

Abstract
We present a novel method to fluorescently label proteins, post-translationally, within live Saccharomycescerevisiae. The premise underlying this work is that fluorescent protein (FP) tags are less disruptive to normal processing and function when they are attached post-translationally, because target proteins are allowed to fold properly and reach their final subcellular location before being labeled. We accomplish this post-translational labeling by expressing the target protein fused to a short peptide tag (SpyTag), which is then covalently labeled in situ by controlled expression of an open isopeptide domain (SpyoIPD, a more stable derivative of the SpyCatcher protein) fused to an FP. The formation of a covalent bond between SpyTag and SpyoIPD attaches the FP to the target protein. We demonstrate the general applicability of this strategy by labeling several yeast proteins. Importantly, we show that labeling the membrane protein Pma1 in this manner avoids the mislocalization and growth impairment that occur when Pma1 is genetically fused to an FP. We also demonstrate that this strategy enables a novel approach to spatiotemporal tracking in single cells and we develop a Bayesian analysis to determine the protein’s turnover time from such data.
Keywords: membrane protein, protein engineering, S. cerevisiae, single cell, SpyCatcher-SpyTag
Introduction
Fluorescent labeling is a powerful strategy with which to study the localization and dynamics of proteins in living cells. Most commonly, labeling is achieved by directly fusing a fluorescent protein (FP), such as Green Fluorescent Protein (GFP) to the protein of interest. A limitation of this approach, however, is the relatively large size of the FP tag (GFP = 27 kDa), which has the potential to interfere with the assembly, localization and function of the protein to which it is fused. Proteome-wide studies in Saccharomycescerevisiae found that ~25% of proteins cannot tolerate a GFP C-terminal fusion. Plasma membrane transporter proteins are particularly sensitive, with only 46 of the 139 putative transporter proteins exhibiting any plasma membrane fluorescence when fused to GFP, and only 20 of those 139 localizing exclusively to the plasma membrane, endoplasmic reticulum or Golgi body (Huh et al., 2003; Brohée et al., 2010).
There are several potential strategies for fluorescently labeling proteins of interest in live cells. One of the most popular is to fuse a protein domain that binds a fluorescent small molecule, such as SNAP tag (Keppler et al., 2003), CLIP tag (Gautier et al., 2008) or HaloTag, (Los et al., 2008) to the target protein. The small molecule binding domain is still relatively large however (19.4, 20.6 and 33 kDa for SNAP tag, CLIP tag and HaloTag, respectively), and therefore susceptible to the same issues as a direct FP fusion. Although these methods have been applied successfully in mammalian cells, they require significant additional manipulations if they are to work in yeast (Lacy et al., 2017). Another labeling approach is to fuse the protein of interest to a short peptide tag that acts as the substrate recognition sequence onto which an enzyme covalently attaches a fluorescent small molecule. Labeling a target protein in the complex intracellular environment is difficult, however. Lipoic acid ligase is one of the few examples where this strategy has been successful intracellularly, in mammalian cells (Uttamapinant et al., 2010; Ho and Tirrell, 2016). To successfully label target proteins, the exogenous small molecule must be cell permeable, minimally cytotoxic, display negligible off-target binding and it must be easy to ‘wash’ the unreacted label from the cell.
A protein can, in principal, be detected in vivo by interaction with a fluorescently labeled protein binding domain—such as a single chain antibody variable fragment—that recognizes the native protein (Riedl et al., 2008; Schmidthals et al., 2010). A limitation of this approach, however, is that a new binding domain must be generated for each target protein. A more widely applicable strategy is to tag the protein of interest with a short peptide and detect the protein of interest by interaction with the fluorescently labeled peptide–binding module. The advantage of this approach is that the same peptide–binding domain pair can be used for different proteins. The disadvantage is that the peptide–binding domain interaction is non-covalent (Pratt et al., 2016). Here we present a new strategy for imaging proteins in live yeast cells that employs an engineered open isopeptide domain (SpyoIPD), a derivative of the SpyCatcher protein (Zakeri et al., 2012). SpyCatcher/SpyTag is a protein–peptide interaction pair that associates and spontaneously forms an intermolecular covalent isopeptide bond and is thus a useful tool for post-translationally linking proteins together. SpyCatcher/SpyTag has been widely used in vitro and in bacteria (Veggiani et al., 2014), but never, to our knowledge, within live eukaryotic cells. The SpyoIPD we develop here is more stable than the original SpyCatcher and exhibits greater reactivity in the yeast cytosol. Our strategy to fluorescently label proteins is to express the target protein fused to SpyTag (13 amino acids) and to separately express SpyoIPD fused to an FP. Reaction between the SpyoIPD and SpyTag post-translationally labels the protein of interest with the FP, thus allowing visualization. Although the final labeled form of the target protein possesses a relatively large modification, we hypothesized that labeling post-translationally would be less disruptive to native function because the target protein is allowed to properly fold and reach its native localization before being modified.
We demonstrate that this labeling strategy can be used to image a variety of proteins, highlighting the plasma membrane proton pump, Pma1. Pma1 is of particular interest because direct fusion of Pma1 to an FP results in its mislocalization to the vacuole, and cells expressing only FP tagged forms of Pma1 exhibit a significant growth defect. We show that labeling Pma1 using SpyoIPD/SpyTag results in neither mislocalization nor a growth defect. We also demonstrate how this method can be adapted to temporally track a protein in a particular subcellular location, and develop a Bayesian analysis to determine the protein’s turnover time from such data.
Materials and methods
Molecular biology
The plasmid containing the original SpyCatcher gene (Zakeri et al., 2012) was obtained from Addgene (Addgene plasmid #35044), EGFP was amplified from the Regan lab vector pPROEX HTa M EGFP-MEEVD (pPROEX HTa M is a modified version of the pPROEX HTa vector) (Cormack et al., 1996) (Invitrogen) and mCherry was amplified from pNAS1b (Addgene plasmid #61968) (Sawyer et al., 2014). SpyoIPD was generated by site-directed mutagenesis of FbaB-CnaB2-Asp556Ala (Hagan et al., 2010) to introduce the Ile552 to Ala mutation. The original SpyCatcher protein contains the point mutations Glu473Ile and Tyr508Met, but these are not included in the SpyoIPD designs. The original SpyCatcher protein also has an additional 20 residues at the N-terminus that are not part of the isopeptide domain fold (Hagan et al., 2010), and two residues at the C-terminus (Arg-Ser), which are not present in FbaB. These 22 residues are not included in SpyoIPD. Typically, genes were inserted into plasmids using circular polymerase extension cloning following published procedures (Speltz and Regan, 2013), and unless stated otherwise, tags were attached to inserts through incorporation within PCR primers. Table I lists all oligonucleotides used in this study and Table II lists all plasmids used.
