

Abstract
Purpose
Targeted next-generation sequencing (NGS) of DNA has become more widely used in the management of patients with lung adenocarcinoma, however no clear mitogenic driver alteration is found in some cases. We evaluated the incremental benefit of targeted RNAseq in the identification of gene fusions and MET exon 14 (METex14) alterations in DNAseq driver-negative lung cancers.
Experimental Design
Lung cancers driver-negative by MSK-IMPACT underwent further analysis using a custom RNAseq panel (MSK-Fusion). Tumor mutation burden (TMB) was assessed as a potential prioritization criterion for targeted RNAseq.
Results
As part of prospective clinical genomic testing, we profiled 2522 lung adenocarcinomas using MSK-IMPACT, which identified 195 (7.7%) fusions and 119 (4.7%) METex14 alterations. Among 275 driver-negative cases with available tissue, 254 (92%) had sufficient material for RNAseq. A previously undetected alteration was identified in 14% (36/254) of cases, 33 of which were actionable (27 in-frame fusions, 6 METex14). Of these 33 patients, 10 then received matched targeted therapy which achieved clinical benefit in 8 (80%). In the 32% (8½54) of DNAseq driver-negative cases with low TMB (0–5 mutations/Megabase), 25 (31%) were positive for previously undetected gene fusions on RNAseq whereas, in 151 cases with TMB >5 mut/Mb, only 7% were positive for fusions (P<0.0001).
Conclusion
Targeted RNAseq assays should be used in all cases that appear driver-negative by DNAseq assays to ensure comprehensive detection of actionable gene rearrangements. Furthermore, we observed a significant enrichment for fusions in DNAseq driver-negative samples with low TMB, supporting the prioritization of such cases for additional RNAseq.
Keywords: Lung adenocarcinoma, kinase fusion, DNA sequencing, RNA Sequencing, TMB
INTRODUCTION
The identification of ALK and ROS1 kinase fusions in non-small-cell lung cancer (NSCLC) has led to the approval of a number of successful targeted therapies, which has revolutionized the treatment of patients whose tumors harbor those fusions [1–9]. Inhibitors targeting lower frequency fusions (NTRK½/3, RET and NRG1) and mutations causing MET exon 14 skipping have also shown dramatic and durable responses in patients enrolled in clinical trials [10–12], FDA approval was recently granted to a TRK inhibitor (Larotrectinib) in patients with tumors harboring an NTRK fusion. Gene fusions are also becoming increasingly important mechanisms of acquired resistance to tyrosine kinase inhibitors in lung adenocarcinomas [13–16]. The widespread clinical implementation of next generation sequencing (NGS), along with technical advances, has resulted in enhanced detection of oncogenic gene fusions and intense interest in their clinical targeting [17–22].
Targeted DNA-based next generation sequencing techniques specifically designed to detect rearrangements in kinases can effectively detect oncogenic kinase fusions with high confidence [23–25]. For instance, the FDA-cleared MSK-IMPACT™ large panel, hybrid capture-based NGS assay [21, 26], is designed to detect many common kinase fusions, including those involving ALK, RET, and ROS1, and MET exon 14 skipping mutations, via tiling of the appropriate introns for hybrid capture. However, there are technical limitations to the ability of such DNA based assays to detect gene fusions [27]. First, such assays can only identify fusions in genes where the genomic rearrangements occur in typically short introns effectively covered in the panel (Figure 1). Some clinically important fusions arise from rearrangements in very long introns, the tiling of which would significantly compromise coverage of the remainder of the genes on the panel. Moreover, some introns harbor repetitive sequence elements also present elsewhere in the genome that therefore cannot be assessed by short-read NGS due to the difficulty in uniquely mapping such reads, resulting in gaps in the coverage of certain introns and hence blind spots in the detection of potential rearrangement breakpoints. Second, DNAseq assays provide no direct evidence that the rearrangement produces a fusion expressed at the mRNA level [28], a particular problem for rearrangements that appear non-canonical at the genomic DNA level. To address this need, our laboratory has validated an RNA-based custom solid tumor Fusion-Panel (MSK-Fusion) [29, 30] that utilizes Archer™ Anchored Multiplex PCR (AMP™) technology [31] to detect gene fusions in carcinomas and sarcomas.
Figure 1. Comprehensive DNASeq and RNASeq for effective detection of gene fusions.
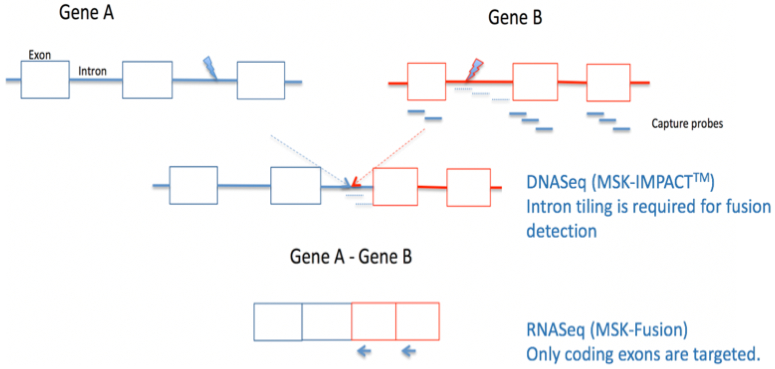
Gene fusion detection in the DNA using next generation sequencing (e.g. MSK-IMPACT™) requires the tiling of intronic regions known to likely harbor the genomic breakpoint. This approach is challenging when 1) the introns are too long to tile 2) the introns contain repetitive elements which don’t sequence very well or 3) genomic breakpoints take place in alternative introns not predicted by the panel design. RNA sequencing offers a more direct approach to fusion detection as the introns are removed by splicing.
Tumor mutation burden (TMB) is an emerging potential biomarker for immunotherapy [32–34]. Nivolumab and Ipilimumab have recently been found to be more effective in extending progression-free survival in patient subsets with higher TMB [35–37]. Recent studies have observed that most tumors with oncogenic kinase driver alterations have low TMB [38, 39]. Our large cohort of prospectively sequenced clinical samples provides the opportunity to more broadly examine the relationship between TMB status and gene fusions in lung cancer, where targetable kinase fusions are frequently detected. Moreover, we reasoned that low TMB could be an indicator of the greater likelihood of occult gene fusions in driver negative tumors that could benefit from RNA sequencing using the MSK-Fusion panel.
