

Abstract
Introduction
Cisplatin has been reported to elicit the DNA damage response (DDR) via activation of the ATR-Chk1 pathway, which in turn contributes to the induction of cisplatin resistance. Inhibition of ATR-Chk1 signaling reverses cisplatin resistance in some cancers. However, the influence of inhibiting ATR-Chk1 signaling on cisplatin resistance in chondrosarcoma cancer has not been reported.
Materials and methods
We compared the expression levels of ATR kinases in human nasopharyngeal carcinoma, choriocarcinoma and chondrosarcoma cell lines. We inhibited ATR kinase function with VE-822, a selective ATR inhibitor, and suppressed ATR kinase expression with shRNA. Western blotting, the CCK-8 assay, cell cycle distribution assay and apoptosis analysis were used to study the influence of inhibiting ATR-Chk1 signaling on reversing cisplatin resistance in chondrosarcoma cell lines.
Results
We found that chondrosarcoma cells expressed very low basal levels of phosphorylated ATR, but cisplatin treatment induced the activation of ATR-Chk1 signaling in a dose- and time-dependent manner, suggesting the induction of DDR. As expected, ATR inhibition with VE-822 reversed cisplatin-induced DDR and enhanced cisplatin-induced activation of H2AX, which is an important marker of DNA damage. Meanwhile, ATR inhibition by RNA interference also reversed DDR and promoted DNA damage. Furthermore, both pharmacological and molecular inhibition of ATR accelerated cisplatin-induced inhibition of cell proliferation and cell death.
Conclusion
Our results suggested that inhibiting ATR activation promoted cisplatin-induced cell death via reversion of DDR, and VE-822 may be a valuable strategy for the prevention of cisplatin resistance in chondrosarcoma.
Keywords: chondrosarcoma, ATR, VE-822, DNA damage response, cell death
Introduction
Chondrosarcoma (25.8%) is the second most frequently diagnosed primary sarcoma of bone following osteosarcoma (35.1%).1 The vast majority (90%) of these cancers are conventional chondrosarcomas, which are subdivided into the central, peripheral and periosteal subgroups.2 The remaining 10% are rare variants of chondrosarcoma, which include dedifferentiated, clear cell and mesenchymal chondrosarcomas.2 The 5-year survival rate of patients with grade I chondrosarcoma is 83%, whereas patients with grade II or III disease have a combined 5-year survival rate of only 53%.3 Due to the limited clinical efficacies of chemotherapy and radiotherapy, broad surgical resection of the tumor is the most efficient and commonly used treatment method. However, after surgical treatment, the tumor tends to recur in subsequent years, and the degree of malignancy often increases over time. Furthermore, surgical treatment is not applicable to metastatic or unresectable disease. Therefore, improving the chemosensitivity and radiosensitivity of chondrosarcoma can diversify treatment programs and significantly improve the survival of patients.
Ataxia telangiectasia mutated (ATM) and ATM- and RAD3-related (ATR) kinase are key regulators of the DNA damage response (DDR) signal pathway.4 Once DNA damage occurs, the ATR-Chk1 and ATM-Chk2 pathways halt cell cycle progression at the G2/M and G1/S checkpoints, respectively, to allow time for DNA repair. Activation of the ATR-Chk1 pathway initiates cell cycle arrest, DNA repair and stabilization of the replication fork.
VE-822, the improved molecule of VE-821, is a novel ATR inhibitor with high selectivity and specificity to prevent signal transduction of the ATR-Chk1 pathway.5 In general, irradiation and chemotherapeutic drugs induce DNA damage, which triggers the activation of the ATR-Chk1 and ATM-Chk2 pathways and renders the cell cycle halted in the G2 and G1 phases, respectively, to facilitate DNA repair. However, ATR inhibition with VE-822 forces cells with DNA damage in G2 phase to rapidly progress into M phase with DNA repair that is severely incomplete. Cells with damaged DNA fail to divide to the next generation and instead shift toward mitotic catastrophe.6 Therefore, VE-822 has a dramatic impact on cells active within the cell cycle, such as cancer cells, leading to a sharp increase in the number of apoptotic cells. However, this treatment is ineffective let alone beneficial for the survival of noncycling cells, such as healthy normal cells.7
Some experimental studies have indicated that VE-822 can enhance the sensitivity of several cancer lines to chemotherapy and irradiation.8–10 However, VE-822 has never been reported to increase the chemosensitivity or radiosensitivity of chondrosarcoma. Our study systematically elaborated the effectiveness of VE-822 to increase the chemosensitivity of chondrosarcoma cells to cisplatin.
