

Abstract
Here, we describe a fourth case of a human with a de novo KCNJ6 (GIRK2) mutation, who presented with clinical findings of severe hyperkinetic movement disorder and developmental delay, similar to Keppen-Lubinsky syndrome but without lipodystrophy. Whole exome sequencing of the patient’s DNA revealed a heterozygous de novo variant in the KCNJ6 (c.512T>G, p.Leu171Arg). We conducted in vitro functional studies to determine if this Leu-to-Arg mutation alters the function of GIRK2 channels. Heterologous expression of the mutant GIRK2 channel alone produced an aberrant basal inward current that lacked G protein activation, lost K+ selectivity and gained Ca2+ permeability. Notably, the inward current was inhibited by the Na+ channel blocker QX-314, similar to the previously reported weaver mutation in murine GIRK2. Expression of a tandem dimer containing GIRK1 and GIRK2(p.Leu171Arg) did not lead to any currents, suggesting heterotetramers are not functional. In neurons expressing p.Leu171Arg GIRK2 channels, these changes in channel properties would be expected to generate a sustained depolarization, instead of the normal G protein-gated inhibitory response, which could be mitigated by expression of other GIRK subunits. The identification of the p.Leu171Arg GIRK2 mutation potentially expands the Keppen-Lubinsky syndrome phenotype to include severe dystonia and ballismus. Our study suggests screening for dominant KCNJ6 mutations in the evaluation of patients with severe movement disorders, which could provide evidence to support a causal role of KCNJ6 in neurological channelopathies.
Keywords: KCNJ6, weaver mouse, movement disorder, channelopathy, inward rectifier, Kir3
Introduction
Genetic neurological channelopathies are a group of heterogeneous disorders, usually with dominant inheritance, and causing some form of paroxysmal neurological disturbances in function, which may become permanent in time (Spillane et al., 2016). For example, ion channelopathies have been implicated in diseases including epileptic encephalopathies, ataxia, paroxysmal dyskinesias, migraine, pain syndromes, skeletal muscle disorders, and periodic paralysis (Spillane et al., 2016). Under normal conditions, ion channels serve an important role of permitting rapid and selective movement of ions across plasma membranes, influencing the excitability of neurons and subsequent signaling in the brain. Channelopathies result from defects in the ion channel function or from changes in trafficking to the plasma membrane. A large number of channelopathies involve potassium channels, which can be divided into three families: voltage-gated K+ channels (KV1–18), two-pore K+ channels (K2P1- K2P18), and inwardly rectifying K+ channels (KIR1-KIR7) (Serratrice et al., 2010). KV channels contribute to the repolarization of the action potential. Defects in this family of channels commonly lead to some form of epilepsy. For example, mutations in KCNQ2 (KV7.2) and KCNA1 (KV1.2) cause benign familial neonatal seizures and myoclonic epilepsy, respectively (Villa and Combi, 2016). Thus, loss of these channels leads to dramatic increase in neuronal excitability. K2P channelopathies have been linked to familial migraine with aura, and have been attributed to have a role in pain sensation (Serratrice et al., 2010).
Inwardly rectifying potassium (KIR) channels play a key role in the maintenance of the resting potential and regulation of cell excitability (Hibino et al., 2010, Lüscher and Slesinger, 2010). These channels lack the voltage-sensitivity of their Kv counterparts and preferentially limit the outward flow of K+ to a voltage range near the resting membrane potential. KIR channels can have high basal activity (constitutively open), such as KIR2 channels, or low basal activity that can be enhanced by ligands such as G proteins and alcohol, such as KIR3 channels. There are seven different KIR families (KIR1-KIR7) that form either homotetramers or heterotetramers. However, KIR channels typically assemble with only members from the same subfamily, for example, KIR3.1 does not coassemble with KIR2.1 but it does co-assemble with KIR3.2 (Hibino et al., 2010, Lüscher and Slesinger, 2010). The constitutively active KIR2 (KCNJ2) has been associated with Andersen syndrome, a human disease that is characterized by periodic paralysis, cardiac arrhythmia, and dysmorphism (Plaster et al., 2001). Point mutations in KIR6.2 (KCNJ11) have been described for the human disease developmental delay, epilepsy and neonatal diabetes mellitus (DEND) syndrome, a treatable channelopathy of the brain and pancreas (Hibino et al., 2010, Lüscher and Slesinger, 2010). The G protein-gated inwardly-rectifying K+ (GIRK) channel belongs to the KIR3 subfamily and is a regulator of cardiac and neuronal excitability (Hibino et al., 2010, Lüscher and Slesinger, 2010). There are four GIRK channel subunits in humans, referred to as KIR3.1 (KCNJ3), KIR3.2 (KCNJ6, Girk2), KIR3.3 (KCNJ9), and KIR3.4 (KCNJ5) (for review, see Hibino et al., 2010, Lüscher and Slesinger, 2010). Mice lacking GIRK channels display altered responses to addictive drugs and in the case of the Girk2 knockout, result in epileptic seizures (for review, see Rifkin et al., 2017). Heretofore, human neurological diseases caused by mutations in GIRK channels have been uncommon.
Recently, dominant mutations in KCNJ6 (GIRK2) have been linked to Keppen-Lubinsky Syndrome (MIM# 614098) (Gorlin RJ, 2001), a disorder characterized by lipodystrophy, severe developmental delay, intellectual disability, hypertonia, hyperreflexia and growth retardation (Gorlin RJ, 2001, Masotti et al., 2015). Three patients were reported to have KCNJ6 mutations near the ion selective pore; one patient with a mutation (p. Gly154Ser) corresponding to the developmentally impaired weaver mouse (Rakic and Sidman, 1973a, b, Goldowitz and Mullen, 1982, Hatten et al., 1986, Goldowitz, 1989, Patil et al., 1995, Hess, 1996) and two patients with deletions in the pore (p.Thr152del). These three patients all show lipodystrophy, which ranges from a generalized loss of adipose tissue in one case, to a restricted loss of facial adipose tissue in two cases (Masotti et al., 2015). (De Brasi et al., 2003, Basel-Vanagaite et al., 2009). Thus, lipodystrophy is a hallmark of Keppen-Lubinsky Syndrome.
Here we report a case study with a de novo KCNJ6 missense mutation [NM_002240: c.512T>G; NP_002231: p. Leu171Arg], without recognizable lipodystrophy, i.e., no obvious absence of facial adipose tissue. Instead, the patient presented with developmental delay, hypotonia, and a severe hyperkinetic movement disorder. Epilepsy was also not present. The p. Leu171Arg mutation was determined to be a gain-of-function, with dramatic changes in G protein activation and ion selectivity. Though, p. Leu171Arg resides in a different region of the protein, the electrophysiological phenotype resembles that described in the murine weaver (wv) mouse, which has extensive developmental defects and also carries a mutation in KCNJ6 (Patil et al., 1995, Hess, 1996).
Experimental Procedures
Whole Exome Sequencing.
Patient and family were enrolled into the TIDEX gene discovery study (UBC IRB approval H12–00067) and provided informed and written consent for data and sample collection, WES, as well as publication of the current case report. WES was performed for the index and her unaffected parents using the Agilent SureSelect kit and Illumina HiSeq 2000 (Perkin-Elmer, USA). Data were analyzed using our semi-automated bioinformatics pipeline (Tarailo-Graovac et al., 2016). Briefly, the sequencing reads were aligned to the human reference genome version hg19 and rare variants were identified and assessed for their potential to disrupt protein function, and subsequently screened under a series of genetic models: homozygous, hemizygous, compound heterozygous and de novo. Sanger sequencing was performed to confirm the presence of the variants, as called by the exome sequencing data.