In the present study, we conducted a retrospective sequencing analysis using the MSK-Fusion panel on lung adenocarcinomas that were previously profiled by MSK-IMPACT and were found to lack an oncogenic driver [40]. We aimed to elucidate the importance of following DNAseq by RNAseq for the comprehensive detection of gene fusions, determine the clinical feasibility of having adequate tissue for both DNA and RNA testing, and explore the possible correlation of TMB with the likelihood of kinase fusion detection via additional RNAseq testing.
PATIENTS AND METHODS
We identified patients with NSCLC who underwent targeted DNAseq using the MSK-IMPACT assay from January 2014 through January 2018. Lung adenocarcinomas cases with DNAseq lacking an oncogenic activating mutation, defined as hotspot mutations in BRAF, EGFR, NRAS, KRAS, ERBB2, MAP2K1, MET; amplification of EGFR, ERBB2, FGFR1, MET; rearrangements involving ALK/RET/ROS, NTRK½/3, NRG1, BRAF were subject to further analysis using the MSK-Fusion panel (RNASeq). This study was performed after Memorial Sloan Kettering Cancer Center (MSK) Institutional Review Board Approval. All patients provided informed written consent for these somatic genomic analyses. The studies were conducted in accordance with Declaration of Helsinki and the U.S. Common Rule. MSK-IMPACT and the MSK-Fusion assay have been approved by the New York State Department of Health as clinical assays. MSK-IMPACT also received FDA clearance as a class 2 in vitro diagnostic test (tumor profiling assay) in November 2017.
Patients identified to have an actionable gene fusion by RNAseq in their tumor were reviewed for treatment outcomes including rate of matching to targeted therapy and overall response rate (ORR). ORR to matched targeted therapy was assessed with RECIST version 1.1 by a dedicated study radiologist.
RNA Extraction and Quality Control
A minimum of 10 unstained slides and 1 H&E stained slide from Formalin Fixed Paraffin-embedded tissue (FFPE) were obtained for each sample and reviewed by a pathologist. Macro-dissection was performed whenever indicated. 10ul of mineral oil was applied to each slide before scraping the tissue and placing it in a 1.5ml Eppendorf tube. An additional 800ul of mineral oil was added to each tube for tissue deparaffinization. RNA extraction was then performed using the standard RNeasy FFPE Kit and protocol (Qiagen, Catalog #73504). To address challenges around limited or unavailable tissue for RNA testing, our laboratory has tested RNA extraction on lysed cell material (lysate) left from DNA extraction and stored at room temperature for one year before they are moved to 4°C. Lysate material is obtained from FFPE tissue scraped and deparaffinized as indicated above. 150ul of Proteinase k and 250ul of lysis buffer is added to the 1.5ml Eppendorf tube and incubated overnight. About 250ul of lysate is obtained, of which 40ul is saved at room temperature for RNA extraction and used whenever the corresponding tissue is exhausted.
Total extracted RNA was quantified using the Qubit Broad Range RNA Assay Kit (Life Tech., Catalog #Q10211) and run on the TapeStation using RNA ScreenTape (Agilent, Catalog #5067–5576). Each RNA sample was tested using the Archer® PreSeq™ RNA QC Assay, a qPCR-based method for assessing RNA quality, prior to library preparation and sequencing. A Ct value [41] >28 indicated a low-quality RNA sample and would be deemed insufficient for RNAseq. Samples with at least 50ng (200ng preferred) of RNA were used for testing.
Sequencing assays and analysis
RNASeq.
cDNA libraries were made using the Archer™ FusionPlex™ standard protocol and supplied reagents including Archer® Universal RNA Reagent Kit for Illumina® (Catalog #AK-0040–8), Archer MBC adapters (Catalog #SA0040–45) and our custom designed Gene Specific Primer (GSP) Pool kit. Fusion unidirectional GSPs have been designed to target specific exons in 62 genes known to be involved in chromosomal rearrangements based on current literature. GSPs, in combination with adapters-specific primers, enrich for known and novel fusion transcripts. The assay includes 346 GSPs ranging from 18 to 39 base pairs in length designed by Archer™ to hybridize in either the 5’or 3’ direction to the relevant exons of each gene. The 62 target genes and their corresponding NCBI RefSeq ID used for gene annotation are listed in Supplementary Table 1.
A detailed description of the Anchored Multiplex Technology is available elsewhere [31]. Briefly, cDNA undergoes end repair, dA-tailing and ligation with half-functional Illumina molecular barcode adapters (MBC). These sequencing adapters contain molecular barcodes that allow for read de-duplication and quantitative analysis. Clean-up after all enzymatic steps are performed using AMPURE XP magnetic beads (Fisher Scientific, Catalog #NC0110018). Cleaned ligated fragments are subject to two consecutive rounds of PCR amplifications using two sets of gene specific primers (GSP1 pool used in PCR1 and a nested GSP2 pool designed 3’ downstream of GSP1, used in PCR2) and universal primers complementary to the Illumina adapters. This allows for the enrichment of fusion transcripts with the knowledge of only one of the gene partners. At the end of the two PCR steps the final targeted amplicons are ready for 2×150bp sequencing on an Illumina MiSeq sequencer. At the end of MiSeq sequencing, fastq files are automatically generated using the MiSeq reporter software (Version 2.6.2.3) and analyzed using the Archer™ analysis software (Version 5.0.4).
DNASeq and Tumor Mutation Burden.
A detailed description of MSK-IMPACT workflow and data analysis is described elsewhere [21, 26]. Tumor mutational burden (TMB) was calculated as the total number of mutations reported for a patient divided by the coding region target territory of MSK-IMPACT and is characterized as the number of somatic base substitution and indel alterations per Mb.
RESULTS
Clinical characteristics and patient demographics
2522 tumors from unique lung adenocarcinoma patients were profiled with DNAseq between January 2014 and January 2018. 589 cases lacked a driver alteration and were considered for further RNA Sequencing analysis. Additional specimens for RNA extraction were available for 275 (46%) of the 589 samples, 21 of which were found to be insufficient for testing due to low quality of RNA resulting in 254 cases amenable for RNA sequencing (Figure 2). The clinical characteristics of the patients tested are described in Supplementary Table 2. The remaining 314 of the 589 cases did not have available tissue for RNA extraction because all submitted material was used for DNA extraction and no additional recuts could be requested as the original block was either exhausted or not available.
Figure 2. Description of the lung adenocarcinomas cohort.
