

Abstract
We previously encountered regulatory processes wherein dihydrotestosterone (DHT) exerted its inhibitory effect on parathyroid hormone‐related protein (PTHrP) gene repression through the estrogen receptor (ER)α, but not the androgen receptor (AR), in breast cancer MCF‐7 cells. Here, we investigated whether such aberrant ligand‐nuclear receptor (NR) interaction is present in prostate cancer LNCaP cells. First, we confirmed that LNCaP cells expressed large amounts of AR at negligible levels of ERα/β or progesterone receptor. Both suppression of PTHrP and activation of prostate‐specific antigen genes were observed after independent administration of 17β‐estradiol (E2), DHT, or R5020. Consistent with the notion that the LNCaP AR lost its ligand specificity due to a mutation (Thr‐Ala877), experiments with siRNA targeting the respective NR revealed that the AR monopolized the role of the mediator of shared hormone‐dependent regulation, which was invariably associated with nuclear translocation of this mutant AR. Microarray analysis of gene regulation by DHT, E2, or R5020 disclosed that more than half of the genes downstream of the AR (Thr‐Ala877) overlapped in the LNCaP cells. Of particular interest, we realized that the AR (wild‐type [wt]) and AR (Thr‐Ala877) were equally responsible for the E2‐AR interactions. Fluorescence microscopy experiments demonstrated that both EGFP‐AR (wt) and EGFP‐AR (Thr‐Ala877) were exclusively localized within the nucleus after E2 or DHT treatment. Furthermore, reporter assays revealed that some other cancer cells exhibited aberrant E2‐AR (wt) signaling similar to that in the LNCaP cells. We herein postulate the presence of entangled interactions between wt AR and E2 in certain hormone‐sensitive cancer cells. J. Cell. Physiol. 230: 1594–1606, 2015. © 2014 The Authors. Journal of Cellular Physiology Published by Wiley Periodicals, Inc.
Abbreviations
- DHT
dihydrotestosterone
- PTHrP
parathyroid hormone‐related protein
- ER
estrogen receptor
- AR
androgen receptor
- NR
nuclear receptor
- PSA
prostate cancer antigen
- wt
wild‐type
- E2
17β‐estradiol
- HHM
humoral hypercalcemia of malignancy
- PR
progesterone receptor
- atRA
all‐trans retinoic acid
- qRT‐PCR
quantitative real‐time PCR
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- siCT
control siRNA
- ARE
AR response element
- GR
glucocorticoid receptor
- Dex
dexamethasone, TSA, trichostatin A
Estrogen and androgen are key regulators of sex steroid‐dependent cancers. The conventional view is that most breast cancers depend on estrogen‐estrogen receptor (ER)α signaling for their development and proliferation, while prostate cancers rely largely on the androgen‐androgen receptor (AR) axis.
Breast cancer is commonly associated with humoral hypercalcemia of malignancy (HHM) (Hickey et al., 1981) due to ectopic production of parathyroid hormone‐related protein (PTHrP) by cancer tissues and its systemic action on bone and kidney (Mundy and Edwards, 2008). Local production of PTHrP in osseous tissues following bone metastasis of primary breast cancer also contributes to deleterious development of hypercalcemia and aggressive bony destruction (Chirgwin and Guise, 2000). On the other hand, prostate cancer is less commonly associated with HHM and local osteolytic lesions. Nonetheless, PTHrP is crucially involved in enhancing cancer cell proliferation, survival, and migration (Dougherty et al., 1999; Asadi and Kukreja, 2005). As such, it is important to understand the regulatory mechanism of the PTHrP gene, and several intriguing signal molecules have been postulated to stimulate PTHrP expression in breast and prostate cancers (Lindemann et al., 2001; Lindemann et al., 2003; Sterling et al., 2006; Gilmore et al., 2008).
Recently, we and others reported that expression of the PTHrP gene is commonly repressed by several steroid hormones including estrogens (Rabbani et al., 2005), androgens (Pizzi et al., 2003), 1,25‐dihydroxyvitamin D3 (Ikeda et al., 1989; Inoue et al., 1993; Endo et al., 1994; Falzon, 1996; Nishishita et al., 1998; Okazaki et al., 2003), glucocorticoids (Lu et al., 1989; Kasono et al., 1991; Liu et al., 1993; Glatz et al., 1994; Rizzoli et al., 1994; Walsh et al., 1995; Ahlstrom et al., 2009), and progesterone (Sugimoto et al., 1999; Kurebayashi et al., 2003). To comprehensively explore these repression processes, we systematically surveyed several cell lines and characterized PTHrP gene regulation in response to a series of steroid hormones mediated by their cognate nuclear receptors (NRs). In our previous report, we described suppression of the PTHrP gene by complexes of administered steroid hormones and their cognate NRs in common, with the exception of the dihydrotestosterone (DHT)‐AR partnership in human breast cancer MCF‐7 cells. Interestingly, DHT repressed PTHrP gene expression through ERα, but not the endogenous and functional AR in these cells (Kajitani et al., 2011).
In this study, we found that such a distorted ligand‐NR interaction is also present in another steroid hormone‐dependent cell line, namely, human prostate cancer LNCaP cells, by examining the repression of PTHrP and the activation of the prostate‐specific antigen (PSA) genes. It is known that the AR in LNCaP cells has a point mutation (Thr‐Ala877) in its ligand‐binding domain (LBD) (Buchanan et al., 2001) that results in the partial loss of the AR's ligand specificity and cross‐reaction with several ligands, including estrogen, pregnenolone, progesterone, and steroidal compounds such as antiandrogens (Veldscholte et al., 1992; Grigoryev et al., 2000). AR (Thr‐Ala877)‐expressing LNCaP was originally established from a metastatic lesion of human prostatic adenocarcinoma (Horoszewicz et al., 1980) and there are several reports describing AR (Thr‐Ala877) is expressed in clinical prostate cancers (Suzuki et al., 1993; Gaddipati et al., 1994; Suzuki et al., 1996; Taplin et al., 2003; Ceraline et al., 2004).