Table I.
Oligonucleotides used in this study
| # | Name | Fwd | Purpose |
|---|---|---|---|
| 1 | PMA1_CT_F | GAGGGTCACGAGAACACC | Checking C-terminus of genomic PMA1 |
| 2 | PMA1_CT_R | GAAAAATTAAACCAGAAAAATCAAGTTG | Checking C-terminus of genomic PMA1 |
| 5 | HTB2_CT_F | GCAAACTCACCCAGACAC | Checking C-terminus of genomic HTB2 |
| 6 | HTB2_CT_R | CCAAACTGCTCAAGATAAGATCG | Checking C-terminus of genomic HTB2 |
| 7 | CDC12_CT_F | GAGGGTCACGAGAACACC | Checking C-terminus of genomic CDC12 |
| 8 | CDC12_CT_R | CAGTTACTTCTGCTGGTTCC | Checking C-terminus of genomic CDC12 |
| 9 | PMA1_ST_F | ATGGCTGCTATGCAAAGAGTCTCTACTCAACACGAAAAGGAAACCGGTGGATCAGGCTCTGG | Attaching SpyTag to genomic PMA1 |
| 10 | PMA1_ST_R | AAAATGTGACAAAAATTATGATTAAATGCTACTTCAACAGGATTATTAGAAAAACTCATCGAGCATC | Attaching SpyTag to genomic PMA1 |
| 13 | HTB2_ST_F | GAAGGTACTAGGGCTGTTACCAAATACTCCTCCTCTACTCAAGCCGGTGGATCAGGCTCTGG | Attaching SpyTag to genomic HTB2 |
| 14 | HTB2_ST_R | GATGCTCGATGAGTTTTTCTAAGTCACTCACTAGGTATTGTGATTTAGTCATGTTTTCTTTTTATTA | Attaching SpyTag to genomic HTB2 |
| 15 | CDC12_ST_F | GAAGAGCAGGTCAAAAGCTTGCAAGTAAAAAAATCCCATTTAAAAGGTGGATCAGGCTCTGG | Attaching SpyTag to genomic CDC12 |
| 16 | CDC12_ST_R | GATGCTCGATGAGTTTTTCTAATGATTAATTAATGTCTTCCTCTTTGTCTCGTCAATTTCAACGCCT | Attaching SpyTag to genomic CDC12 |
| 17 | PMA1_Chr_F | ATGGCTGCTATGCAAAGAGTCTCTACTCAACACGAAAAGGAAACCGCATCCGTGAGCAAGGG | Attaching mChery to genomic PMA1 |
| 18 | PMA1_Chr_R | AAAATGTGACAAAAATTATGATTAAATGCTACTTCAACAGGATTATTAGAAAAACTCATCGAGCATC | Attaching mChery to genomic PMA1 |
| 19 | GAL2_SpG_F | GGAGAAAAAACCCCGGATTCATGTCGTACTACCATCACCATC | Inserting SpyoIPD-GFP at GAL2 locus |
| 20 | GAL2_SpG_R | CCGCTGCCGCTGCCGCCAGCAGCAACCATGACAGC | Inserting SpyoIPD-GFP at GAL2 locus |
| 21 | GAL1_Sp_F | GGAGAAAAAACCCCGGATTCATGTCGTACTACCATCACCATC | Transfering SpyoIPD to GFP pFA6His3, to make SpyoIPD-GFP |
| 22 | Sp_GAL1_R | CCGCTGCCGCTGCCGCCAGCAGCAACCATGACAGC | Transfering SpyoIPD to GFP pFA6His3, to make SpyoIPD-GFP |
| 23 | FA6LnkCYCF | GCTGAAGCTTCGTACGCT GGTGGATCAGGCTCTGGTTTGCAA TAA GTCATGTAATTAGTTATGTCACGC | Inserting Linker-CYC1 into pFA6 KanMX6 |
| 24 | CYC_pFA6_R | CTGGCGCGCCTTAATTAACCGCAAATTAAAGCCTTCGAGC | Inserting Linker, ST, and mCherry-CYC1 into pFA6 KanMX6 |
| 25 | Lnk_ST_CYCR | GTAAGCGTGACATAACTAATTACATGATCATTATTTCGTCGGTTTATACGCATCCACCATGACAATGTGAGCTTGCAAACCAGAGCCTG | Inserting ST into pFA6 His3 L |
| 26 | FA6_F | GGTTATTGTCTCATGAGCGG | Inserting ST into pFA6 His3 L |
| 27 | PFA6_mCher_F | GCTGAAGCTTCGTACGCTGCATCCGTGAGCAAGGG | Inserting mCherry into pFA6 KanMX6 |
| 28 | mCher_CYC_R | CATAACTAATTACATGACTTATCACTTGTACAGCTCGTCC | Inserting mCherry into pFA6 KanMX6 |
| 29 | mCher_CYC_F | GCTGTACAAGTGATAAGTCATGTAATTAGTTATGTCACGC | Inserting mCherry into pFA6 KanMX6 |
| 30 | Gal_SC_F | CGGATTCTAGAACTAGTGGATCCATGCGTCTGTCGTACTACCATCAC | Transferring Original SpyCatcher into p424 Gal1 |
| 31 | SC_Gal_R | GAGTCATGTAATTAGTTATGTCACGCGTGACGCTCATATTAGATCTTAGTGA | Transferring Original SpyCatcher into p424 Gal1 |
| 32 | 424_Sp_F | GGAGAAAAAACCCCGGATTC ACTTTAAGAAGGAGATATACATATGTCG | Transferring SpyoIPD to p424 Gal1 |
| 33 | IPD_424_R | GCTTGATATCGAATTCCTGCAGAGCTCGAATTCCGGATCC | Transferring SpyoIPD to p424 Gal1 |
| 34 | 415_V5_GFP_F | GGATCCACTAGTTCTAGATCCGATGGGTAAACCAATTCCAAATCCATTGTTGGGTTTGGATTCTACTGGTTCTAGTAAAGGAGAAGAACTTTTCACTG | Attaching V5 epitope to GFP and transferring to pcu415CUP1 |
| 35 | GFP_ST_415_R | CTATTAAAGCTTATCGATACCGTCGACCTTAGTAGGTTTATAAGCATCAACCATAACAATATGAGCAGAACCTTTGTATAGTTCATCCATGCCATG | Attaching ST to GFP and transferring to pcu415CUP1 |
Table II.