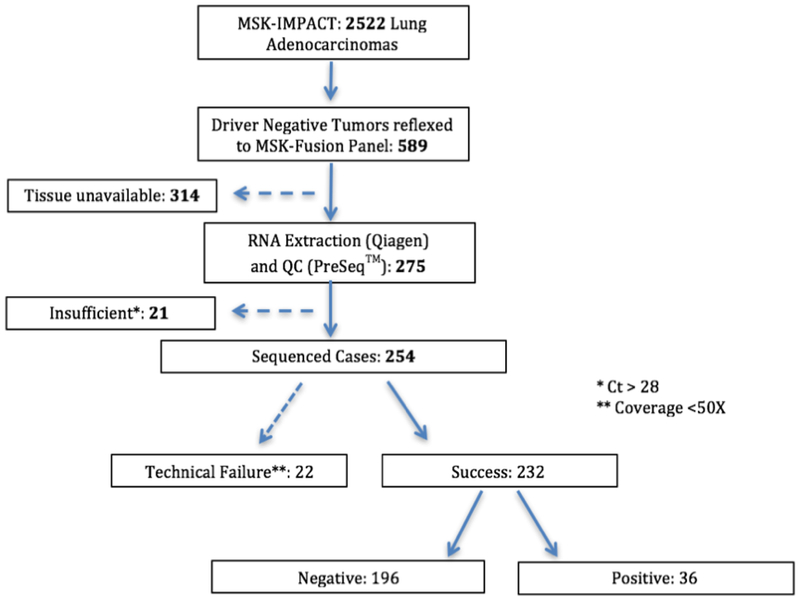
A total of 2,522 unique lung adenocarcinomas were submitted for MSK-IMPACT sequencing between January 2014 and December 2017. 589/2,522 cases were negative for driver alterations and submitted for MSK-Fusion testing. Adequate tissue for RNA extraction was only available in 275 samples. 21 cases were deemed insufficient due to low RNA quality based on the PreSeq™ assay. Out of the 254 sequenced samples, 22 failed based on their unique sequencing coverage (<50X). In total, 232 MSK-IMPACT driver negative cases had successfully undergone RNASeq using the MSK-Fusion panel for an assay success rate of 91%.
RNASeq in MSK-IMPACT Driver-Negative Lung Adenocarcinomas
Among the 2522 unique lung adenocarcinomas profiled by MSK-IMPACT, 1933 (77%) were positive for oncogenic drivers as previously defined [40]. KRAS (785) and EGFR (643) were the 2 genes with the most commonly detected oncogenic mutations in 31% and 25% of the patients, respectively, in a mutually exclusive fashion. Other known mitogenic driver alterations were also identified and included mutations in BRAF (56), ERBB2 (61), MET exon14 (55), NRAS (19), MAP2K1 (17) or gene fusions involving ALK (84), ROS1 (47), RET (42), BRAF (6), FGFR3 (5), NTRK1 (4), NRG1 (3), FGFR1 (1), FGFR2 (1), or high level, genomically focal amplification of MET (18) and ERBB2 (13), most of which represent actionable or potentially actionable alterations classified as OncoKB Levels 1 to 3 events in lung adenocarcinoma [42] (Figure 3A).
Figure 3. Driver alteration profile by comprehensive DNASeq and RNA Seq.

(A) 1,933/2,522 cases were positive for oncogenic drivers using MSK-IMPACT. RNA from the available driver negative cases (n=232) was tested using the MSK-Fusion panel. Gene fusions (n=29), METex14 mutations (n=6) and EGFRviii (n=1) were detected. (B) Schematic representation of the 15/29 gene fusions that were not expected to be detected by custom DNA-based sequencing due to panel design (C) Schematic representation of the 14/29 expected to be detected by DNASeq but only a fusion transcript was detected in the RNA.
254 cases where a driver alteration was not detected by MSK-IMPACT (DNAseq) were subject to further analysis using the RNA-based MSK-Solid Fusion panel (RNASeq). 22 cases failed sequencing due to low coverage defined as the average number of unique RNA reads per targeted region (<50X). Among the 232 (91%) successfully sequenced samples, 196 samples remained driver negative by both DNA and RNA sequencing and 36 were positive for mitogenic driver alterations (Figure 2, 3A). Among the 36 driver positive cases, 33 showed actionable in-frame fusions involving METex14 skipping (n=6) or one of the following kinase genes: 28% ROS1 (n=10), 13.8% NRG1 (n=5), 11% ALK (n=4), 8% RET (n=3), 5% NTRK3 (n=2), 2.7% BRAF (n=1), 2.7% FGFR2 (n=1), 2.7% NTRK2 (n=1).
The gene fusions identified represent a diverse landscape of fusion partners (Figure 3 B/C), some of which are novel. For example, Chromobox 5 (CBX5) and Striatin (STRN) are novel fusion partners for FGFR2 and NTRK2, respectively. Additionally, some of the identified gene fusions have not been previously observed in lung adenocarcinomas: RNA Binding Protein, MRNA Processing Factor (RBPMS) and Sequestosome 1 (SQSTM1) were the gene fusion partners involved in NTRK3 fusions. Both of these fusions were previously detected in papillary thyroid carcinoma [43–45]. More details about these fusions including the exons involved and Refseq IDs are included in Supplementary Table 3. A novel in-frame fusion involving the first 2 exons of Histone Deacetylase 5 (HDAC5) and exons 1 through 22 of Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA) was also detected. Gene fusions involving the full length of PIK3CA were previously reported and are potentially actionable [46]. An additional novel fusion involving YWHAE (Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Epsilon) and the tumor suppressor gene SMYD4 (SET and MYND Domain Containing 4) was identified. Gene fusions involving tumor suppressor genes were identified in different tumor types including lung adenocarcinomas and showed a trend for a decreased tumor suppressor expression [19].
RNASeq fusions not detected by MSK-IMPACT due to panel design
52% (15/29) gene fusions detected by RNASeq were not expected to be called by MSK-IMPACT due to the lack of coverage of introns inferred to be the site of the genomic breakpoints (Figure 3B): SLC34A2-ROS1 (n=5), SLC3A2-NRG1 (n=2), CD74-NRG1 (n=2), SDC4-ROS1, SDC4-NRG1, SQSTM1-NTRK3, RBPMS-NTRK3, HDAC5-PIK3CA and YWHAE-SMYD4. All SLC34A2-ROS1 and SDC4-ROS1 fusions involved Exon 32 of ROS1, which predict, although not unequivocally, the possibility of ROS1 intron 31 to be involved in the genomic breakpoint. As previously described [47], intron 31 of ROS1 is known to harbor repetitive elements; most of this intron is excluded from the MSK-IMPACT hybrid capture bait design as the reads would be difficult or impossible to reliably map to the genome. Thus, intronic repetitive regions are not covered by MSK-IMPACT. A small portion of the 5’ region of intron 31 is covered by MSK-IMPACT (Supplementary Figure 1) and unless the genomic breakpoint occurs in that specific region, a rearrangement would not be detected by DNAseq.