Materials and methods
Cell culture
SW1353, and JEG-3 cells were purchased from the American Type Culture Collection (American Type Culture Collection) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (BI Inc.), 100 units/ml penicillin and 100 μg/ml streptomycin (BI Inc.). CS-1 is a human chondrosarcoma cell line, kindly provided by Dr. Zhenfeng Duan (Sarcoma Biology Laboratory, Center for Sarcoma and Connective Tissue Oncology, Massachusetts General Hospital).11 5-8F is a nasopharyngeal carcinoma cell line,12 kindly provided by Dr. H.M. Wang (Cancer Center, Sun Yat-sen University, China).13 The use of all these cell lines was approved by the Ethics Committee of Changsha Central Hospital and the Ethics Committee of the Second Xiangya Hospital. All cells were maintained at 37°C in a humidified incubator with 5% CO2. The medium was changed every two or three days.
Cell viability assay
Cell viability after cisplatin treatment combined with VE-822 was determined using CCK-8 reagent purchased from Dojindo (Kumamoto, Japan). In brief, chondrosarcoma cells (1×104 per well) were seeded into 96-well plates and cultured overnight at 37°C in a humidified incubator. Compared with the control wells, the experimental wells were treated with cisplatin and/or VE-822. All treatments were performed in triplicate. After 24 h of treatment, 10 μl of CCK-8 reagent was added per well (containing 100 µl of DMEM) according to the manufacturer’s instructions. Following an incubation at 37°C, the absorbance at 450 nm was measured using a microplate reader (ELx800, BioTek Inc., USA).
Western blot assay
Cells were lysed with prechilled radioimmunoprecipitation (RIPA) buffer containing phenylmethylsulfonyl fluoride (PMSF) and disrupted 60 times in icy water for 5 s each with an Ultrasonic Cell Disruption System at intervals of 5 s. After centrifugation at 12,000 rpm/min at 4°C for 10 min, supernatants were collected, and protein concentrations were determined with a BCA protein assay kit (Pierce, Rockford, IL, USA). After the samples were mixed thoroughly with loading buffer, they were heated at 100°C for 5 min. Equal amounts of total protein (20–100 μg) were separated by SDS-PAGE on a 10% gel and transferred onto a PVDF membrane. Following blocking by incubation in 3% dry nonfat milk at room temperature for 1 h, the membrane was incubated with primary antibodies at 4°C overnight. Next, the membrane was rinsed three times with PBS-T (containing 0.01% Tween 20), incubated for 1.5 h with secondary antibodies conjugated to HRP and rinsed three times with PBS-T. The protein bands were detected with SuperSignal chemiluminescent substrate-stable peroxide solution (Pierce Rockford, IL, USA), and reactions in the protein bands were developed on BIOMAX-MR film (Eastman Kodak Co., Rochester, NY, USA).
Apoptosis analysis
Cells were harvested and washed twice with PBS and subsequently suspended in 200 μl of annexin V binding buffer. After the suspension was incubated with Annexin V-FITC in the dark for 15 min at 4°C, propidium iodide (PI) was added to the samples and incubated in the dark for 5 min at 4°C. The samples were analyzed on a BD Accuri C6 using FACSuite analysis software (BD Bioscience).