First, we quantified the expression of the AR, ERα, ERβ, and progesterone receptor (PR) genes. After performing quantitative real‐time (qRT)‐PCR to examine transcriptional regulation of the PTHrP and PSA genes by several steroid hormones, we investigated the knockdown effect of several NRs on gene expression in LNCaP cells. Then, we carried out microarray experiments to explore whether or not hormonal cross‐reactivity mediated by the AR (Thr‐Ala877) was widespread in these cells. To determine whether the aberrant ligand‐NR interaction in the LNCaP cells was a direct consequence of this AR mutation, we next employed AR (wild‐type [wt]) and AR (Thr‐Ala877) expression‐based reporter assays to determine whether or not this AR mutation leads to the distorted ligand‐NR interaction. Finally, we examined the AR nuclear translocation in response to these hormones by confocal immunofluorescence microscopy.
Materials and Methods
Cell cultures and hormones
Prostate cancer LNCaP cells and Rv22 cells, gifts from Dr. Shigeo Horie (Department of Urology, Teikyo Medical School, Japan), and breast cancer MCF‐7 cells, T47D cells, and MDA‐MB‐453 cells, kindly provided by Dr. Shunji Takahashi (Cancer Institute Hospital of the Japanese Foundation for Cancer Research, Japan), were maintained in monolayer cultures in RPMI‐1640 phenol red free medium (Life Technologies, Carlsbad, CA) supplemented with 5% (v/v) fetal bovine serum and antibiotics (Life Technologies) in humidified 5% CO2–95% air at 37°C. The following chemicals were obtained from commercial sources: 17β‐estradiol (E2) (Wako Pure Chemical, Osaka, Japan), DHT (Sigma Aldrich, St. Louis, MO), all‐trans retinoic acid (atRA) (Sigma Aldrich), R5020 (Thermo Fisher Scientific, Waltham, MA), TSA (Sigma Aldrich), and MDV3100 (Selleckchem, Houston, TX). At 24 h before hormone treatment, serum‐free media were used, and the cells were exposed to the indicated hormones with serum‐free media for another 24 h. As shown in Supplemental Fig. 1, the use of these serum‐free media minimally affected the cell growth and the cell number, indicating intensive apoptosis did not occur. Forty‐eight to 72 h serum starvation for examining the hormonal effect in LNCaP cells has been employed by others (Perry and Tindall, 1996; Shanmugam et al., 2007).
The ligand concentration was 2.0 × 10−9 M, 2.0 × 10−8 M, and 2.0 × 10−7 M, respectively. MDV3100, TSA and atRA were dissolved in DMSO, and the others in ethanol.
siRNA transfection
Control siRNA and mixtures of siRNAs for the ERα, ERβ, AR, and PR genes were obtained from Dharmacon (Thermo Fisher Scientific). Forward transfection of siRNA was performed with LipofectamineRNAiMAX (Life Technologies) according to the manufacturer's instructions.
Quantitative real‐time PCR
qRT‐PCR was performed as described previously (Kajitani et al., 2011). Relative gene expression was evaluated by the ΔCT method. The primer sets used in this study are shown in Supplemental Table 1. Data are normalized to glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) expression and presented relative to the vehicle (%). Values are expressed as means ± SD (n = 4, unless otherwise stated).
Western blotting
Cells were dissolved in SDS buffer containing 62.5 mM Tris‐HCl (pH 6.8), 2% SDS, 10% glycerol, and 100 mM dithiothreitol for whole cell protein preparation. For further cell fractionation, cells were suspended in buffer A (50 mM Hepes pH7.4, 0.1% Traiton‐X100, 10 mM NaCl, 20% glycerol, 1 mM EDTA, 2 mM EGTA, 2 mM MgCl2, 1 mM DTT, 1 mM NaVO4, 10 mM NaF and protease inhibitors (Roche, Basel, Switzerland). After incubation on ice for 20 min, lysates were centrifuged at 500 g for 3 min. Pellets were washed with buffer A and stored as nuclear pellets. The supernatants were stored as cytoplasmic fraction. The cytoplasmic fraction and the nuclear pellets were dissolved in SDS buffer and used as cytoplasmic protein and nuclear protein, respectively. Each protein was separated on 7.5% SDS‐polyacrylamide gel, followed by blotting to a Hybond‐LFP polyvinylidene fluoride membrane (GE Healthcare, Inc., Piscataway, NJ). After washing in PBS containing 0.2% (v/v) Tween 20 (PBST), the membrane was incubated with blocking solution composed of 5% (w/v) skim milk in PBST for 1 h. The membrane was reacted with rabbit antibody against human AR (H‐280; Santa Cruz Biotechnology, Santa Cruz, CA) diluted at 1:1,000, rabbit antibody against human AR (C‐19; Santa Cruz Biotechnology) diluted at 1:100, rabbit antibody against human ERα (HC‐20; Santa Cruz Biotechnology) diluted at 1:500, rabbit antibody against human PR (C‐20; Santa Cruz Biotechnology) diluted at 1:500, rabbit antibody against mouse Pol II (N‐20; Santa Cruz Biotechnology) diluted at 1:100, goat antibody against human LAMIN A/C (N‐18; Santa Cruz Biotechnology) diluted at 1:2,000 or mouse monoclonal antibody against α‐tubulin (T5168; Sigma Aldrich) diluted at 1:10,000 in the blocking solution overnight at 4°C. After washing in PBST, the membrane was reacted for 1 h at room temperature in blocking solution containing goat anti‐rabbit or anti‐mouse IgG, or donkey anti‐goat IgG antibody conjugated with Cy5 (Jackson ImmunoResearch, West Grove, PA) diluted at 1:2,000. Then, the membrane was washed in PBST, and the immunoreacted proteins were visualized using Typhoon (GE Healthcare).
Immunocytochemistry
LNCaP cells were fixed with 4% paraformaldehyde in PBS for 20 min. The primary antibody was rabbit polyclonal antibody against human AR (H‐280) diluted 1:2,000 in PBST overnight. After washing in PBS, cells were incubated with a secondary antibody, Alexa 546‐labeled anti‐mouse goat IgG (Molecular Probes, Eugene, OR) in PBST for 2 h. Cells were washed in PBS, and their nuclei were stained with DAPI (Life Technologies) followed by immunofluorescence microscopy. Fluorescent images were obtained using an A1 Confocal Microscope System (Nikon, Tokyo, Japan).
