

Abstract
Targeting of nanoparticles to tumors can potentially improve specificity of imaging and treatments. We have developed a multicompartmental pharmacokinetic model in order to analyze some of the factors that control efficiency of targeting to intravascular (endothelium) and extravascular (tumor cells and stroma) compartments. We make the assumption that transport across tumor endothelium is an important step for subsequent nanoparticle accumulation in the tumor (area-under-the-curve, AUC) regardless of entry route (interendothelial and transendothelial routes) and study this through a multicompartmental simulation. . Our model reveals that increasing endothelial targeting efficiency has a much stronger effect on the AUC than increasing extravascular targeting efficiency. Furthermore, our analysis reveals that both extravasation and intratumoral diffusion rates need to be increased in order to significantly increase the AUC of extravascular-targeted nanoparticles. Increasing the nanoparticle circulation half-life increases the AUC independently of extravasation and intratumoral diffusion. Targeting the extravascular compartment leads to a buildup in the first layer surrounding blood vessels at the expense of deeper layers (binding site barrier). This model explains some of the limitations of tumor targeting, and provides important guidelines for the design of targeted nanomedicines.
Keywords: modeling, pharmacokinetics, nanoparticle, targeting, tumor, extravasation, diffusion
BACKGROUND
Significant progress has been made in the synthesis and characterization of engineered nanoparticles for imaging and treatment of cancers, with several promising candidates in clinical trials (1). At the same time, clinical applications of nanoparticulate systems are still limited by poor penetration, diffusion and retention in solid tumors (2, 3). The phenomenon of passive accumulation of nanoparticles via enhanced permeability and retention “EPR” (4), previously considered to be a universal phenomenon, is now believed to be highly variable and limited to fast-growing tumors with leaky vasculature (5, 6). Nevertheless, it has been difficult to trace nanoparticle transport through interendothelial fenestrations in tumors, and as such the operation of pore-dependent mechanism of extravasation has been questioned (7, 8). Recently, we discussed that weak/moderate nanoparticle interaction with endothelium may be a prerequisite for successful erythrocyte-mediated forced extravasation (in a scenario analogous to the “endothelial massaging” hypothesis in liver sinusoids (8). Contrary to interendothelial route, there is limited morphological evidence that support the operation of transendothelial route in some tumors (9–11). Again, the efficacy of this process may still depend on the extent of nanoparticle-endothelium interaction (8). Furthermore, there has been increasing interest in designing nanoparticles conjugated to tumor-specific ligands. Indeed, nanoparticles targeted to the intravascular compartment, e.g., to angiogenic endothelium receptors or to intravascular fibrin clots, showed enhanced accumulation compared to non-targeted formulations (12–16). On the other hand, while some studies have shown a modest benefit of targeting the extravascular compartment compared to non-targeted particles [approximately a 1.5- to 2.0-fold increase (17–20)], other studies showed no increase in accumulation in tumors, with a concomitant decrease in penetration into deeper tumors layers (20–22).
Considering the enormous effort invested in development of targeted nanoparticles, there is a need in better understanding of factors that control targeting of intravascular and extravascular compartments of solid tumors. A significant amount of research has been done on developing physiological models of tumor accumulation as a function of carrier size and morphology, tumor density, vascularity, and permeability (20, 23–26). These studies revealed important limitations of nanoparticle penetration and diffusion (20, 27). Modeling of nanoparticle accumulation in tumors is intrinsically complex as it requires knowledge of difficult-to-measure parameters such as tumor flux and permeability, non-linear diffusion rates in a viscous tumor interstitium, blood vessel margination efficiency and stochastic intratumoral blood flow rates (20, 23–26), and can become even more complex for targeted particles, since targeting efficiency is difficult to measure experimentally.
Here we have developed a physiologically based multi-compartmental pharmacokinetic model of nanoparticle targeting to tumor compartments. Next, we derived extravasation and intratumoral diffusion rates and targeting efficiencies by fitting the available tumor accumulation data (percent of injected dose per gram tumor (%ID/g) over time) and blood elimination half-life for non-targeted and tumor vasculature-targeted single-wall carbon nanotubes (SWNTs) (12) into the model. Then, we performed simulations to understand parameters that play a role in tumor accumulation and tumor penetration for these nanoparticles. The choice of SWNT was due to the availability of the full set of data required by the model. There are only a handful of publications that provide both %ID/g in tumor over time and blood elimination half-life. Despite the fact that this study uses the data for non-translational nanoparticles, it represents an important step towards understanding the tumor accumulation of other tumor targeted nanomedicines.
METHODS
Physiologically based multicompartmental model of tumor targeting
According to Fig. 1, the model is based on the following compartments: blood, endothelium and tumor. In addition, the extravascular tumor compartment is arbitrarily divided into layers in order to model diffusion inside the tumor compartment (akin to theoretical plates in chromatography). The concentrations of nanoparticles in each compartment (bound and unbound fractions) can be described by a set of differential equations (only 2 tumor layers are shown):
Capital letters designate the compartment (B for blood, E for endothelium, and T for tumor, with subscript either Bound or Unbound), kel is the blood elimination rate, kEI is the extravasation-intravasation rate (hereafter EIR), and kID is the intratumoral diffusion rate (hereafter ITDR). For simplicity, EIR and ITDR are assumed to be the same in both directions, but in reality they could be different due to active transport across the endothelium (8, 10). kIVB and kEVB are intravascular and extravascular binding rates (hereafter IVB and EVB), respectively. When multiplied by 100 these rates are referred to as IVB and EVB efficiencies throughout the text, or percentage of nanoparticles that binds to a compartment per minute. A recursive method to compute nanoparticle concentration in each compartment was used. The recursive calculations are based on incremental addition or subtraction of a mass of nanoparticles that has moved between compartments each minute, divided by volume of the compartment. The graphical user interface and the code were written and implemented in MATLAB R2013a version (MathWorks, Natick, MA). The program is available from our laboratory upon signing Material Transfer Agreement with the University of Colorado.