Likewise, DNAseq of introns is not an effective modality to detect NTRK3 fusions because the NTRK3 introns involved in recurrent genomic breakpoints, introns 13 and 14, respectively span 93 and 92 Kb. Tiling such large introns would result in a significant increase to the overall DNA panel size. Only NTRK3 fusions with ETV6 as the 5’ partner are expected to be detected by MSK-IMPACT because the panel captures ETV6 intronic regions known to be involved in fusions. As with NTRK3, NRG1 relevant introns are not captured by MSK-IMPACT due to their large size. CD74 intron 6 is tiled in the MSK-IMPACT panel. However, the CD74-NRG1 fusion detected by RNASeq involved exon 8 indicating the possibility that the genomic breakpoint of this fusion took place in intron 7, which isn’t captured by the DNA panel. HDAC5-PIK3CA and YWHAE-SMYD4 fusions are novel fusions without specific intronic tiling in the DNA Sequencing panel.
RNASeq-only fusions expected to be called by MSK-IMPACT
Nearly half (48%; 14/29) of the additional gene fusions identified by RNAseq would have been expected to be detected by the MSK-IMPACT panel based on its design (Figure 3C): CD74-ROS1 (n=3), EML4-ALK (n=2), KIF5B-RET (n=2), KIF5B-ALK (n=1), CLTC-ALK (n=1), AGK-BRAF(n=1), FGFR2-CBX5 (n=1), STRN-NTRK2 (n=1), CCDC6-RET (n=1), and LRIG3-ROS1 (n=1). The above fusions involved ROS1 exons 34 and 35, ALK exons 20 and 19, RET exon 12, BRAF exon 8, NTRK2 exon 16 and FGFR2 exon 17. For all of these, the corresponding introns (ROS1 introns 33/34, ALK introns19/18, RET intron11, BRAF intron 7, and NTRK2 intron15) are effectively tiled in the MSK-IMPACT DNASeq panel (Supplementary Table 4). Upon manual review in Integrative Genomics Viewer (IGV) [48, 49], those specific intronic regions had sufficient sequencing coverage except for two fusion positive samples, CD74-ROS1 (exon34) and EML4-ALK (exon20), where the DNA quantity was sub-optimal and resulted in low sequencing coverage and for the KIF5B-RET positive tumor where intron 11 had lower coverage. This could have led to less sensitivity for the detection of fusions involving this particular intron. (Supplementary Table 5). Additionally, both the samples positive for CLTC-ALK and CCDC6-RET fusions by RNASeq had low tumor purity (<20%) which was assessed by a pathologist but also confirmed by the fact that no DNA mutations were called in the sample including silent mutations (Supplementary Table 5). This demonstrates the ability of RNASeq to detect fusion events even in specimens with a proportion of tumor cells that is suboptimal or inadequate for DNAseq, presumably because high expression of the fusion mRNA can “compensate” for low tumor content. Finally, in the six samples positive for AGK-BRAF, CD74-ROS1, KIF5B-RET, KIF5B-ALK, EML4-ALK and FGFR2-CBX5 fusions, a structural variant involving one of the fusion partners was detected by DNASeq. In these cases, it is likely that the oncogenic fusion was caused by one or more complex DNA rearrangements that couldn’t be fully captured by our DNA panel (Supplementary Table 5). This further illustrates the advantage of RNASeq in detecting gene fusions that are challenging to capture by targeted DNASeq assay designs.
METex14 Skipping
RNAseq identified 6 samples positive for METex14 skipping that were not noted on DNAseq, including canonical MET splice mutations. Upon further manual review of DNAseq variants in MET introns 13 and 14, non-canonical MET deletions involving intronic nucleotide sequences up to 26 base pairs from the splice site were detected in 5/6 samples (Supplementary Table 6). One sample was negative for MET mutations by DNASeq possibly indicating a different mechanism leading to METex14 skipping.
Low Tumor Mutation Burden in cases with kinase fusions
TMB was assessed for all MSK-IMPACT cases that were positive for a driver alteration including hotspot mutations, amplifications and gene fusions (n=1933). TMB median was calculated and compared between all fusion positive (1.97 mutations/Mb, IQ range: 0.88 – 3.51) and fusion negative samples (5.58 mutations/Mb, IQ range: 2.95– 8.85), representing a significant difference in TMB (p<0.00001, Mann-Whitney test) (Figure 4A) and indicating an enrichment for kinase fusions in low TMB samples. To see if a TMB cutoff could be used to identify cases in which additional RNAseq testing would be the most fruitful and therefore of highest priority, TMB was assessed in the 232 DNAseq driver negative cases that successfully underwent RNASeq; in this subset, 81 cases had low TMB (0–5 mut/Mb) of which 31% were fusion positive. In contrast, in the 151 cases with higher TMB (>5 mut/Mb), only 7% of the cases were positive for fusions (P<0.0001, Mann-Whitney test) further supporting the notion that gene fusions are enriched in low TMB samples Figure 4B.
Figure 4. Assessment of Tumor Mutation Burden in fusion positive and negative tumors.
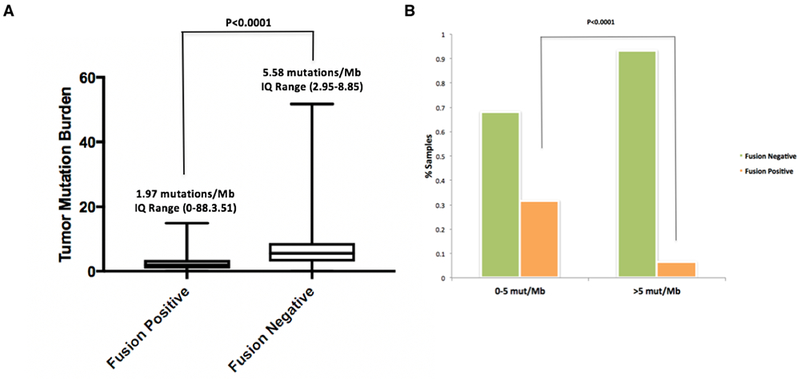
(A) The tumor mutation burden (TMB) of 1,933 MSK-IMPACT driver-positive lung adenocarcinomas was compared between fusion positive (n=195, left) and negative (n=1,738, right) samples. Median TMB and IQ range are indicated on top of each plot. The median TMB of Fusion positive samples was significantly lower than the fusion negative ones (Mann Whitney, p<0.0001). 4 outlier TMB values were excluded from the plot but not from the statistical analysis. (B) A cohort of 232 MSK-IMPACT driver negative lung adenocarcinomas is organized by low TMB (0-5 mut/Mb, left) and higher TMB (>5 mut/Mb, right). The percentage of patients whose tumors are fusion negative and positive by RNASeq (MSK-Fusion) is indicated. The percentage of fusion-positive samples with low TMB (31%) was significantly higher when compared with the percentage of fusion-positive ones with high TMB (7%), (Mann Whitney, p<0.0001).