Microarray analysis
cDNA microarray analysis was performed using an authorized custom analysis service provider (APRO Life Science, Tokushima, Japan). Total RNA from the LNCaP cells transfected with control siRNA (siCT) or siRNA for AR (siAR) followed by 10−7 M of DHT, E2, or R5020 exposure for another 24 h was isolated using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA samples were quantified using an ND‐1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE), and their quality was confirmed using an Experion System (Bio‐Rad Laboratories, Hercules, CA). The cRNA was amplified and labeled using a Low Input Quick Amp Labeling Kit (Agilent Technologies, Inc., Santa Clara, CA), followed by hybridization to a SurePrint G3 Human GE Microarray 8 × 60K v2 (Agilent Technologies, Inc.) according to the manufacturer's instructions. All hybridized microarray slides were scanned using an Agilent scanner. Relative hybridization intensities and background hybridization values were calculated using Agilent Feature Extraction Software (9.5.1.1). Raw signal intensities and flags for each probe were calculated from the hybridization intensities (gProcessedSignal) and spot information (e.g., gIsSaturated) according to the procedures recommended by Agilent (Flag criteria in GeneSpring Software: Absent (A), “Feature is neither positive or significant” or “Feature is not above background”; Marginal (M), “Feature is not uniform”, “Feature is saturated”, or “Feature is a population outlier”; and Present (P), other). In addition, the raw signal intensities were log2‐transformed and normalized using the quantile algorithm in the ‘preprocessCore' library package (Bolstad et al., 2003) of the Bioconductor software (Gentleman et al., 2004). GEO accession number is GSE58615. We selected the probes that call the ‘P' flag in the siCT‐vehicle sample. The signals of each probe on the microarray were expressed as the signal/median value divided by the median value of all probes. To identify up‐ or down‐regulated genes, we calculated ratios (non‐log scaled fold‐change) for comparisons between each vehicle and hormone treatment sample. Then, we selected hormone target genes mediated by the LNCaP AR by applying criteria satisfying (siCT) ≥5.0 and (siAR) 1.0–2.0 for gene activation or (siCT) ≤0.2 and (siAR) 0.75–1.0 for gene repression.
Plasmid construction and luciferase assay
After confirming that the AR cDNA obtained from the LNCaP cells had only one mutation (Thr‐Ala877) as previously reported, it was ligated into the mammalian expression vector pcDNA3.1Zeo+ (pcDNA3.1, Life Technologies). We then separately introduced the mutant AR or wt AR expression vector (a gift from Dr. Sigeaki Kato, then at The University of Tokyo). As a reporter plasmid, three copies of the AR response element (ARE; GGAACAgtaTGTTCT) (Roche et al., 1992) were ligated into the upstream region of the thymidine kinase promoter of the pGL4‐TK vector (Promega, Madison, WI). At 23 h after seeding approximately 2 × 104 cells/100 μl/well onto 96‐well plates, the media were changed to serum‐free RPMI‐1640 with antibiotics for 1 h followed by the introduction of 50 ng DNA (20 ng ARE/pGL4‐TK, 10 ng of either AR expression vector, and 20 ng phRL‐TK (Promega)/0.3 μl FuGeneHD (Promega)/well) into the LNCaP, MDA‐MB‐453, Rv22, and MCF‐7 cells. After incubation for 24 h, the cells were treated with each steroid hormone for another 24 h, and luciferase activity was assayed using the Dual‐luciferase Reporter Assay System (Promega). Means ± SD were calculated after quadruplicate transfection.
Localization analysis by enhanced green fluorescent protein (EGFP)‐AR fusion protein
To produce EGFP‐AR (wt) and EGFP‐AR (Thr‐Ala877) expression vectors, the coding sequence of each AR cDNA was ligated into multiple cloning sites of the pEGFP‐C1 vector (Clontech, Palo Alto, CA). At 23 h after seeding approximately 2 × 104 cells/100 μl/well onto a multiple‐chamber slide, LNCaP cells were transfected with 50 ng of each AR/pEGFP‐C1 in 0.3 μl FuGeneHD (Promega)/well in a manner similar to the introduction of the AR expression vectors. Then, 24 h later, 2.0 × 10−8 M of each steroid hormone was administrated to the cells which were incubated for another 24 h. The cells were fixed by 4% paraformaldehyde, and intracellular localization of EGFP‐AR was examined using the Nikon A1 Confocal Microscope System. Nuclei were stained with DAPI.
Statistical analysis
Data are expressed as means ± SD of four independent experiments, unless otherwise stated. Statistical analysis was carried out using Student's t‐test (Fig. 1C, F, G, and H) or one‐factor ANOVA and Tukey's test (Figs. 1B, E, I, J, K, 2, 3B, 5, 6A, and 7) (Supplemental Fig. 2, 4A, B and 5).
Figure 1.
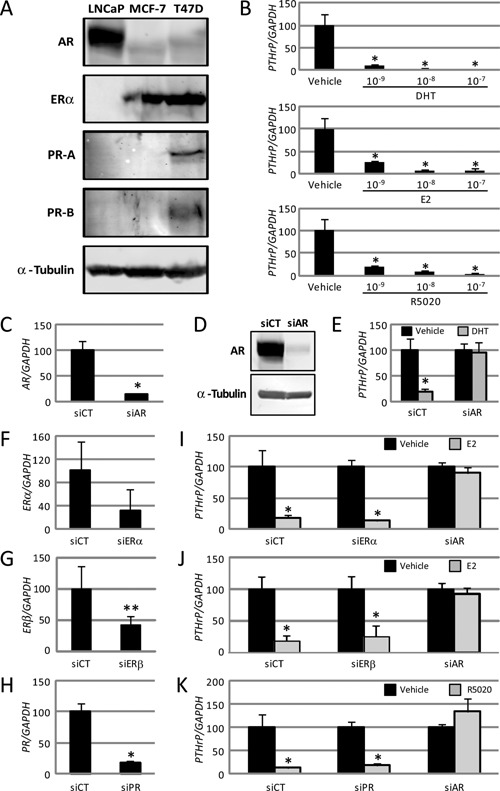
The androgen receptor (AR) exclusively mediates parathyroid hormone‐related protein gene repression by several steroid hormones in LNCaP cells. (A) Western blotting of AR, estrogen receptor (ER)α, progesterone receptor (PR)‐A and PR‐B proteins in LNCaP, MCF‐7, and T47D cells using AR (H‐280), ERα, and PR (reacted with both isoforms of PR‐A and PR‐B) antibodies. Anti‐α‐tubulin antibody was used as the loading control. (B) Parathyroid hormone‐related protein (PTHrP) mRNA in LNCaP cells treated with dihydrotestosterone (DHT), 17β‐estradiol (E2), or R5020 was analyzed by quantitative real‐time (qRT)‐PCR. The concentrations of the used hormones were 10−9, 10−8, and 10−7 M, respectively. Data are normalized to GAPDH mRNA levels and represented relative to the vehicle (%). * P < 0.01 compared with the vehicle. (C–E) Control siRNA (siCT) and AR siRNA (siAR) were introduced into the respective LNCaP cells. At 24 h after transfection, AR mRNA (C) and protein (D) were analyzed. (C) AR mRNA in the transfected