Figure 1. Physiologically based multicompartmental model of tumor targeting:
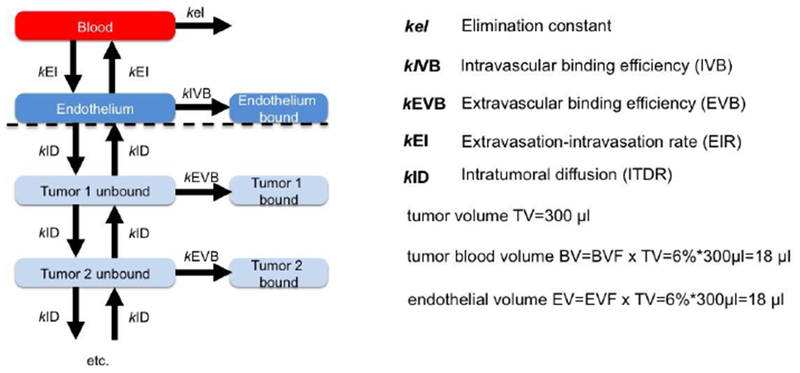
The model consists of blood, endothelium and extravascular compartments and respective rates (k). The full list of differential equations is provided in the Methods section. The extravascular compartment consists of multiple layers, and the diffusion rates are assumed to be equal in all directions. The number of extravascular layers in the model is arbitrarily set to 4 in the model (only 2 are shown here) and does not affect net tumor accumulation. The volumes of compartments were calculated based on the values reported in the literature (see Results).
For calculation of the endothelium volume fraction, values were determined from previously published CD31 immunostained tumor images of U85MG tumors (28) as follows. The fluorescent images were analyzed with ImageJ for the percent of area occupied by CD31 positive cells (determined with the color thresholding tool). It was assumed that the volume fraction is equal to the area fraction of a section; accordingly a 5% area represents a 5% volume fraction.
The absolute injected dose (ID) of nanoparticles was not reported in the original work and therefore was chosen arbitrarily. We must note that %ID/g tissue does not depend on the ID. On the other hand, the AUCT value does depend on the ID, therefore AUCT values were presented as arbitrary units rather than μg x h/mL. All the numerical data including %ID/g in tumor and blood were extracted from Liu et al. (12) using WebPlotDigitizer software. The blood half-life of nanoparticles was determined by plotting %ID/g in blood over time in Prism (GraphPad, San Diego, USA) and fitting the profile to monoexponential decay.
RESULTS
Physiologically based multicompartmental model of tumor targeting
We developed a physiologically-based multicompartmental tumor model that consists of blood, endothelial (intravascular) and tumor (extravascular) compartments (Fig. 1 and Methods). This model is similar to the one previously described for polymeric nanoparticles (26), except that the extravascular compartment consists of multiple layers. The model is based on non-saturated Fickian diffusion kinetics, i.e., the rate of diffusion in all compartments is proportional to the concentration gradient. The model was developed based on the assumption that there is no saturation of targeting/uptake due to the low dose of intravenously injected nanoparticles. The validity and limitations of this assumption will be discussed below. Under this model, accumulation in the tumor is determined by several rates (Fig. 1A): a) blood elimination rate (kel) a) extravasation-intravasation rate (kEI, hereafter EIR), which is the same in both directions; b) intratumoral diffusion rate (kID, hereafter ITDR), which is the same in all directions; c) intravascular (endothelial) binding rate (kIVB, hereafter IVB) and extravascular (tumor cells and stroma) binding rate (kEVB, hereafter EVB). According to the Fickian diffusion law for the transport of nanoparticles under non-saturation conditions (29), the targeting efficiency corresponds to the fraction of nanoparticles that becomes associated with the compartment per minute, which in turn is determined by receptor binding and dissociation rates, endocytosis and exocytosis rates.
Data for non-targeted and tumor-targeted nanoparticles can be described with the model
IVB, EVB, EIR and ITDR are unknown parameters, but can be estimated from known tumor accumulation profiles (%ID per gram of tumor at different time points) for non-targeted and targeted nanoparticles. We used data published by Liu et al. (12) in which the authors used PEGylated single-wall carbon nanotubes (SWNTs) to target U85MG glioblastoma xenograft tumors. The authors used non-targeted and ligand (RGD) modified SWNT-PEG5000. Cyclic RGD is a ligand that targets αvβ3 and αvβ5 integrins on angiogenic blood vessels (30). We selected these data for modeling for the following reasons: a) the authors conveniently reported %ID/g for targeted and non-targeted control nanotubes (Table 1); b) the authors used positron emission tomography (PET) imaging of Cu-64 labeled nanotubes, which enabled accurate quantification of the tumor accumulation in vivo while using a small, non-saturating injection dose; c) targeting was confirmed using rigorous controls, including receptor-negative tumors and free ligand blocking; d) SWNTs showed no measurable elimination from the tumor in the observed time course, which obviates the need to add tumor elimination via degradation or lymphatics to the model (31). The physiological model inputs (Fig. 1) were total blood volume (1.7 mL for a 20 g mouse), tumor volume TV [300 mm3 per Liu et al. (12)], tumor blood volume fraction BVF [6% for the U85MG xenograft tumor (32)], and endothelial volume fraction EVF (6% based on image analysis of CD31 stained sections of U85MG tumors, see Methods). From these parameters, the volumes of the endothelium, intravascular and extravascular compartments were calculated (Fig. 1B). Based on the blood elimination profile reported by Liu et al. (12), the nanotube half-life was determined to be 100 min for SWNT-PEG5000. Since the blood clearance profile of SWNT-PEG5000-RGD was not provided, we assumed the value determined for SWNT-PEG5000.
Table 1.