ATR knockdown in SW1353 cells
Small hairpin (sh)-RNA targeting ATR was integrated into the pLent-U6-GFP-Puro vector. The sequence targeting ATR was as follows: 5ʹ-GCCAAAGTATTTCTAGCCTATCTTCAAGAGAGATAGGCTAGAAATACTTTGGCTTTTTT-3ʹ.14 Vectors with or without the shRNA targeting ATR were transfected into SW1353 cells using Lipofectamine 3000 (Invitrogen Life Technologies) according to the manufacturer’s protocols. Subsequently, monoclonal cells were selected using complete medium supplemented with 1 μg/ml puromycin.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from treated cells using TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. For the detection of the mRNA levels, complementary DNA primed by oligo-dT was synthesized with a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Pittsburgh, PA, USA). Subsequently, PCR was performed for 20–30 cycles under the following cycling conditions: denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 68°C for 40 s. PCR products were separated on a 1% agarose gel and then stained with GelRed. The following primers were used:
ATR-forward: 5ʹ-TCCCTTGAATACAGTGGCCTA-3ʹ, and ATR-reverse: 5ʹ-TCCTTGAAAGTACGGCAGTTC-3ʹ; CHK1-forward: 5ʹ-ATATGAAGCGTGCCGTAGACT-3ʹ, and CHK1-reverse: 5-TGCCTATGTCTGGCTCTATTCTG-3ʹ; and GAPDH-forward: 5ʹ-AATCCCATCACCATCTTCCA-3ʹ, and GAPDH-reverse: 5-CCTGCTTCACCACCTTCTTG-3ʹ.
Quantitative real-time RT-PCR (qRT-PCR)
cDNA was synthesized as described previously. Real-time PCR amplification was performed with specific primers on a Roche LightCycler 96 Sequence Detection System. The reactions were prepared using SYBR Green reaction mix from Roche. Using the 2−ΔΔCt method, the expression levels of the relative mRNAs were normalized to those of GAPDH. The primers used for RT-PCR were used in these experiments.
PI staining of dead cells
SW1353 cells and the above mentioned monoclonal cells transfected with plasmids with or without ATR shRNA were treated with 10 μM cisplatin for 48 h. Then, 1 μg/ml PI was directly added into the control and treated wells, and the cells were incubated at room temperature for 30 min. The bright field and fluorescence images in the same field were captured with fluorescence microscopy (Leica).
Cell cycle analysis
Cells treated with VE-822 and/or cisplatin were collected and rinsed once with PBS. Then, they were fixed with 70% ethanol at 4°C overnight and rinsed once with PBS. After the cells were suspended with 1 ml PBS per tube, 50 μg/ml RNase was added. Next, 5 μg/ml PI was incubated with the samples in the dark for 30 min at room temperature, after which the cell cycle distribution was analyzed on a BD Accuri C6 using FACSuite analysis software (BD Bioscience). The percentage of cells in the different phases of the cell cycle was counted and compared. The experiments were repeated in triplicate.
Statistical analysis
All experiments were repeated three times independently. The results are shown as the mean ± SD (standard deviation). Either Student’s t-test or one-way ANOVA was used to analyze the data with SPSS software. P-values <0.05 were considered to be statistically significant.
Results
The treatment of chondrosarcoma cells with cisplatin induces the DNA damage response
It is well known that the clinical effects of cisplatin treatment are relatively ideal for nasopharyngeal carcinoma and endometrial carcinoma compared with chondrosarcoma. We measured the basal levels of phosphorylated ATR kinase in SW1353, CS-1, 5-8F, and JEG-3 cells. The results showed that the chondrosarcoma CS-1 cell line and SW1353 cells expressed very low levels of phosphorylated ATR at baseline. As experimental controls, 5-8F and JEG-3 expressed moderate levels of phosphorylated ATR (Figure 1A). Then, we focused on SW1353 and CS-1 cells to investigate the impact of cisplatin on the levels of activated ATR and Chk1, which are two important markers of the DNA damage response (DRR). The phosphorylated ATR kinases in both chondrosarcoma cell lines were significantly upregulated by cisplatin treatment in a dose- and time-dependent manner (Figure 1B and C). Cisplatin treatment also induced the activation of Chk1, which is a downstream molecule of ATR, in both SW1353 and CS-1 cells in a dose- and time-dependent manner (Figure 1B and C). These results suggest that cisplatin elicits the DDR in chondrosarcoma.