LNCaP cells was analyzed by real‐time PCR. Data are normalized to GAPDH and represented as the ratio of AR mRNA in the siAR groups to that in the siCT groups (%). * P < 0.01 compared with siCT. (D) Western blotting with the AR antibody. Anti‐α‐tubulin antibody was used as the loading control. (E) Real‐time PCR assay of PTHrP mRNA. LNCaP cells with or without siAR were treated with 10−7 M DHT for 24 h. Black and gray bars indicate the effect of vehicle and DHT, respectively. Data are shown relative to the siCT of the vehicle (%). * P < 0.01 compared with each vehicle. (F–K) siCT and siRNAs for each NR (siERα, siERβ, and siPR) were introduced into the respective LNCaP cells. Data are normalized to GAPDH expression. (F–H) At 24 h after transfection, the mRNA expression of ERα (F), ERβ (G), and PR (H) in the LNCaP cells was analyzed by real‐time PCR. Data are presented as ratios relative to siCT (%). * P < 0.01 and ** P < 0.05 compared with the siCT lane. (I–K) At 24 h after transfection of each siRNA, LNCaP cells were treated with 10−7 M E2 (I, J) or R5020 (K) for another 24 h. Expression of the PTHrP gene was analyzed by real‐time PCR. Black and gray bars indicate the results after vehicle and steroid hormone treatment, respectively. Data are shown relative to the vehicle (%). * P < 0.01 compared with each vehicle. (B–C and E–K) Data are presented as means ± SD (n = 4).
Results
Several steroid hormones repressed PTHrP expression in LNCaP cells
To explore the effects of E2, DHT, and R5020 on PTHrP expression in prostate cancer LNCaP cells, we first examined the cognate NR mRNAs of ERα, AR, and PR in LNCaP cells by qRT‐PCR. We also analyzed the expression of NRs in prostate cancer Rv22 and breast cancer MCF‐7, T47D, and MDA‐MB‐453 cells. The expression of NRs was presented relative to that of GAPDH. Considerable amounts of AR expression were confirmed in both LNCaP and Rv22 cells, and excessive levels were found in the MDA‐MB‐453 cells (Table 1). MCF‐7 and T47D cells also expressed AR mRNA, but the expression levels seemed notably lower in these cells than in the prostate cancer cells. On the other hand, expression of ERα and PR was negligible in the LNCaP cells. ERα and PR expression was recognized only in the breast cancer cell lines (Table 1). We found that the amount of ERβ mRNA in LNCaP cells was extremely low compared to that of ERα in MCF‐7 or LNCaP cells (Table 1), although some reports argued that LNCaP cells express fairly abundant amounts of ERβ protein (Lau et al., 2000; Bektic et al., 2004).
Table 1.
Expression of NR mRNAs in prostate cancer and breast cancer cell lines
| Relative expressions toward GAPDH (%) | |||||
|---|---|---|---|---|---|
| Cell | Origin | ERα | ERβ | AR | PR |
| LNCaP | Prostate cancer | <0.0 | <0.0 | 100.0 ± 0.7 | <0.0 |
| Rv22 | Prostate cancer | <0.0 | <0.0 | 60.0 ± 8.2 | <0.0 |
| MCF‐7 | Breast cancer | 100.0 ± 6.1 | <0.0 | 14.2 ± 1.0 | 3.4 ± 0.4 |
| T47D | Breast cancer | 146.9 ± 17.0 | 0.6 ± 0.1 | 30.2 ± 1.0 | 100.0 ± 11.8 |
| MDA‐MB‐453 | Breast cancer | <0.0 | <0.0 | 17.8 ± 24.9 | <0.0 |
ERα and ERβ values are shown relative to the ERα in the MCF‐7 cells 100(%). AR values are shown relative to the AR in the LNCaP cells (%). PR values are shown relative to the PR in the T47D cells (%). Data are expressed as means ± SD of four independent experiments.
Using western blotting, we analyzed the expression of the AR, ERα, and two PR isoforms, PR‐A and PR‐B proteins, in LNCaP cells along with MCF‐7 and T47D cells as positive controls. As shown Fig. 1A, the LNCaP cells, unlike the other cells, expressed a massive amount of AR, while the expression of ERα and PRs was negligible, demonstrating that the LNCaP cells expressed only AR protein among the NRs examined in this study.
We next investigated the effects of these steroid hormones on PTHrP expression. Endogenous PTHrP mRNA expression was repressed by 10−9 M DHT to 11% of the vehicle (Fig. 1B). Higher concentrations of DHT showed further dose‐dependent repression (Fig. 1B). Similarly, in the LNCaP cells, E2 and R5020 repressed endogenous PTHrP mRNA expression in a dose‐dependent manner (Fig. 1B), while the cognate NR mRNAs or proteins were minimally expressed in the LNCaP cells.
PTHrP gene repression by steroid hormones was abrogated on knockdown of the AR, but not other NRs
To test whether or not the repression of PTHrP by the steroid hormones was mediated through the cognate NRs, we performed knockdown of NR expression in the LNCaP cells by transfection with siRNA specific to each NR. First, we employed AR‐specific siRNA, which decreased the AR mRNA to 13% (Fig. 1C), and the AR protein level dropped to <10% that of the control (Fig. 1D). As shown in Fig. 1E, knockdown of the AR completely counteracted the DHT‐dependent repression of PTHrP expression. These results imply that the AR in the LNCaP cells is required for DHT‐mediated PTHrP mRNA repression.
Next, we performed a series of knockdown experiments using siRNAs specific to ERα and PR. Efficient further knockdown of the scarce expression of each NR was confirmed by qRT‐PCR; ERα to 32% (Fig. 1F), ERβ to 43% (Fig. 1G) and PR to 19% (Fig. 1H). Surprisingly, the individual reduction of each NR failed to reverse the PTHrP repression mediated by the respective cognate hormone (Fig. 1I–K), suggesting that the NRs, except for the AR, were dispensable for the steroid hormone‐dependent suppression of the PTHrP gene. We then examined whether the AR was involved in the repression of PTHrP induced by E2 and R5020. As shown in Fig. 1I–K, knockdown of the AR completely restored the E2‐ and R5020‐induced reductions in PTHrP mRNA, indicating that the AR in LNCaP cells was required for repression of the PTHrP gene mediated not only by DHT, but also by E2 and R5020.