Tumor accumulation values (perfused values – blood pool subtracted) derived from Liu et al (12). The values were extracted from graphs using Plot Digitizer software.
| SWNT-PEG5000 | SWNT-PEG5000-RGD | ||
|---|---|---|---|
| Time (min) | %ID per gram tumor | Time (min) | %ID per gram tumor |
| 25 | 1.29 | 27 | 4.73 |
| 96 | 2.68 | 123 | 9.31 |
| 329 | 3.99 | 348 | 12.00 |
| 1012 | 4.36 | 1012 | 13.36 |
| 1365 | 4.53 | 1414 | 13.37 |
Given the fact that the limited number of reported %ID/g data points makes simultaneous extraction of all rates (EIR, ITDR, IVB and EVB) challenging, we used a rational step-wise approach. Because the flow of nanoparticles is from the blood vessel towards the tumor layers, we set the EIR to a positive value and then sequentially added ITDR and binding efficiencies to the model (Fig. 2A). First, we used data for non-targeted SWNT-PEG5000 (Table 1). As shown in Fig. 2B, left, the EIR value of 0.022 determines the initial rate of tumor accumulation. However, in the absence of intratumoral diffusion, the concentrations in the tumor decrease in parallel with a decrease in normal tissue concentrations (Fig. 2B, black dotted line). Setting the ITDR to 0.015 increased the retention at later time points compared with the normal tissue (Fig. 2B, center), which is the essence of the EPR effect (4, 33). Still, the plateau observed for SWNT-PEG5000 was not reached due to the nanotubes being washed out from the tumor. To increase the retention in the tumor, we set the EVB efficiency to 2% (even though these were non-targeted SWNTs, there is likely a non-specific binding and uptake by the extravascular compartment, for example by stroma and macrophages, respectively). Addition of extravascular binding achieved a good correlation (R2=0.959) to the actual data (Fig. 2B, right). We performed additional manual iterations of EIR, ITDR and EVB, and found that a set of parameters (EIR = 0.018; ITDR = 0.022; EVB = 0.082) results in a better R2 of 0.984 (Supplemental Fig. S1).
Figure 2. Algorithm for describing pharmacokinetic profile of non-targeted nanoparticles with the model:
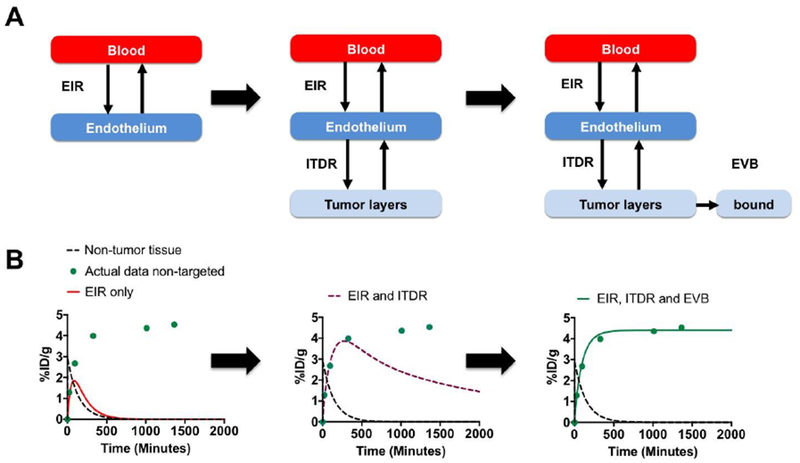
A) simplified schematic of the PK model (based on Fig. 1) shows compartments used in each step; B) actual data for targeted nanoparticles (Table 1) were plotted as %ID/g over time (green circles). Each step corresponds to the schematic above. The tumor profile cannot be described by EIR only, as nanoparticles rapidly wash out (left graph, red line); addition of ITDR improves the profile, but nanoparticles are still being washed out from the tumor (center graph, purple dashed line); addition of EVB into the model achieves a good correlation with the data (right graph, green line).
Next, we used %ID/g data of endothelium-targeted SWNT-PEG5000-RGD (Table 1). Compared with non-targeted SWNT-PEG5000, endothelium-targeted SWNT-PEG5000-RGD achieved much higher accumulation in tumors, reaching 14 %ID/g. We used EIR, ITDR and EVB for non-targeted nanotubes (Fig. 3A); with an IVB value of 1.7, a good correlation with the experimental data was achieved (Fig. 3B, blue trace, R2=0.8820). Interestingly, intravascular targeted nanoparticles showed faster rate of accumulation in the tumor than non-targeted nanoparticles (Fig. 3B, blue trace vs. green trace), which corroborates previous reports that intravascular targeting leads to rapid tumor accumulation of nanoparticles (12–16).
Figure 3. Algorithm for describing pharmacokinetic profile of endothelium-targeted nanoparticles with the model:
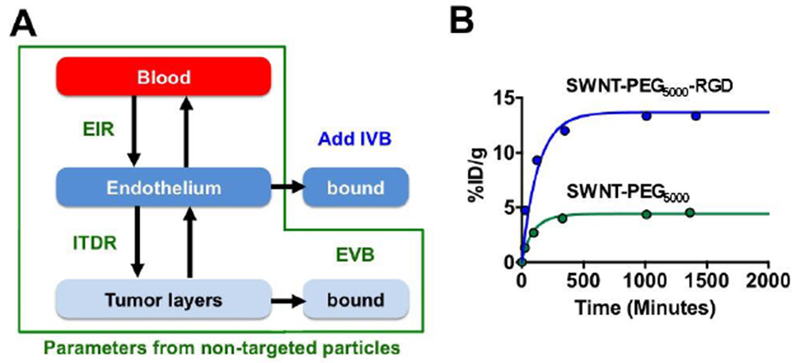
A) Profile for endothelium-targeted nanoparticles was described by using EIR, ITDR and EVB for non-targeted nanoparticles (Fig. 2B) and adding IVB; B) actual and simulated profiles for non-targeted and targeted nanoparticles. Note that intravascular targeting increases the rate of nanoparticle accumulation in the tumor.