Figure 1.
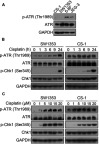
Cisplatin activates the ATR-Chk1 pathway in chondrosarcoma cells. (A) Western blot analysis of the total and phosphorylated levels of basal ATR and Chk1 in untreated CS-1, SW1353, 5-8F, and JEG-3 cells. GAPDH protein levels were measured as loading controls. (B) Western blot analysis of the levels of total and phosphorylated ATR and Chk1 in CS-1 and SW1353 cells treated with 10 μM cisplatin for the indicated time periods. (C) Western blot analysis of the levels of total and phosphorylated ATR and Chk1 in CS-1 and SW1353 cells treated with the indicated concentrations of cisplatin for 24 h.
Inhibition of ATR promotes cisplatin-induced DNA damage
DNA damage is a key driver of the therapeutic mechanism of chemotherapy of cancers. The ATR-Chk1 pathway can be activated by DNA damage and is essential for the survival of many cancer cells in response to multiple anticancer drugs.15 Therefore, ATR-Chk1 activation contributes to the induction of drug resistance.16 To determine the roles of the ATR-Chk1 pathway in the induction of cisplatin resistance in chondrosarcoma, we tested the effect of the inhibition of ATR on DNA damage induced by cisplatin. We found that cisplatin activated the ATR-Chk1 pathway in both SW1353 and CS-1 chondrosarcoma cells (Figure 2A and B). Pharmacological inhibition of ATR with VE-822 dose dependently reversed the activation of ATR-Chk1 signaling induced by cisplatin (Figure 2A and B). Cisplatin had little effect on the activation of H2AX, suggesting that cisplatin alone induced a limited amount of DNA damage (Figure 2C and D). However, VE-822 significantly upregulated the activation of H2AX in a dose-dependent manner in both SW1353 and CS-1 cells (Figure 2C and D), suggesting increased DNA damage. Meanwhile, molecular inhibition of ATR via RNA interference downregulated the expression of ATR (Figure 2E), inhibited the activation of the ATR-Chk1 pathway, and promoted DNA damage induced by cisplatin (Figure 2F and G). These results suggested that ATR inhibition promoted cisplatin-induced DNA damage and may function to reverse cisplatin resistance in chondrosarcoma.
Figure 2.

The inhibition of ATR reverses cisplatin-induced DDR. (A) Western blot and semiquantitative analysis of the levels of phosphorylated ATR and Chk1 in SW1353 cells treated with the indicated concentrations of VE-822 alone or VE-822 plus 10 μM cisplatin for 24 h. (B) Western blot and semiquantitative analysis of the levels of phosphorylated ATR and Chk1 in CS-1 cells treated with the indicated concentrations of VE-822 alone or VE-822 plus 10 μM cisplatin for 24 h. (C) Western blot and semiquantitative analysis of the levels of phosphorylated H2AX in SW1353 cells treated with the indicated concentrations of VE-822 alone or VE-822 plus 10 μM cisplatin for 24 h. (D) Western blot and semiquantitative analysis of the levels of phosphorylated H2AX in CS-1 cells treated with the indicated concentrations of VE-822 alone or VE-822 plus 10 μM cisplatin for 24 h. (E) Western blot and quantitative RT-PCR analysis of the levels of ATR in SW1353 cells transfected with ATR shRNA. ***P<0.001 compared with the control group. (F) Western blot and semiquantitative analysis of the levels of phosphorylated ATR and Chk1 in SW1353 cells transfected with ATR shRNA and cultured in the presence or absence of 10 μM cisplatin for 24 h. (G) Western blot and semiquantitative analysis of the levels of phosphorylated H2AX in SW1353 cell, transfected with ATR shRNA and cultured in the presence or absence of 10 μM cisplatin for 24 h.