Role of the AR in PSA gene activation
We next focused on the transcriptional regulation of the PSA gene as an AR target in the LNCaP cells. Expression of the PSA gene was significantly stimulated by DHT in a dose‐dependent manner (Fig. 2A), and knockdown of the AR completely counteracted the DHT‐dependent activation of PSA gene expression (Fig. 2D). Subsequently, we investigated the effects of other steroid hormones on PSA gene expression. As shown in Fig. 2B, C, E, F, and G, E2 and R5020 also stimulated PSA gene expression, but further knockdown of their cognate NRs did not abolish their respective stimulatory effects. Nevertheless, as seen in PTHrP gene regulation (Fig. 1), simultaneous activation of the PSA gene by each hormone was completely abolished by single knockdown of the AR (Fig. 2D–G). These results strongly imply that the AR in the LNCaP cells was solely responsible for PTHrP gene repression, as well as the PSA gene activation mediated concurrently by the two steroid hormones in addition to DHT. In separate experiments, AR‐driven PTHrP and PSA gene regulation was confirmed using the AR‐specific antagonist MDV3100 (Scher et al., 2010; Ha et al., 2013). Co‐treatment of all the used steroid hormones with 10−5 M MDV3100 antagonized the gene regulation of PTHrP and PSA (Supplemental Fig. 2).
Figure 2.
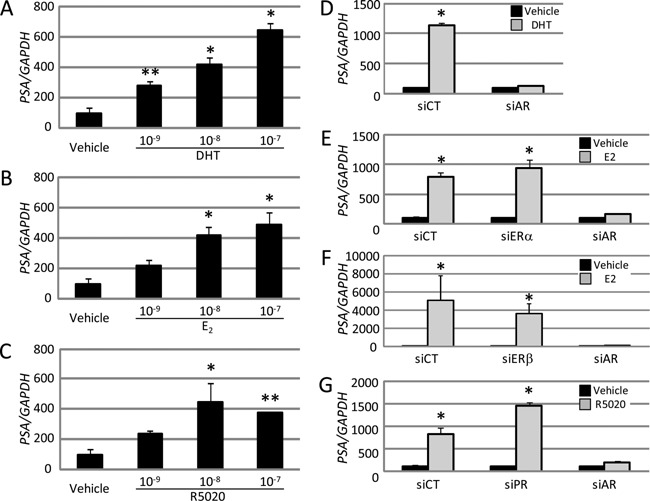
AR as an exclusive mediator of prostate‐specific antigen activation by several steroid hormones in LNCaP cells. Levels of prostate‐specific antigen (PSA) mRNA in LNCaP cells treated with DHT (A, D), E2 (B, E, F), and R5020 (C, G) were analyzed by qRT‐PCR. The concentrations of the hormones were 10−9, 10−8, and 10−7 M for A–C, and 10−7 M for D‐G, respectively. (D–G) The effects of siCT and siNRs on PSA gene activation are shown. Data are normalized to GAPDH expression and presented as means ± SD (n = 4). Data are shown relative to the vehicle (%). * P < 0.01 and ** P < 0.05 compared with the vehicle.
Several steroid hormones induced nuclear localization of the endogenous AR (Thr‐Ala877) in LNCaP cells
Mutation of the AR (Thr‐Ala877) in LNCaP cells was reported to result in the partial loss of the AR's ligand specificity and cross‐reaction with several steroid ligands including E2 and progesterone (Veldscholte et al., 1992). We analyzed the intracellular localization of the endogenous AR (Thr‐Ala877) by immunocytochemistry. At 24 h after 10−7 M hormone treatment, the LNCaP cells were fixed with 4% paraformaldehyde, followed by immunocytochemistry with AR (H‐280) of which the epitope corresponds to amino acids 91–370 of human AR. Consistent with a previous study (Ni et al., 2013), we noticed that nuclear, but not cytoplasmic staining, of the AR (Thr‐Ala877) was a characteristic feature, irrespective of the hormone status in the LNCaP cells. In vehicle‐treated LNCaP cells, the nuclear AR (Thr‐Ala877) was observed at a low frequency (Fig. 3A, arrow). In contrast, the AR (Thr‐Ala877) treated with DHT showed well‐demarcated nuclear localization with notably stronger signals at a higher frequency, which was reproducibly observed in both the E2‐ and R5020‐treated cells (Fig. 3A). Real‐time PCR (Fig. 3B) and western blotting using whole cell protein (Fig. 3C) showed that these three hormones actually enhanced AR expression. Western blotting using fractionated proteins revealed that these hormones increased the amount of AR (Thr‐Ala877) in the nuclear fraction (Fig. 3C). Notably, even in vehicle‐treated LNCaP cells, AR (Thr‐Ala877) was present both in cytoplasm and nucleus, indicating hormone‐independent nuclear localization of the mutant AR (Thr‐Ala877) (Fig. 3C). Western blotting (Fig. 3C) seemed to conform to those of immunocytochemistry (Fig. 3A). The hormone‐dependent nuclear accumulation and the hormone‐independent nuclear localization of the endogenous AR (Thr‐Ala877) in LNCaP was reconfirmed with another AR antibody, AR (C‐19), whose epitope corresponds to amino acids 869–919 of AR of human origin (Supplemental Fig. 3).
Figure 3.