In order to apply the model to different nanoparticles in a different tumor model, we used the results from Goel et al. (34) on Zr-89 labeled mesoporous silica nanoparticles conjugated to anti-CD105 antibody targeted to tumor vasculature in a 4T1 breast cancer xenograft model, using the %ID/g for non-targeted and targeted nanoparticles (Supplemental Table 1). The relevant physiological parameters were obtained from the literature, and EVF for 4T1 tumors was determined experimentally (Supplemental Fig. S2). Using the same approach as in Fig. 2, we were able to simulate the actual tumor profile data (Supplemental Fig. S3) for non-targeted and targeted nanoparticles using EIR = 0.011, ITDR = 0.0052, EVB = 0.14, and IVB = 0.59 (R2=0.75 and R2=0.62 for non-targeted and targeted nanoparticles, respectively). Interestingly, while EIR and IVB for silica particles in the 4T1 tumor model were similar to SWCNT in the U85MG tumor model, ITDR was 5-fold less for silica nanoparticles, suggesting significant differences in intratumoral diffusion, which could be due to differences in tumor stroma and nanoparticle shape/surface characteristics.
Effect of EIR, ITDR, EVB, IVB and circulation half-life on tumor AUC
We performed a sensitivity analysis where the where EIR and ITDR determined SWNTs were changed in 2-fold steps (one parameter at a time) and the % change in AUCT was calculated. The analysis showed that AUCT is relatively insensitive to the changes in the lower range of values (Fig. 4A, EIR<0.22; ITDR<0.09), but is sensitive to the changes in the higher range of values (Fig 4A, EIR>0.22; ITDR>0.09). Moreover, in order to achieve significant increase in AUCT (2-5 fold), both EIR and ITDR need to be significantly increased. This result suggests that for tumors with low intratumoral diffusion, an increase in extravasation alone cannot increase tumor accumulation; similarly, with low extravasation, the increase in intratumoral diffusion cannot increase tumor accumulation.
Figure 4. Sensitivity of the AUC to changes in EIR, ITDR, EVB and IVB:
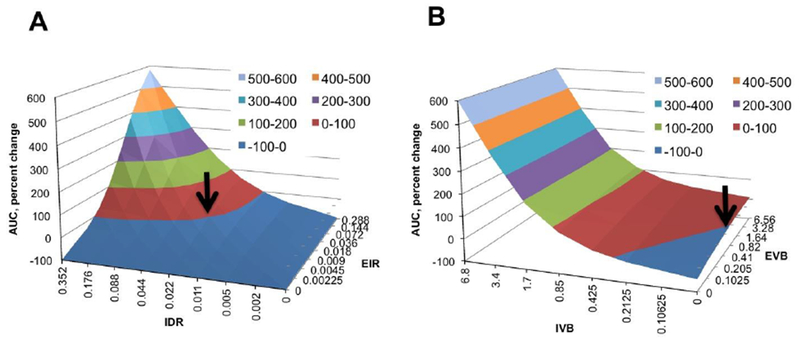
The parameters were changed one at a time and the percent change of AUCT was plotted on the Y-axis. A) Effect of EIR and ITDR on AUCT. EVB was set to 0.82 and IVB was set to 0. Arrow points to the set of parameters for non-targeted SWNT-PEG5000 found in Fig. 2. The model is not very sensitive in the range of lower EIR and ITDR values, but becomes sensitive in the range of higher values; B) Effect of EVB and IVB on AUCT. EIR and ITDR were set to 0.018 and 0.022, respectively. Arrow points to the set of parameters for non-targeted SWNT-PEG5000. The model is not sensitive to changes in EVB but very sensitive to changes in IVB, suggesting that intravascular targeting is a more efficient process than extravascular targeting.
Next, we performed another sensitivity analysis where the values of EIR and ITDR found above were kept constant, but the values of IVB and EVB were changed in 2-fold steps. According to Fig. 4B, the model is not sensitive to changes in EVB in all ranges of values (less than 2-fold change upon 64-fold change in EVB), but sensitive to the changes in IVB in all ranges of values (over 6-fold change upon 32-fold change in IVB). This simulation suggests that increasing extravascular binding efficiency of SWNT-PEG5000 will not dramatically increase the amount of nanotubes accumulating to the tumor. At the same time, increasing endothelium binding efficiency of SWNT-PEG5000-RGD will significantly increase the amount of nanotubes accumulating in the tumor.
Since the AUCT was not sensitive to changes in EVB value using the original EIR, ITDR and EVB values for non-targeted SWNT-PEG5000, we performed simulations where EIR or ITDR (or both) were increased by 10-fold, and EVB was increased by 5-fold (from 0.82 to 4.0). As shown in Fig. 5A, increasing either EIR or ITDR alone only marginally increases AUCT (1.5-2-fold). Increasing EVB by 5-fold led to an additional 1.3-fold increase in AUCT. However, increasing both EIR and ITDR by 10-fold leads to over 5-fold increase in AUCT. Increasing EVB to 4.0 further increased the AUCT 1.5-fold. These simulations suggest that extravascular targeting is limited by the extravasation and diffusion rates rather than by targeting efficiency, and increasing both extravasation and intratumoral diffusion is necessary to gain any benefit from extravascular targeting. Thus, tumors with high stroma density and poor vascular permeability may only modestly benefit from improvements in targeting avidity of nanomedicines, and enhancement of extravasation (for example by using tumor penetrating compounds or interstitial-pressure-lowering drugs (35)) combined with agents that improve intratumoral diffusion [for instance hyaluronidase (36)] is needed to realize targeting potential.