ATR inhibition sensitizes chondrosarcoma cells to cisplatin-induced inhibition of cell proliferation
The ATR inhibitor VE-822 has been reported to enhance cisplatin chemosensitivity in esophageal squamous cell carcinoma.9 In this study, we detected the effect of VE-822 on cell proliferation in chondrosarcoma cells. The results showed that VE-822 alone did not affect the proliferation of either SW1353 or CS-1 cells (Figure 3A). When cells were cotreated with VE-822 and cisplatin, the inhibition of cell proliferation was increased (Figure 3B and C). Meanwhile, we found that ATR downregulation by RNA interference promoted cisplatin-induced inhibition of cell proliferation in chondrosarcoma cells (Figure 3D).
Figure 3.

The inhibition of ATR reverses cisplatin-induced inhibition of cell proliferation. (A) CCK-8 analysis of the proliferation of SW1353 and CS-1 cells treated with the indicated concentrations of VE-822 for 24 h. (B) CCK-8 analysis of the proliferation of SW1353 and CS-1 cells treated with the indicated concentrations of VE-822 and cisplatin for 24 h. (C) CCK-8 analysis of the proliferation of SW1353 cells that were untreated or treated with VE-822 (0.2 μM, 48 h), cisplatin (20 μM, 48 h), VE-822 (0.2 μM, 48 h) + cisplatin (20 μM, 24 h), or cisplatin (20 μM, 48 h) + VE-822 (0.2 μM, 24 h). (D) CCK-8 analysis of the proliferation of SW1353 cells that were untreated or treated with cisplatin (10 μM, 48 h) or cisplatin (10 μM, 48 h) plus VE-822 (0.05 μM, 48 h) and of SW1353 cells transfected with empty vector (E.V.) and treated with cisplatin (10 μM, 48 h) or transfected with ATR shRNA and treated with cisplatin (10 μM, 48 h). *P<0.05, **P<0.01, ***P<0.001.
ATR inhibition sensitizes chondrosarcoma cells to cisplatin-induced cell death
Cisplatin has been reported to induce cell cycle arrest at G2/M phase.17 To determine the effect of VE-822 on cisplatin-induced cell cycle arrest in chondrosarcoma cells, SW1353 cells were treated with VE-822 alone, cisplatin alone, or VE-822 and cisplatin together. PI staining results showed that VE-822 alone had little effect on the percentage of cells in the G2 and M phases (Figure 4A and B), but cisplatin alone induced significant cell cycle arrest at the G2/M checkpoint (Figure 4A and B). Moreover, VE-822 treatment combined with cisplatin significantly decreased the number of cells in G2/M phase but increased the number of cells in G1 phase. These results suggested that cisplatin treatment induced cell cycle arrest at the G2/M checkpoint to signal DNA repair via activation of the ATR-Chk1 pathway, and the ATR inhibitor VE-822 blocked this mechanism by promoting cell cycle progression from G2/M to G1 phase.
Figure 4.

Inhibition of ATR promotes apoptosis induced by cisplatin. (A) PI analysis of cell cycle progression of SW1353 cells treated with the indicated concentrations of VE-822 either alone or in combination with 10 μl cisplatin for 24 h. (B) The quantitative data from A. (C) PI/FITC-Annexin V staining analysis of apoptosis of SW1353 cells treated with the indicated concentrations of VE-822 alone or in combination with 10 μM cisplatin for 24, 48, or 72 h. (D) The quantitative data from C. (E) PI staining analysis of cell death in SW1353 cells, empty vector (E.V.) transfected SW1353 cells, and ATR shRNA transfected cells that were cultured in the presence or absence of 10 μM cisplatin for 24 h. (F) The quantitative data from E. (G) Western blot analysis of the levels of cleaved caspase 3 in untreated SW1353 cells, cells transduced with empty vector (E.V.) or cells transduced with ATR shRNA, treated with 10 μM cisplatin for 24 h.
