

Abstract
Insoluble amyloid plagues are likely cytoprotective, but the cellular mechanism remains less known. To model β-amyloid we use a small peptide derivative to generate cytotoxic nanofibrils that cause the death of model neuron cells (i.e., PC12). The use of supramolecular interaction effectively converts the nanofibrils to nanoparticles that are innocuous to cells. This approach also removes the cytotoxicity of the fibrils to other mammalian cells (e.g., HeLa). Preliminary mechanistic study reveals that, in contrast to the fibrils, the particles promote the expression of TNFR2, a cell survival signal, and decrease the expression of TNFR1 and DR5, two extrinsic cell death receptors. As the first use of ligand-receptor interaction to abrogate the cytotoxicity of nanoscale assemblies of small molecules, this work illustrates an effective way to use supramolecular interaction to control the morphology of supramolecular assemblies for modulating their biological activity.
Graphical Abstract
This communication describes the use of ligand-receptor interaction to convert cytotoxic molecular nanofibrils of small molecules to innocuous particles, illustrating that control over aggregate morphology is an effective approach to reduce the toxicity of molecular nanofibrils. Recent advances suggest that soluble β-amyloid (Aβ) oligomers are the most neurotoxic species.1 Emerging studies also suggest that the early assemblies of misfolded non-disease-associated proteins2,3 and oligomers of disease-associated proteins (e. g., Aβs)4 exhibit similar inherent cytotoxicity, suggesting a common mechanism of the cytotoxicity of the molecular aggregates.5 These new insights imply that the insoluble plaques of Aβ in Alzheimer’s disease are neuroprotective6 and have led to efforts to control the morphology of the molecular aggregates for reducing the cytotoxicity of amyloids.7–9 For example, Shoichet et al. have demonstrated that colloidal aggregates of small molecules physically sequester proteins or early assemblies of the proteins to nonspecifically inhibit amyloid formation, implying that morphological control of colloids formed by molecular self-assembly may be relevant in more biological milieus.10,11
Encouraged by the above development, we design a small molecule 1 containing D-Ala-D-Ala that self-assembles12,13 in water to result in molecular nanofibrils and to form hydrogels. Below its minimal gelation concentration (mgc), 1 forms nanofibrils that inhibit the proliferation of PC12 cells, a model of neuron cells.14 Upon the addition of vancomycin (Van)—a well-established ligand of D-Ala-D-Ala,13,15,16 ligand-receptor interaction effectively converts the fibrils of 1 into particles (Fig. 1). The resulting particles are innocuous to PC12 cells and other mammalian cells. Preliminary mechanistic examination indicates that, while fibrils of 1 suppress the expression of decoy cell death receptors (DcR217 and DcR318) and increase the expression of cell death receptors (TNFR119, DR520), particles made of 1 and Van promote the expression of a cell survival signal (TNFR221) and decrease the expression of cell death receptors (TNFR1, DR5). As the first use of ligand-receptor interaction to abrogate the cytotoxicity of molecular nanofibrils of small molecules via morphological modulation, this work illustrates a new approach to tune the morphology of supramolecular assemblies for modulating their biological activities, and contributes useful insights for the exploration of biofunctional assemblies of small molecules,10,22–24 an underexplored subject that is increasingly significant in biology and medicine.25,26
Figure 1.

(A) Molecular structures of the ligand (Van), the receptor (a derivative of D-Ala-D-Ala (1)). (B) Illustration of the ligand-receptor interaction that transforms the cytotoxic fibrils, formed by self-assembly (SA) of 1, to innocuous particles made of Van and 1.