Plasmids used in this study
| Name | Parent | Description | Source |
|---|---|---|---|
| pFA6His3MX6 | His3 | Longtine et al. (1998) | |
| pFA6His3MX6-SpyoIPD-GFP | pFA6His3MX6 | His3, SpyoIPD-GFP | This study |
| pFA6KanMX6 | KanR | Longtine et al. (1998) | |
| pFA6KanMX6L | pFA6KanMX6 | KanR, Linker | This study |
| pFA6KanMX6 LST | pFA6KanMX6 | KanR, Linker-SpyTag | This study |
| pFA6KanMX6 LC | pFA6KanMX6 | KanR, Linker-mCherry | This study |
| p424gal1 | 2 μ/Trp, GAL1, empty vector | Mumberg et al. (1994) | |
| p424gal1-SpyCatcher | p424gal1 | 2 μ/Trp, GAL1, SpyCatcher | This study |
| p424gal1-SpyoIPD | p424gal1 | 2 μ/Trp, GAL1, SpyoIPD | This study |
| pcu415CUP1 | CEN/Leu, CUP1, empty | Mumberg et al. (1994) | |
| pcu415CUP1-GFPST | pcu415CUP1 | CEN/Leu, CUP1, GFP-SpyTag | This study |
Constructs for testing SpyoIPD reactivity in yeast: His6 tagged SpyCatcher and SpyoIPD were amplified from their bacterial expression vectors (see above) and inserted into p424GAL1 (Mumberg et al., 1994), a yeast shuttle vector containing the strong galactose-inducible GAL1 promoter, a high-copy number 2 μ replication origin and a TRP1 selection marker. The gene encoding EGFP was tagged at the 5' end with a sequence coding for the V5 epitope, and the 3' end with DNA coding for SpyTag, and inserted into pCu415CUP1 (Labbé and Thiele, 1999), a yeast shuttle vector that contains the intermediate strength, copper-inducible CUP1 promoter (Lee et al., 2015), a low copy number CEN replication origin and a LEU2 selectable marker.
Constructs for Spycatcher/SpyTag imaging in yeast: SpyoIPD was attached to the N-terminus of EGFP via an 8-residue linker (GGSGSGLQ), and inserted into p424GAL1. The promoter, gene fusion and CYC1 terminator (CYC1T) were then amplified from this vector and inserted into pFA6a-His3MX6 (Longtine et al., 1998), a yeast insertion vector that contains a HIS3 selectable marker. For tagging a protein of interest with SpyTag, oligonucleotides were used to amplify CYC1T from p424GAL1 and attach a linker (GGSGSGLQ) upstream of CYC1T. This fragment was inserted into pFA6KanMX6 (Longtine et al., 1998), a yeast insertion vector with a kanamycin selectable marker (KanR). This construct was then used as a template for making linker-SpyTag (GGSGSGLQAHIVMVDAYKPTK), by amplifying the linker and a portion of the pFA6KanMX6 vector, attaching SpyTag in the process. This product was then inserted back into pFA6KanMX6 Linker (L) to create pFA6KanMX6 SpyTag (LST). mCherry was also inserted into pFA6a-KanMX6 using the same strategy as used for pFA6KanMX6 Linker.
Characterization of SpyCatcher and its variants
NMR: uniformly 15N-labeled samples of SpyCatcher and SpyoIPD were produced and purified using established protocols (Hagan et al., 2010). NMR samples typically contained 0.1 mM protein in phosphate buffered saline, pH 7.4, 2% (v/v) D2O. 1H-15N HSQC spectra were recorded on a Bruker Ascend 700 MHz spectrometer equipped with a Prodigy TCI probe at 22°C. A standard Bruker pulse sequence for gradient-enhanced HSQC including WATERGATE water suppression and water flip-back pulse was used, with 2 transients recorded at spectral resolutions of 12.8 Hz and 31.0 Hz in the direct and indirect dimension, respectively. Spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed with CCPN Analysis 2 (Vranken et al., 2005).
Differential scanning fluorimetry: fluorescence at 570 nm (excitation 480 nm) of SYPRO© Orange in the presence of 5 μM protein was recorded using a real-time PCR instrument. The samples were heated at a rate of 1°C per minute, between 25°C and 95°C. These denaturation transitions are irreversible, so it is inappropriate to calculate a Tm. We show the raw data.
Yeast strain construction
Unless noted otherwise, standard techniques and growth media were used for cultivating and genetically manipulating yeast strains (Fink, 2002). Dropout media was prepared using purchased amino acid dropout mixes (Clontech). Table III lists the yeast strains used in this study. All genomic insertions were verified by PCR using primers that anneal outside the insert cassette, and sequencing the resulting PCR products.
Table III.
Yeast strains used in this study
| Name | Parent | Genotype | Reference |
|---|---|---|---|
| JY102 | MATalpha ura3-52 lys2-801 trp1-D63 his3-D200 leu2-D1 | Hochstrasser Lab | |
| PC | JY102 | MATalpha ura3-52 lys2-801 trp1-D63 his3-D200 leu2-D1 PMA1-mCherry::KanMX6 | This study |
| PLCG | JY102 | MATalpha ura3-52 lys2-801 trp1-D63 his3-D200 leu2-D1 gal2Δ::His3MX6 SpyoIPD-GFP PMA1-linker::KanMX6 | This study |
| PSTSG | JY102 | MATalpha ura3-52 lys2-801 trp1-D63 his3-D200 leu2-D1 gal2Δ::His3MX6 SpyoIPD-GFP His PMA1-SpyTag::KanMX6 | This study |
| HSTSG | JY102 | MATalpha ura3-52 lys2-801 trp1-D63 his3-D200 leu2-D1 gal2Δ::His3MX6 SpyoIPD-GFP HTB2-SpyTag::KanMX6 | This study |
| CSTSG | JY102 | MATalpha ura3-52 lys2-801 trp1-D63 his3-D200 leu2-D1 gal2Δ::His3MX6 SpyoIPD-GFP CDC12-SpyTag::KanMX6 | This study |
| PG | ATCC 201388 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 PMA1-GFP::His3MX | Huh et al. (2003) |
| HG | ATCC 201390 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 HTB2-GFP::His3MX | Huh et al. (2003) |
| CG | ATCC 201391 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 CDC12-GFP::His3MX | Huh et al. (2003) |
Target proteins were tagged at the C-terminus with DNA coding for linker-SpyTag or linker alone in the yeast strain MHY2587 (an Ade+ variant of YPH499) by amplifying the desired tag and the KanR selectable marker from the appropriate template vector using primers that also attached 45 bp homology arms. Homology arms were designed so to match the final 45 bp of the target protein and the 45 bp immediately following the stop codon.
To insert SpyoIPD-EGFP, the fusion gene was amplified from vector pFA6His3MX6—SpyoIPD-EGFP, along with the upstream GAL1 promoter and downstream HIS3 selectable marker. Homology arms were attached that matched a 45 bp sequence 700 bp upstream of the GAL2 gene, and 45 downstream of the GAL2 stop codon.