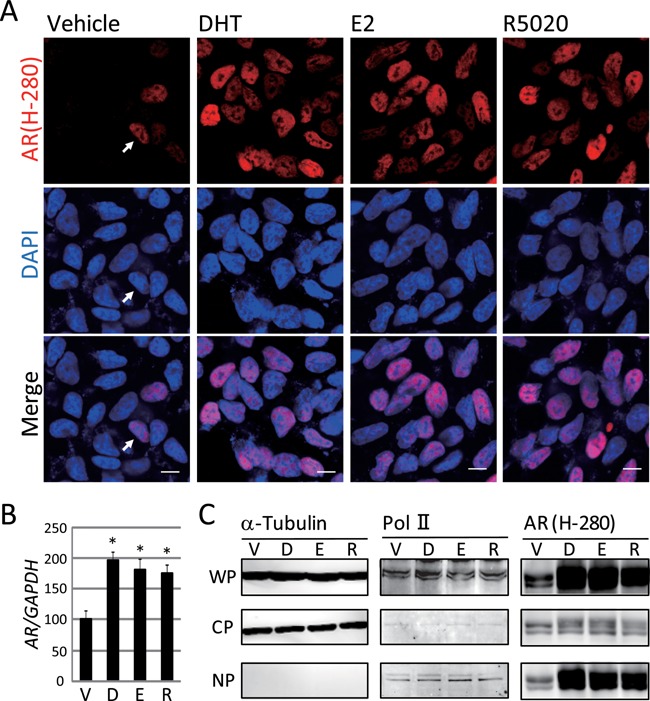
Nuclear translocation of the AR triggered by various steroid hormones in LNCaP cells. (A) Immunocytochemical analysis with AR antibody, AR (H‐280) was performed. At 24 h after treatment with each steroid hormone, LNCaP cells fixed by 4% paraformaldehyde were incubated with the anti‐AR antibody. Each hormone was added at a dose of 10−7 M. (Upper panel) AR visualized with the Alexa 546‐labeled secondary antibody (red). (Middle panel) Nuclear staining with DAPI (blue). (Lower panel) Merged images. Modest staining of the nuclear AR, even in the absence of hormones, is indicated by the arrow. Scale bar = 10 μm. (B) AR mRNA in LNCaP cells treated with DHT, E2, or R5020 was analyzed by quantitative real‐time PCR. The concentrations of the used hormones were 10−7 M, respectively. Data are normalized to GAPDH mRNA levels and represented relative to the vehicle (%). * P < 0.01 compared with the vehicle. (C) Western blotting of AR (H‐280) using fractionated LNCaP cells protein was performed. Anti‐α‐tubulin antibody was used as the loading control for whole protein (WP). Anti‐α‐tubulin antibody was used as the cell fraction control for cytoplasm protein (CP) and anti‐Pol II antibody were used as that for nuclear protein (NP), respectively. We also used anti‐Lamin A/C antibody as another NP fraction control (Supplemental Fig. 7C). V, D, E, and R indicated Vehicle, DHT, E2 and R5020, respectively.
These results indicate that the three hormones used in this study provoked expression and nuclear translocation of the AR (Thr‐Ala877) in parallel. However, treatment of LNCaP cells with atRA had no influence on the expression of PTHrP or PSA, and no nuclear localization of the AR (Thr‐Ala877) was convincingly confirmed (Supplemental Fig. 4A–C).
Exhaustive gene expression profiles regulated by cross‐reactive hormones via AR (Thr‐Ala877)
To examine whether hormonal cross‐reactivity mediated by the AR (Thr‐Ala877) was widespread in the LNCaP cells, we performed cDNA microarray analysis. The comprehensive gene expression profile was determined by assessing the total RNA from the LNCaP cells after transfection with either control siRNA (siCT) or siRNA for AR (siAR) followed by 10−7 M of DHT, E2, or R5020 administration (GEO accession number: GSE58615). The signals derived from each probe were expressed as the signal/median value divided by the median value of all the probes, and the fold increment of the activities after each hormone treatment was calculated. To isolate the putative target genes of the AR (Thr‐Ala877) stimulated by each hormone, we focused on the probes whose values were drastically changed by hormone administration using siCT (≥5.0 or ≤0.2) with minimal changes in siAR (1.0–2.0 and 0.75–1.0). As shown in Fig. 4A, 547, 426, and 394 probes were target probes for the DHT‐, E2‐, and R5020‐AR, respectively. Overall, 51.9% and 49.5% of the DHT‐AR target genes were shared with the E2‐AR and R5020‐AR targets, respectively. Similarly, 66.7% and 68.3% of the E2‐AR target genes were shared with the DHT‐AR and R5020‐AR probes, and 68.8% and 73.9% of the R5020‐AR target genes overlapped with the DHT‐AR and E2‐AR probes, respectively. These results led us to conclude that more than half of the genes were common targets among the AR (Thr‐Ala877) responders regulated by the three hormones (Fig. 4B).
Figure 4.
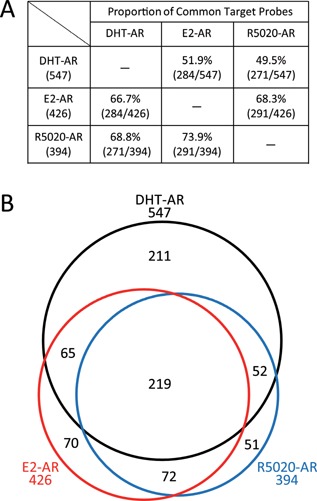
Comparison of DHT‐AR, E2‐AR, and R5020‐AR target gene profiles in LNCaP cells. Microarray analysis was performed to investigate the exhaustive target gene profiles of DHT, E2, and R5020 via the AR (Thr‐Ala877) in LNCaP cells. (A) The proportion of common target probes between each group is listed. The percentage and number of probes in each group is shown. (B) Venn diagram showing the distribution of the DHT‐AR (black), E2‐AR (red), and R5020‐AR (blue) target probes. The numbers outside the Venn diagram indicate the total number of each probe and those inside the Venn diagram indicate the number of probes in each group.
To demonstrate the interaction of AR with some cofactors, we administrated trichostatin A (TSA) as an HDAC inhibitor. TSA is expected to abolish gene repression if HDACs are recruited to down‐regulate PTHrP expression. On the other hand, TSA is also reported to inhibit hormone‐dependent recruitment of co‐activators such as p300 and SRC1 in gene activation (Welsbie et al., 2009). Thus, TSA might cancel hormone‐dependent PSA gene activation. As shown in Fig. 5, PTHrP gene repression and PSA gene activation both mediated by the three hormones in LNCaP cells were lost in common after TSA treatment, implying that the co‐factors such as HDACs for PTHrP repression and, p300 and SRC1 for PSA activation were recruited to the hormone‐ and AR‐dependent transcription machinery.
Figure 5.