Chk1 inhibition has been reported to enhance cisplatin cytotoxicity.18 In this study, we tested the effect of ATR inhibition on cisplatin-induced cell death. The results showed that both cisplatin and the ATR inhibitor VE-822 induced very low levels of cell death in SW1353 cells (Figure 4C and D). However, cotreatment of cells with cisplatin and VE822 significantly increased cell death in SW1353 cells (Figure 4C and D). Meanwhile, ATR inhibition by RNA interference significantly promoted cell death induced by cisplatin (Figure 4E and F). Western blot results showed that ATR inhibition upregulated the activation of caspase 3 induced by cisplatin (Figure 4G). These results suggested that ATR inhibition enhanced cisplatin cytotoxicity in chondrosarcoma cells.
Discussion
In this study, we found that cisplatin treatment of chondrosarcoma cells induced the activation of the ATR-Chk1 pathway, suggesting the induction of the DDR, which may contribute to cisplatin resistance in chondrosarcoma. The ATR inhibitor VE-822 and ATR shRNA reversed cisplatin-induced DDR and promoted DNA damage. Meanwhile, ATR inhibition increased the inhibition of the proliferation of chondrosarcoma cells induced by cisplatin and upregulated cisplatin-induced cell death, suggesting that inhibiting ATR may be a strategy for enhancing the chemosensitivity of chondrosarcoma treated with cisplatin.
Cisplatin was first approved for the treatment of testicular cancer and remains one of the most effective antitumor agents in a wide variety of solid tumors, such as head and neck, lung, bladder, ovarian, and cervical neoplasms.19 Although surgical resection is the first treatment option for chondrosarcoma patients, chemotherapy including cisplatin is an additional option.20 However, one crucial obstacle encountered often in cisplatin chemotherapy is drug resistance.19 Several strategies have been reported to sensitize chondrosarcoma to cisplatin.21–24
Cisplatin binds DNA to form intra- and interstrand crosslinks, resulting in DNA double helix disruption and further blockage of DNA replication and transcription. In response to the formation of cisplatin-DNA crosslinks and adducts, DNA damage is induced, which can be sensed by Ataxia telangiectasia mutated (ATM) and Ataxia telangiectasia and Rad-3 related (ATR) kinases, leading to the induction of the DNA damage response.25 ATM and ATR phosphorylate downstream kinases, such as checkpoint kinase 1 (Chk1) and checkpoint kinase 2 (Chk2), and regulate multiple cellular responses, including DNA repair, cell cycle arrest, and cell death.25 In this study, we found that chondrosarcoma cells at rest did not express phosphorylated ATR or Chk1. However, cisplatin treatment induced the phosphorylation of both ATR and Chk1, suggesting that cisplatin induces the DNA damage response in chondrosarcoma cells.
ATR is activated by DNA damaging agents and regulates cisplatin-induced cell death.
ATR activation facilitates chemotherapy resistance to cisplatin,16,26,27 and ATR inhibition enhances cisplatin chemosensitivity.9,28,29 In this study, we found that blocking ATR with the inhibitor VE-822 or with ATR shRNA upregulated the phosphorylation of H2AX, a biomarker of DNA damage,30,31 indicating that ATR inhibition promoted cisplatin-induced DNA damage. As expected, ATR inhibition promoted cisplatin-induced inhibition of cell proliferation and cell death in chondrosarcoma cells, suggesting that VE-822 mediated inhibition of ATR is a valuable strategy for chemotherapy of chondrosarcoma with cisplatin.
Acknowledgments
This study was supported by grants from the Natural Science Foundation of Hunan Province (2018JJ2577) and the Key Research Program of Hunan Province (2018SK2074). We thank Dr. Hui-Min Wang for kindly providing the 5-8F cell line, and Dr. Zhenfeng Dua for kindly providing the CS-1 cell line.
Highlights
Cisplatin induces ATR activation in chondrosarcoma cells.
ATR inhibition reverses the cisplatin-induced DNA damage response and promotes DNA repair.
Pharmacological and molecular inhibition of ATR signaling sensitizes chondrosarcoma to cisplatin-induced inhibition of cell proliferation.