Our analysis indicates that the volume of aggregates rather than the numbers of individual molecules dictates the cytotoxicity of molecular aggregates, implying that these aggregates cause cell death with rather promiscuous interactions.5,27 Particularly, our recent work shows that ligand-receptor interaction catalyzes the formation of aggregates of Fmoc-D-Ala-D-Ala to inhibit the proliferation of cells at 24h.28 Considering that 1, like Fmoc-D-Ala-D-Ala, contains the D-Ala-D-Ala segment, we expect that Van would catalyze the aggregation of 1 to inhibit cell proliferation. According to this rationale, we synthesize 1 and test its cytotoxicity without and with the addition of Van. As shown in Fig. 2, 1 exhibits steep cytotoxicity at concentrations above 400 μM (with an IC90 between 400 and 500 μM against PC12 cells29). This threshold cytotoxicity (Fig. 2A) indicates the formation of aggregates of 1 that inhibit cell proliferation, agreeing with reports on the cytotoxicity of nanoscale assemblies.5,23,24,30–32
Figure 2.

(A) Cell viability of PC12 incubated with 1 and 1+Van after 5 days, [1]0 : [Van]0 = 1 : 1. (B) IC50 values of 1/Van against PC12 cells after 5 days at different conditions: 1 incubated with PC12 cells for 12 h, either adding Van or changing medium (↔) abrogates the cytotoxicity of the nanofibrils. TEM images of (C) solution of 1 and (D) mixture of 1 and Van, inset are corresponding optical images under UV irradiation, [1]0 = [Van]0 = 500 μM (enlarged Fig. 2B in Fig. S2).
Surprisingly, in contrast to our previous report that Van catalyzes the aggregation of Fmoc-D-Ala-D-Ala to inhibit the cells, the addition of Van completely abrogates the cytotoxicity of the nanofibrils of 1 (Fig. 2A). This result, in fact, is similar with the induced formation of plaques to decrease Aβ-associated toxicity33 and neuronal death occurring at regions (entorhinal cortex and hippocampus) with few Aβ plaques.34 Optical images under UV irradiation clearly show the transition from a clear solution to a suspension (the insets of Fig. 2C and 2D, the bright spots in the bottom of the vial are particles excited by UV). According to visual inspection, the ligand-receptor interaction between Van and 1 results in precipitates, suggesting the formation of particles(Fig. S3). Furthermore, transmission electron microscopy (TEM) reveals drastically different morphology of the assemblies of 1 before and after the addition of Van. As shown in Fig. 2C, the molecules of 1 self-assemble to form nanofibrils at a concentration of 500 μM (215 μg/mL), which is consistent with light scattering results (Fig. S5), implying that the resulting nanofibrils exhibit cytotoxicity. In contrast, the addition of Van completely converts the fibrils to particles (Fig. 2D), which are innocuous. This result indicates that ligand-receptor interactions among the small molecules (Van and 1) convert fibrils of 1 to particles made of 1 and Van, thus abrogating the cytotoxicity caused by assemblies of 1.
To verify that the elimination of cytotoxicity is due to the ligand-receptor interaction between Van and 1, we examine the cell viability of PC12 cells incubated with 1+Van under three control conditions. As shown in Fig. 2B, after incubation of PC12 cell with 1 for 12h, extending the incubation time for another five days without any change, the assemblies of 1 are able to inhibit the proliferation of PC12 cells, with IC50 value of 357 μM. In contrast, the addition of Van (at same concentration (i.e., 500 μM) as that of 1) effectively abrogates the cytotoxicity of assemblies of 1. Furthermore, after replacing the medium with fresh culture medium, the IC50 of 1 against PC12 cells is larger than 500 μM after 5 days, suggesting that the cytotoxicity unlikely originates from cellular uptake of 1, but rather results from assemblies of 1 outside cells. These results further demonstrate that ligand-receptor interactions between Van and 1 are able to abrogate the cytotoxicity of fibrils of 1.