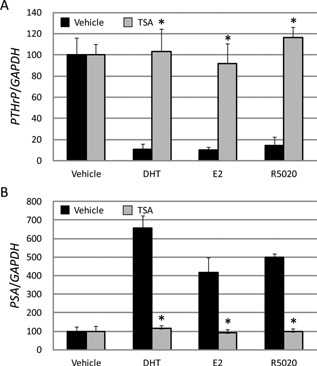
TSA commonly abandoned DHT, E2, and R5020 dependent repression of PTHrP as well as activation of PSA. The concentration of DHT, E2, and R5020 was 10−7 M. TSA was used concomitantly at the dose of 10−6 M for 24 h. The expression levels of PTHrP (A) and PSA mRNAs (B) in LNCaP cells were analyzed by quantitative real‐time PCR. Black and gray bars indicate treatment of Vehicle and TSA, respectively with each steroid hormone. * P < 0.01 compared with the vehicle.
E2‐dependent gene regulation by the AR was caused not by the mutation of the AR (Thr‐Ala877)
To verify whether the skewed crosstalk of the AR (Thr‐Ala877) with the used steroid hormones originated from its LBD mutation, we expressed AR (Thr‐Ala877) and AR (wt) separately in the LNCaP cells wherein we confirmed the endogenous existence of the mutated AR (Thr‐Ala877) (data not shown). We then performed a luciferase reporter assay using the canonical and widely appreciated ARE (AR response element; GGAACAgtaTGTTCT) (Roche et al., 1992) along with either of the AR expression vectors in the LNCaP cells. This ARE sequence is closely related to the native ARE in the 5' flanking region of the PSA gene (Cleutjens et al., 1996) and is considered to represent the bona fide AREs in various cell types (Roche et al., 1992) with high binding affinity for the AR and high transcription activity by means of hormone‐stimulated AR.
As shown in Fig. 6A, DHT treatment in the absence of exogenous AR hardly enhanced ARE promoter activity. In contrast, when either the AR (wt) or AR (Thr‐Ala877) was expressed, we observed marked reporter activation by DHT in a dose‐dependent manner. Treatment with 10−9, 10−8, and 10−7 M DHT conferred a 6.8‐, 8.7‐, and 10.5‐fold stimulation, respectively on the reporter activity after co‐transfection with the AR (wt) in the LNCaP cells, while co‐transfection with the AR (Thr‐Ala877) yielded weaker but significantly enhanced reporter activity (Fig. 6A). In this experiment, disproportionally excessive amounts of the ARE were co‐introduced in our reporter assay (Fig. 6A). We reasoned that the reporter activity in response to DHT failed to increase in the presence of the endogenous AR (Thr‐Ala877) alone, probably because of depletion or squelching of the transcriptional coregulators after these procedures (Fig. 6A).
Figure 6.
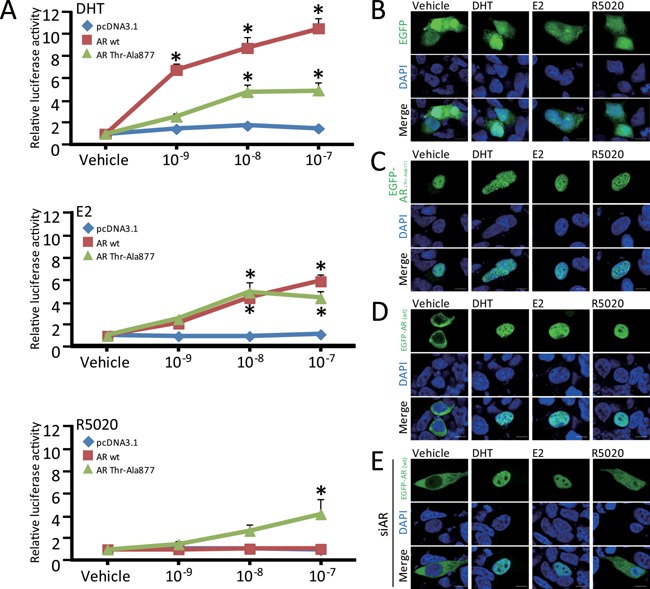
Steroid hormone‐dependent promoter activation and nuclear translocation of the exogenously introduced AR (wild‐type) and AR (Thr‐Ala877) in LNCaP cells. (A) The plasmid pcDNA3.1‐based expression vectors, AR (wild‐type; wt) and AR (Thr‐Ala877), as well as an empty vector, were introduced into LNCaP cells together with the ARE × 3/pGL4‐TK reporter. At 24 h after transfection, the cells were treated with 10−9, 10−8, and 10−7 M of DHT, E2, and R5020, respectively for 24 h followed by luciferase assay. The blue, red, and green lines in each graph denote the response induced by the introduction of pcDNA3.1, AR (wt), and AR (Thr‐Ala877), respectively. Data are expressed as fold changes relative to the vehicle. * P < 0.01 compared with the vehicle. (B–E) At 24 h after transfection with enhanced green fluorescent protein (EGFP) (B), EGFP‐AR (Thr‐Ala877) (C), or EGFP‐AR (wt) (D and E), the indicated steroid hormones (10−7 M) were included for another 24 h. In E, 24 h before transfection with EGFP‐AR (wt), AR siRNA was introduced concurrently with cell seeding to knockdown the endogenous mutant AR (Thr‐Ala877). (Upper panels) EGFP (B), EGFP‐AR (Thr‐Ala877) (C), and EGFP‐AR (wt) (D and E) (green). (Middle panels) DAPI (blue). (Lower panels) Merged images. Note that the stained cells in each upper panel and the double stained cells in each lower panel originated from the DAPI‐positive cells shown in the middle panels, reflecting a transfectional efficiency of around 5%. Scale bar = 10 μm.
We then examined the effects of other steroid hormones on the ARE reporter activity after separate introduction of each AR. Similar to DHT, we observed that both the AR (wt) and AR (Thr‐Ala877) noticeably responded to E2 in a dose‐dependent manner.
In response to the progesterone agonist R5020, the AR (Thr‐Ala877), but not the AR (wt), stimulated the reporter activity in a dose‐dependent manner (Fig. 6A), implying crosstalk between R5020 and the AR (Thr‐Ala877), as reported previously (Veldscholte et al., 1992). We therefore assumed that the AR (Thr‐Ala877), unlike the AR (wt), responds to R5020.
