimmune induced hypertension in the rat by lymph node cells., Tex. Rep. Biol. Med 25 (1967) 257–64. http://www.ncbi.nlm.nih.gov/pubmed/6040652 (accessed January 21, 2019). [PubMed] [Google Scholar]
- [200].Olsen F, Inflammatory cellular reaction in hypertensive vascular disease., Acta Pathol. Microbiol. Scand. Suppl. 220 (1971) Suppl 220:1–76. http://www.ncbi.nlm.nih.gov/pubmed/5002833 (accessed January 21, 2019). [PubMed] [Google Scholar]
- [201].Svendsen UG, The importance of thymus for hypertension and hypertensive vascular disease in rats and mice., Acta Pathol. Microbiol. Scand. Suppl. (1978) 1–15. http://www.ncbi.nlm.nih.gov/pubmed/352093 (accessed January 21, 2019). [PubMed] [Google Scholar]
- [202].Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B, Mycophenolate Mofetil Treatment Improves Hypertension in Patients with Psoriasis and Rheumatoid Arthritis, J. Am. Soc. Nephrol 17 (2006) S218–S225. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- [203].Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG, Role of the T cell in the genesis of angiotensin II–induced hypertension and vascular dysfunction, J. Exp. Med 204 (2007) 2449–2460. doi: 10.1084/JEM.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Crowley SD, Song Y-S, Lin EE, Griffiths R, Kim H-S, Ruiz P, Lymphocyte responses exacerbate angiotensin II-dependent hypertension, Am. J. Physiol. Integr. Comp. Physiol 298 (2010) R1089–R1097. doi: 10.1152/ajpregu.00373.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H, Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage, Am. J. Physiol. Integr. Comp. Physiol 304 (2013) R407–R414. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Theuer J, Dechend R, Muller DN, Park J-K, Fiebeler A, Barta P, Ganten D, Haller H, Dietz R, Luft FC, Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats., BMC Cardiovasc. Disord. 2 (2002) 3. doi: 10.1186/1471-2261-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK, Brain Microglial Cytokines in Neurogenic Hypertension, Hypertension. 56 (2010) 297–303. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS, Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation, J. Clin. Invest 125 (2015) 1189–1202. doi: 10.1172/JCI76327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL, Hypertension Caused by Angiotensin II Infusion Involves Increased Superoxide Production in the Central Nervous System, Circ. Res 95 (2004) 210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- [210].Chapleau MW, Abboud FM, Neuro-cardiovascular regulation: from molecules to man. Introduction., Ann. N. Y. Acad. Sci 940 (2001) xiii–xxii. http://www.ncbi.nlm.nih.gov/pubmed/11458711 (accessed January 24, 2019). [PubMed] [Google Scholar]
- [211].Paton JFR, Waki H, Abdala APL, Dickinson J, Kasparov S, Vascular-brain signaling in hypertension: role of angiotensin II and nitric oxide., Curr. Hypertens. Rep 9 (2007) 242–7. http://www.ncbi.nlm.nih.gov/pubmed/17519132 (accessed January 24, 2019). [DOI] [PubMed] [Google Scholar]
- [212].Menani JV, Vieira AA, Colombari DSA, De Paula PM, Colombari E, De Luca LA, Preoptic–Periventricular Integrative Mechanisms Involved in Behavior, Fluid–Electrolyte Balance, and Pressor Responses, 2014. http://www.ncbi.nlm.nih.gov/pubmed/24829988 (accessed January 24, 2019). [PubMed]
- [213].Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG, Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension., Circ. Res 107 (2010) 263–70. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J, Osborn JW, Itani HA, Harrison DG, Renal denervation prevents immune cell activation and renal inflammation in Angiotensin II-induced hypertension, Circ. Res 117 (2015) 547–557. doi: 10.1161/CIRCRESAHA.115.306010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Foss JD, Wainford RD, Engeland WC, Fink GD, Osborn JW, A novel method of selective ablation of afferent renal nerves by periaxonal application of capsaicin, Am. J. Physiol. Integr. Comp. Physiol 308 (2015) R112–R122. doi: 10.1152/ajpregu.00427.