

Abstract
Pulmonary arterial hypertension (PAH) is an uncommon disease characterized progressive remodeling of pulmonary vasculature. Although treatment for PAH have improved in last two decades but the outcome remains fatal. Currently, the therapies for PAH target three well-established pathways the nitric oxide (NO) pathway, endothelin receptors, and prostanoids. There are multiple potential targets for development of newer drugs in PAH which requires meticulous research and clinical trials.
Keywords: pulmonary arterial hypertension, therapeutic, novel, phosphodiesterase, nitric oxide, prostaglandins, endothelin receptor
Pulmonary arterial hypertension (PAH) is defined as an increase in mean pulmonary arterial pressure (mPAP) of 25 mm Hg or greater at rest, as assessed by right heart catheterization. It is classified into five main subgroups based on clinical and hemodynamic criteria. 1 2 Despite multiple strategies and therapies available, PAH remains an incurable disease with a dismal prognosis. 2 The mPAP greater than 20 mm Hg is above the upper normal limits. Thus, 6th WSPH (World Symposia on Pulmonary Hypertension) Task Force held at Nice in 2018 proposes to include pulmonary vascular resistance ≥ 3Wood Units in the definition of all forms of precapillary pulmonary hypertension (PH) associated with mPAP > 20 mm Hg. But this will require prospective trials in PAH patients whether inclusion of patients with borderline PAH (21–24 mm Hg) will alter outcomes or not. 3 In patients with systemic sclerosis, chronic thromboembolic pulmonary hypertension (CTEPH) and chronic lung disease even a mild elevation of PAP (21–24 mm Hg) is associated with poor prognosis. 4 5 6 Despite multiple strategies and therapies available, PAH remains an incurable disease with a dismal prognosis. 2 Whether to hit early in the course of disease will be beneficial or not have to be confirmed in prospective trials. However, clinically PAH can be classified according to response to vasodilators like inhaled nitric oxide (NO), intravenous epoprostenol, or adenosine responders are rare among patients with PAH but those with vasoreactive are good candidates for treatment with calcium channel blockers. An acute response is defined as decrease in mPAP by at least 10 mm Hg to an absolute mPAP value lower than 40 mm Hg in setting of unchanged or increased cardiac output. Currently, three well established targets for PAH involve the NO-sGC-cGMP pathway, endothelin receptor antagonists, and prostanoids ( Fig. 1 ). However, there is lack of comparative data for the currently available drugs. There is also need for newer therapies which are more specific and may confer a mortality benefit, as the annual mortality with current classes of drugs still remains high, at approximately 10% per year.
Fig. 1.
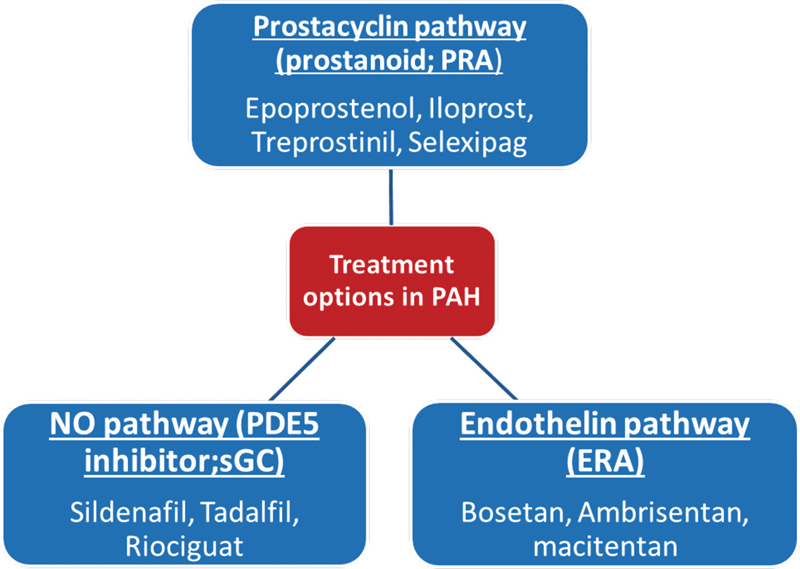
Pathobiological pathways in PAH. PAH, pulmonary arterial hypertension.
Pathobiological Pathways in PAH
NO-sGC-cGMP Pathway
PAH severity correlates with reduction in NO production. Inhaled NO is effective in neonates with persistent PH but has a limited use due to short half-life, tolerability, and rebound withdrawal.