Complete landscape of fusions in lung adenocarcinomas
Next, we used the combined NGS data to provide the most complete and accurate assessment to date of the prevalence of known kinase fusions in lung adenocarcinoma in our patient population. Comprehensive DNA and RNA sequencing in 2522 unique lung adenocarcinomas identified 223 high-confidence in-frame and targetable gene fusions (Figure 5) involving NRG1 (0.32%) and the following kinase genes: ALK (3.44%), ROS1 (2.26%), RET (1.78%), BRAF (0.28%), FGFR3 (0.20%), NTRK1 (0.16%), NTRK3 (0.08%), FGFR2 (0.08%), FGFR1 (0.04%), NTRK2 (0.04%), PIK3CA (0.04%), MET (0.04%) and EGFR (0.04%). Additionally, our analysis also provides further evidence of the promiscuity of certain 5’ partner genes, that are found to recombine with multiple kinase genes. For example, KIF5B is a common upstream fusion partner to RET (n=31) but also to ALK (n=3) and EGFR (n=1). Similarly, CD74 and SDC4 partner with both NRG1 (n=5 and n=1) and ROS1 (n=29 and n=8), respectively, to form fusion transcripts.
Figure 5. Gene fusion landscape in 2,522 lung adenocarcinomas.
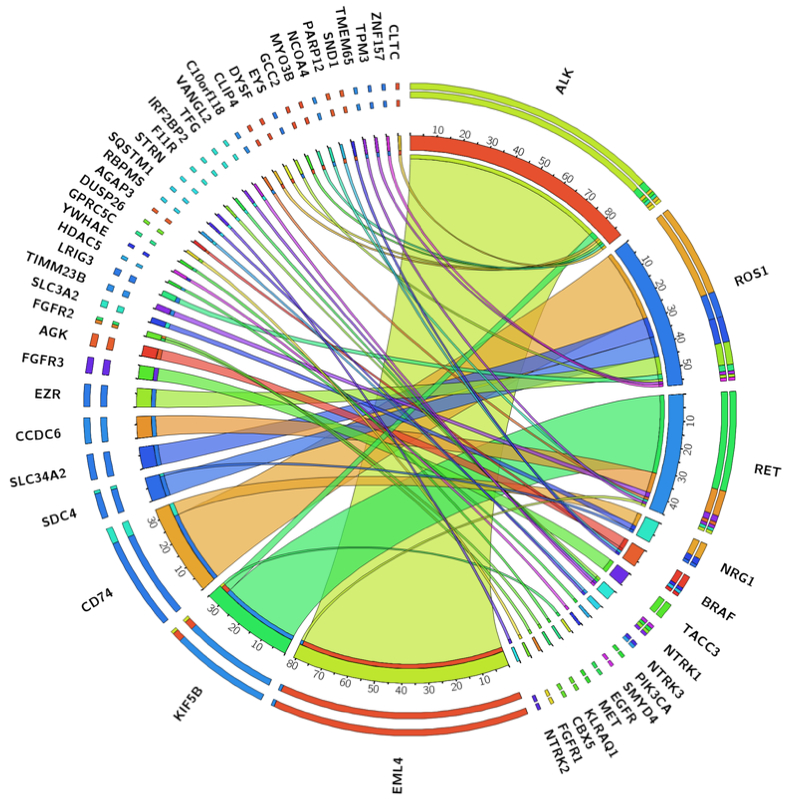
Illustration of all gene fusions detected by comprehensive DNASeq (MSK-IMPACT) and RNASeq (MSK-Fusion).
Clinical outcomes of patients with RNAseq fusion positive DNAseq negative tumors
Of the 33 RNAseq positive (27 with kinase gene fusions and 6 with METex14) DNAseq negative patients, 10 went on to be matched to targeted therapy. Alterations in the tumors from these 10 patients included 1 ALK fusion, 4 ROS1 fusions, 2 NTRK fusions, 2 NRG1 fusions, and 1 METex14 skipping alteration. Treatment and response to therapy, as defined by RECIST v1.1, is outlined in Figure 6 which shows that 8 patients (80%) had clinical benefit from the matched targeted therapy identified thanks to the additional RNAseq testing. The other 23 patients did not receive targeted therapy for a variety of reasons: 6 did not have metastatic disease, 4 were on active surveillance with stable disease after prior treatment modalities, 6 were already on other systemic therapy at the time of the result, 1 patient was lost to follow up, and in 5 retrospective patients the RNAseq results were only available post-mortem.
Figure 6. Study summary and clinical findings.

RNASeq-based matched therapy for 10/33 patients whose tumors were found to be positive for gene fusions (9) or METex14 (1). 80% of the patients have shown clinical benefit. PR: partial response, SD: stable disease; PD: progression of disease. * Response assessment by RECIST v1.1. ** Confirmed PR
DISCUSSION
The number of kinase inhibitors successfully targeting oncogenic gene fusions and rearrangements is increasing, providing better disease management options for patients with cancer [19, 50–55]. Therefore, the accurate detection and characterization of those events is clinically essential. Targeted DNA-based sequencing offers a comprehensive tool to detect all types of oncogenic alterations including some structural variants. However, due to the frequent complexity of DNA rearrangements and assay design limitations, it is plausible that some important gene fusions and rearrangements are not accurately detected by DNA-based sequencing techniques. In this study, we have used a clinically validated targeted RNA Sequencing assay (MSK-Fusion) to test lung adenocarcinomas lacking oncogenic driver alterations by DNA Sequencing (MSK-IMPACT). We have demonstrated that 14% (n=36) of the tested DNAseq negative cases were positive for fusions or rearrangements by RNAseq. Additionally, a clinical benefit was achieved in 80% of the patients whose tumors were positive for fusions or METex14 skipping and who received matched targeted therapy. Importantly, as we have previously found [38, 39], tumors with low TMB are enriched for the presence of a targetable oncogenic driver. This may reflect the fact that most major oncogenic alterations driving MAPK signaling in lung adenocarcinoma (with the exception of KRAS G12C mutations [56] , MAP2K1 mutations [57], some non-V600E BRAF mutations [58–60]) are typically seen in never smokers whose tumors therefore do not show the elevated TMB consequent to smoking-induced mutagenesis. Based on this observation, we found that, in more resource-limited settings, the yield of additional RNAseq testing could be increased by focusing on cases that are driver-negative by DNAseq and show low TMB.