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Banek CT, Knuepfer MM, Foss JD, Fiege JK, Asirvatham-Jeyaraj N, Van Helden D, Shimizu Y, Osborn JW, Resting Afferent Renal Nerve Discharge and Renal Inflammation, Hypertension. 68 (2016) 1415–1423. doi: 10.1161/HYPERTENSIONAHA.116.07850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Banek CT, Gauthier MM, Baumann DC, Van Helden D, Asirvatham-Jeyaraj N, Panoskaltsis-Mortari A, Fink GD, Osborn JW, Targeted afferent renal denervation reduces arterial pressure but not renal inflammation in established DOCA-salt hypertension in the rat, Am. J. Physiol. Integr. Comp. Physiol 314 (2018) R883–R891. doi: 10.1152/ajpregu.00416.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Asirvatham-Jeyaraj N, Fiege JK, Han R, Foss J, Banek CT, Burbach BJ, Razzoli M, Bartolomucci A, Shimizu Y, Panoskaltsis-Mortari A, Osborn JW, Renal Denervation Normalizes Arterial Pressure with No Effect on Glucose Metabolism or Renal Inflammation in Obese Hypertensive Mice, Hypertension. 68 (2016) 929–936. doi: 10.1161/HYPERTENSIONAHA.116.07993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Osborn JW, Banek CT, Catheter-Based Renal Nerve Ablation as a Novel Hypertension Therapy, Hypertension. 71 (2018) 383–388. doi: 10.1161/HYPERTENSIONAHA.117.08928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Lori A, Perrotta M, Lembo G, Carnevale D, The Spleen: A Hub Connecting Nervous and Immune Systems in Cardiovascular and Metabolic Diseases, (n.d.). doi: 10.3390/ijms18061216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Carnevale D, Pallante F, Fardella V, Fardella S, Iacobucci R, Federici M, Cifelli G, De Lucia M, Lembo G, The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension., Immunity. 41 (2014) 737–52. doi: 10.1016/j.immuni.2014.11.002. [DOI] [PubMed] [Google Scholar]
- [222].Foss JD, Fink GD, Osborn JW, Reversal of genetic salt-sensitive hypertension by targeted sympathetic ablation, Hypertension. 61 (2013) 806–811. doi: 10.1161/HYPERTENSIONAHA.111.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Méndez-Ferrer S, Battista M, Frenette PS, Cooperation of β2- and β3-adrenergic receptors in hematopoietic progenitor cell mobilization, Ann. N. Y. Acad. Sci 1192 (2010) 139–144. doi: 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Scheiermann C, Kunisaki Y, Lucas D, Chow A, Jang J-E, Zhang D, Hashimoto D, Merad M, Frenette PS, Adrenergic Nerves Govern Circadian Leukocyte Recruitment to Tissues, Immunity. 37 (2012) 290–301. doi: 10.1016/j.immuni.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Grisanti LA, Evanson J, Marchus E, Jorissen H, Woster AP, DeKrey W, Sauter ER, Combs CK, Porter JE, Pro-inflammatory responses in human monocytes are β1-adrenergic receptor subtype dependent, Mol. Immunol 47 (2010) 1244–1254. doi: 10.1016/j.molimm.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Emeny RT, Gao D, Lawrence DA, Beta1-adrenergic receptors on immune cells impair innate defenses against Listeria., J. Immunol 178 (2007) 4876–84. http://www.ncbi.nlm.nih.gov/pubmed/17404268 (accessed January 20, 2019). [DOI] [PubMed] [Google Scholar]
- [227].Ahmari N, Schmidt JT, Krane GA, Malphurs W, Cunningham BE, Owen JL, Martyniuk CJ, Zubcevic J, Loss of bone marrow adrenergic beta 1 and 2 receptors modifies transcriptional networks, reduces circulating inflammatory factors, and regulates blood pressure, Physiol. Genomics 48 (2016) 526–536. doi: 10.1152/physiolgenomics.00039.2016. [DOI] [PubMed] [Google Scholar]
- [228].Flierl M, Rittirsch D, Huber-Lang M, Sarma JV, Ward PA, Catecholamines—Crafty Weapons in the Inflammatory Arsenal of Immune/Inflammatory Cells or Opening Pandora’s Box?, Mol. Med 14 (2008) 1. doi: 10.2119/2007-00105.Flierl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Gage JR, Fonarow G, Hamilton M, Widawski M, Martinez-Maza O, Vredevoe DL, Beta Blocker and Angiotensin-Converting Enzyme Inhibitor Therapy Is Associated with Decreased Th1/Th2 Cytokine Ratios and Inflammatory Cytokine Production in Patients with Chronic Heart Failure, Neuroimmunomodulation. 11 (2004) 173–180. [DOI] [PubMed] [Google Scholar]