Yeast strains expressing target proteins fused at the C-terminus to EGFP were obtained from the Yeast EGFP Clone Collection (Thermo Fisher), originally created and described in Huh et al. (2003).
SpyoIPD-EGFP sequence:
MSYYHHHHHHDCDIPTTENLYFQGAMVDSATHIKFSKRDIDGKELAGATMELRDSSGKTISTWISDGQVKDFYLMPGKYTFVETAAPDGYEVATAITFTVNEQGQVTVNGKATKGDAHAVMVAAGGSGSGLQSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTLVTTLTYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNTPIGDGPVLSPDNHYLSTQSKLSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
Linker: GGSGSGLQ
Linker-SpyTag: GGSGSGLQAHIVMVDAYKPTK
Western blot analysis
To assess the in vivo activity of SpyCatcher and its variants, yeast colonies were picked and grown overnight in synthetic defined medium (Leu−/Trp−) containing 0.1% glucose, 2% galactose and 100 μM CuSO4. The next day, cultures were diluted to an OD600 of 0.2 into fresh selection medium containing 2% galactose and 100 μM CuSO4, and grown to an OD600 between 1.0 and 2.0 (usually ~20 h at 30°C). At this point, 10 OD600 equivalents were pelleted, washed once with H2O and stored at −80°C for later analysis.
Yeast pellets (from 10 OD600 equivalents) were lysed using the alkali lysis procedure (Kushnirov, 2000) and final pellets were resuspended in 50 μL of 1×SDS-PAGE buffer. Lysate (10 μl) was loaded onto 10% or 15% SDS-PAGE gels, transferred to nitrocellulose, and probed using appropriate primary antibodies. Mouse anti-His6 (GenScript (Piscataway, NJ), Cat. # A00186-100), and mouse antiV5 (Invitrogen (Carlsbad, CA), Cat. # 46–0705) primary antibodies were each diluted for use 1:1000 in Tris Buffered Saline with 0.1% Tween (TBST) and 5% w/v nonfat dry milk. For all immunoblots, the secondary antibody used was sheep anti-mouse IgG (diluted 1:10 000 in 5% milk/TBST, GE (Little Chalfont, UK), Product code NXA931) conjugated to horse radish peroxidase. Immunoblots were visualized by enhanced chemiluminscence, using ClarityTM ECL Western Blotting substrate (BioRad) and imaged using a GBox-Chemi 16 Bio Imaging System (Syngene).
Microscopy
For imaging experiments, single colonies were picked and grown overnight in non-inducing His−/G418+ synthetic defined medium (2% sucrose/1% raffinose). The next day, overnight cultures were diluted to an OD600 of 0.05 into fresh His−/G418+ synthetic defined medium (2% sucrose/1% raffinose), supplemented with the desired concentration of galactose. Cultures were grown 8 h before imaging.
For pulse-chase experiments, glucose was added to a final concentration of 2% w/v after 8 h of induction. The OD600 was sampled at regular intervals following glucose addition, and kept below 2.0 throughout the experiment by diluting with prewarmed medium (that exactly matched the original growth medium).
Fluorescent images were collected using Olympus IX-71 microscope with a 100 × 1.4 NA Plan Apo lens (Olympus) and a CSU-X1 (Andor Technology) confocal spinning-disk confocal system equipped with an iXON-EMCCD camera (Andor Technology).
Microfluidics experiments
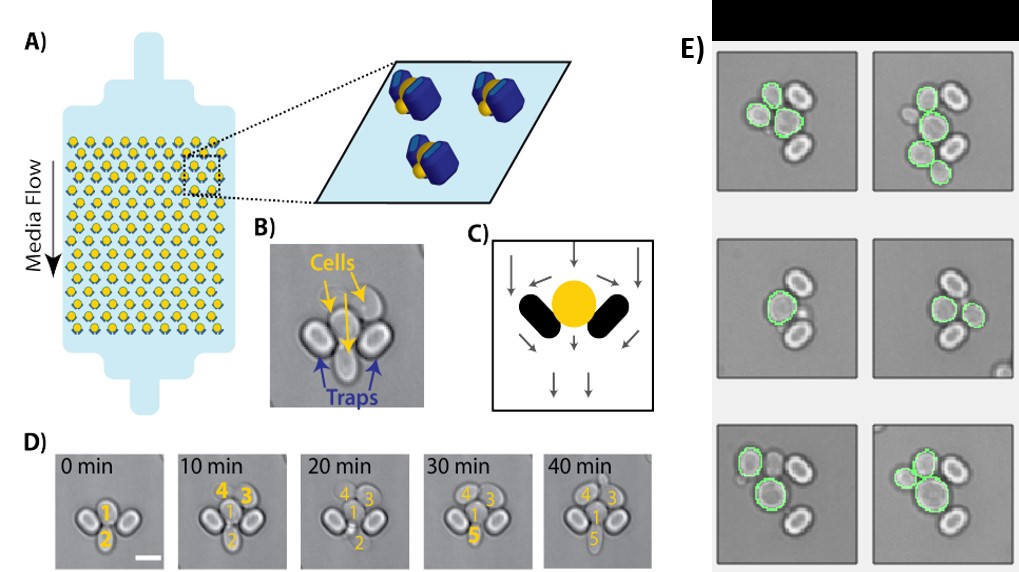
Microfluidic devices were fabricated with polydimethylsiloxane using standard techniques (Crane et al., 2014). Single colonies were inoculated into a liquid culture of synthetic complete medium (2% raffinose, 0.5% galactose) and grown overnight at 30°C. The following day, cells were loaded into a prewarmed (30°C) microfluidic device and incubated in the synthetic complete medium (2% raffinose, 0.25% galactose) for 1 h before switching to glucose (0.1%). Throughout the experiment, the device was perfused with fresh medium at a flow rate of 4 μl/min, controlled by syringe pumps (World Precision Instruments), and temperature was maintained at a constant environment of 30°C using a temperature controlled incubation chamber (Okolabs).
Time-lapse image acquisition was performed on a Nikon Eclipse Ti inverted microscope, with a 60 × 1.4 NA oil immersion objective (Nikon). The experiment was controlled using a custom Matlab script (Mathworks) written for Micromanager (Edelstein et al., 2010). Images were taken in brightfield and fluorescence, using a filter set appropriate for EGFP. Exposure intensities (LED lamp, 4 V), exposure times (30 ms) and imaging intervals (0.5 h−1) were set to avoid photobleaching. Data analysis was performed using image segmentation, cell tracking and data extraction using custom Matlab script (Crane et al., 2014). To determine the ratio of membrane to cytosol signal, median membrane pixel intensities for each cell were extracted from images using the cell outline generated during cell identification.