Riociguat : it is an oral sGC stimulator with dual mode of action. It sensitizes endogenous NO by stabilizing NO-sGC complex and also directly stimulates sGC independently of NO, resulting in increased cGMP levels which relaxes the vascular smooth muscles. It is the only drug approved for both CTEPH and PAH in World Health Organization (WHO) functional class (FC) II and III. Based on the results of patent-1 trial, riociguat 2.5 mg thrice daily improved the 6-minute-walk distance, NT-proBNP levels, functional class, and time to clinical worsening at 12 weeks irrespective of whether patients were receiving other therapies for PAH or not. 7 Riociguat and phosphodiesterase type 5 (PDE5) inhibitors act on separate targets within the NO pathway but their combination is best avoided as it will increase risk of hypotension. Syncope was the most frequent adverse effect noticed (4–7%). Patients with PAH associated human immunodeficiency virus (HIV), schistosomiasis, and chronic hemolytic anemia were excluded. Patent-2 which was an open label extension study included 324 patients, which were followed-up for up to 1 year, showed sustained improvement in functional capacity. 8
PDE5 Inhibitors : sildenafil and tadalafil are oral PDE5 inhibitors (type 5A) with the latter being with longer half-life and once daily dosing. PDE5 inhibitors block the breakdown of cGMP, increases the levels of cGMP, which relaxes the smooth muscles and causes vasorelaxation. Sildenafil improved the functional status even at 3 years in SUPER-1 (sildenafil use in pulmonary arterial hypertension-1) and SUPER-2 trial with maximum dose of 80 mg thrice daily. 9 Tadalafil in doses up to 40 mg once daily has shown benefit in 6-minute-walk distance in PHIRST-1 (pulmonary arterial hypertension and response to tadalafil) and PHIRST-2 studies for up to 52 weeks. 10 Most common adverse effects observed were headache and flushing.
Advances : PHACeT trial in (phase-1) tested the tolerability of culture-derived endothelial progenitor cells transfected with eNOS (endothelial nitric oxide synthase) in seven PAH patients unresponsive to PAH-specific therapies and observed hemodynamic as well as functional benefit. 11
Prostacyclin Pathway
Prostacyclins (IP) act on IP receptor in the vascular smooth muscles and have vasodilatory, antiproliferative, and antiapoptotic effect on pulmonary smooth muscles via cyclic adenosine monophosphate (cAMP) protein kinase A (PKA) pathway and potential for reverse remodelling in the pulmonary vasculature. 12
Epoprostenol : only treatment shown to reduce mortality in PAH. It is endogenous prostacyclin with half-life of 6 minutes. It is stable at room temperature for 8 hours but recently a thermostable form is also approved. It requires intravenous infusion in dose of 25 to 40 ng/kg/min and is approved for WHO functional class III and IV. 13 Use of closed hub system is useful in preventing catheter-related bacterial infection which is a major concern of this therapy. Other side effects are flushing, headache, hypotension, diarrhea due to elevated cyclic AMP, and thrombocytopenia.
Prostacyclin analogues : trepoprostinil, beraprost, and iloprost are prostacyclin analogues which are relatively chemically stable.
Trepoprostinil : it is administered subcutaneously at a dose of 1 to 2 ng/kg/min. Most common side is infusion site pain which is not related with dose. Thrombocytopenia is less frequent as compared with epoprostenol. 14 TRIUMPH I has studied efficacy of inhaled trepoprostinil and FREEDOM-M has studied the efficacy of oral trepoprostinil over 12 weeks and has shown improvement in 6-minute-walk distance. 15 16
Beraprost : it is an oral analogue and only approved in japan. 17 Arterial Pulmonary Hypertension and Beraprost European (ALPHABAT) study was an European trial with over 100 patients and demonstrated improvement in 6-minute-walk test and dyspnea. 18
Iloprost : it is only inhalational prostacyclin analogue with half-life of 20 to 30 minutes which requires six to nine times inhalation in a day with the advantage of much smaller dose and fewer side effects. 19 AIR study with around 200 patients showed that inhalational iloprost improved 6-minute-walk distance and functional class as compared with placebo.
Selexipag : it is an oral nonprostanoid molecule which acts as agonist on IP receptors. It does not act on other prostanoid receptors, like in gastrointestinal tract, and hence an improved safety profile. It is initiated in doses of 200 mcg twice daily to a maximum of 1,600 mcg twice daily. After the benefits in phase-2 study, GRIPHON phase-3 trial which was event driven study involving more than 1,100 patients observed that death or complication related to PAH were significantly lowered in patients who received selexipag as compared with placebo. 20 21 However, there was no difference in mortality between two groups. Selexipag got the Food and Drug Administration (FDA) approval in the year 2015 for treatment of PAH.