Protein modeling.
The sequence of full-length human GIRK2 protein (NP_002231.1) was submitted for automated modeling and template selection using the SWISS-MODEL web interface (Arnold et al., 2006, Guex et al., 2009, Kiefer et al., 2009, Biasini et al., 2014). This software returned a model structure (in PDB format) for GIRK2 residues 53–378 based on the template PDB 3SYO, chain A (>99% sequence identity). The modeling of mutated sequence was performed using the same template. The models of wild-type and mutated proteins were visualized and inspected by the UCSF (University of California, San Francisco) Chimera visualization system (http://www.cgl.ucsf.edu/chimera) (Pettersen et al., 2004).
Molecular Biology and Cell Culture.
A single point mutation L173R was introduced into murine GIRK2c (referred to as GIRK2) using site-directed mutagenesis (QuikChange II XL, Agilent Technology) and confirmed by automatic DNA sequencing. Human Embryonic Kidney 293T (HEK293T) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) Fetal Bovine Serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 1X Glutamax (ThermoFisher) at 37° C and in a 5% CO2 humidified atmosphere. Cells were plated onto poly-D-lysine (100 μg/ml; SigmaAldrich) coated 12-mm glass coverslips in 24-well plates and transiently transfected with cDNA using Lipofectamine 2000 (ThermoFisher). HEK293T cells were transfected with GIRK channel cDNAs (GIRK2 (0.4 μg) or GIRK2(L173R) (0.4 μg), GIRK1-GIRK2 dimer (0.2 μg), or GIRK1-GIRK2(L173R) dimer (0.5 μg) along with GABAB1b (0.2 μg), GABAB2 (0.2 μg) and eYFP cDNA (0.02 μg; to identify transfected cells). In some experiments, GIRK1 (0.2 μg) was transfected with GIRK2 (0.2 μg) or GIRK2(L173R) (0.2 μg). QX-314 (100 μM) was routinely added to the culture medium to promote survival of transfected cells (Kofuji et al., 1996, Slesinger, 2001).
Electrophysiology.
Whole-cell patch-clamp recordings were made as described previously (Bodhinathan and Slesinger, 2013). Borosilicate glass electrodes (Warner Instruments) of 3–6 MΩ were filled with an intracellular solution containing 130 mM KCl, 20 mM NaCl, 5 mM EGTA, 5.46 mM MgCl2, 2.56 mM K2ATP, 0.3 mM Li2GTP and 10 mM HEPES (pH 7.4, ~313 mOsm). The extracellular ‘20K’ solution contained 20 mM KCl, 140 mM NaCl, 0.5 mM CaCl2, 2 mM MgCl2, and 10 mM HEPES (pH 7.4, ~318 mOsm). The extracellular ‘5K’ and ‘2K’ solution contained otherwise identical compositions but different concentrations of KCl and NaCl (5K: 5mM KCl, 155mM NaCl; 2K: 2mM KCl, 158mM NaCl). Currents were elicited at 0.5 Hz with a voltage step to −120 mV from a holding potential of −40 mV, followed by a ramp voltage protocol (−120 mV to +50 mV). K+ currents were measured at −120 mV with either extracellular solution or drug solution. Drugs were diluted into extracellular solution with a final concentration of 100 μM Baclofen, 100 μM QX-314, 100 mM Propanol, and 1 mM BaCl2. The percentage change in current was calculated by measuring the amplitude of the basal (Ibasal) and drug-induced current (Idrug) using the equation %I = (Idrug – Ibasal) / Ibasal. The voltage-dependent activation was measured in response to a voltage-step from −40 mV to −120 mV. The reversal potential was measured by examining the zero current potential for the Ba2+-inhibited and QX-314-inhibited currents for wild-type and L173R, respectively. The basal current density was measured at −70 mV (i.e. approximating the resting potential of a neuron) with the ‘5K’ solution. The rectification ratio was calculated by measuring the current at −20 mV + Erev (inward) and dividing by the current at +20 mV + Erev (outward) with the ‘5K’ solution.
Ca2+ imaging.
HEK293T cells stably expressing a genetically encoded Ca2+ sensor (Twitch 2B) (Thestrup et al., 2014) were transiently transfected with GABAB1b and GABAB2 receptor cDNAs, and either GIRK2 wild-type or GIRK2(L173R) cDNA. Transfected cells were incubated with 100 μM QX-314 to suppress basal leak current prior to imaging. One day after transfection, cells were plated on poly-D-lysine-coated 96-well plates, incubated for 1h, and then the media was replaced with 100 μl artificial cerebral spinal fluid (ACSF; 125 mM NaCl, 5 mM KCl, 10 mM D-glucose, 10 mM HEPES, 3.1 mM CaCl2, and 1.3 mM MgCl2, pH 7.4) with or without 100 μM QX-314 to inhibit GIRK2(L173R). Plates were loaded into the FlexStation3 (Molecular Devices), and basal FRET ratio was measured at 37°C using 430nm excitation and emission at 480nm for CFP, and 530nm for YFP. Increases in intracellular Ca2+ level lead to higher basal FRET ratios.
Statistical tests.
Significant differences with respect to current density and rectification ratio between wild-type and L173R mutant channel were determined by unpaired t test. In calcium imaging experiment, significant differences among groups were determined by one-way ANOVA followed by Tukey’s test. P < 0.05 was considered significant.
Results
Clinical findings
The patient is a 4-year old girl from non-consanguineous parents, with an unremarkable pregnancy and term delivery. Her birth weight was 3210 g (30th percentile), and there were no neonatal complications. The first concerns arose when she did not achieve head control by the age of 6 months. Her mother also noticed posturing of her legs, abnormal movements of her arms, and arching of her back. The patient had frequent episodes in which her eyes rolled up. These movements at times were quite prominent, causing her significant discomfort and a neurologist assessed her for the possibility of seizures. An EEG showed background dysrhythmia but no epileptiform discharges (Figure 1). At 8 months she was diagnosed with global developmental delay, hypotonia and a hyperkinetic movement disorder. MRI brain performed at 9 months was normal, with normal myelination for age.
Figure 1: EEG samples in hyperkinetic movements.
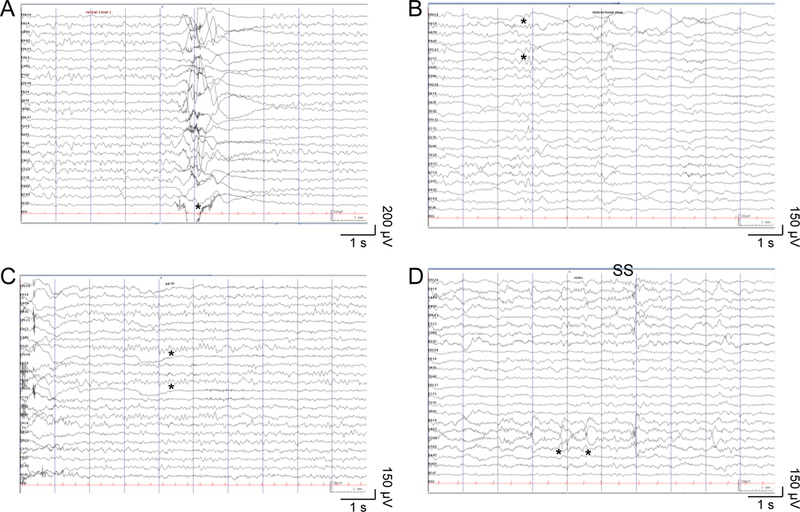
(A) Paroxysmal hyperkinetic movement (*) with no ictal epileptiform electrographic correlate. (B) Occasional poorly-formed bilateral frontal sharp waves (*) in non-REM sleep. (C) Normal 9 Hz alpha posterior dominant rhythm (*) during relaxed wakefulness with eyes closed. (D) Normal symmetric and synchronous sleep spindles (SS) and vertex waves (*) in non-REM sleep. Ceegraph EEG: anterior-posterior bipolar montage, sensitivity 15 μV/mm, time scale 30 mm/s, LFF 0.05 Hz, HFF 70 Hz.