To further confirm that the morphological modulation by the ligand-receptor interaction is a cell line independent process, we examine the cytotoxicity of 1 with or without the addition of Van against three mammalian cell lines. As shown in Fig. 3A, 1 inhibits the proliferation of HeLa, T98G, and HT1080 cells, with IC50 values of 293 μM, 321 μM, and 280 μM, respectively. Their corresponding mass concentrations are 126 μg/mL, 138 μg/mL, and 120 μg/mL, respectively, which are comparable to the mass concentration of amyloids (e.g. Aβ42: 45 μg/mL)8,35 in most of studies. After mixing with Van in equal molar amounts, 1 becomes innocuous to HeLa, T98G, and HT1080 cells at concentrations much higher than the IC50 values.
Figure 3.

(A) IC50 of 1 without and with the addition of Van ([1]0 : [Van]0 = 1:1) against HeLa, T98G, and HT1080 cells for 48 h. (B) At 24 h, the viability of HeLa cells incubated with 1 ([1]0 = 500 μM) and varying amounts of Van (from 71 μM to 500 μM). (C) IC50 of 1+Van against HeLa cells at different conditions: 1 (or Van), at concentrations of 100 μM to 500 μM, incubated with HeLa cells for 12 h, with (or without) changing the medium (↔), then adding Van (or 1). TEM images of 1 ([1]0 = 500 μM) and varying amounts of Van (D: 71 μM, E: 100 μM, F: 167 μM), and corresponding optical images under UV irradiation.
We synthesize a control molecule (2) by replacing D-Ala-D-Ala with L-Ala-L-Ala (Fig.S1). As shown in Fig.4, 2 inhibits the proliferation of HeLa cell with or without the addition of Van, with similar IC50 values (Fig. S7), suggesting that the addition of Van hardly affects the cytotoxicity of 2. Meanwhile, the IC50 of 2 with or without addition of Van against PC12 cells are 303 μM and 285 μM after 5 days, such little difference further confirms Van hardly reduces the cytotoxicity of 2. Moreover, TEM images of solution of 2 and 2+Van show similar long, flexible nanofibrils (Fig. S8). These results agree with negligible interactions between 2 and Van, thus further supporting that the ligand-receptor interaction modulates the morphology and the cytotoxicity of the fibrils of 1. To quantify the ligand-receptor interaction, we use isothermal titration calorimetry (ITC) to measure the binding between 1 and Van (Fig. S9). Upon the titration of Van (8.0 mM) into a solution of 1 (0.7 mM) in PBS buffer, fitting the data using independent model gives a dissociation constant (Kd) to be 20.7 μM and stoichiometry (n, Van/1) to be about 1. The dissociation constant agrees with the binding constants of Van and D-Ala-D-Ala determined by other methods,36–38 suggesting high affinity between Van and 1 and 1:1 binding of Van and 1. The control molecule (2) barely binds with Van, resulting in an affinity too weak to be measured by ITC (Fig. S10). The above results imply the generality that ligand-receptor interaction modulates the morphology of molecular assemblies to abrogate the cytotoxicity of the nanofibrils of small molecules.
Figure 4.

Cell viability of HeLa cells at 48h, and PC 12 cells at 120h incubated with 2 and 2+Van.