Pharmacological and molecular inhibition of ATR signaling sensitizes chondrosarcoma to cisplatin-induced apoptosis.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Dorfman HD, Czerniak B. Bone cancers. Cancer. 1995;75(1 Suppl):203–210. doi: [DOI] [PubMed] [Google Scholar]
- 2.Polychronidou G, Karavasilis V, Pollack SM, Huang PH, Lee A, Jones RL. Novel therapeutic approaches in chondrosarcoma. Future Oncol. 2017;13(7):637–648. doi: 10.2217/fon-2016-0226 [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg AE. WHO classification of soft tissue and bone, fourth edition: summary and commentary. Curr Opin Oncol. 2013;25(5):571–573. doi: 10.1097/01.cco.0000432522.16734.2d [DOI] [PubMed] [Google Scholar]
- 4.Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16(2):81–104. doi: 10.1038/s41571-018-0114-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fokas E, Prevo R, Pollard JR, et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012;3:e441. doi: 10.1038/cddis.2012.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32(33):3789–3797. doi: 10.1038/onc.2012.556 [DOI] [PubMed] [Google Scholar]
- 7.Kemp MG, Sancar A. ATR kinase inhibition protects non-cycling cells from the lethal effects of DNA damage and transcription stress. J Biol Chem. 2016;291(17):9330–9342. doi: 10.1074/jbc.M116.719740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitt A, Knittel G, Welcker D, et al. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res. 2017;77(11):3040–3056. doi: 10.1158/0008-5472.CAN-16-3398 [DOI] [PubMed] [Google Scholar]
- 9.Shi Q, Shen LY, Dong B, et al. The identification of the ATR inhibitor VE-822 as a therapeutic strategy for enhancing cisplatin chemosensitivity in esophageal squamous cell carcinoma. Cancer Lett. 2018;432:56–68. doi: 10.1016/j.canlet.2018.06.010 [DOI] [PubMed] [Google Scholar]
- 10.Leszczynska KB, Dobrynin G, Leslie RE, et al. Preclinical testing of an Atr inhibitor demonstrates improved response to standard therapies for esophageal cancer. Radiat Oncol. 2016;121(2):232–238. doi: 10.1016/j.radonc.2016.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu P, Shen JK, Hornicek FJ, Liu F, Duan Z. Wnt inhibitory factor 1 (WIF1) methylation and its association with clinical prognosis in patients with chondrosarcoma. Sci Rep. 2017;7(1):1580. doi: 10.1038/s41598-017-01763-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song Q, Wang G, Chu Y, et al. TNF-α up-regulates cellular inhibitor of apoptosis protein 2 (c-IAP2) via c-Jun N-terminal kinase (JNK) pathway in nasopharyngeal carcinoma. Int Immunopharmacol. 2013;16(2):148–153. doi: 10.1016/j.intimp.2013.03.034 [DOI] [PubMed] [Google Scholar]
- 13.Song LB, Yan J, Jian SW, et al. Molecular mechanisms of tumorgenesis and metastasis in nasopharyngeal carcinoma cell sublines. Ai Zheng. 2002;21(2):158–162. [PubMed] [Google Scholar]
- 14.Di Micco R, Sulli G, Dobreva M, et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat Cell Biol. 2011;13(3):292–302. doi: 10.1038/ncb2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0 [DOI] [PubMed] [Google Scholar]
- 16.Meng Y, Chen CW, Yung MMH, et al. DUOXA1-mediated ROS production promotes cisplatin resistance by activating ATR-Chk1 pathway in ovarian cancer. Cancer Lett. 2018;428:104–116. doi: 10.1016/j.canlet.2018.04.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossmann KF, Brown JC, Moses RE. Cisplatin DNA cross-links do not inhibit S-phase and cause only a G2/M arrest in Saccharomyces cerevisiae. Mutat Res. 1999;434(1):29–39. doi: 10.1016/s0921-8777(99)00011-7 [DOI] [PubMed] [Google Scholar]