Biochemical investigations were initiated at age of 14 months according to the Treatable Intellectual Disabilities (TIDE) protocol (van Karnebeek et al., 2014). These included normal plasma amino acids, acylcarnitine profile, ammonia, lactate, liver enzymes, CK, copper, ceruloplasmin, urine organic acids, and purines and pyrimidines. CSF glucose (including CSF/plasma glucose ratio), amino acids, and lactate were normal, but there was a mildly low level of homovanillic acid (dopamine metabolite) of 214 nmol/L (reference range: 294–1115 nmol/L), with normal levels of 5HIAA (serotonin metabolite), folate and biopterin metabolites. Genetic testing included molecular analysis of TH (encoding tyrosine hydroxylase); mutation analysis and deletion/duplication screening were both negative. Chromosome microarray was unremarkable.
At 18 months, the patient showed severe global developmental delay; she had poor head control, was unable to sit or roll, did not hold or reach for objects, and was not crawling. She could recognize her parents, was smiling and cooing, but not babbling. She did not gain developmental milestones over the next year, but also did not experience any regression. Her growth parameters were between 25–50th percentile for weight, 85–97th percentile for height, and just below the 50th percentile for head circumference.
She had feeding difficulties with two episodes of aspiration pneumonia. At the age of 2 years, she had a gastrostomy feeding tube placed, and was dependent on this for feeds. At the age of 4 years, her movement disorder can be described as dystonic posturing of her arms and legs, opisthotonus, ballismus, and choreoatethosis. She also has some violent mycolonic jerks involving her arms, and torso, mostly when she is excited, afraid, or before falling asleep (Videos 1, 2, 3). These movements can wake her up from sleep. All movements were recorded with 24-hours video-EEG monitoring with no associated epileptiform activity; only occasional bifrontal, sharply contoured waveforms were seen during the recording. She is quite irritable most of the time. Most recently she had brief episodes of oculogyric crisis.
For possible treatments, L-dopa therapy, starting with a small dose with gradual titration, was tried but the patient experienced a severe exacerbation of her hyperkinetic movements, even with the lowest dose, requiring discontinuation. She was also tried on a small dose of dopamine agonist (Pramipexole), which she did not tolerate. She had minor improvement in her movements with initiation of Clonazepam, but she had increased salivation, which limited its use. Other medications were gradually introduced, including Trihexyphenidyl, Gabapentin and finally, Tetrabenazine. This combination has decreased the severity, amplitude and frequency of the movements, although she continues to have severe exacerbations on a daily basis. These exacerbations are mainly associated with excitement, anxiety or frustration. When these movements continue for more than half an hour, parents can give a small dose of Clonidine, which usually helps her movements to calm down.
Genetic Analysis
To identify the genetic basis of her neurologic condition, the family was enrolled in the TIDEX discovery research program for trio whole exome sequencing (WES) (Tarailo-Graovac et al., 2016). The WES analyses revealed 8 candidate genes affected by rare variants: eight compound heterozygous (AMER2, DNAH3, GAS2L2, MGAM2, SRGAP1, TEX15 and VWDE) and one de novo variant (KCNJ6); there were no homozygous or hemizygous candidate variants. Of these 8 candidates (Table 1), a de novo variant located on chromosome 21 in KCNJ6 (MIM 600877) was considered a functional candidate: g. 39086948A>C [NM_002240: c. 512T>G; NP_002231: p. Leu171Arg] (Figure 2A). This variant is novel; it has not been observed in dbSNP (Build 147), Exome Aggregation Consortium (ExAC) (Lek et al., 2016), or our in-house genome database (comprising of more than 400 exome and genome sequencing results). The variant is predicted to be deleterious by multiple tools: high 25.8 CADD (Combined Annotation Dependent Depletion) score (Kircher et al., 2014), a “damaging” SIFT (Kumar et al., 2009) (Sorting Tolerant From Intolerant) prediction score of 0.001 (cutoff = 0.05) and PolyPhen2 “probably damaging score” of 1.000 (Adzhubei et al., 2013). Sanger sequencing confirmed that the proband was heterozygous for the variant, while the parents were homozygous wild-type. Similar to the three other reported patients (Masotti et al., 2015), the missense variant is located in exon 3. Using the atomic resolution structure of GIRK2 (Whorton and MacKinnon, 2013), we investigated the molecular interactions around the p.L171R using a structural model of human GIRK2. The mutation p.Leu171Arg allows p.Arg171 to form H-bonds with Glu148 in the adjacent subunit (Figures 2B and 2C). This interaction could lead to the formation of a more stable structure and limit conformational movements. Therefore, this mutant conformation may prevent a correct opening/closing of the channel and/or impair its ion selectivity.
Table 1:
Candidate genes from the trio-WES data
| Gene | Description | OMIM disease | Nucleotide Change (hg19, cDNA) |
Protein Change (AA, transcript) |
Inheritance | In silico prediction scores | gnomAD frequency |
Exclusion |
|---|---|---|---|---|---|---|---|---|
| AMER2 | APC membrane recruitment protein 2 | NONE | chr13: 25743858 T>C c.1900A>G chr13: 25745168 A>G c.590T>C | p.Met634Val NP_689917 p.Leu197Pro NP_689917 | Compound heterozygous | CADD (0.001) SIFT (Tolerated; 1.000) PolyPhen2(BENIGN; 0.000) CADD (24.4) SIFT (DAMAGING; 0.003) PolyPhen2(P. DAMAGING; 0.986) | 0 0 |
Limited knowledge of gene function + one variant is predicted benign |
| DNAH3 | dynein axonemal heavy chain 3 | NONE | chr16: 20975457 T>C c.9749A>G chr16: 20959830 T>C c.11318A>G | p.Lys3250Arg NP_060009 p.Tyr3773Cys NP_060009 | Compound heterozygous | CADD (15.46) SIFT (DAMAGING; 0.001) PolyPhen2(BENIGN; 0.109) CADD (17.34) SIFT (DAMAGING; 0.044) PolyPhen2(BENIGN; 0.053) | 0.001627 4 Hom 0.004640 44 Hom | Does not fit limited knowledge of gene function + one variant observed in 44 homozygotes in gnomAD |