One of the challenges of DNA-based gene fusion detection is that most genomic breakpoints that produce fusion genes take place in introns, which cannot always be fully covered by hybrid capture-based NGS either because they contain repetitive elements [61, 62] or they are too long for targeted panel assays. For example, 34% (10/29) of the fusion transcripts not detected by DNA sequencing included the ROS1 gene. Six of those fusions involved ROS1 Exon 32 predicting that the genomic breakpoint site may have possibly taken place in intron 31. This intron is known to include numerous repetitive elements. These can be present at many other sites in the genome and inclusion of baits for these regions would simply result in unmappable reads; therefore, such repetitive regions are not covered in hybrid capture-based NGS assays and hence genomic breaks in these regions are usually missed. Additionally, several introns that are known to be involved in genomic breakpoints tend to be very long. For example, each of introns 13 and 14 of the kinase gene NTRK3 or intron 5 of NRG1 are close to 100Kb in length (UCSC Genome Browser), which is close to 10% of the total size of MSK-IMPACT. Tiling such introns is not only technically challenging but also not practical in terms of overall sequencing throughput and cost, for high volume clinical laboratories that have to make optimal use of resources and limited sequencing capacity.
Nearly half (48%) of the gene fusions not detected by DNASeq involved exons where the presumably involved introns are well covered by the DNA panel. It is possible that the genomic breakpoint causing the rearrangement simply took place in a non-predicted intron that was not tiled by the panel. The second possible reason from missing gene fusions by DNA Sequencing is low tumor purity. Although our cutoff for DNA Sequencing is 20% tumor content by histologic assessment, the true proportion of tumor cells in the sample can be lower when estimated using somatic mutation variant frequencies. For example, two samples in our cohort were positive for CLTC-ALK and CCDC6-RET fusions by RNASeq whereas in both cases no DNA mutations, including silent ones, were detected in the sample. This indicates that the true tumor purity of those samples is likely very low (<5%) and highlights of the advantages of RNA Sequencing where a highly expressed event can still be detected in the context of low tumor purity. Finally, suboptimal gDNA quality/quantity leading to low quality sequencing reads can also interfere with gene fusion detection. DNASeq did not detect a CD74-ROS1 and EML4-ALK in two separate samples with low gDNA input. in both cases, poor DNA quality was confirmed by further QC of the sequencing results.
In 6 cases, METex14 skipping events were detected by RNASeq but no MET exon 14 splice mutations were called in the corresponding DNA by MSK-IMPACT. Upon further visual inspection of reads in IGV [48, 49] we identified intronic MET mutations located up to 40 base pairs away from the splice site in intron 13 (Supplementary table 6). The MSK-IMPACT pipeline was not originally configured to call mutations that far into the intron. Because of this finding, any sample with a putative splicing mutation in MET detected in intron 13 or 14 by MSK-IMPACT is now reflexed to RNASeq to confirm the presence of METex14 skipping at the RNA level.
One of our study limitations is that out of the 589 driver negative lung adenocarcinoma cases that were candidate for RNA Sequencing follow up, only 275 (47%) had available tissue for RNA extraction. Given the rate of detection in the subset of cases with adequate material (14%), the total number of fusions detected in this study would have been significantly higher if material was available for all cases. This highlights the fact that, in the real-world setting, additional material for RNA extraction is unavailable in many cases. Of the lung adenocarcinomas submitted for DNASeq, 70% were from very limited samples, such as small lung biopsies (53%) or cytology (17%) specimens. Often, all of the unstained FFPE sections are used up for DNA extraction with little left for RNA extraction. Additionally, recuts from the original FFPE block are often not possible due to exhaustion of tumor material. In order to circumvent the challenge around limited material in our clinical laboratory, we have validated RNA extraction on limited amounts of lysate remaining after automated DNA extraction and stored at room temperature (Supplementary figure 2). This has allowed us to have immediate access to adequate material for RNA extraction and to enable comprehensive DNA and RNA sequencing for most of the eligible cases in our clinical laboratory. Clinical requests for RNASeq testing can occur up to approximately 1–2 months after the cell lysate is originally preserved at room temperature. Although the extracted RNA quality and quantity are compatible with downstream sequencing for the majority of the cases, this approach has not been systematically evaluated on lysate material saved at room temperature for longer timeframes.
Another study limitation is that RNA Sequencing was performed using a targeted amplicon-based panel, which included a limited number of genes. Additionally, a primer design was only included for the canonical exons known to be involved in gene fusions or isoforms. It is also possible that our targeted RNASeq panel assay has missed as yet undescribed but possibly clinically important gene fusions that could be detected with other sequencing approaches including targeted hybridization-capture based RNA sequencing [23, 25] or whole transcriptome sequencing [63].
It has been previously noted that tumors positive for gene fusions contained a low number of mutations [64]. In this study, we have also demonstrated that, in a driver positive lung adenocarcinomas cohort assessed by DNASeq, the fusion positive samples had a significantly lower TMB than the fusion negative ones. Additionally, among the driver negative samples tested by RNASeq, those with low TMB were enriched for gene fusions when compared to the ones with higher TMB. These results indicate that driver negative tumors with low TMB are more likely to harbor fusions than the ones with higher TMB. In a clinical setting, such patients should be prioritized for RNASeq for the potential detection of targetable gene fusions, although our results do not support limiting additional RNAseq testing to this patient subset. Indeed, we recommend that all patients whose tumors are driver-negative by DNASeq go on to RNAseq to ensure that no driver alterations are missed. Overall, we find that a rational, algorithmic approach to the use of targeted RNA-based NGS to complement increasingly routine large panel DNA-based NGS testing can be a highly effective strategy to comprehensively uncover targetable gene fusions or oncogenic isoforms not just in lung adenocarcinomas but also more generally across different tumors types.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVANCE.