Endothelin Receptor Antagonist Pathway
Endothelin-1 (ET-1) is a peptide secreted by endothelial cells which act via ET A and ET B receptors on smooth muscles to cause vasoconstriction, migration, inflammation, fibrosis, and proliferation. ET B receptors in lungs and kidneys also act as clearance receptors for ET-1 and generate NO, PGI 2 through endothelial cells. 22
There are three oral endothelin receptor antagonists available for treatment of PAH: bosentan, ambrisentan, and macitentan ( Table 1 ). 23 24 25 Only SERAPHIN study with macitentan involving 742 patients was the first study to reduce morbidity and mortality in patients with PAH. The primary end point of this study was the time to first occurrence of composite end point of death, atrial septostomy, lung transplantation, or worsening of PAH. Studies with bosentan and ambrisentan had primary end point like 6-minute-walk distance, time to clinical worsening. 26
Table 1. Pharmacological properties of endothelin receptor antagonists.
| FDA approval | Bosentan | Ambrisentan | Macitentan |
|---|---|---|---|
| 2001 | 2007 | 2013 | |
| Pharmacological group | Sulfonamide | Carboxylic | Sulfonamide |
| Selectivity ET A :ET B | 30:1 | 4,000:1 | 50:1 |
| Half-life (h) | 6 | 15 | 18 |
| Dosing range (mg) | 62.5–125 | 5–10 | 10 |
| Dosing frequency | Twice daily | Once daily | Once daily |
| Metabolism | CytochromeP450, CYP2C9, CYP3A4 | Glucuronidation, lesser extent by cytochrome |
CYP3A4 |
| Drug interactions | Warfarin, ketoconazole, glibenclamide | Cyclosporin A | Rifampicin, ketoconazole, ritonavir |
| Major trials | Study-351, BREATHE-1, BREATHE-2, BREATHE-5, EARLY | ARIES-1, ARIES-2, ARIES-E, AMBITION | SERAPHIN |
Abbreviations: ET, endothelin receptor; FDA, Food and Drug Administration.
Treatment with endothelin receptor antagonist (ERA) is associated with side effects like flushing, headache, hypotension, and peripheral edema due to vasodilatory properties. Most worrisome adverse is hepatic dysfunction up to the magnitude of 10% annually with bosentan. Monitoring of liver enzymes in follow-up and before dose escalation is recommended. 27 Macitentan appears to be relatively safe regarding hepatic dysfunction. Anemia has been observed in patients receiving ERA and hemoglobin monitoring is recommended as anemia can aggravate symptoms of PAH. All the endothelin receptor antagonists are teratogenic.
Treatment Approach
The current treatment approach for PAH patient is shown in Fig. 2 . 28
Fig. 2.
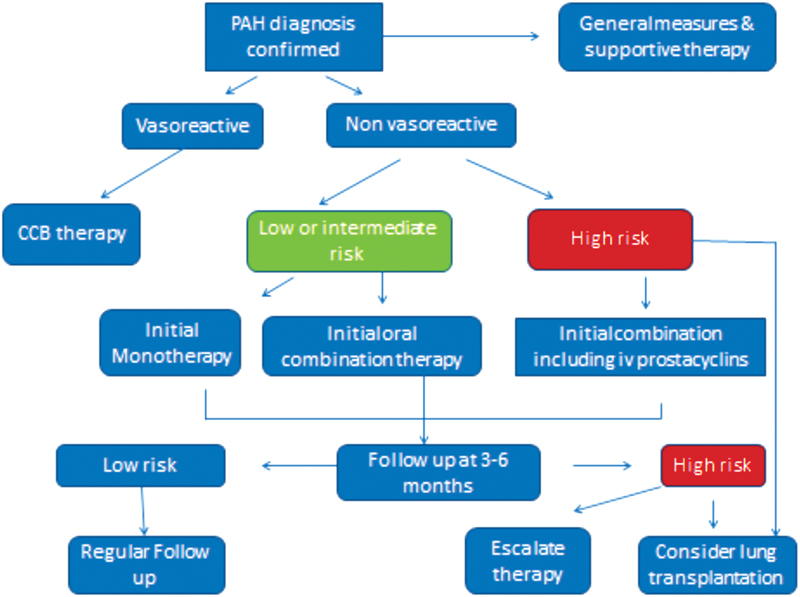
Algorithm for treatment–naive PAH patient. CCB, calcium channel blocker; PAH, pulmonary arterial hypertension.