| GAS2L2 | growth arrest specific 2 like 2 | NONE | chr17: 34074164 G>T c.956C>A chr17: 34079730 C>T c.140G>A | p.Pro319His NP_644814 p.Gly47Glu NP_644814 | Compound heterozygous | CADD (24.2) SIFT (DAMAGING; 0.001) PolyPhen2(P. DAMAGING; 1.000) CADD (24.5) SIFT (Tolerated; 0.192) PolyPhen2(P. DAMAGING; 0.884) | 4.063e-6 0 Hom 0.0006211 0 Hom | Does not fit gene function |
| MGAM2 | maltase-glucoamylase 2 (putative) | NONE | chr7: 141840614 T>A c.1099T>A chr7: 141831870 G>A c.560G>A | p.Ser367Thr NP_001280555 p.Ser187Asn NP_001280555 | Compound heterozygous | CADD (23.1) SIFT (Tolerated; 0.642) PolyPhen2(BENIGN; 0.026) CADD (23.8) SIFT (DAMAGING; 0.050) PolyPhen2(BENIGN; 0.010) | 0.0001769 0 Hom 0.003703 16 Hom | Does not fit gene function + one variant was observed in 16 homozygotes in gnomAD |
| SRGAP1 | SLIT-ROBO Rho GTPase activating protein 1 | {Thyroid cancer, nonmedullary, 2}, AD, 188470 | chr12: 64458925 G>A c.1051G>A chr12: 64502714 A>C c.1816A>C chr12: 64521811 A>G c.2711A>G | p.Val351Ile NP_065813 p.Ile606Leu NP_065813 p.Asn904Ser NP_065813 | Compound heterozygous | CADD (23.6) SIFT (Tolerated; 0.051) PolyPhen2(BENIGN; 0.007) CADD (10.05) SIFT (Tolerated; 1.000) PolyPhen2(BENIGN; 0.045) CADD (0.006) SIFT (Tolerated; 0.770) PolyPhen2(BENIGN; 0.000) | 8.160e-6 0 Hom 0.0001111 0 Hom 0.0002742 1 Hom | Does not fit phenotype or mode of inheritance + two variants are predicted benign |
| TEX15 | testis expressed 15, meiosis, and synapsis associated | NONE | chr8: 30703949 C>T c.2585G>A chr8: 30705135 A>C c.1399T>G | p.Arg862His NP_112561 p.Trp467Gly NP_112561 | Compound heterozygous | CADD (5.474) SIFT (Tolerated; 1.000) PolyPhen2(BENIGN; 0.000) CADD (0.857) SIFT (Tolerated; 0.329) PolyPhen2(BENIGN; 0.000) | 2.897e-5 0 Hom 2.858e-5 0 Hom | Does not fit gene function + both variants are predicted benign |
| VWDE | von Willebrand factor D and EGF domains | NONE | chr7: 12370825 T>C chr7: 12412800 G>A c.1390C>T | NA p.Arg464Cys NP_001129396 | Compound heterozygous | CADD (21.7) SPLICE_ACCEPTOR VARIANT CADD (34) SIFT (DAMAGING; 0.000) PolyPhen2(P. DAMAGING; 1.000) | 3.366e-5 0 Hom 0.0001387 0 Hom | Does not fit the limited knowledge of gene function |
| KCNJ6 | potassium voltage-gated channel subfamily J member 6 | Keppen-Lubinsky syndrome, AD, 614098 | chr21: 39086948 A>C c.512T>G | p.Leu171Arg NP_002231 | Heterozygous De novo | CADD (25.8) SIFT (DAMAGING; 0.001) PolyPhen2(P. DAMAGING; 1.000) | 0 | Fits with reported phenotype and predicted to be damaging |
Figure 2: Identification of mutation in human KCNJ6.
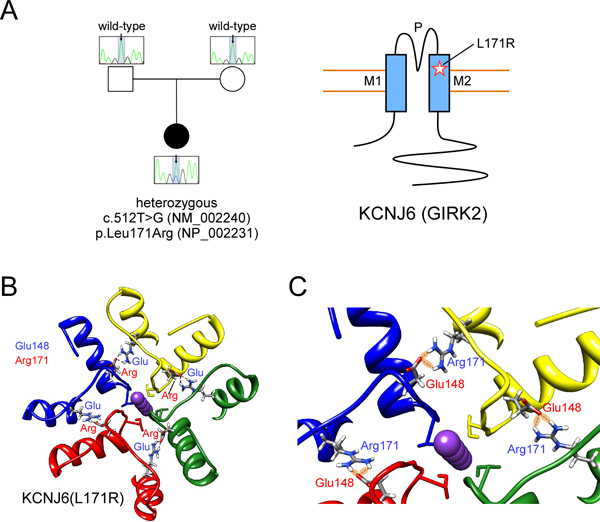
(A) Sequencing of patient and parent DNA revealed a mutation in KCNJ6. Cartoon shows position of L171R mutation in GIRK2 ion channel (M1, M2: transmembrane domains; P: pore). (B) 3D structural model of human GIRK2 obtained by homology modeling from mouse GIRK2 channel (PDB: 3SYO). The mutation L171R allows Arg171 to potentially form H-bonds with Glu148 in the adjacent subunit. This electrostatic interaction could produce a more stable structure and impair conformational movements, i.e., opening/closing of the channel, or alter ion selectivity. (C) A detail of the 3D structural model of human KCNJ6 shows the network of interactions (H-bonds) that stabilize the conformation of the outer part (the selectivity filter) of the KCNJ6 channel.
Functional studies on mutant GIRK2 channel
To study the functional effect of the human L171R mutation in GIRK2, we used a heterologous expression system (i.e., HEK-293T cells) and expressed the wild-type or mutant murine GIRK2 channels with the GABAB1b/GABAB2 receptors. Murine and human GIRK2 are identical except for the first two amino acids, thus murine GIRK2(L173) is homologous to human GIRK2(L171) (see Figure 3A). We examined the effect of the GABAB receptor agonist, Baclofen, as well as activation with alcohol, e.g., propanol (PrOH), a G protein-independent activator (Mutneja et al., 2005, Bodhinathan and Slesinger, 2013). In contrast to wild-type GIRK2, which showed large Baclofen- and PrOH-induced currents (Figure 3B), we observed little change with Baclofen and a small inhibition with PrOH with the GIRK2(L173R) mutant channel (Figure 3C). Thus, GIRK2(L173R) channels exhibit impairment of both G protein- and alcohol-dependent activation. In the brain, GIRK2 commonly coassembles with the GIRK1 subunit to form heterotetramers (Kofuji et al., 1995, Liao et al., 1996). We therefore examined the effect of coexpressing the GIRK1 subunit with GIRK2(L173R). Like GIRK2(L173R), GIRK1 + GIRK2(L173R) co-expression showed no response to Baclofen and inhibition with PrOH (Figure 3D,E).
Figure 3: Change in gating and ion selectivity of mouse GIRK2(L173R) channels.
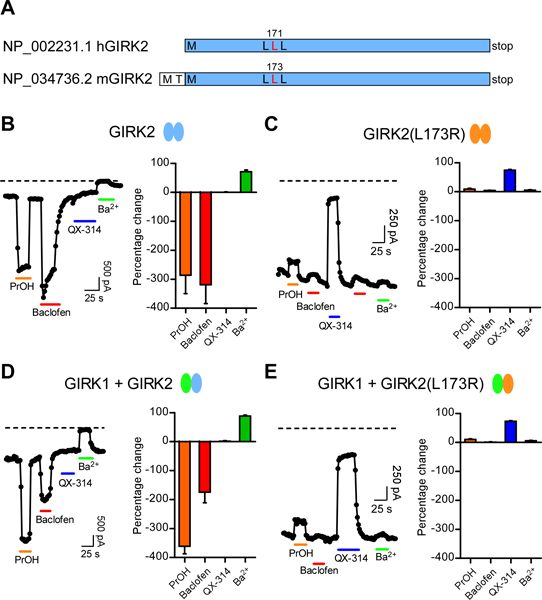
(A) Cartoon shows similarity between human and mouse GIRK2 protein, with homologous Leu highlighted (red). (B,C) Whole-cell patch-clamp currents are plotted as a function of time for wild-type GIRK2 (B) and mutant GIRK2(L173R) (C) channels. Currents were measured at −120 mV from HEK293T cells transfected with GABAB1b/B2 receptor cDNA and either GIRK1, GIRK2 and/or GIRK2(L173R) cDNAs. Plots show the response to bath application of PrOH (100 mM, orange bar), Baclofen (100 μM, red bar), the sodium channel inhibitor QX-314 (100 μM, blue bar) and the Kir potassium channel inhibitor Ba2+ (1 mM, green bar). Note that the mutant GIRK2(L173R) channel shows no activation with Baclofen or PrOH, lack sensitivity to inhibition with Ba2+, but is inhibited with QX-314 (100 μM). Similar results were observed for HEK293T cells expressing heterotetramers of GIRK1+GIRK2 (D) or GIRK1+GIRK2(L173R). (E) channels. Dashed line indicates zero current level. Bar graphs show average percentage change (± SEM) in current with indicated compounds (N=8–9). A negative percentage change indicates activation.