To investigate the effectiveness of the ligand-receptor interaction to minimize the cytotoxicity of nanofibrils, we examine the cell viability of HeLa cells incubated with a fixed amount of 1 (500 μM), but with varying amounts of Van (from 71.4 μM to 500 μM). As shown in Fig. 3B, when the concentration of Van is above 250 μM ([1]0 : [Van]0 = 2 : 1), the HeLa cells become almost 100% viable at 24h. This result confirms that the ligand-receptor interaction between Van and 1 can effectively eliminate the cytotoxicity of the fibrils of 1. Meanwhile, when the concentration of Van increases (from 71.4 μM to 166.7 μM), the semi-transparent solution of 1 gradually becomes a suspension (as shown in the insets of Fig. 3D, 3E, and 3F). Moreover, TEM images of the solutions and suspensions reveal the co-existence of fibrils and particles when the [Van]0 is less than 166.7 μM, implying that the remaining fibrils of 1 are able to inhibit the proliferation of HeLa cells. To further verify that the ligand-receptor interaction abrogates the cytotoxicity of extracellular fibrils of 1, we measure the cell viability of HeLa cells treated by 1+Van under three different conditions (Fig. 3C). Even after incubation of 1 with the cells for 12h, equal molar amounts of Van are still able to completely eliminate the cytotoxicity of the fibrils of 1, similar to the result with PC12 cells. To further establish the generality of ligand and receptor interaction for abrogating the cytotoxicity caused by nanofibrils, we synthesize 3 and 4 by inserting Gly-Gly into 1 and 2, respectively (Fig.S1). Similar to 1, 3 inhibits HeLa cells, with IC50 value of 382 μM. Upon the addition of Van at same mole ratio, the resulting particles are innocuous at concentrations as high as 500 μM (Fig. S11). Similar to 2, molecule 4 inhibits the proliferation of HeLa cells with or without the addition of Van (Fig. S12). The nearly identical morphology of assemblies of 4 before and after addition of Van also agrees with negligible interaction between 4 and Van (Fig. S13).
To further confirm that ligand-receptor interaction controls morphologies of supramolecular assemblies that interact with cells, we used a 7-nitrobenzofurazan derivatized vancomycin (NBD-Van) to visualize the aggregates of (1+Van) in PBS buffer. After mixing of 1 (500 μM), Van (490 μM), and NBD-Van (10 μM), many yellow spots appeared on and away from the cells, indicating the formation of particles (Fig. S14) and agreeing with TEM result. When 2 replaces 1, few yellow spots appear. This result agrees with 2 barely binding with Van. Upon incubation of HeLa cells with 1 and Van, with 1 μM of NBD-Van to visualize the aggregation process, we observe considerable amount of aggregates formed away from cells, in addition to the yellow spots on the HeLa cell surface (Fig. S15, also see the movie of 1&Van _HeLa cell). After we extend the incubation time to 8 h and wash the cells with PBS buffer for 3 times, yellow dots still adhered to cells. There is little cell inhibition indicates that the aggregates of 1+Van on the cells are likely innoucous. As expected, no particles appeared around the cells in the case of 2 (Fig. S15 and S16, also see the movie of 2&Van _HeLa cell), indicating that the fibrils of 2 remain unchanged and still inhibit the cells. Additionally, similar phenomenon appeared in PC12 cells after same treatment (Fig. S17. and Fig.S18). Moreover, we used FITC-annexin V and propidium iodide (PI) to stain the PC12 cells. After incubation with the fibrils of 1 for 12h, most of PC12 cells are only stained by FITC-annexin V (Fig. S19), which is different from necrotic cells induced by the treatment of 10% DMSO for 8h (Fig. S20). This result indicated that the PC12 cells enter the early stage of apoptosis.39 However, upon treatment by the same stains, HeLa cells exhibit green and red fluorescence after treatment with 1 (500 μM) for 12h (Fig. S21), suggesting that fibrils of 1 cause the necroptosis of the HeLa cells.40 Furttrher studies found that pretreatment with necrostatin-141 is unable to reduce the cytotoxicity of fibrils of 1(Shown in Fig. S22), suggesting that an alternative cell death pathway, other than necroptosis, is responsible for the observed cytotoxicity.