Newer Targets
There is no scarcity of targets for newer drugs in pulmonary arterial hypertension.
Genetics
BMPR2 TGF β-pathway is impaired in patients with familial PAH. Tacrolimus activates BMPR2 signaling by binding to FKBP12 has been used in clinical trials. 29 Another approach is to target TGF β-activity by inhibiting it with molecules like sotatercept, an activin receptor fusion protein. 30 HDAC (histone deacetylase inhibitors), inhibitors of PARP (poly ADP ribose polymerase) to regulate DNA damage and miRNA (miR-124/143/145/204) have been used in animal models. 31 32 33
Mitochondrial Metabolism and Oxidative Stress
Pharmacological targets targeting apoptosis via inhibition of apoptosis signal regulation kinase-1 (ASK-1) are in phase-2 trials. 34 Nuclear erythroid 2-related factor (Nrf2) induction by bardoxolone methyl suppresses inflammation are in phase-3 study. 35 Dichloroacetate inhibits pyruvate dehydrogenase (PDK) prevents pyruvate from entering curb cycle. This drug has shown to reduce smooth muscle proliferation and apoptosis in pulmonary vasculature in patients with PAH by switching metabolism from oxidative phosphorylation to glycolysis. 36 peroxisome proliferator-activated receptors (PPARs) γ is a downstream target of BMPR2 which is important transcription factor for proliferation of smooth muscles in pulmonary vasculature can be a potential molecule for reverse remodeling. 37 Aplein is a peptide which is linked to PPARγ and BMP pathway. Levels of aplein are decreased in patients with PAH. As iron deficiency is common in patients of PAH, intravenous ferric carboxymaltose which have shown benefit in heart failure is being tried in two open label trials in patients with PAH. 38
Growth Factors
Epidermal growth factor (EGF) pathway is also involved in proliferation of smooth muscles in pulmonary vasculature but therapies with EGF inhibitors have not shown improvement in right ventricular hemodynamics. 39 Imatinib which also targets platelet derived growth factor (PDGF) in addition to tyrosine kinase receptor has shown to improve 6-minute-walk distance and reduction in pulmonary vascular resistance in IMPRESS trial but serious concerns of subdural hematoma. 40 Rho kinase imbalance is also involved in endothelial dysfunction in PAH. This hypothesis led to studies with fasudil, a rho kinase inhibitor, which are in early phase. 41
Humoral Modulation
Incidence of PAH is higher in female patients of post puberty age group, but there is a paradox, as female patients with PAH have better survival as compared with male patients which might suggest that estrogens may be beneficial in PAH. 42 There are early on-going phase-2 trials with anastrazole.
Serotonin receptor uptake inhibitors have a causal association with PAH. Studies with serotonin receptor antagonist did not result in significant improvement. 43
Vasoactive intestinal peptide is being used in phase-2 trials based on data in animals. 44 There is activation of sympathetic, as well as renin angiotensin system, in patients with PAH but drugs that are beneficial in systemic circulation will act on pulmonary circulation is doubtful and will require further studies. 45 Even the scope of antifibrogenic properties of mineralocorticoid antagonist is being studied.
Inflammation and Immunosuppressant
Inflammation is an inherent pathological finding especially in PAH associated with connective tissue disorder. There are preclinical studies with interleukin-6 (IL-6), CD20 antagonists. 46
Stem Cells
Endothelial progenitor cells are thought to differentiate into endothelium to repair the pulmonary vasculature. Although some results are encouraging but they have to be confirmed. 47
Conclusion
There is a better understanding of pathogenesis of PAH in the last two decades with many new molecular targets in pipeline. Being such a fatal disease, there is need of robust event driven studies to find out new molecules that can provide symptomatic as well as mortality benefits.
Footnotes
Conflict of Interest None declared.