Figure 5. Simulations of tumor accumulation efficiency of non-targeted versus extravascular-targeted nanoparticles:
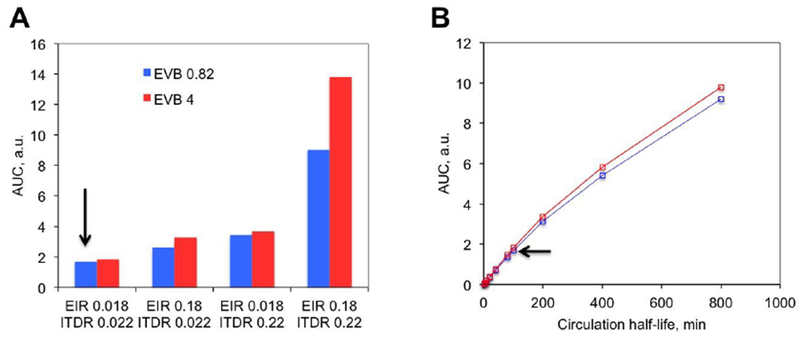
A) Extravascular targeting was simulated as an increase in EVB from 0.82 to 4.0. Arrow points to the original settings of non-targeted SWNT-PEG5000. Increasing either EIR or ITDR alone minimally improves the accumulation of non-targeted and targeted nanotubes, while increasing both EIR and ITDR increases the accumulation of non-targeted and targeted SWNTs; B) Increasing circulation half-life has a major effect on AUCT of non-targeted and extravascular-targeted nanotubes. Red line: EVB 4.0; blue line: EVB 0.82. Arrow points to the original settings of non-targeted SWNT-PEG5000.
A significant (e.g., 10-fold) increase in EIR, ITDR and EVB may be difficult to achieve with existing pharmacological approaches and nano-engineering advances. On the other hand, blood circulation half-life of nanoparticles could be prolonged by various means (37). We set out to test the effect of blood half-life using the original EIR, ITDR values for non-targeted SWNT-PEG5000. As shown in Figure 5B, the circulation half-life has a major effect on the accumulation of nanoparticles. Thus, the increase of half-life from 100 min to 400 min increases AUCT 3-fold. The same result was obtained using settings for extravascular-targeted nanoparticles (e.g., changing EVB from 0.82 to 4.0, Fig. 5B). These results also suggest that prolongation of circulation half-life has a much greater effect on AUCT than adding extravascular targeting to nanoparticles.
Role of EIR, ITDR, EVB, IVB in tumor penetration
Finally, we analyzed the effect of EIR, ITDR and EVB on the profile of nanoparticle distribution in the extravascular tumor layers (4 layers). With the EIR, ITDR and EVB values set for non-targeted SWNT-PEG5000 (IVB set to 0%), the concentration in each layer increased over time until it reached the plateau at approximately 300 min post-injection (Fig. 6A). At 2500 min post-injection, the layers 2, 3 and 4 contained 54%, 30% and 16% of nanoparticles in layer 1, respectively (Fig. 6B). This pattern of distribution around blood vessels was demonstrated for many types of nanoparticles (20, 38). Increasing EVB 5-fold while keeping the same EIR and ITDR changed the distribution in the layers such that the concentration in layers 2, 3 and 4 became 27%, 8% and 2% of nanoparticles in layer 1, respectively. This phenomenon [described initially for tumor-targeted antibodies and referred to as the “binding site barrier” (39, 40)] is due to the fact that the first layer acts as ‘sink’ that decreases the amount of material available to diffuse into the tumor. In order to understand how changes in EIR and ITDR affect the accumulation in different layers, we performed simulations where the increased EIR and/or ITDR 10-fold and increased EVB 5-fold. As seen in Fig. 6C, increasing both EIR and ITDR 10-fold leads to higher concentration of nanoparticles in all layers and better penetration into tumors. Furthermore, increasing EVB 5-fold leads to a much less dramatic binding site barrier effect than for tumors with low EIR and ITDR.
Fig. 6. Simulation of distribution of nanoparticles between tumor layers as function of EIR, ITDR and EVB:
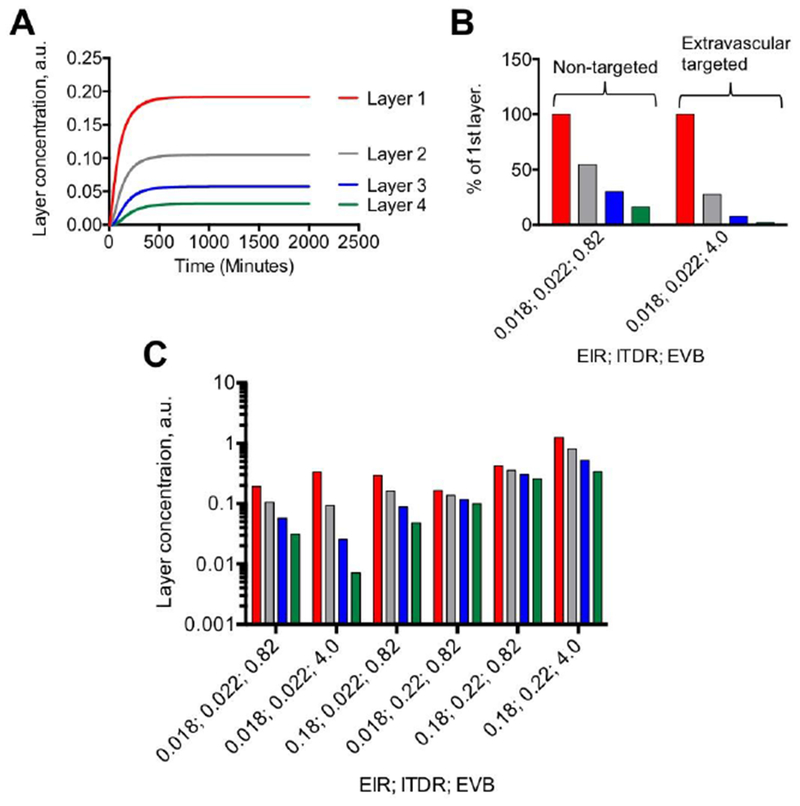
A) Accumulation profile of nanoparticles in tumor layers (concentration in arbitrary units) using settings for non-targeted SWNT-PEG5000; B) Relative concentrations of non-targeted (EVB 0.82) and extravascular-targeted (EVB 4.0) nanoparticles in tumor layers at 2500 min. Legend is the same as in A. Targeted nanoparticles showed decreased tumor penetration (binding site barrier), likely due to the first layer serving a “trap” for nanoparticles; C) Combined effect of EIR, ITDR and EVB on accumulation in tumor layers. At low extravasation and diffusion rates, increasing EVB decreases concentration in deeper layers. Increasing both EIR and ITDR 10-fold increases the concentration of nanoparticles in all layers. Note that increasing EVB 5-fold further increases accumulation in al layers, but the binding site effect is still there.