Photobleaching
We investigated the possible contribution of photobleaching to fluorescence decay by comparing the fluorescence of cells irradiated multiple times at each time-point to cells irradiated once at each time-point. We observed no significant difference in the cellular fluorescence over time between the two sets of cells, indicating that photobleaching does not contribute significantly to the fluorescent decay observed (see Fig. S4).
Results
Design of the post-translational imaging strategy
Our method for imaging proteins in living cells is illustrated schematically in Fig. 1. The genomic copy of the gene encoding the target protein is tagged at the 3' end with a DNA sequence encoding SpyTag. Although in principle, the SpyTag sequence could be placed anywhere in the target protein, for consistency and convenience we typically place SpyTag at the C-terminus. Note that the gene encoding the target protein fused to SpyTag replaces the wild-type copy and is expressed from the target protein’s endogenous promoter. SpyoIPD fused to FP is expressed from the GAL1 promoter, addition of galactose induces expression of SpyoIPD-FP, which reacts with SpyTag and covalently labels the target protein with an FP. Integrating SpyoIPD-EGFP at the GAL2 locus simultaneously deletes the Gal2 permease, making expression from the GAL1 promoter linear with respect to galactose concentration (Hawkins and Smolke, 2006), and enabling finer control of the intracellular concentration of the SpyoIPD-FP fusion protein.
Fig. 1.

Schematic illustration of the labeling strategy. The genomic copy of the gene encoding the target protein (Target) is fused at the 3' end to a sequence encoding the SpyTag (ST), replacing the chromosomal copy of the target’s gene. Expression is from the target’s endogenous promoter. DNA encoding a fusion of SpyoIPD (Spy) and FP (FP) is integrated at the GAL2 locus. Expression is from the GAL1 promoter (pGAL1). Shown to the right are the expressed proteins and the labeling product, with ST illustrated as the small circle fused to Target.
We first tested whether the original SpyCatcher/SpyTag pair is active in yeast, which had not been previously reported. We created a yeast strain that coexpressed SpyCatcher and SpyTag fused to the C-terminus of EGFP. For this experiment, EGFP serves simply as a convenient handle to increase the mass of SpyTag, making it easy to identify in an SDS gel. Because SpyCatcher forms a covalent bond to SpyTag, the conjugate species is resistant to SDS denaturation and can be detected as a higher molecular weight species in a Western Blot. Using this assay, only a small amount of the EGFP-SpyTag-SpyCatcher conjugate was observed (Fig. 2D). In addition, despite the strong promoter and high-copy number plasmid used to drive SpyCatcher expression, we never observed unconjugated SpyCatcher in Western blots. We hypothesized that SpyCatcher expression and therefore labeling efficiency could be improved by SpyCatcher derivatives with increased structural stability.
Fig. 2.

Design and properties of SpyoIPDs in vitro and in vivo. (A) To improve stability of the SpyCatcher protein (large rectangle with cutouts), we reintroduced portions of the C-terminal β-strand (thin rectangular ‘overhang’ attached to SpyCatcher) that was originally removed to make SpyTag. The reactive Asp on this extension was mutated to Ala (D556A) to prevent reaction with the Lys in the SpyCatcher region, and the appended sequence was also mutated (I552A) to weaken its interaction with the rest of the domain, allowing SpyTag (thin rectangle) to displace the reintroduced β-strand and react with SpyCatcher. (B) Comparison of the C-terminal sequences of SpyTag, SpyCatcher and SpyoIPD. The highlighted Ala second from the C-terminus replaces the isopeptide bond-forming Asp. The highlighted Ala six residues from the C-terminus replaces Ile 552 to weaken binding between SpyCatcher and the reintroduced sequence. (C) Differential scanning fluorimetry traces of SpyCatcher (solid circles) and SpyoIPD (hollow circles). (D) Comparison of the in vivo activity of SpyCatcher and SpyoIPD. SpyTag was expressed as a fusion to EGFP from a medium strength promoter on a low copy number plasmid. An N-terminal V5 epitope was also fused to EGFP to facilitate easy detection. A Western blot, probing for the V5 epitope, is shown. Lanes and bands are as labeled. The lower molecular weight band corresponds to unreacted EGFP-ST (filled triangle), and the higher molecular weight band to the covalent EGFP-ST-SC or EGFP-ST-SpyoIPD conjugate (hollow triangle).
SpyoIPD, a more stable derivative of the SpyCatcher protein
The second CnaB domain of streptococcal surface protein FbaB contains a covalent isopeptide bond between a Lys on the N-terminal β-strand and an Asp on the C-terminal β-strand (Hagan et al., 2010). Splitting this domain gave rise to the SpyCatcher/SpyTag system, where the β-strand containing the Asp residue (SpyTag) is expressed separately from the remainder of the protein (SpyCatcher). SpyTag associates and reacts with SpyCatcher to form the isopeptide bond between the Asp and Lys sidechains in trans, so that each β-strand now comes from a separate protein (Zakeri et al., 2012). We sought to create a more stable derivative of this system by designing an ‘open’ isopeptide domain (SpyoIPD) that retains the C-terminal β-strand that was removed to create SpyCatcher. The reintroduced strand was mutated to remove the reactive Asp (Asp556Ala), and to weaken the interaction between the reintroduced strand and the rest of the protein (Ile552Ala). The goal of this design was to increase the stability of the domain, but not so much that reactivity would be inhibited (see Fig. 2A and B). 1H–15N HSQC NMR spectra show that both SpyCatcher and SpyoIPD are folded in solution, even in the absence of SpyTag (Fig. S1). Differential scanning fluorimetry (DSF) analyses, however, indicate that SpyoIPD is more thermally stable than SpyCatcher (Fig. 2C).
Testing the in vivo activity of SpyoIPD
To compare the in vivo activity of SpyoIPD with SpyCatcher, we again used the western blot assay described above and compared yeast strains that expressed either SpyoIPD or SpyCatcher, together with SpyTag fused to the C-terminus of EGFP. It is clearly evident in Fig. 2D that there is greater formation of the EGFP-SpyTag-SpyoIPD conjugate than the EGFP-SpyTag-SpyCatcher conjugate. We therefore used SpyoIPD in all subsequent imaging applications (Fig. 2D).