Inhibitors targeting kinase fusions have shown dramatic and durable responses in lung cancer patients, making their comprehensive detection critical. Here, we evaluated the incremental benefit of targeted RNAseq in the identification of gene fusions in patients where no clear mitogenic driver alteration is found by DNAseq-based panel testing. We found actionable alterations (kinase fusions or METex14 skipping) in 13% of cases apparently driver-negative by previous DNAseq testing. Among the driver negative samples tested by RNASeq, those with low TMB were significantly enriched for gene fusions when compared to the ones with higher TMB. In a clinical setting, such patients should be prioritized for RNASeq. Thus, a rational, algorithmic approach to the use of targeted RNA-based NGS to complement large panel DNA-based NGS testing can be highly effective in comprehensively uncovering targetable gene fusions or oncogenic isoforms not just in lung cancer but also more generally across different tumors types.
ACKNOWLEDGEMENTS
We gratefully acknowledge J. Keith Killian for his expert technical advice.
FINANCIAL SUPPORT
This research was supported in part by the National Cancer Institute of the National Institutes of Health (P01 CA 129243, T32 CA009207, P30 CA008748).
Footnotes
Conflict of Interest Statement:
Ryma Benayed has received a grant from ArcherDx.
Michael Offin has received consulting fees from PharmaMar.
Charles Rudin is a consultant for Abbvie, Amgen, Ascentage, AstraZeneca, Bicycle, Celgene, Chugai, Daiichi Sankyo, Genentech/Roche, GI Therapeutics, Loxo, Novartis, Pharmamar, and Seattle Genetics, and serves on the Scientific Advisory Boards of Elucida and Harpoon.
David Hyman: Consulting or Advisory Role: AstraZeneca, Atara Biotherapeutics, Chugai Pharma, CytomX Therapeutics, Boehringer Ingelheim, Pfizer, Genetech, Bayer, Debiopharm, ArQule. Research Funding: Loxo Oncology, AstraZeneca, and Puma Biotechnology
Maria Arcila received speaker’s fees from Raindance Technologies.
Mark G. Kris has received consulting fees from AstraZeneca, Pfizer, and Regeneron. He has received honoraria for participation in educational programs from WebMD, OncLive, Physicians Education Resources, AstraZeneca, and Research to Practice. Dr. Kris is an employee of Memorial Sloan Kettering. Memorial Sloan Kettering has received research funding from Genentech Roche and PUMA Biotechnology for trials conducted by Dr. Kris. Memorial Sloan Kettering has a collaboration for the development of Watson for Oncology with IBM and receives royalties from IBM for this activity.
Alex Drilon: HONORARIA/ADVISORY BOARDS: Ignyta, Loxo, TP Therapeutics, AstraZeneca Pfizer, Blueprint Medicines, Genentech/Roche, Takeda/Ariad/Millenium, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Bayer, Tyra Biosciences. RESEARCH: Foundation Medicine. ASSOCIATED RESEARCH PAID TO INSTITUTION: Pfizer, Exelixis, GlaxoSmithKlein, Teva, Taiho. ROYALTIES: Wolters Kluwer. CME HONORARIA: Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice. OTHER: Merck - Food/Beverage, MORE Health - Consulting
Michael Berger has received advisory board compensation from Roche and research support from Illumina.
Marc Ladanyi has received advisory board compensation from AstraZeneca, Bristol-Myers Squibb, Takeda, and Bayer, and research support from LOXO Oncology and Helsinn Healthcare.
All other authors declare no potential conflict of interest.
REFERENCES
- 1.Facchinetti F, et al. , Oncogene addiction in non-small cell lung cancer: Focus on ROS1 inhibition. Cancer Treat Rev, 2017. 55: p. 83–95. [DOI] [PubMed] [Google Scholar]
- 2.Holla VR, et al. , ALK: a tyrosine kinase target for cancer therapy. Cold Spring Harb Mol Case Stud, 2017. 3(1): p. a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drilon A, et al. , Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372–001 and STARTRK-1). Cancer Discov, 2017. 7(4): p. 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suehara Y, et al. , Identification of KIF5B-RET and GOPC-ROS1 fusions in lung adenocarcinomas through a comprehensive mRNA-based screen for tyrosine kinase fusions. Clin Cancer Res, 2012. 18(24): p. 6599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies KD, et al. , Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res, 2012. 18(17): p. 4570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw AT, et al. , Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol, 2017. 18(12): p. 1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sholl LM, et al. , Multi-institutional Oncogenic Driver Mutation Analysis in Lung Adenocarcinoma: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol, 2015. 10(5): p. 768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aisner DL, et al. , The Impact of Smoking and TP53 Mutations in Lung Adenocarcinoma Patients with Targetable Mutations-The Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res, 2018. 24(5): p. 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kris MG, et al. , Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA, 2014. 311(19): p. 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drilon A, et al. , Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N Engl J Med, 2018. 378(8): p. 731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drilon A, et al. , Response to ERBB3-Directed Targeted Therapy in NRG1-Rearranged Cancers. Cancer Discov, 2018. 8(6): p. 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subbiah V, et al. , Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol, 2018. 29(8): p. 1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Offin M, et al. , Acquired ALK and RET Gene Fusions as Mechanisms of Resistance to Osimertinib in EGFR-Mutant Lung Cancers. JCO Precision Oncology, 2018(2): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCoach CE, et al. , Resistance Mechanisms to Targeted Therapies in ROS1(+) and ALK(+) Non-small Cell Lung Cancer. Clin Cancer Res, 2018. 24(14): p. 3334–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrock AB, et al. , Receptor Tyrosine Kinase Fusions and BRAF Kinase Fusions are Rare but Actionable Resistance Mechanisms to EGFR Tyrosine Kinase Inhibitors. J Thorac Oncol, 2018. 13(9): p. 1312–1323. [DOI] [PubMed] [Google Scholar]
- 16.Vojnic M, et al. , Acquired BRAF rearrangements induce secondary resistance to EGFR therapy in EGFR-mutated lung cancers. J Thorac Oncol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mertens F, et al. , The emerging complexity of gene fusions in cancer. Nat Rev Cancer, 2015. 15(6): p. 371–81. [DOI] [PubMed] [Google Scholar]
- 18.Mitelman F, Johansson B, and Mertens F, The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer, 2007. 7(4): p. 233–45. [DOI] [PubMed] [Google Scholar]