For both GIRK2(L173R) and GIRK1/GIRK2(L173R) transfected cells, we observed a large agonist-independent basal current, which could not be blocked by the potassium channel inhibitor Ba2+ (Figure 3C,E). Interestingly, the developmentally impaired weaver mouse has a mutation in GIRK2 (GIRK2wv) that impairs G protein activation and produces insensitivity to extracellular Ba2+ (Patil et al., 1995, Slesinger et al., 1996). Previously, it was found that the Na+ channel inhibitor QX-314 could inhibit the agonist-independent basal current of GIRK2wv channels (Kofuji et al., 1996). We therefore examined the effect of direct application of QX-314 on the basal currents, and observed inhibition of currents for GIRK2(L173R) as well as for GIRK1+GIRK2(L173R), but not of currents from wild-type channels (Figure 3B–E).
The co-expression of GIRK1 with GIRK2(L173R) led to currents that were similar to those of GIRK2(L173R) alone, suggesting that the either the heterotetramer adopts the properties of the GIRK2(L173R) channel, or that GIRK1 assembles with GIRK2(L173R) but is not functional. To address this, we compared the kinetics of channel activation for the basal current. Previous studies have shown that the presence of the GIRK1 subunit in a heterotetramers slows the voltage-dependent activation (Kubo et al., 1993). With a voltage step from −40 to −120 mV, we observed the typical slow activation kinetics, i.e., downward increase in current, with GIRK1+GIRK2 but not with GIRK2 channels (Slesinger et al., 1996). By contrast, the kinetics of voltage-dependent activation of GIRK1+GIRK2(L173R) heterotetramers was fast, similar to GIRK2 channels alone (Figure 4A,B). To further examine whether GIRK1 and GIRK2(L173R) form functional heterotetramers, we created tandem dimers of wild-type (GIRK1-GIRK2) (Zhou et al., 2001) and mutant (GIRK1-GIRK2(L173R)) channels. Like co-expression of GIRK1+GIRK2, wild-type GIRK1-GIRK2 channels showed large Baclofen-induced current, Ba2+-sensitive basal current, and no inhibition with QX-314 (Figure 4C). By contrast, there was no effect of Baclofen, QX-314 or Ba2+ on cells expressing the GIRK1-GIRK2(L173R) dimer (Figure 4C). In 12 cells, expression of GIRK1-GIRK2(L173R) cDNA did not produce any basal currents that were inhibited by QX-314 or Ba2+, or activated by Baclofen or Propanol (Figure 4D). These results suggest the heterotetramers containing the GIRK2(L173R) are non-functional, suggesting a dominant-negative effect of GIRK2(L173R) with other subunits.
Figure 4: GIRK1 and GIRK2(L173R) form dominant negative heterotetramers.
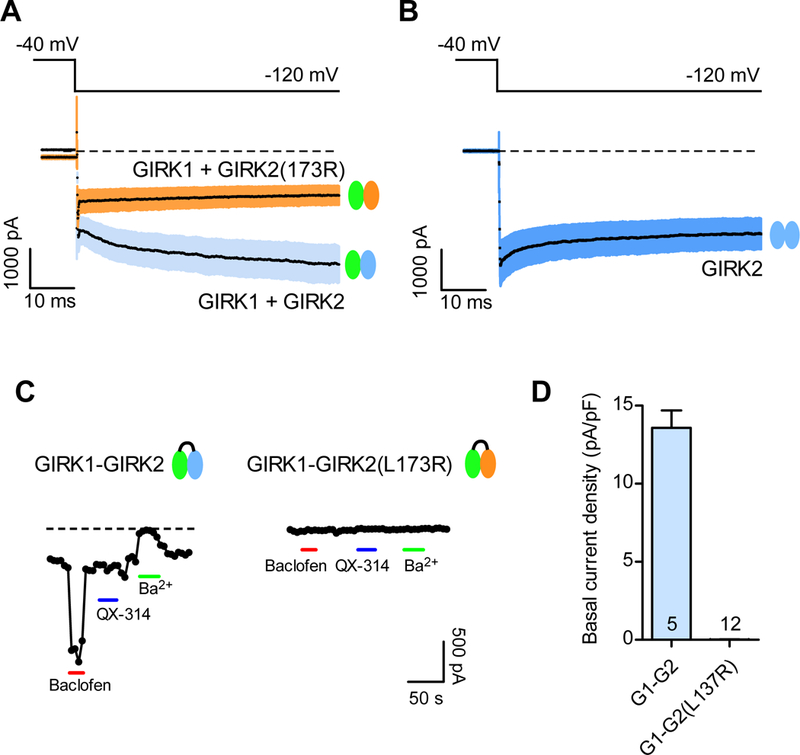
(A,B) Basal currents were elicited with a voltage step from −40 mV to −120 mV for GIRK1+GIRK2 channels (Ba2+-sensitive, light blue), GIRK2 channels (Ba2+-sensitive, blue) and mutant GIRK1+GIRK2(L173R) (QX-314-sensitive, pink). The mean current (black line) is shown with ± SEM (N=9). Dashed line indicates zero current level. Note similarity in kinetics for GIRK1+GIRK2(L173R) and GIRK2 alone. (C) Plots show the response to bath application of PrOH (100 mM, orange bar), Baclofen (100 μM, red bar), QX-314 (100 μM, blue bar) and Ba2+ (1 mM, green bar) for HEK293T cells expressing the GIRK1-GIRK2 dimer and GIRK1-GIRK2(L173R) dimer. Note the mutant heterotetramers show no activation or inhibition. (D) Bar graph shows average basal current density (± SEM) for GIRK1-GIRK2 dimer (Ba2+-sensitive) and GIRK1-GIRK2(L173R) dimer (QX-314 sensitive).
The inhibition of GIRK2wv channels with QX-314 but not with Ba2+ suggested a defect in the channel pore that altered ion selectivity, converting the channel from K+ selective to cationic non-selective (Kofuji et al., 1996, Slesinger et al., 1996, Slesinger, 2001). If GIRK2(L173R) channels also lose K+ selectivity and become non-selective, we would expect the reversal potential (i.e., zero current potential) for GIRK2(L173R) channels to shift to depolarized potentials. To examine this possibility, we examined the current-voltage relationship for wild-type and mutant channels under different extracellular K+ concentrations (Figure 5). In these experiments, the extracellular solution contained either 2mM, 5mM, or 20 mM KCl. For WT channels, the currents reverse at different membrane potentials, close to the calculated Nernst potential for K+. By contrast, the currents for GIRK2(L173R) channels reversed at ~ 0 mV regardless of different external [K+], indicating a loss of K+ selectivity (Figure 5A). To quantify this change in selectivity, we measured the reversal potentials (Erev) for the QX-314-inhibited basal currents for GIRK2(L173R) channels, and compared them to the Ba2+-inhibited currents for wild-type channels. We plotted reversal potentials as a function of external [K+]. GIRK2(L173R) channels showed a shift in the reversal potential toward 0 mV, which is independent to extracellular K+ concentration (Figure 5B). We next examined whether GIRK2(L173R) channels were permeable to Ca2+. To test this, we transiently transfected HEK293T cells stably expressing a genetically encoded Ca2+ sensor Twitch 2B (Thestrup et al., 2014), along with the GABAB1b/B2 receptor and either GIRK2 or GIRK2(L173R). Twitch 2B generates a FRET signal when intracellular Ca2+ levels increase (Thestrup et al., 2014). We plotted the normalized FRET ratios for different groups. L173R expressing cells showed significantly higher basal FRET compared to cells expressing wild-type channels, suggesting an increase in intracellular Ca2+ levels. Moreover, addition of QX-314 significantly inhibited increased FRET ratio in L173R group, suggesting the increase in Ca2+ occurred in part through conductance through GIRK2(L173R) channels (Figure 5C).