Fluorescently labeling proteins in vivo with SpyoIPD
For initial experiments, target proteins were chosen that are abundant, not known to have an inaccessible C-terminus, and which localize to a distinct region of the cell. Here we present data on three proteins from different subcellular compartments: the plasma membrane (Pma1), the nucleus (Htb2), and the bud neck (Cdc12). For each imaging experiment, cells were grown overnight in non-inducing medium, diluted the next day into medium containing galactose, and imaged after an additional 8 h of growth. All three proteins show a clearly localized signal (Fig. 3). When Htb2, a histone protein, is tagged with SpyTag and coexpressed with SpyoIPD-EGFP, spheres of fluorescence corresponding to the nucleus are observed, indicating that the majority of SpyoIPD-EGFP is bound to Htb2-SpyTag. A similar result is observed when Htb2 is fused directly to EGFP. Cdc12, a component of the septin ring, localizes to the bud neck (Madden and Snyder, 1998) and is also readily visualized by the SpyoIPD/SpyTag imaging system. Cells expressing Cdc12-ST and SpyoIPD-EGFP produce tight rings of fluorescence around the bud neck—the same localization pattern observed when native Cdc12 is visualized by immunofluorescence in fixed and permeabilized cells (Haarer and Pringle, 1987). A similar pattern is observed in cells expressing Cdc12 directly fused to EGFP, although ~5% of cells expressing the fusion protein show a distorted morphology, indicating that direct fusion to an FP can interfere with normal Cdc12 function. This phenotype is not observed in cells expressing Cdc12-ST and SpyoIPD-EGFP. Pma1, an essential plasma membrane proton pump naturally, localizes to the plasma membrane (Mason et al., 2014). Pma1 imaged with the SpyoIPD/SpyTag system produces a ring of fluorescence around the cell periphery, consistent with labeling of Pma1 in the plasma membrane. By contrast, cells expressing Pma1 directly fused to EGFP show strong vacuolar fluorescence (Figs 3 and 4B). For comparison, also shown in Fig. 3 is a cell expressing SpyoIPD-EGFP and no SpyTagged protein. A diffuse, non-localized fluorescence is observed.
Fig. 3.

A comparison of direct fusion to EGFP with SpyoIPD/SpyTag labeling. Brightfield (DIC) and fluorescence (Fluor) images are shown for direct fusions of a target protein to EGFP (EGFP fusion) and for labeling of the target protein using the SpyoIPD/SpyTag strategy (SpyoIPD/ST). The identity of the target protein is given to the left of the images. The no SpyTag strain expresses SpyoIPD-EGFP, but no SpyTagged protein. CDC12: direct fusion to EGFP results in a distorted cell morphology in ~5% of CDC12-EGFP expressing cells.; HTB2: direct fusion to EGFP and the SpyoIPD/SpyTag strategy both show the expected labeling of the nucleus; PMA1: direct fusion to EGFP results in significant vacuolar mislocalization.
Fig. 4.

PMA1 localization and function. (A) Immuno-staining of fixed yeast cells (using anti-HA antibodies) in a strain expressing Pma1 fused to the HA peptide. This ‘native’ Pma1 localizes exclusively to the plasma membrane, with none evident in the vacuole. Reproduced with permission from Mason et al. (2014). (B) Live cell imaging of yeast expressing a Pma1-EGFP fusion protein, expressed from the endogenous Pma1 promoter. A significant amount of fluorescence is observed in the vacuole in addition to that present at the plasma membrane. (C) Comparison of the growth of yeast expressing untagged Pma1 (1), Pma1 C-terminally tagged with mCherry (2), or Pma1 C-terminally tagged with SpyTag (3). Strains are streaked on media containing 2% galactose, so the Pma1-ST expressing strain is also expressing SpyoIPD-EGFP. For both tagged strains, Pma1 is expressed under control of its native promoter, and the tagged copy of the strain is the only copy of the protein present. The strain expressing Pma1-mCherry fusion exhibits a significant growth defect, whereas the other two strains grow equally well.
The effects of different labeling strategies on Pma1 function
Pma1 is an essential plasma membrane proton pump in yeast that is responsible for maintaining cytosolic pH and the membrane potential (Serrano et al., 1986). Pma1 has been proposed to play a role in cell aging (Henderson et al., 2014) and has been used to study protein quality control pathways in the secretory system (Ferreira et al., 2001). Immunofluorescent labeling of epitope-tagged Pma1 in fixed cells shows native Pma1 localizes exclusively to the plasma membrane (Fig. 4) (Mason et al., 2014). By contrast, when Pma1 is directly fused to a FP, fluorescence is observed both at the plasma membrane and the vacuole, indicating that this method of labeling Pma1 interferes with normal protein maturation and localization (Fig. 4). A yeast strain expressing Pma1 directly fused to an FP also exhibits compromised cell growth (Fig. 4). By contrast, cells expressing Pma1-ST and SpyoIPD-EGFP exhibit neither mislocalization to the vacuole nor a growth defect (Figs 3 and 4).
Improving signal to noise when labeling Pma1-SpyTag with SpyoIPD-EGFP
We investigated the effect of reducing the concentration of galactose used to induce SpyoIPD-EGFP expression on signal to background fluorescence when labeling Pma1-SpyTag. At low concentrations of galactose, the plasma membrane is clearly labeled and well resolved relative to the cytosolic signal, presumably because the majority of the SpyoIPD-EGFP has reacted with Pma1 (Fig. 5). Increasing the concentration of galactose increases the intensity of plasma membrane signal, but also increases the diffuse cytosolic background, presumably because the higher expression levels of SpyoIPD-EGFP are in excess of Pma1 (Fig. 5). A distinctive characteristic of Pma1’s spatial distribution is that it is retained by mother cells during cell division, so that little to no Pma1 is inherited by daughter cells. Because of this asymmetric division, the irreversible nature of the SpyoIPD-SpyTag interaction and the long half-life of Pma1 (vide infra), we predicted that unbound cytosolic signal could be cleared and plasma membrane signal retained if new SpyoIPD-EGFP expression were turned off following labeling. Since GAL1 is inhibited by glucose, we performed experiments in which SpyoIPD-EGFP expression was first induced with galactose and then shut off with glucose. Samples were imaged at different times after addition of glucose (Fig. 5). This strategy increased the ratio of membrane signal to cytosolic background in Pma1-SpyTag expressing yeast. The amount of time required to clear cytosolic signal depends on the concentration of galactose used to induce SpyoIPD-EGFP expression, with higher concentrations taking longer to clear (data not shown).
Fig. 5.

Labeling Pma1-SpyTag using SpyoIPD-EGFP: (A) Fluorescent images of yeast cells expressing Pma1-SpyTag, in which SpyoIPD-EGFP expression is induced by the indicated concentration of galactose. Note that due to the large difference in SpyoIPD-EGFP expression levels, it was necessary to image samples with different exposure times (1000 ms for [Gal] = 0%; 200 ms for [Gal] = 0.05%; 200 ms for [Gal] = 0.25% and 20 ms for [Gal] = 4.0%). (B) Yeast expressing Pma1-SpyTag and SpyoIPD-EGFP were induced for 8 h in 0.25% galactose, and then chased for an additional 2 h with glucose. Glucose inhibits the GAL1 promoter, turning off new synthesis of SpyoIPD-EGFP (100 ms exposure). Remaining cytosolic SpyoIPD-EGFP either reacts with Pma1-SpyTag, is degraded or is partitioned into the daughter cell. Cytosolic signal is significantly reduced, and the asymmetric segregation of Pma1 is visible, with no plasma membrane signal observed in the budding daughter cell.