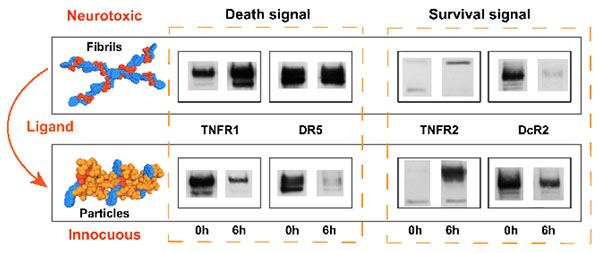
In a preliminary mechanistic study, we examine the expression of proteins in the extrinsic pathway of apoptosis of HeLa cells incubated with 1 or 1+Van (500 μM). As shown in Fig. 5, the fibrils of 1 increase the expression of TNFR1 and DR5, two extrinsic cell death receptors, decrease the expression of DcR2, a decoy of death receptor, and hardly affect the expression of TNFR2, a cell survival signal, and DcR3, another decoy of death receptor. This observation suggests that the fibrils of 1 likely act as a new type of cell death signal, which is consistent with the down-stream fragmentation of RIP and the cytotoxicity of the fibrils of 1. In contrast, the particles made of 1 and Van decrease the expression of TNFR1 and DR5, increase the expression of TNFR2, and maintain the expression of DcR2 and DcR3. This observation agrees with the decreased fragmentation of RIP and the cytocompatibility of the particles of 1+Van. These results suggest that morphological change of the assemblies of small molecules is able to modulate cell death signaling, thus controlling cell fate. Since the particles of 1+Van and aggregates of FmKGGaa and Van show dramatically different cellular responses, we also monitor the changes of apoptotic cell signals (Fig.S24) over time. The aggregates of FmKGGaa and Van, also decrease the expression of TNFR1, increase the expression of TNFR2, and maintain the expression of DcR2, DcR3, and DR5. Notably, the aggregates of FmKGGaa and Van significantly increase the expression of Fas and also induce more and more expression of TNFR2 over time. Since Fas is a death receptor and TNFR2 sensitizes cells for cell death, it is reasonable that the aggregates of FmKGGaa and Van lead to cell death. This result indicates that assemblies of FmKGGaa and Van interact with different death receptors from those of 1+Van, thus resulting in different cell behaviour. In addition, we used Z-VAD-FMK, a pan-caspase inhibitor, to treat the HeLa cells in the presence of the fibrils of 1. The inhibitor only slightly reduces the cytotoxicity of the fibrils of 1 (Fig. S25), suggesting that the fibrils of 1 largely induce the caspase-independent cell death (e.g. necroptosis42), as recently reported by Wells et al.23
Figure 5.

Western blot analysis of the cell lysate of the HeLa cells treated with 1 or 1&Van at 500 μM for different durations.
In this work, one Van binds with one Py-D-Ala-D-Ala (1) to convert the toxic fibrils to innocuous particles; in our previous study, one Van binds with two Fmoc-D-Ala-D-Ala to turn innocuous monomers to toxic fibrils. Such a small difference (between Py and Fmoc) in the receptors results in dramatically different phenotypes of cells. This observation, indeed, provides insights for the paradoxical feature of the molecular aggregates in cellular environment. For example, proteins or oligopeptides bearing Phe and Trp may behave completely different. The use of enantiomers as the control compounds, in fact, side steps the problems of polymorphism associated with the aggregates, thus providing more definitively molecular evidence and cellular mechanism for asserting Van’s role in abrogating the cytotoxicity of fibrils of 1.
Despite the fact that there are many structural characterizations of amyloids (e.g., Aβ42) or aggregates, few investigates the cellular mechanisms. The molecular cell biology characterization of the detoxification of nanofibrils of 1, in this work, thus provides a much needed understanding of the toxicity of molecular aggregates. Since aggregates of small molecules share many common features with amyloids formed by aberrant proteins or peptides43 and associated with unsolved and intriguing problems, such as neurodegenerative diseases, the use of ligand-receptor interaction44,45 effectively converts the neurotoxic nanofibrils to innocuous nanoparticles, which may ultimately, lead to approaches that promote the controlled formation of plaques for treating diseases such as Alzheimer’s disease.
Supplementary Material
{kind=link}
{kind=link}
ACKNOWLEDGMENT
This work was partially supported by NIH (R01CA142746), the W.M. Keck Foundation.