DISCUSSION
Several studies previously modeled factors that affect targeting of nanoparticles to tumors, both at cellular (41, 42) and tumor (20, 24, 25) levels. Most of these studies relied on estimated or measured parameters such as margination efficiency, tumor permeability, pore size and diffusion, intratumoral diffusion, receptor affinity and binding probability. Here we took a different approach, where we used a pharmacokinetic model to derive global parameters that encompass a large number of the sub-parameters mentioned above. Thus, EIR is determined by the margination efficiency, tumor pore permeability and pore diffusion, which are in turn determined by blood flow rate, vessel diameter, particle size, pore size and density, and intraporous viscosity. ITDR is determined by tumor composition, matrix viscosity and porosity, and particle size. EVB is a complex parameter determined by ligand affinity, binding probability, association and dissociation rates, endocytosis and exocytosis rates. After obtaining these global parameters, we performed simulations that allowed us to address important issues on the limitations and opportunities for targeted nanoparticles.
Our simulations demonstrate that targeting of the intravascular compartment has a much stronger effect on tumor accumulation than targeting of the extravascular compartment. Previous studies demonstrated successful targeting of the tumor endothelium (12–16), and one study demonstrated the limited accessibility of extravascular receptors for targeting (24). In accord with these reports, we demonstrate that extravascular targeting is dominated by extravasation and diffusion, which are rate-limiting steps of the process. Moreover, the simulation data suggest that extravascular targeting leads to nanoparticle buildup in the first layers of the tumor, and concomitantly decreases accumulation in the deeper layers (binding site barrier). In some instances, this barrier could be the layer of perivascular fibroblasts (43) or immune cells that are known to take up nanoparticles in the tumor (44, 45). In view of this phenomenon, increasing nanoparticle avidity for tumor cells by increasing the ligand density may not be a very effective strategy to boost nanoparticle accumulation.
Based on the modeling, we suggest that increasing EIR and ITDR of targeted nanoparticles can overcome the binding site barrier. This effect has been achieved, to some extent, by: a) employing tumor penetration strategies (35, 36, 46), or b) disrupting perivascular stroma (43). Several classical studies have demonstrated enhanced accumulation of antibodies and nanoparticles in tumor cells after applying peptides that increase vascular permeability and improve intratumoral diffusion (30, 47, 48). On the nanoparticle side, controlling the size, shape, surface properties and mechanical stiffness of nanoparticles (so-called 4S parameters (49) can also achieve better tumor penetration. It must be noted that changes in 4S parameters inevitably lead to changes in permeability and diffusion parameters as well as binding affinity and clearance time (20, 23–26, 50–52); therefore the overall effect is difficult to compute and therefore was not explored here. An additional strategy to improve tumor penetration of nanoparticles (in combination with the above-mentioned approaches) could be to temporarily decrease the avidity of nanoparticles for tumor receptors, for example by shielding targeting ligands with PEG, and then triggering the binding deep in the tumor by certain intratumoral stimuli, such as pH or tumor-specific proteases.
Our simulations suggest that increasing EVB efficiency together with increasing EIR and ITDR can increase the amount of nanoparticles in the tumor. Practically, a significant improvement in EVB efficiency may not be achievable without affecting non-specific macrophage uptake, extravasation, and diffusion rates. For example, adding multiple targeting ligands can trigger complement activation and immune recognition, as well as increase the size of nanomedicines (53). On the other hand, increasing IVB efficiency, even at the expense of shortening circulation half-life, can result in a significant improvement in delivery of nanoparticles. Indeed, rapid homing of nanoparticles to the vasculature and fast elimination from blood is critical for targeted imaging applications that require a high tumor-to-background ratio within reasonable time. Previous reports with vascular-targeted (anti-VEGFR or anti-CD105 decorated), short-circulating nanoparticles suggest that efficient tumor accumulation within a short time is feasible (14, 54). Endothelial targeting of nanoparticles can be improved by modifying shape, size, and ligand density (55), and recently it was found that discoid micron-sized structures are optimal for margination and tumor endothelial binding (49). Therefore, development of short circulating intravascular-targeted imaging agents with enhanced avidity for tumor endothelium is a promising strategy for nano-sized imaging agents.
There are several limitations with our current model. First, the model assumes homogenous distribution of tumor vasculature and stroma. Therefore, the parameters that were derived in this work are “averaged” over the entire tumor volume. In reality, blood vessel fraction and receptor density are lower in large tumors (56) and angiogenesis is usually concentrated in “hot spots”. In addition, macrophages and other stroma cells could be unevenly distributed throughout the tumor (57). Therefore the “local” rates may be different in different parts of the tumor. In order to derive these rates, the model would need to be further compartmentalized. Second, our model does not consider saturation of tumor uptake. Current simulations suggest that by increasing the IVB efficiency of SWNT-PEG5000-RGD to 12.8%, the accumulation of 60% ID/g can be theoretically achieved. However, values exceeding 10-20% ID/g tumor in mouse models have rarely been reported for targeted nanoparticles (12, 58–60). It is possible that in vivo saturation limits tumor accumulation. The conditions used for the analysis are presumed to be non-saturating due to low doses of nanoparticles (usually picomolar amounts for PET imaging). The issue of in vivo saturation of tumor uptake is extremely important, and to our knowledge this has not been investigated.
Future versions of the model will need to take into account tumor heterogeneity, uptake saturation and nanoparticle parameters such as size, shape, and binding avidity. Furthermore, bigger sets of data, including half-life for both targeted and non-targeted particles, and more data points, would be required for more accurate compartmental modeling. It is important to obtain these sets of data in untreated animals as well as in animals treated with different therapeutic agents. For example, tumors treated with anti-VEGF agents can dramatically affect transport properties (vascular normalization (27)) as well as receptor expression in neovasculature. We invite laboratories to share with us sets of data on nanoparticle accumulation in different tumors in order to evolve the model. Ultimately, the value of any model is measured in its ability to predict experimental results from a variety of nanoparticle types and especially in human subjects. For instance, based on human imaging data, the model can be potentially used to design nanocarriers with improved drug delivery and performance. This will accelerate development of personalized nanomedicines tailored to individual patient’s tumor physiology.