References
- 1.Hoeper M M, Humbert M, Souza R et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4(04):306–322. doi: 10.1016/S2213-2600(15)00543-3. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin V V, Archer S L, Badesch D B et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009;119(16):2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Montani D, Celermajer D Set al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension Eur Respir J 201953011.801913E6[https://doi.org/10.1183/13993003.01913-2018] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bishop J M, Cross K W. Physiological variables and mortality in patients with various categories of chronic respiratory disease. Bull Eur Physiopathol Respir. 1984;20(06):495–500. [PubMed] [Google Scholar]
- 5.Pepke-Zaba J, Delcroix M, Lang I et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation. 2011;124(18):1973–1981. doi: 10.1161/CIRCULATIONAHA.110.015008. [DOI] [PubMed] [Google Scholar]
- 6.Coghlan J G, Wolf M, Distler O et al. Incidence of pulmonary hypertension and determining factors in patients with systemic sclerosis. Eur Respir J. 2018;51(04):1.701197E6. doi: 10.1183/13993003.01197-2017. [DOI] [PubMed] [Google Scholar]
- 7.Ghofrani H A, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013;369:330–340. doi: 10.1056/NEJMoa1209655. [DOI] [PubMed] [Google Scholar]
- 8.Rubin L J, Galiè N, Grimminger F et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2) Eur Respir J. 2015;45(05):1303–1313. doi: 10.1183/09031936.00090614. [DOI] [PubMed] [Google Scholar]
- 9.Galiè N, Ghofrani H A, Torbicki A et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 10.Galiè N, Brundage B H, Ghofrani H Aet al. Tadalafil therapy for pulmonary arterial hypertension Circulation 2009119222894–2903.Circulation 2011;124(10):e279 [DOI] [PubMed] [Google Scholar]
- 11.Granton J, Langleben D, Kutryk M B et al. Endothelial NO-synthase gene-enhanced progenitor cell therapy for pulmonary arterial hypertension: the PHACeT trial. Circ Res. 2015;117(07):645–654. doi: 10.1161/CIRCRESAHA.114.305951. [DOI] [PubMed] [Google Scholar]
- 12.Falcetti E, Hall S M, Phillips P G et al. Smooth muscle proliferation and role of the prostacyclin (IP) receptor in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(09):1161–1170. doi: 10.1164/rccm.201001-0011OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akagi S, Nakamura K, Matsubara H et al. Epoprostenol therapy for pulmonary arterial hypertension. Acta Med Okayama. 2015;69(03):129–136. doi: 10.18926/AMO/53519. [DOI] [PubMed] [Google Scholar]
- 14.Simonneau G, Barst R J, Galie N et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(06):800–804. doi: 10.1164/ajrccm.165.6.2106079. [DOI] [PubMed] [Google Scholar]
- 15.McLaughlin V V, Benza R L, Rubin L J et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol. 2010;55(18):1915–1922. doi: 10.1016/j.jacc.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 16.Jing Z C, Parikh K, Pulido T et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: a randomized, controlled trial. Circulation. 2013;127(05):624–633. doi: 10.1161/CIRCULATIONAHA.112.124388. [DOI] [PubMed] [Google Scholar]
- 17.Barst R J, McGoon M, McLaughlin V et al. Beraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2003;41(12):2119–2125. doi: 10.1016/s0735-1097(03)00463-7. [DOI] [PubMed] [Google Scholar]
- 18.Galiè N, Humbert M, Vachiéry J L et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2002;39(09):1496–1502. doi: 10.1016/s0735-1097(02)01786-2. [DOI] [PubMed] [Google Scholar]
- 19.Olschewski H, Simonneau G, Galiè N et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347(05):322–329. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- 20.Galiè N, Channick R, Chin Ket al. Effect of selexipag on morbidity/mortality in pulmonary arterial hypertension: results of the GRIPHON study J Am Coll Cardiol 201534(4, Suppl.):S163 [Google Scholar]
- 21.Sitbon O, Channick R, Chin K M et al. Selexipag for the treatment of pulmonary artery hypertension. N Engl J Med. 2015;373(26):2522–2533. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 22.Dupuis J, Stewart D J, Cernacek P, Gosselin G. Human pulmonary circulation is an important site for both clearance and production of endothelin-1. Circulation. 1996;94(07):1578–1584. doi: 10.1161/01.cir.94.7.1578. [DOI] [PubMed] [Google Scholar]
- 23.Clozel M, Breu V, Burri Ket al. Pathophysiological role of endothelin revealed by the first orally active endothelin receptor antagonist Nature 1993365(6448):759–761. [DOI] [PubMed] [Google Scholar]
- 24.Galié N, Badesch D, Oudiz R et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(03):529–535. doi: 10.1016/j.jacc.2005.04.050. [DOI] [PubMed] [Google Scholar]