Footnotes
Supporting Information
Details of the synthesis, cytotoxicity, and confocal images. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Oda T; Pasinetti GM; Osterburg HH; Anderson C; Johnson SA; Finch CE Biochem. Biophys. Res. Commun 1994, 204, 1131. [DOI] [PubMed] [Google Scholar]
- (2).Bucciantini M; Giannoni E; Chiti F; Baroni F; Formigli L; Zurdo JS; Taddei N; Ramponi G; Dobson CM; Stefani M Nature 2002, 416, 507. [DOI] [PubMed] [Google Scholar]
- (3).Dobson CM Nature 2003, 426, 884. [DOI] [PubMed] [Google Scholar]
- (4).Kayed R; Head E; Thompson JL; McIntire TM; Milton SC; Cotman CW; Glabe CG Science 2003, 300, 486. [DOI] [PubMed] [Google Scholar]
- (5).Zhou R; Xu B PLoS One 2014, 9, e95759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kinghorn KJ; Crowther DC; Sharp LK; Nerelius C; Davis RL; Chang HT; Green C; Gubb DC; Johansson J; Lomas DA J. Biol. Chem 2006, 281, 29268. [DOI] [PubMed] [Google Scholar]
- (7).Ehrnhoefer DE; Bieschke J; Boeddrich A; Herbst M; Masino L; Lurz R; Engemann S; Pastore A; Wanker EE Nat. Struct. Mol. Biol 2008, 15, 558. [DOI] [PubMed] [Google Scholar]
- (8).Bieschke J; Herbst M; Wiglenda T; Friedrich RP; Boeddrich A; Schiele F; Kleckers D; Lopez del Amo JM; Grüning BA; Wang Q et al. Nat. Chem. Biol 2012, 8, 93. [DOI] [PubMed] [Google Scholar]
- (9).Cao P; Raleigh DP Biochemistry 2012, 51, 2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Feng BY; Toyama BH; Wille H; Colby DW; Collins SR; May BCH; Prusiner SB; Weissman J; Shoichet BK Nat. Chem. Biol 2008, 4, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McGovern SL; Caselli E; Grigorieff N; Shoichet BK J Med Chem 2002, 45, 1712. [DOI] [PubMed] [Google Scholar]
- (12).Zhang Y; Gu H; Yang Z; Xu B J Am Chem Soc 2003, 125, 13680. [DOI] [PubMed] [Google Scholar]
- (13).Zhang Y; Yang Z; Yuan F; Gu H; Gao P; Xu B J Am Chem Soc 2004, 126, 15028. [DOI] [PubMed] [Google Scholar]
- (14).Martin TFJ; Grishanin RN In Method Cell Biol; Academic Press Inc: San Diego, 2003. [Google Scholar]
- (15).Hu Y; Helm JS; Chen L; Ye X-Y; Walker S J Am Chem Soc 2003, 125, 8736. [DOI] [PubMed] [Google Scholar]
- (16).Rao J; Lahiri J; Isaacs L; Weis RM; Whitesides GM Science 1998, 280, 708. [DOI] [PubMed] [Google Scholar]
- (17).Marsters SA; Sheridan JP; Pitti RM; Huang A; Skubatch M; Baldwin D; Yuan J; Gurney A; Goddard AD; Godowski P et al. Curr Biol 1997, 7, 1003. [DOI] [PubMed] [Google Scholar]
- (18).Pitti RM; Marsters SA; Lawrence DA; Roy M; Kischkel FC; Dowd P; Huang A; Donahue CJ; Sherwood SW; Baldwin DT et al. Nature 1998, 396, 699. [DOI] [PubMed] [Google Scholar]
- (19).Tartaglia LA; Weber RF; Figari IS; Reynolds C; Palladino MA; Goeddel DV Proc. Natl. Acad. Sci. U.S.A 1991, 88, 9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).MacFarlane M; Ahmad M; Srinivasula SM; Fernandes-Alnemri T; Cohen GM; Alnemri ES J Biol Chem 1997, 272, 25417. [DOI] [PubMed] [Google Scholar]
- (21).Aggarwal BB Nat. Rev. Immunol 2003, 3, 745. [DOI] [PubMed] [Google Scholar]