Figure 5: Altered ion selectivity of GIRK2(L173R) channels.
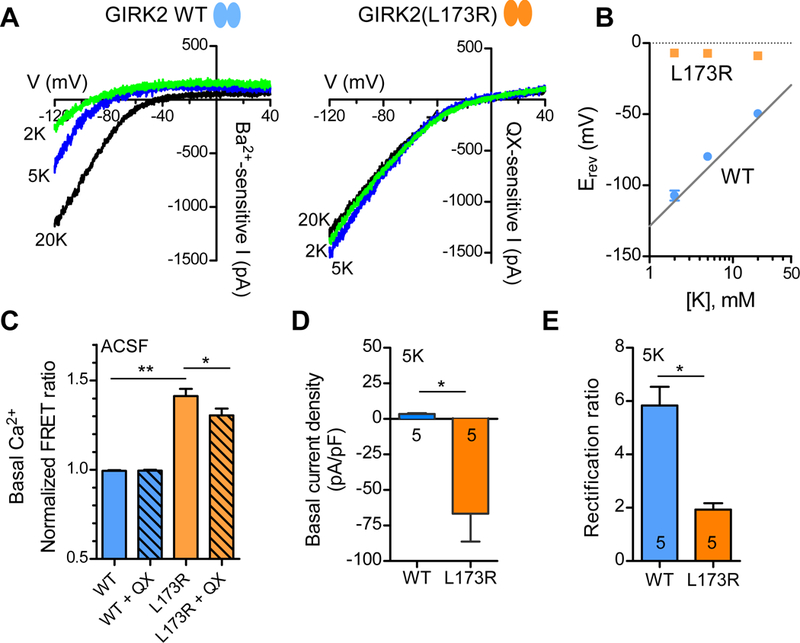
(A) Current-voltage plots (I-V) show Ba2+-sensitive basal currents for wild-type channels and QX-314-sensitive basal current for GIRK2(L173R) channels with ‘2K’ (green trace), ‘5K’ (blue trace), and ‘20K’ (black trace) solutions. (B) Reversal potentials for wild-type (blue circles) and mutant (orange squares) GIRK2 channels are plotted as a function of extracellular [K+] (mean ± SEM, N=5). (C) Bar graph shows normalized FRET ratio measured in a fluorometric plate reader with ACSF for wild-type and L173R channels (± SEM, N=48). QX: 100 μM QX-314 (**P < 0.001; *P < 0.05, One-way ANOVA with Tukey’s post hoc test). HEK293T cells coexpressing FRET-based Ca2+ detector, Twitch 2B. (D) Bar graph shows average basal current density for wild-type (Ba2+-sensitive) and mutant (QX-314 sensitive) channels measured at −70 mV using ‘5K’ solution (± SEM). Outward current indicated as positive number and inward current as negative number (*P < 0.01, Student’s t test). (E) Bar graph shows average rectification ratio for wild-type and mutant channels with ‘5K’ solution. Rectification ratio was calculated as inward current (−20 mV + Erev / outward current (+20 mV + Erev) (± SEM) (*P < 0.001, Student’s t test).
We next compared the basal currents of wild-type GIRK2 and GIRK2(L173R) channels with 5K solutions. The basal Ba2+-sensitive current density measured at −70 mV (near a cell’s resting membrane potential) was outward in the ‘5K’ solution for wild-type channels, (Figure 5D). By contrast, the basal QX-314-sensitive current was larger and inwardly for the mutant L173R channel (Figure 5D). For possible changes in the extent of inward rectification between wild-type and mutant channels, we calculated the ratio of inward current (−20 mV + Erev) to outward current (+20 mV + Erev), i.e., rectification ratio, with the ‘5K’ solution. L173R channels showed a significant decrease in the rectification ratio compared to wild-type, indicating a reduction of inward rectification with L173R channels (Figure 5E).
Discussion
Here, we report a gain-of-function mutation in the KCNJ6 (Girk2) gene contributes to a human neurological disease that has many similarities to Keppen-Lubinsky syndrome. Whole exome sequencing of the patient and her parents revealed a de novo variant, p.Leu171Arg, located on chromosome 21 in KCNJ6 of the patient. Expression of the GIRK2 mouse homologue with the mutation, GIRK2(L173R), alone or in the presence of the GIRK1 subunit, revealed several significant changes in channel properties. First, the Leu-to-Arg mutation appears to disrupt both GABAB receptor-dependent activation and G protein-independent activation with alcohol (i.e., PrOH). In addition, the Leu-to-Arg mutation significantly reduces K+ selectivity, allowing Na+ and Ca2+ to permeate the channel. In neurons, this latter effect would be expected to convert the GIRK response from an inhibitory to excitatory channel, i.e., depolarizing. Moreover, GIRK2(L173R) mutant channels exhibited a significantly larger basal inward current, indicating mutant channels are constitutively active and depolarizing. The co-expression of wild-type GIRK1 with GIRK2(L173R) could not ameliorate the effect of the mutant GIRK2(L173R), and GIRK1-GIRK2(L173R) heterotetramers appear to be not functional. Together, these results suggest the homotetramer is depolarizing while the heterotetramers are functionally silent. Thus, different ratios of GIRK subunits could determine the consequence for a neuron expressing GIRK2(L173R).
Collectively, the effects of the p.Leu173Arg mutation on channel function are remarkably similar to those described in the weaver mouse (Slesinger et al., 1996). The weaver (wv) mouse, first described over 40 years ago (Rakic and Sidman, 1973a, b), was determined to be caused by a G156S mutation in the Girk2 gene (Patil et al., 1995). The p.Gly156Ser mutation in Girk2 produced a channel that was constitutively active, exhibited reduced G protein activation, and became Na+ permeable (Kofuji et al., 1996, Slesinger et al., 1996, Slesinger, 2001). The loss of G protein activation and appearance of basal activity were attributed to the Na+-dependent influx and activation of the channel (Silverman et al., 1996). The depolarizing inward current through the GIRK2wv channel was inhibited by the Na channel blocker QX-314 (Kofuji et al., 1996, Slesinger, 2001). Similarly, GIRK2(L173R) channels exhibited defects in gating, changes in ion selectivity, and inhibition with QX-314. Thus, the functional properties of these two mutant channels, GIRK2(L173R) and GIRK2wv-G156S, are remarkably similar, even though these mutations are located in different regions of the channel protein. Protein structural modeling of L171R in the atomic structure of GIRK2 illustrates how the mutation could potentially change ion selectivity.