Supplementary Material
ACKNOWLEDGEMENTS
The study was funded by the University of Colorado Denver startup fund and NIH grants EB022040 and CA194058-01A1 to DS, and the Danish Agency for Science, Technology and Innovation (Det Strategiske Forskningsråd), reference 09-065746 as well as RiboBio Co. Ltd. (Guangzhou, China) to SMM. Jim Hesson of AcademicEnglishSolutions.com proofread the text.
ABBREVIATIONS USED
- AUC
area-under-the-curve
- EPR
enhanced permeability and retention
- %ID/g
percent of injected dose per gram tissue
- EIR
extravasation-intravasation rate
- ITDR
intratumoral diffusion rate
- IVB
intravascular binding rate
- EVB
extravascular binding rate
- SWNTs
single-wall carbon nanotubes
- BVF
blood volume fraction
- EVF
endothelial volume fraction
Footnotes
DECLARATION OF INTEREST
Authors declare no conflicts of interest.
REFERENCES
- 1.Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv Drug Deliv Rev 2017;108:25–38. [DOI] [PubMed] [Google Scholar]
- 2.Grodzinski P, Farrell D. Future opportunities in cancer nanotechnology--NCI strategic workshop report. Cancer Res 2014;74:1307–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hull LC, Farrell D, Grodzinski P. Highlights of recent developments and trends in cancer nanotechnology research--view from NCI Alliance for Nanotechnology in Cancer. Biotechnol Adv 2014;32:666–78. [DOI] [PubMed] [Google Scholar]
- 4.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986;46:6387–92. [PubMed] [Google Scholar]
- 5.Simberg D Opening Windows into Tumors. ACS nano 2015;9:8647–50. [DOI] [PubMed] [Google Scholar]
- 6.Lammers T, Kiessling F, Hennink WE, Storm G. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release 2012;161:175–87. [DOI] [PubMed] [Google Scholar]
- 7.Smith BR, Kempen P, Bouley D, Xu A, Liu Z, Melosh N, et al. Shape matters: intravital microscopy reveals surprising geometrical dependence for nanoparticles in tumor models of extravasation. Nano Lett 2012;12:3369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moghimi SM, Simberg D. Nanoparticle transport pathways into tumors. J Nanopart Res 2018;20:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest 1998;101:1401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu X, Lin P, Perrett I, Lin J, Liao YP, Chang CH, et al. Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer. J Clin Invest 2017;127:2007–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dvorak AM, Feng D. The vesiculo-vacuolar organelle (VVO). A new endothelial cell permeability organelle. J Histochem Cytochem 2001;49:419–32. [DOI] [PubMed] [Google Scholar]
- 12.Liu Z, Cai W, He L, Nakayama N, Chen K, Sun X, et al. In vivo biodistribution and highly efficient tumour targeting of carbon nanotubes in mice. Nature nanotechnology 2007;2:47–52. [DOI] [PubMed] [Google Scholar]
- 13.Goel S, Chen F, Hong H, Valdovinos HF, Hernandez R, Shi SX, et al. VEGF(121)-Conjugated Mesoporous Silica Nanoparticle: A Tumor Targeted Drug Delivery System. ACS applied materials & interfaces 2014;6:21677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen F, Hong H, Shi S, Goel S, Valdovinos HF, Hernandez R, et al. Engineering of hollow mesoporous silica nanoparticles for remarkably enhanced tumor active targeting efficacy. Scientific reports 2014;4:5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunjachan S, Pola R, Gremse F, Theek B, Ehling J, Moeckel D, et al. Passive versus active tumor targeting using RGD- and NGR-modified polymeric nanomedicines. Nano Lett 2014;14:972–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang L, et al. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci U S A 2007;104:932–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ElBayoumi TA, Torchilin VP. Tumor-targeted nanomedicines: enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin Cancer Res 2009;15:1973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, Peng XH, Wang YA, Wang X, Cao Z, Ni C, et al. Receptor-targeted nanoparticles for in vivo imaging of breast cancer. Clin Cancer Res 2009;15:4722–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang X, Peng X, Wang Y, Wang Y, Shin DM, El-Sayed MA, et al. A reexamination of active and passive tumor targeting by using rod-shaped gold nanocrystals and covalently conjugated peptide ligands. ACS nano 2010;4:5887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sykes EA, Chen J, Zheng G, Chan WC. Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS nano 2014;8:5696–706. [DOI] [PubMed] [Google Scholar]
- 21.Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release 2011;153:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi CH, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci U S A 2010;107:1235–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt MM, Wittrup KD. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Molecular cancer therapeutics 2009;8:2861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hussain S, Rodriguez-Fernandez M, Braun GB, Doyle FJ 3rd, Ruoslahti E. Quantity and accessibility for specific targeting of receptors in tumours. Scientific reports 2014;4:5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frieboes HB, Wu M, Lowengrub J, Decuzzi P, Cristini V. A computational model for predicting nanoparticle accumulation in tumor vasculature. PLoS One 2013;8:e56876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schluep T, Hwang J, Hildebrandt IJ, Czernin J, Choi CH, Alabi CA, et al. Pharmacokinetics and tumor dynamics of the nanoparticle IT-101 from PET imaging and tumor histological measurements. Proceedings of the National Academy of Sciences of the United States of America 2009;106:11394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chauhan VP, Stylianopoulos T, Martin JD, Popovic Z, Chen O, Kamoun WS, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nature nanotechnology 2012;7:383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Gao H, Guo N, Niu G, Ma Y, Kiesewetter DO, et al. Site-Specific Labeling of scVEGF with Fluorine-18 for Positron Emission Tomography Imaging. Theranostics 2012;2:607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng X, Zhang Y, Nystrom AM. Endocytic uptake and intracellular trafficking of bis-MPA-based hyperbranched copolymer micelles in breast cancer cells. Biomacromolecules 2012;13:3814–22. [DOI] [PubMed] [Google Scholar]