- (22).Jadhav A; Ferreira RS; Klumpp C; Mott BT; Austin CP; Inglese J; Thomas CJ; Maloney DJ; Shoichet BK; Simeonov A J Med Chem 2009, 53, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Julien O; Kampmann M; Bassik MC; Zorn JA; Venditto VJ; Shimbo K; Agard NJ; Shimada K; Rheingold AL; Stockwell BR et al. Nat. Chem. Biol 2014, 10, 969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zorn JA; Wille H; Wolan DW; Wells JA J Am Chem Soc 2011, 133, 19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Frenkel YV; Clark AD; Das K; Wang YH; Lewi PJ; Janssen PAJ; Arnold EJ Med. Chem 2005, 48, 1974. [DOI] [PubMed] [Google Scholar]
- (26).Owen SC; Doak AK; Ganesh AN; Nedyalkova L; McLaughlin CK; Shoichet BK; Shoichet MS ACS Chem. Biol 2014, 9, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nobeli I; Favia AD; Thornton JM Nat Biotech 2009, 27, 157. [DOI] [PubMed] [Google Scholar]
- (28).Shi J; Du X; Huang Y; Zhou J; Yuan D; Wu D; Zhang Y; Haburcak R; Epstein IR; Xu B J Am Chem Soc 2015, 137, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rukenstein A; Rydel RE; Greene LA J Neurosci 1991, 11, 2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shoichet BK J Med Chem 2006, 49, 7274. [DOI] [PubMed] [Google Scholar]
- (31).Kuang Y; Xu B Angew. Chem. Int. Ed 2013, 52, 6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kuang Y; Shi J; Li J; Yuan D; Alberti KA; Xu Q; Xu B Angew. Chem. Int. Ed 2014, 53, 8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kinghorn KJ; Crowther DC; Sharp LK; Nerelius C; Davis RL; Chang HT; Green C; Gubb DC; Johansson J; Lomas DA J Biol Chem 2006, 281, 29268. [DOI] [PubMed] [Google Scholar]
- (34).Musiek ES; Holtzman DM Nat Neurosci 2015, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fukuda T; Matsumoto E; Onogi S; Miura Y Bioconj. Chem 2010, 21, 1079. [DOI] [PubMed] [Google Scholar]
- (36).Williams DH; Cox JPL; Doig AJ; Gardner M; Gerhard U; Kaye PT; Lal AR; Nicholls IA; Salter CJ; Mitchell RC J Am Chem Soc 1991, 113, 7020. [Google Scholar]
- (37).Wright GD; Walsh CT Accounts Chem Res 1992, 25, 468. [Google Scholar]
- (38).Rao J; Whitesides GM J Am Chem Soc 1997, 119, 10286. [Google Scholar]
- (39).Lincz LF Immunol Cell Biol 1998, 76, 1. [DOI] [PubMed] [Google Scholar]
- (40).Galluzzi L; Vitale I; Abrams JM; Alnemri ES; Baehrecke EH; Blagosklonny MV; Dawson TM; Dawson VL; El-Deiry WS; Fulda S et al. Cell Death Differ 2012, 19, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Christofferson DE; Yuan J Curr Opin Cell Biol 2010, 22, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Ofengeim D; Yuan J Nat. Rev. Mol. Cell Biol 2013, 14, 727. [DOI] [PubMed] [Google Scholar]
- (43).Liang C; Ni R; Smith JE; Childers WS; Mehta AK; Lynn DG J Am Chem Soc 2014, 136, 15146. [DOI] [PubMed] [Google Scholar]
- (44).Qiu Z; Yu H; Li J; Wang Y; Zhang Y Chem. Commun 2009, 3342. [DOI] [PubMed] [Google Scholar]
- (45).Shi J; Du X; Yuan D; Haburcak R; Wu D; Zhou N; Xu B Chem. Commun 2015, 51, 4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.