Despite the difference in species, there are some similarities between the clinical phenotype in the present case and the weaver mice. The weaver mouse is characterized by severe ataxia, hyperactivity and tremor, implying cerebellar involvement. Similarly, the patient presented with myoclonic jerks and choreiform movements, suggestive of deep cerebellar nuclei involvement (Rakic and Sidman, 1973a, b, Goldowitz and Mullen, 1982, Hatten et al., 1986, Goldowitz, 1989). On the other hand, the weaver cerebellum is notably smaller than littermate controls, due to the apoptotic loss of cerebellar granule cells (Goldowitz and Mullen, 1982, Hatten et al., 1986), as well as some Purkinje cells during development (Rakic and Sidman, 1973b, Goldowitz and Mullen, 1982). The patient had a normal MRI at 9 months of age, though no further imaging studies were performed to assess possible progressive cerebellar involvement.
In addition to the cerebellar defects, the homozygote weaver mice also show a progressive loss of SNc dopamine neurons during postnatal development (Roffler-Tarlov and Graybiel, 1984, Richter et al., 1992), similar to the defect in Parkinson’s disease. Notably, the GIRK2 subunit is uniquely expressed in SNc dopamine neurons (Inanobe et al., 1999). Dopaminergic cell death is selective and not driven by apoptosis, but rather most likely by inflammation, and can be rescued by the anti-inflammatory minocycline (Peng et al., 2006). Weaver mice also exhibit locomotor hyperactivity, similar to our patient’s phenotype and suggestive of abnormal dopaminergic neurotransmission. It is possible the chronic depolarization and loss of GPCR-dependent inhibitory control of dopamine neuron firing could lead to elevated dopamine levels in meso-limbic and meso-cortical structures. This could contribute to the patient’s dystonic posturing, opisthotonus, ballismus, and choreoatethosis, perhaps explaining the response to tetrabenazine. We cannot rule out the possibility of some dopaminergic neuronal cell death. However, the presence of one wild-type GIRK2 allele might mitigate some of the toxic effects of p.Leu171Arg. On the other hand, the low level of dopamine metabolite HVA in the patient’s CSF is suggestive of a dopamine deficiency. This would perhaps explain the brief episodes of oculogyric crisis. We noted that the patient did not tolerate treatment with L-dopa, experiencing more severe and increased hyperkinetic movements. Perhaps this is due to a degenerating but over-active dopaminergic network. Further research is needed to explore the mechanisms of dopamine deficiency and the possible treatments targeting the dysfunctional KCNJ6 channel.
What might be the consequence of the p.Leu171Arg mutation on GIRK2 function in the human brain? In rodents, GIRK2 is widely-expressed throughout the brain, including the cerebellum, hippocampus, mid-brain, habenula, amygdala, pontine nuclei, and olfactory bulb (Karschin et al., 1996). GIRK2 is commonly coexpressed with another GIRK subunit, such as GIRK1 or GIRK3 (Karschin et al., 1996, Lüscher and Slesinger, 2010). By contrast, GIRK2 is expressed alone in the substantia nigra compacta (SNc) dopamine neurons (Inanobe et al., 1999, Reyes et al., 2012). The functional studies with heterologously expressed channels demonstrated that the Leu-to-Arg mutation produces constitutively active channels with altered ion selectivity, i.e., non-selective channels that are open in the absence of neurotransmitter. Thus, we predict wide-spread effects on multiple-types of different neurons, both excitatory and inhibitory, that express p.Leu171Arg GIRK2. Notably, GIRK2(L173R) and GIRK1 form dominant negative heterotetramers. Thus, expression of GIRK2 WT, or other GIRK subunits, could mitigate some of the deleterious effects of the mutant channel in brain regions that express different GIRK subunits. However, SNc dopamine cells only express GIRK2 subunit, which may explain why the patient exhibits clinical signs of DA dysfunction. The conversion of a normally G protein-dependent inhibitory K channel to a constitutively active, excitatory ion channel, and the gain of Ca2+ permeability, could lead to changes in neurotransmitter release, cell death and/or developmental abnormalities. Expression of p.Leu171Arg GIRK2 in the cortex could lead to changes in cognitive function. In midbrain DA neurons, p.Leu171Arg GIRK2 could alter both nigral-striatal and meso-limbic functions, perhaps relating to the hypersensitivity of the patient to dopaminergic drugs (i.e., L-dopa, Pramipexole).
The p.Leu171Arg is the fourth patient reported with a dominant KCNJ6 mutation. Previously, two other mutations, p.Gly154Ser and p.Thr152del, were identified in three Keppen-Lubinsky patients (Masotti et al., 2015). One notable difference is the extent of lipodystrophy, ranging from no change in our case study, to absence of facial adipose in two subjects, and to generalized absence of subcutaneous fat in one subject with Keppen-Lubinsky syndrome (Masotti et al., 2015). Recently, a low level KCNJ6 expression was reported in human adipose tissue-derived stem cells (Soheilifar et al., 2017), raising the possibility that mutant GIRK2 channels could alter adipose production. However, the lack of penetrance suggests that other genes may mitigate some of the deleterious effects of KCNJ6 mutations. Also, we completely cannot rule out the contribution of other genes in our patient. Nonetheless, mutant GIRK2 channels likely contribute to the clinical findings reported here. Thus, we propose expanding the Keppen-Lubinsky phenotype to include severe dystonia and ballismus. The current report suggests screening for KCNJ6 mutations in the evaluation of patients with severe movement disorders.
Supplementary Material
Dystonic posturing followed by hyperkinetic choreiform movements and ballismus
Dystonic posturing of lower limbs with striatal toes
Hyperkinetic movements triggered by excitement
Acknowledgements:
We gratefully acknowledge the patient and family for their participation in this study; Dr. G. Sinclair and Dr. H. Vallance for interpretation of biochemical data; Ms. E. Aisenberg for creating the mouse L173R mutation in Girk2, Ms. X. Han for Sanger sequencing; Ms. A. Ghani for consenting and data management; Mrs. M. Higginson for DNA extraction, sample handling and technical data; Mr. D. Arenillas and Mr. M. Hatas for systems support, Ms E. Lomba and Mrs. D. Pak for research management support (University of British Columbia, Vancouver, CA).
This work was supported by grants from the National Institutes of Health (DA037170 to PAS; AA018734 to PAS); the B.C. Children’s Hospital Foundation (“1st Collaborative Area of Innovation” www.tidebc.org); Genome British Columbia (SOF-195); the Canadian Institutes of Health Research (#301221); NeuroDevNet (Strategic Opportunity Grant); and informatics infrastructure supported by Genome British Columbia and Genome Canada (ABC4DE Project). Dr. van Karnebeek is a recipient of the Michael Smith Foundation for Health Research Scholar Award.
Footnotes
Conflict of interest: The authors have no conflict(s) of interest to declare.
Video consent: the authors received a signed release form for the patient videotaped, authorizing the offline and/or online distribution of this video material.