- 19.Gao Q, et al. , Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep, 2018. 23(1): p. 227–238 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stransky N, et al. , The landscape of kinase fusions in cancer. Nat Commun, 2014. 5: p. 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zehir A, et al. , Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med, 2017. 23(6): p. 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farago AF, et al. , Clinicopathologic Features of Non-Small-Cell Lung Cancer Harboring an NTRK Gene Fusion. JCO Precis Oncol, 2018. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He J, et al. , Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood, 2016. 127(24): p. 3004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross JS, et al. , The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer, 2016. 138(4): p. 881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reeser JW, et al. , Validation of a Targeted RNA Sequencing Assay for Kinase Fusion Detection in Solid Tumors. J Mol Diagn, 2017. 19(5): p. 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng DT, et al. , Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn, 2015. 17(3): p. 251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lang UE, Yeh I, and McCalmont TH, Molecular Melanoma Diagnosis Update: Gene Fusion, Genomic Hybridization, and Massively Parallel Short-Read Sequencing. Clin Lab Med, 2017. 37(3): p. 473–484. [DOI] [PubMed] [Google Scholar]
- 28.Kumar-Sinha C, Kalyana-Sundaram S, and Chinnaiyan AM, Landscape of gene fusions in epithelial cancers: seq and ye shall find. Genome Med, 2015. 7: p. 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hechtman JF, et al. , Pan-Trk Immunohistochemistry Is an Efficient and Reliable Screen for the Detection of NTRK Fusions. Am J Surg Pathol, 2017. 41(11): p. 1547–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu G, et al. , Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod Pathol, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Z, et al. , Anchored multiplex PCR for targeted next-generation sequencing. Nat Med, 2014. 20(12): p. 1479–84. [DOI] [PubMed] [Google Scholar]
- 32.Goodman AM, et al. , Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther, 2017. 16(11): p. 2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hendriks LE, Rouleau E, and Besse B, Clinical utility of tumor mutational burden in patients with non-small cell lung cancer treated with immunotherapy. Transl Lung Cancer Res, 2018. 7(6): p. 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heeke S and Hofman P, Tumor mutational burden assessment as a predictive biomarker for immunotherapy in lung cancer patients: getting ready for prime-time or not? Transl Lung Cancer Res, 2018. 7(6): p. 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hellmann MD, et al. , Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell, 2018. 33(5): p. 853–861 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hellmann MD, et al. , Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med, 2018. 378(22): p. 2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samstein RM, et al. , Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Offin M, et al. , Tumor Mutation Burden and Efficacy of EGFR-Tyrosine Kinase Inhibitors in Patients with EGFR-Mutant Lung Cancers. Clin Cancer Res, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sabari JK, et al. , PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol, 2018. 29(10): p. 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jordan EJ, et al. , Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov, 2017. 7(6): p. 596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wittwer CT, et al. , Continuous fluorescence monitoring of rapid cycle DNA amplification. Biotechniques, 1997. 22(1): p. 130–1, 134–8. [DOI] [PubMed] [Google Scholar]
- 42.Chakravarty D, et al. , OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol, 2017. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iyama K, et al. , Identification of Three Novel Fusion Oncogenes, SQSTM1/NTRK3, AFAP1L2/RET, and PPFIBP2/RET, in Thyroid Cancers of Young Patients in Fukushima. Thyroid, 2017. 27(6): p. 811–818. [DOI] [PubMed] [Google Scholar]
- 44.Amatu A, Sartore-Bianchi A, and Siena S, NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open, 2016. 1(2): p. e000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu Z, et al. , Targeted next generation sequencing identifies somatic mutations and gene fusions in papillary thyroid carcinoma. Oncotarget, 2017. 8(28): p. 45784–45792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson D, et al. , Integrative Clinical Genomics of Advanced Prostate Cancer. Cell, 2015. 162(2): p. 454. [DOI] [PubMed] [Google Scholar]
- 47.Davies KD, et al. , Comparison of Molecular Testing Modalities for Detection of ROS1 Rearrangements in a Cohort of Positive Patient Samples. J Thorac Oncol, 2018. 13(10): p. 1474–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson JT, et al. , Integrative genomics viewer. Nat Biotechnol, 2011. 29(1): p. 24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thorvaldsdottir H, Robinson JT, and Mesirov JP, Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform, 2013. 14(2): p. 178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schram AM, et al. , Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol, 2017. 14(12): p. 735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cerrato A, Visconti R, and Celetti A, The rationale for druggability of CCDC6-tyrosine kinase fusions in lung cancer. Mol Cancer, 2018. 17(1): p. 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim HS, et al. , EGFR and HER3 signaling blockade in invasive mucinous lung adenocarcinoma harboring an NRG1 fusion. Lung Cancer, 2018. 124: p. 71–75. [DOI] [PubMed] [Google Scholar]
- 53.Gay ND, et al. , Durable Response to Afatinib in Lung Adenocarcinoma Harboring NRG1 Gene Fusions. J Thorac Oncol, 2017. 12(8): p. e107–e110. [DOI] [PubMed] [Google Scholar]
- 54.Fernandez-Cuesta L and Thomas RK, Molecular Pathways: Targeting NRG1 Fusions in Lung Cancer. Clin Cancer Res, 2015. 21(9): p. 1989–94. [DOI] [PubMed] [Google Scholar]
- 55.Drilon A, et al. , Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat Rev Clin Oncol, 2018. 15(3): p. 150. [DOI] [PubMed] [Google Scholar]
- 56.Dogan S, et al. , Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res, 2012. 18(22): p. 6169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arcila ME, et al. , MAP2K1 (MEK1) Mutations Define a Distinct Subset of Lung Adenocarcinoma Associated with Smoking. Clin Cancer Res, 2015. 21(8): p. 1935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Litvak AM, et al. , Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol, 2014. 9(11): p. 1669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tissot C, et al. , Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer, 2016. 91: p. 23–8. [DOI] [PubMed] [Google Scholar]
- 60.Marchetti A, et al. , Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol, 2011. 29(26): p. 3574–9. [DOI] [PubMed] [Google Scholar]
- 61.Treangen TJ and Salzberg SL, Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet, 2011. 13(1): p. 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alkan C, Coe BP, and Eichler EE, Genome structural variation discovery and genotyping. Nat Rev Genet, 2011. 12(5): p. 363–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson DR, et al. , Integrative clinical genomics of metastatic cancer. Nature, 2017. 548(7667): p. 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoshihara K, et al. , The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene, 2015. 34(37): p. 4845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.