Spatiotemporally tracking Pma1 in living cells
The labeling strategy that we present can also be used to follow a protein’s spatiotemporal dynamics in a single cell. Because the protein of interest is labeled post-translationally, only protein that is present when SpyoIPD-FP is expressed will be labeled. Thus, turning off expression of SpyoIPD-FP allows one to follow the fate of only the protein that was present during the labeling phase. We used this strategy to transiently label Pma1 and thus determine the half-life of Pma1 at the cell membrane. To follow individual cells over many hours, we used a microfluidic device that holds individual mother cells in place, but allows daughter cells to be washed away by medium flow (Crane et al., 2014). Cells were first grown overnight, in the presence of galactose, to induce expression of SpyoIPD-EGFP and label Pma1-SpyTag. Cells were loaded into the microfluidic device then, and after a short equilibration period, switched to medium containing glucose to inhibit expression of SpyoIPD-EGFP. Cells were tracked for many hours following the switch to glucose-containing medium (Fig. 6).
Fig. 6.

Using SpyoIPD-EGFP to study Pma1 temporal dynamics in single cells. Each trace in the plot corresponds to the total cellular fluorescence versus time for a single yeast cell, with t = 0 h corresponding to the time of glucose addition. Fluorescence decreases over time as SpyoIPD-EGFP is cleared from the trapped mother cell through cell division, SpyoIPD-EGFP turnover and Pma1 turnover. See Supplementary Information for a detailed description of how these data are analyzed to calculate the in-membrane half-life of fluorescently labeled Pma1. Inset: The posterior probability for the half-life of Pma1, , found by integrating the probability corresponding to the surface in Fig.S5.
We estimated the half-life of in-membrane PMA1 in two complementary ways. In the first method, we assumed that the total cellular fluorescence of an individual cell expressing Pma1-SpyTag and SpyoIPD-EGFP is comprised of (i) fluorescence from SpyoIPD-EGFP covalently bound to Pma1-SpyTag in the membrane; (ii) fluorescence from unreacted cytosolic SpyoIPD-EGFP and (iii) cellular auto-fluorescence. We developed a novel Bayesian analysis that integrates the data across all cells to infer a half-life for the Pma1 in the plasma membrane. Details of this analysis are given in the Supplementary Information. We verified our Bayesian approach with a second ad hoc method that requires data from cells expressing Pma1-SpyTag and SpyoIPD-EGFP and also from cells expressing untagged Pma1 and SpyoIPD-EGFP. Comparing the change in fluorescence intensity over time in the membrane region of these two strains allowed us to extract the signal from Pma1 at the plasma membrane. The two approaches give similar estimates of the in-membrane half-life of Pma1—11.5 and 10.2 h, respectively, agreeing with each other and the previously reported half-life of Pma1 of 11 h, determined by cyclohexamide inhibition of translation (Benito et al., 1991).
Discussion
We present a novel method for fluorescently labeling proteins, post-translationally, within living yeast using SpyoIPD/SpyTag, a derivative of the SpyCatcher/SpyTag interaction pair. Our method requires only a small modification to the protein of interest and is directly compatible with any FP. A recently introduced method that uses split GFP in labeling has similar strengths, but requires significant engineering and testing to work with different colored FPs (Kamiyama et al., 2016). The recent development of the orthogonal SnoopCatcher/SnoopTag covalent interaction pair (Veggiani et al., 2016) also allows for the future extension of this strategy to multicolor labeling. While our method has only been tested in S. cerevisiae, it should be compatible with a wide range of model organisms including mammalian systems.
Previous work has used SpyCatcher fused to a FP to label proteins fused to SpyTag in the extracellular environment (Bedbrook et al., 2015; Walden et al., 2015) and in fixed and permeabilized cells (Pessino et al., 2017). To our knowledge, however, our work is the first to use this approach to fluorescently label and image proteins within living eukaryotic cells, and to demonstrate that labeling proteins in this manner can be less disruptive to protein function than a direct FP fusion.
SpyCatcher/SpyTag is a highly versatile tool for post-translationally linking proteins together in a number of applications (Veggiani et al., 2014). The more spyoIPD presented here provides a useful derivative for applications that require increased stability and expression. Interestingly, when the original SpyCatcher was fused to EGFP and coexpressed with Pma1-SpyTag, plasma membrane labeling was also observed (data not shown). We speculate that attaching EGFP stabilizes the original SpyCatcher, because this result was unexpected given SpyCatcher’s low in vivo expression and reactivity when not fused to EGFP (Fig. 2D). The availability and different properties of the original SpyCatcher, SpyoIPD and SnoopCatcher will allow researchers to choose the variant that is most appropriate for their particular application. Not all the proteins we tested showed clear labeling using SpyoIPD-EGFP. We speculate that inaccessibility of the SpyTag is the most likely explanation for this lack of signal, although we did not investigate this idea nor try different tag locations.
The ability to label Pma1 in living cells without vacuolar mislocalization is a significant result. Pma1 is an essential plasma membrane proton pump that plays an important role in a variety of processes (Ferreira et al., 2001; Henderson et al., 2014). Prior to this work, the only established method for imaging Pma1 in live cells was via a direct fusion to a FP, which causes mislocalization to the vacuole and a significant growth defect. The lack of either a growth defect or vacuolar mislocalization supports our hypothesis that the post-translational nature of our labeling that makes it less disruptive, despite the large size of the final label. To the best of our knowledge, this is the first work that shows fluorescent labeling of Pma1 in live cells in a nondisruptive manner.
Finally, because the method we present involves irreversible covalent labeling, it can be used to study a variety of time-dependent changes, including protein turnover rates, accumulation of post-translational modifications, exchange of protein interaction partners and the selective labeling of organelles, subcellular membraneless compartments and even entire cells in an age dependent manner. Here we show that SpyoIPD-EGFP can be used to study protein turnover in individual cells. Traditional biochemical methods for quantifying protein turnover rates use translational inhibitors (e.g. cycloheximide) that block the translation of all proteins in the cell and are therefore generally disruptive to cellular function, or radioactive labels, which require specialized safety protocols. The SpyoIPD-EGFP method we present suffers from neither of these limitations, and therefore represents a useful tool for labs interested in studying the turnover of proteins, particularly in individual cells.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The yeast vectors and strains we started with were a gift from Mark Hochstrasser. We thank the Hochstrasser lab for valuable advice and gifts of reagents throughout the project. We thank members of the Regan lab for advice and for helpful comments on the manuscript. We thank members of the Swain lab for their advice and technical insights, especially Ivan Clark and Elco Bakker for help with data analysis and numerous discussions.