References
- Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. [DOI] [PubMed] [Google Scholar]
- Basel-Vanagaite L, Shaffer L, Chitayat D (2009) Keppen-Lubinsky syndrome: Expanding the phenotype. Am J Med Genet A 149A:1827–1829. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhinathan K, Slesinger PA (2013) Molecular mechanism underlying ethanol activation of G-protein-gated inwardly rectifying potassium channels. Proc Natl Acad Sci USA 110:18309–18314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Brasi D, Brunetti-Pierri N, Di Micco P, Andria G, Sebastio G (2003) New syndrome with generalized lipodystrophy and a distinctive facial appearance: confirmation of Keppen-Lubinski syndrome? Am J Med Genet A 117A:194–195. [DOI] [PubMed] [Google Scholar]
- Goldowitz D (1989) The weaver granuloprival phenotype is due to intrinsic action of the mutant locus in granule cells: evidence from homozygous weaver chimeras. Neuron 2:1565–1575. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Mullen RJ (1982) Granule cell as a site of gene action in the weaver mouse cerebellum: evidence from heterozygous mutant chimeras. J Neurosci 2:1474–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlin RJCM, Hennekam RCM (2001) Keppen-Lubinski syndrome. New York: Oxford University Press. . [Google Scholar]
- Guex N, Peitsch MC, Schwede T (2009) Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: a historical perspective. Electrophoresis 30 Suppl 1:S162–173. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Liem RK, Mason CA (1986) Weaver mouse cerebellar granule neurons fail to migrate on wild-type astroglial processes in vitro. J Neurosci 6:2676–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess EJ (1996) Identification of the weaver mouse mutation: The end of the beginning. Neuron 16:1073–1076. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90:291–366. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Yoshimoto Y, Horio Y, Morishige K-I, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, Kurachi Y (1999) Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci 19:1006–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin C, Dissmann E, Stuhmer W, Karschin A (1996) IRK(1–3) and GIRK(1–4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J Neurosci 16:3559–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res 37:D387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Davidson N, Lester HA (1995) Evidence that neuronal G-protein-gated inwardly rectifying K+ channels are activated by Gβγ subunits and function as heteromultimers. Proc Natl Acad Sci USA 92:6542–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Hofer M, Millen KJ, Millonig JH, Davidson N, Lester HA, Hatten ME (1996) Functional analysis of the weaver mutant GIRK2 K+ channel and rescue of weaver granule cells. Neuron 16:941–952. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Reuveny E, Slesinger PA, Jan YN, Jan LY (1993) Primary structure and functional expression of a rat G-protein-coupled muscarinic potassium channel. Nature 364:802–806. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, Exome Aggregation C (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YJ, Jan YN, Jan LY (1996) Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J Neurosci 16:7137–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Slesinger PA (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat Rev Neurosci 11:301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masotti A, Uva P, Davis-Keppen L, Basel-Vanagaite L, Cohen L, Pisaneschi E, Celluzzi A, Bencivenga P, Fang M, Tian M, Xu X, Cappa M, Dallapiccola B (2015) Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am J Hum Genet 96:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutneja M, Berton F, Suen K-F, Luscher C, Slesinger PA (2005) Endogenous RGS proteins enhance acute desensitization of GABAB receptor-activated GIRK currents in HEK-293T cells. Pflügers Arch -- Eur J Physiol 450:61–73. [DOI] [PubMed] [Google Scholar]
- Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS (1995) A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nature Genetics 11:126–129. [DOI] [PubMed] [Google Scholar]
- Peng J, Xie L, Stevenson FF, Melov S, Di Monte DA, Andersen JK (2006) Nigrostriatal dopaminergic neurodegeneration in the weaver mouse is mediated via neuroinflammation and alleviated by minocycline administration. J Neurosci 26:11644–11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr., Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ (2001) Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105:511–519. [DOI] [PubMed] [Google Scholar]
- Rakic P, Sidman RL (1973a) Organization of cerebellar cortex secondary to deficit of granule cells in weaver mutant mice. J Comp Neurol 152:133–161. [DOI] [PubMed] [Google Scholar]
- Rakic P, Sidman RL (1973b) Weaver mutant mouse cerebellum: defective neuronal migration secondary to abnormality of Bergmann glia. Proc Natl Acad Sci USA 70:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes S, Fu Y, Double K, Thompson L, Kirik D, Paxinos G, Halliday GM (2012) GIRK2 expression in dopamine neurons of the substantia nigra and ventral tegmental area. J Comp Neurol 520:2591–2607. [DOI] [PubMed] [Google Scholar]
- Richter JA, Stotz EH, Ghetti B, Simon JR (1992) Comparison of alterations in tyrosine hydroxylase, dopamine levels, and dopamine uptake in the striatum of the weaver mutant mouse. Neurochem Res 17:437–441. [DOI] [PubMed] [Google Scholar]
- Rifkin RA, Moss SJ, Slesinger PA (2017) G Protein-Gated Potassium Channels: A Link to Drug Addiction. Trends Pharmacol Sci 38:378–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roffler-Tarlov S, Graybiel AM (1984) Weaver mutation has differential effects on the dopamine-containing innervation of the limbic and nonlimbic striatum. Nature 307:62–66. [DOI] [PubMed] [Google Scholar]
- Serratrice G, Pellissier JF, Serra-Trice J, Weiller PJ (2010) [Potassium channelopathies and Morvan’s syndromes]. Bulletin de l’Academie nationale de medecine 194:391–406; discussion 406–397. [PubMed] [Google Scholar]
- Silverman SK, Kofuji P, Dougherty DA, Davidson N, Lester HA (1996) A regenerative link in the ionic fluxes through the weaver potassium channel underlies the pathophysiology of the mutation. Proc Natl Acad Sci USA 93:15429–15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA (2001) Ion selectivity filter regulates local anesthetic inhibition of G protein-gated inwardly rectifying K+ channels. Biophys J 80:707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA, Patil N, Liao YJ, Jan YN, Jan LY, Cox DR (1996) Functional effects of the mouse weaver mutation on G protein-gated inwardly rectifying K+ channels. Neuron 16:321–331. [DOI] [PubMed] [Google Scholar]
- Soheilifar MH, Javeri A, Amini H, Taha MF (2017) Generation of dopamine-secreting cells from human adipose tissue-derived stem cells in vitro. Rejuvenation Res. [DOI] [PubMed] [Google Scholar]
- Spillane J, Kullmann DM, Hanna MG (2016) Genetic neurological channelopathies: molecular genetics and clinical phenotypes. J Neurol Neurosurg Psychiatry 87:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, Zhang LH, Bhavsar AP, Lee JJ, Drogemoller BI, Abdelsayed M, Alfadhel M, Armstrong L, Baumgartner MR, Burda P, Connolly MB, Cameron J, Demos M, Dewan T, Dionne J, Evans AM, Friedman JM, Garber I, Lewis S, Ling J, Mandal R, Mattman A, McKinnon M, Michoulas A, Metzger D, Ogunbayo OA, Rakic B, Rozmus J, Ruben P, Sayson B, Santra S, Schultz KR, Selby K, Shekel P, Sirrs S, Skrypnyk C, Superti-Furga A, Turvey SE, Van Allen MI, Wishart D, Wu J, Wu J, Zafeiriou D, Kluijtmans L, Wevers RA, Eydoux P, Lehman AM, Vallance H, Stockler-Ipsiroglu S, Sinclair G, Wasserman WW, van Karnebeek CD (2016) Exome Sequencing and the Management of Neurometabolic Disorders. N Engl J Med 374:2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thestrup T, Litzlbauer J, Bartholomaus I, Mues M, Russo L, Dana H, Kovalchuk Y, Liang Y, Kalamakis G, Laukat Y, Becker S, Witte G, Geiger A, Allen T, Rome LC, Chen TW, Kim DS, Garaschuk O, Griesinger C, Griesbeck O (2014) Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods 11:175–182. [DOI] [PubMed] [Google Scholar]
- van Karnebeek CD, Shevell M, Zschocke J, Moeschler JB, Stockler S (2014) The metabolic evaluation of the child with an intellectual developmental disorder: diagnostic algorithm for identification of treatable causes and new digital resource. Mol Genet Metab 111:428–438. [DOI] [PubMed] [Google Scholar]
- Villa C, Combi R (2016) Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front Cell Neurosci 10:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton MR, MacKinnon R (2013) X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature 498:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Arrabit C, Choe S, Slesinger PA (2001) Mechanism underlying bupivacaine inhibition of G protein-gated inwardly rectifying K+ channels. Proc Natl Acad Sci USA 98:6482–6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dystonic posturing followed by hyperkinetic choreiform movements and ballismus
Dystonic posturing of lower limbs with striatal toes
Hyperkinetic movements triggered by excitement