- 30.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009;16:510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol 2010;7:653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kording F, Weidensteiner C, Zwick S, Osterberg N, Weyerbrock A, Staszewski O, et al. Simultaneous Assessment of Vessel Size Index, Relative Blood Volume, and Vessel Permeability in a Mouse Brain Tumor Model Using a Combined Spin Echo Gradient Echo Echo-Planar Imaging Sequence and Viable Tumor Analysis. J Magn Reson Imaging 2014;40:1310–8. [DOI] [PubMed] [Google Scholar]
- 33.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotech 2007;2:751–60. [DOI] [PubMed] [Google Scholar]
- 34.Goel S, Chen F, Luan SJ, Valdovinos HF, Shi SX, Graves SA, et al. Engineering Intrinsically Zirconium-89 Radiolabeled Self-Destructing Mesoporous Silica Nanostructures for In Vivo Biodistribution and Tumor Targeting Studies. Adv Sci 2016;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a Tumor-Penetrating Peptide Enhances the Efficacy of Cancer Drugs. Science 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou H, Fan Z, Deng J, Lemons PK, Arhontoulis DC, Bowne WB, et al. Hyaluronidase Embedded in Nanocarrier PEG Shell for Enhanced Tumor Penetration and Highly Efficient Antitumor Efficacy. Nano Lett 2016;16:3268–77. [DOI] [PubMed] [Google Scholar]
- 37.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 2001;53:283–318. [PubMed] [Google Scholar]
- 38.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett 2009;9:1909–15. [DOI] [PubMed] [Google Scholar]
- 39.Carter P Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 2001;1:118–29. [DOI] [PubMed] [Google Scholar]
- 40.Saga T, Neumann RD, Heya T, Sato J, Kinuya S, Le N, et al. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. Proceedings of the National Academy of Sciences of the United States of America 1995;92:8999–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramezanpour M, Leung SS, Delgado-Magnero KH, Bashe BY, Thewalt J, Tieleman DP. Computational and experimental approaches for investigating nanoparticle-based drug delivery systems. Biochim Biophys Acta 2016;1858:1688–709. [DOI] [PubMed] [Google Scholar]
- 42.Duncan GA, Bevan MA. Computational design of nanoparticle drug delivery systems for selective targeting. Nanoscale 2015;7:15332–40. [DOI] [PubMed] [Google Scholar]
- 43.Miao L, Newby JM, Lin CM, Zhang L, Xu F, Kim WY, et al. The Binding Site Barrier Elicited by Tumor-Associated Fibroblasts Interferes Disposition of Nanoparticles in Stroma-Vessel Type Tumors. ACS nano 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weissleder R, Nahrendorf M, Pittet MJ. Imaging macrophages with nanoparticles. Nat Mater 2014;13:125–38. [DOI] [PubMed] [Google Scholar]
- 45.Dai Q, Wilhelm S, Ding D, Syed AM, Sindhwani S, Zhang Y, et al. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano 2018;12:8423–35. [DOI] [PubMed] [Google Scholar]
- 46.Choi I-K, Strauss R, Richter M, Yun C-O, Lieber A. Strategies to Increase Drug Penetration in Solid Tumors. Frontiers in Oncology 2013;3:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roth L, Agemy L, Kotamraju VR, Braun G, Teesalu T, Sugahara KN, et al. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene 2012;31:3754–63. [DOI] [PubMed] [Google Scholar]
- 48.Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010;328:1031–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Key J, Palange AL, Gentile F, Aryal S, Stigliano C, Di Mascolo D, et al. Soft Discoidal Polymeric Nanoconstructs Resist Macrophage Uptake and Enhance Vascular Targeting in Tumors. ACS Nano 2015;9:11628–41. [DOI] [PubMed] [Google Scholar]
- 50.Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm 2008;5:496–504. [DOI] [PubMed] [Google Scholar]
- 51.Anselmo AC, Mitragotri S. Impact of particle elasticity on particle-based drug delivery systems. Adv Drug Deliv Rev 2016. [DOI] [PubMed] [Google Scholar]
- 52.Doshi N, Mitragotri S. Macrophages recognize size and shape of their targets. PLoS One 2010;5:e10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang G, Griffin JI, Inturi S, Brenneman B, Banda NK, Holers VM, et al. In Vitro and In Vivo Differences in Murine Third Complement Component (C3) Opsonization and Macrophage/Leukocyte Responses to Antibody-Functionalized Iron Oxide Nanoworms. Front Immunol 2017;8:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goel S, Chen F, Hong H, Valdovinos HF, Hernandez R, Shi S, et al. VEGF(1)(2)(1)-conjugated mesoporous silica nanoparticle: a tumor targeted drug delivery system. ACS applied materials & interfaces 2014;6:21677–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin A, Sabnis A, Kona S, Nattama S, Patel H, Dong JF, et al. Shear-regulated uptake of nanoparticles by endothelial cells and development of endothelial-targeting nanoparticles. Journal of biomedical materials research Part A 2010;93:833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Imoukhuede PI, Popel AS. Quantitative fluorescent profiling of VEGFRs reveals tumor cell and endothelial cell heterogeneity in breast cancer xenografts. Cancer Med 2014;3:225–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun 2015;6:8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dawidczyk CM, Russell LM, Searson PC. Nanomedicines for cancer therapy: state-of-the-art and limitations to pre-clinical studies that hinder future developments. Front Chem 2014;2:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liang M, Liu X, Cheng D, Liu G, Dou S, Wang Y, et al. Multimodality Nuclear and Fluorescence Tumor Imaging in Mice Using a Streptavidin Nanoparticle. Bioconjugate Chem 2010;21:1385–8. [DOI] [PubMed] [Google Scholar]
- 60.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, et al. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials 2016;1:16014. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.