- 25.Iglarz M, Binkert C, Morrison K et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327(03):736–745. doi: 10.1124/jpet.108.142976. [DOI] [PubMed] [Google Scholar]
- 26.Pulido T, Adzerikho I, Channick R N et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(09):809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 27.Benedict N, Seybert A, Mathier M A. Evidence-based pharmacologic management of pulmonary arterial hypertension. Clin Ther. 2007;29(10):2134–2153. doi: 10.1016/j.clinthera.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Galiè N, Channick R N, Frantz R P et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(01):1.801889E6. doi: 10.1183/13993003.01889-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spiekerkoetter E, Sung Y K, Sudheendra D et al. Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur Respir J. 2017;50(03):1.602449E6. doi: 10.1183/13993003.02449-2016. [DOI] [PubMed] [Google Scholar]
- 30.Komrokji R, Garcia-Manero G, Ades L et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol. 2018;5(02):e63–e72. doi: 10.1016/S2352-3026(18)30002-4. [DOI] [PubMed] [Google Scholar]
- 31.Boucherat O, Chabot S, Paulin R et al. HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci Rep. 2017;7(01):4546. doi: 10.1038/s41598-017-04874-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chun H J, Bonnet S, Chan S Y. Translational advances in the field of pulmonary hypertension. Translating microRNA biology in pulmonary hypertension. It will take more than “miR” words. Am J Respir Crit Care Med. 2017;195(02):167–178. doi: 10.1164/rccm.201604-0886PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meloche J, Pflieger A, Vaillancourt M et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation. 2014;129(07):786–797. doi: 10.1161/CIRCULATIONAHA.113.006167. [DOI] [PubMed] [Google Scholar]
- 34.Budas G R, Boehm M, Kojonazarov B et al. ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2018;197(03):373–385. doi: 10.1164/rccm.201703-0502OC. [DOI] [PubMed] [Google Scholar]
- 35.Lu H I, Huang T H, Sung P H et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharmacol Sin. 2016;37(05):589–603. doi: 10.1038/aps.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piao L, Fang Y H, Cadete V J et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl) 2010;88(01):47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansmann G, de Jesus Perez V A, Alastalo T P et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118(05):1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Viethen T, Gerhardt F, Dumitrescu D et al. Ferric carboxymaltose improves exercise capacity and quality of life in patients with pulmonary arterial hypertension and iron deficiency: a pilot study. Int J Cardiol. 2014;175(02):233–239. doi: 10.1016/j.ijcard.2014.04.233. [DOI] [PubMed] [Google Scholar]
- 39.Dahal B K, Cornitescu T, Tretyn A et al. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am J Respir Crit Care Med. 2010;181(02):158–167. doi: 10.1164/rccm.200811-1682OC. [DOI] [PubMed] [Google Scholar]
- 40.Hoeper M M, Barst R J, Bourge R C et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation. 2013;127(10):1128–1138. doi: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 41.Mouchaers K T, Schalij I, de Boer M A et al. Fasudil reduces monocrotaline-induced pulmonary arterial hypertension: comparison with bosentan and sildenafil. Eur Respir J. 2010;36(04):800–807. doi: 10.1183/09031936.00130209. [DOI] [PubMed] [Google Scholar]
- 42.Benza R L, Miller D P, Gomberg-Maitland M et al. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL) Circulation. 2010;122(02):164–172. doi: 10.1161/CIRCULATIONAHA.109.898122. [DOI] [PubMed] [Google Scholar]
- 43.Ghofrani H A, Al-Hiti H, Vonk-Noordegraaf A et al. Proof-of-concept study to investigate the efficacy, hemodynamics and tolerability of terguride vs. placebo in subjects with pulmonary arterial hypertension: results of a double blind, randomised, prospective phase IIa study. Am J Respir Crit Care Med. 2012;185:A2496. [Google Scholar]
- 44.Said S I.Vasoactive intestinal peptide in pulmonary arterial hypertension Am J Respir Crit Care Med 201218507786, author reply 786 [DOI] [PubMed] [Google Scholar]
- 45.van Campen J S, de Boer K, van de Veerdonk M C et al. Bisoprolol in idiopathic pulmonary arterial hypertension: an explorative study. Eur Respir J. 2016;48(03):787–796. doi: 10.1183/13993003.00090-2016. [DOI] [PubMed] [Google Scholar]
- 46.Tamura Y, Phan C, Tu L et al. Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J Clin Invest. 2018;128(05):1956–1970. doi: 10.1172/JCI96462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diller G P, van Eijl S, Okonko D O et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117(23):3020–3030. doi: 10.1161/CIRCULATIONAHA.108.769646. [DOI] [PubMed] [Google Scholar]