Supplementary Data
Supplementary data are available at Protein Engineering, Design and Selection online.
Funding
This work was funded by the Raymond and Beverley Sackler Institute for Biological, Physical, and Engineering sciences [to L.R.]; the National Institute of Health [Grant nos. GM118528 and CA209992 to M. H. and L. R.]; the Medical Research Council [Grant no. MR/K001485 to U.S.L. and J. M. E.]; a Leverhulme Trust Visiting Professorship [to L. R.]; and Royal Society of Edinburgh [Caledonian Scholarship to O.K.M.].
References
- Bedbrook C.N., Kato M., Kumar S.R., Lakshmanan A., Nath R.D., Sun F., Sternberg P.W., Arnold F.H. and Gradinaru V. (2015) Chem. Biol., 22, 1108–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito B., Moreno E. and Lagunas R. (1991) Biochim Biophys. Acta, 1063, 265–268. [DOI] [PubMed] [Google Scholar]
- Brohée S., Barriot R., Moreau Y. and André B. (2010) Biochim Biophys. Acta, 1798, 1908–1912. [DOI] [PubMed] [Google Scholar]
- Cormack B.P., Valdivia R.H. and Falkow S. (1996) Gene, 173, 33–38. [DOI] [PubMed] [Google Scholar]
- Crane M.M., Clark I.B., Bakker E., Smith S. and Swain P.S. (2014) PLoS ONE, 9 10.1371/journal.pone.0100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J. and Bax A. (1995) J. Biomol. NMR, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Edelstein A., Amodaj N., Hoover K., Vale R. and Stuurman N. (2010) Curr. Protoc. Mol. Biol., 92, 14.20.11–14.20.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira T., Mason A.B. and Slayman C.W. (2001) J. Biol. Chem., 276, 29613–29616. [DOI] [PubMed] [Google Scholar]
- Fink G. (2002) Methods Enzymol., 350, 21. [Google Scholar]
- Gautier A., Juillerat A., Heinis C., Corrêa I.R., Kindermann M., Beaufils F. and Johnsson K. (2008) Chem. Biol., 15, 128–136. [DOI] [PubMed] [Google Scholar]
- Haarer B.K. and Pringle J.R. (1987) Mol. Cell. Biol., 7, 3678–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan R.M., Björnsson R., McMahon S.A., Schomburg B., Braithwaite V., Bühl M., Naismith J.H. and Schwarz‐Linek U. (2010) Angewand. Chem., 122, 8599–8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins K.M. and Smolke C.D. (2006) J. Biol. Chem., 281, 13485–13492. [DOI] [PubMed] [Google Scholar]
- Henderson K.A., Hughes A.L. and Gottschling D.E. (2014) Elife, 3. doi 10.7554/eLife.03504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.H. and Tirrell D.A. (2016) J. Am. Chem. Soc., 138, 15098–15101. [DOI] [PubMed] [Google Scholar]
- Huh W.-K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S. and O’Shea E.K. (2003) Nature., 425, 686–691. [DOI] [PubMed] [Google Scholar]
- Kamiyama D., Sekine S., Barsi-Rhyne B. et al. (2016) Nat. Commun., 7 10.1038/ncomms11046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler A., Gendreizig S., Gronemeyer T., Pick H., Vogel H. and Johnsson K. (2003) Nat. Biotechnol., 21, 86–89. [DOI] [PubMed] [Google Scholar]
- Kushnirov V.V. (2000) Yeast., 16, 857–860. [DOI] [PubMed] [Google Scholar]
- Labbé S. and Thiele D.J. (1999) Method Enzymol., 306, 145–153. [DOI] [PubMed] [Google Scholar]
- Lacy M.M., Baddeley D. and Berro J. (2017) Mol. Biol. Cell., 28, 2251–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.E., DeLoache W.C., Cervantes B. and Dueber J.E. (2015) ACS Synthet. Biol., 4, 975–986. [DOI] [PubMed] [Google Scholar]
- Longtine M.S., McKenzie A III, Demarini D.J., Shah N.G., Wach A., Brachat A., Philippsen P. and Pringle J.R. (1998) Yeast, 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Los G.V., Encell L.P., McDougall M.G. et al. (2008) ACS Chem. Biol., 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Madden K. and Snyder M. (1998) Annu. Rev. Microbiol., 52, 687–744. [DOI] [PubMed] [Google Scholar]
- Mason A.B., Allen K.E. and Slayman C.W. (2014) Eukaryotic Cell, 13, 143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D., Müller R. and Funk M. (1994) Nucleic Acids Res., 22, 5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessino V., Citron Y.R., Feng S. and Huang B. (2017) Chembiochem. 10.1002/cbic.201700177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt S.E., Speltz E.B., Mochrie S.G. and Regan L. (2016) Chembiochem, 17, 1652–1657. [DOI] [PubMed] [Google Scholar]
- Riedl J., Crevenna A.H., Kessenbrock K. et al. (2008) Nat. Methods, 5, 605–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer N., Gassaway B.M., Haimovich A.D., Isaacs F.J., Rinehart J. and Regan L. (2014) ACS. Chem. Biol., 9, 2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidthals K., Helma J., Zolghadr K., Rothbauer U. and Leonhardt H. (2010) Anal. Bioanal. Chem., 397, 3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano R., Kielland-Brandt M.C. and Fink G.R. (1986) Nature, 319, 689–693. [DOI] [PubMed] [Google Scholar]
- Speltz E.B. and Regan L. (2013) Prot. Sci., 22, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttamapinant C., White K.A., Baruah H., Thompson S., Fernández-Suárez M., Puthenveetil S. and Ting A.Y. (2010) Proc. Natl. Acad. Sci. USA, 107, 10914–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veggiani G., Nakamura T., Brenner M.D., Gayet R.V., Yan J., Robinson C.V. and Howarth M. (2016) Proc. Natl. Acad. Sci. USA, 113, 1202–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veggiani G., Zakeri B. and Howarth M. (2014) Trends Biotechnol., 32, 506–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken W.F., Boucher W., Stevens T.J. et al. (2005) Proteins, 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Walden M., EdwarPds J.M., Dziewulska A.M. et al. (2015) Elife, 4 10.7554/eLife.06638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakeri B., Fierer J.O., Celik E., Chittock E.C., Schwarz-Linek U., Moy V.T. and Howarth M. (2012) Proc. Natl. Acad. Sci. USA, 109, E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.