- 18.Hsu WH, Zhao X, Zhu J, et al. Checkpoint kinase 1 inhibition enhances cisplatin cytotoxicity and overcomes cisplatin resistance in SCLC by promoting mitotic cell death. J Thorac Oncol. 2019;14:1032–1045. doi: 10.1016/j.jtho.2019.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics. 2018;73(suppl 1):e478s. doi: 10.6061/clinics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riedel RF, Larrier N, Dodd L, Kirsch D, Martinez S, Brigman BE. The clinical management of chondrosarcoma. Curr Treat Options Oncol. 2009;10(1–2):94–106. doi: 10.1007/s11864-009-0088-2 [DOI] [PubMed] [Google Scholar]
- 21.Zhu Z, Wang CP, Zhang YF, Nie L. MicroRNA-100 resensitizes resistant chondrosarcoma cells to cisplatin through direct targeting of mTOR. Asian Pac J Cancer Prev. 2014;15(2):917–923. doi: 10.7314/apjcp.2014.15.2.917 [DOI] [PubMed] [Google Scholar]
- 22.Song YD, Zhang KF, Liu D, et al. Inhibition of EGFR-induced glucose metabolism sensitizes chondrosarcoma cells to cisplatin. Tumour Biol. 2014;35(7):7017–7024. doi: 10.1007/s13277-014-1902-4 [DOI] [PubMed] [Google Scholar]
- 23.Huang K, Chen J, Yang MS, Tang YJ, Pan F. Inhibition of Src by microRNA-23b increases the cisplatin sensitivity of chondrosarcoma cells. Cancer Biomark. 2017;18(3):231–239. doi: 10.3233/CBM-160102 [DOI] [PubMed] [Google Scholar]
- 24.Chen C, Zhou H, Xu L, et al. Recombinant human PDCD5 sensitizes chondrosarcomas to cisplatin chemotherapy in vitro and in vivo. Apoptosis. 2010;15(7):805–813. doi: 10.1007/s10495-010-0489-5 [DOI] [PubMed] [Google Scholar]
- 25.Zhu S, Pabla N, Tang C, He L, Dong Z. DNA damage response in cisplatin-induced nephrotoxicity. Arch Toxicol. 2015;89(12):2197–2205. doi: 10.1007/s00204-015-1633-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Cui B, Du J, et al. ATR activated by EB virus facilitates chemotherapy resistance to cisplatin or 5-fluorouracil in human nasopharyngeal carcinoma. Cancer Manag Res. 2019;11:573–585. doi: 10.2147/CMAR.S187099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Chen X, Xie G, et al. Cdc6 contributes to cisplatin-resistance by activation of ATR-Chk1 pathway in bladder cancer cells. Oncotarget. 2016;7(26):40362–40376. doi: 10.18632/oncotarget.9616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XQ, Ren J, Chen P, et al. Co-inhibition of Pol eta and ATR sensitizes cisplatin-resistant non-small cell lung cancer cells to cisplatin by impeding DNA damage repair. Acta Pharmacol Sin. 2018;39(8):1359–1372. doi: 10.1038/aps.2017.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang HG, Chen P, Su JY, et al. Knockdown of REV3 synergizes with ATR inhibition to promote apoptosis induced by cisplatin in lung cancer cells. J Cell Physiol. 2017;232(12):3433–3443. doi: 10.1002/jcp.25792 [DOI] [PubMed] [Google Scholar]
- 30.Rao VA, Agama K, Holbeck S, Pommier Y. Batracylin (NSC 320846), a dual inhibitor of DNA topoisomerases I and II induces histone gamma-H2AX as a biomarker of DNA damage. Cancer Res. 2007;67(20):9971–9979. doi: 10.1158/0008-5472.CAN-07-0804 [DOI] [PubMed] [Google Scholar]
- 31.Alipoor A, Fardid R, Sharifzadeh S. Evaluating gamma-H2AX expression as a biomarker of DNA damage after X-ray in angiography patients. J Biomed Phys Eng. 2018;8(4):393–402. [PMC free article] [PubMed] [Google Scholar]