Nuclear localization of the exogenous EGFP‐AR (wt) after E2 or DHT treatment of LNCaP cells
In order to prove the responsibility of not only the AR (Thr‐Ala877), but also the AR (wt) for the skewed crosstalk of the steroid hormones, we analyzed the influence of each steroid hormone on the subcellular localization of the AR (wt) in the LNCaP cells. We utilized the EGFP‐AR (wt) or EGFP‐AR (Thr‐Ala877) fusion protein expression vector to visualize intracellular localization of either type of AR in the LNCaP cells with or without steroid hormones. At 24 h after introducing one of the EGFP‐ARs, we added 10−7 M of each steroid hormone for another 24 h and then performed confocal fluorescence microscopy analysis. The hormones used in this experiment had no influence on the intracellular behavior of EGFP when the AR was absent from the transfection (Fig. 6B). Remarkably, treatment of the LNCaP cells with vehicle alone exhibited distinct cytoplasmic retention of the exogenously introduced EGFP‐AR (wt) (Fig. 6D), while the extrinsic EGFR‐AR (Thr‐Ala877) remained exclusively in the nucleus in this condition and continued remaining in the nucleus after each hormone treatment (Fig. 6C). Such AR (Thr‐Ala877) behavior was reminiscent of the endogenous nuclear AR (Thr‐Ala877) retention pattern observed irrespective of hormonal status (Fig. 3). Molecular mechanisms of this AR (Thr‐Ala877) behavior are still unclear. The AR Thr‐Ala877 mutation might affect the interdomain interaction of AR or change its interaction with some import factor(s). Most importantly, E2 treatment forced the cytoplasmic EGFP‐AR (wt) to move into the nuclear compartment as completely as with the DHT treatment (Fig. 6D). R5020 also seemed to convert the intracellular EGFP‐AR (wt) in the nuclear compartment. Treatment with atRA had no influence on the localization of the EGFP‐AR (wt) (Supplemental Fig. 4D).
To examine whether the endogenous AR (Thr‐Ala877) affects the subcellular localization of the introduced AR (wt), we expressed the EGFP‐AR (wt) in the LNCaP cells after knockdown of the endogenous AR (Thr‐Ala877) with siRNA. As shown in Fig. 6E, R5020 changed the EGFP‐AR (wt) location from nuclear (Fig. 6D) to both compartments (Fig. 6E), implying that the endogenous AR (Thr‐Ala877), unless silenced, somehow interfered with the cytoplasmic retention of the introduced EGFP‐AR (wt) (Fig. 6D). However, addition of E2 or DHT after introduction of the EGFP‐AR (wt) into the AR (Thr‐Ala877)‐disrupted cells essentially did not alter EGFP‐AR (wt) nuclear localization (Fig. 6E).
Along with the reporter gene assay (Fig. 6A), these subcellular microscopic experiments strongly validate our hypothesis that E2, as well as DHT, utilize the AR (wt) to regulate the AR (wt)‐responsive genes in LNCaP cells independently of the AR mutation, such as AR (Thr‐Ala877).
MDA‐MB‐453 cells, unlike LNCaP cells, did not exhibit E2‐dependent ARE promoter activation via the wild‐type AR
Of note, our concern regarding the subtle contamination of any used hormones with other hormone reagents was eliminated by the experiment using the MDA‐MB‐453 breast cancer cell line, which is ERα‐negative but AR‐positive. Reportedly, the AR in MDA‐MB‐453 cells acquired a substitution of Glu‐His865 in its LBD, resulting in the altered sensitivity to DHT and medroxyprogesterone acetate, but not to non‐androgenic ligands (Moore et al., 2012). We confirmed that expression of the FKBP51 gene, a DHT target in the MDA‐MB‐453 cells, was stimulated by DHT in these cells, but not by E2 or R5020 (Supplemental Fig. 5), validating the purity of our hormone preparations.
We next employed the MDA‐MB‐453 cells for the ARE reporter assay (Fig. 7A). In these cells with no overexpression of AR, three hormones including DHT failed to stimulate ARE promoter activity. However, when excessive amounts of the AR (wt) were exogenously introduced to counteract the relative lack of AR over the massive level of the ARE, the luciferase activity driven by the ARE‐containing reporter gene was stimulated by DHT, but again not by E2 or R5020 (Fig. 7A). These results indicate that the MDA‐MB‐453 cells, unlike the LNCaP cells, failed to stimulate E2‐dependent reporter activity via the AR (wt). On the other hand, when the AR (Thr‐Ala877) alone was co‐transfected, E2 and R5020, in addition to DHT, gained the ability to stimulate the promoter activities (Fig. 7A), again reproducing the partial loss of the AR's ligand specificity after the substitution of Thr‐Ala877 (Veldscholte et al., 1992; Grigoryev et al., 2000), even in the MDA‐MB‐453 cells.
Figure 7.
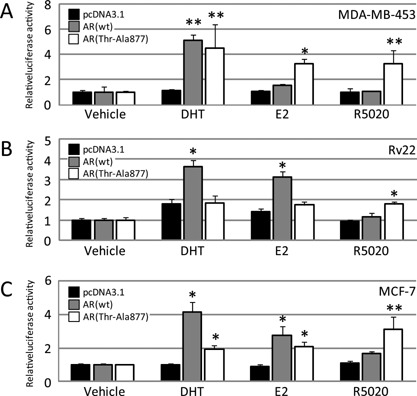
Steroid hormone‐dependent activation of the AR response element containing reporter activity in other prostate and breast cancer cells. Luciferase assay after introducing the empty pcDNA3.1, AR (wt) expression vector, or AR (Thr‐Ala877) expression vector together with the AR response element (ARE) × 3/pGL4‐TK vector in MDA‐MB‐453 (A), Rv22 (B), and MCF‐7 (C) cells. Cells were treated and assays were performed as described in Fig. 5. The black, gray, and white lines show the values for pcDNA3.1, AR (wt), and AR (Thr‐Ala877), respectively. Data are expressed as fold changes relative to the vehicle. * P < 0.01 and **P < 0.05 compared with pcDNA3.1 in each group, respectively.
MCF‐7 and Rv22 cells, like the LNCaP cells, showed DHT‐ and E2‐dependent ARE promoter activation via the wild‐type AR
To verify whether or not the E2‐dependent ARE promoter activation via the wt AR was an LNCaP cell‐specific event, we carried out the ARE reporter assay in prostate cancer Rv22 cells and breast cancer MCF‐7 cells. Interestingly, in both cell lines, unlike in the MDA‐MB‐453 cells, the exogenous AR (wt) enhanced not only the DHT‐, but also the E2‐dependent ARE promoter activities, implying that these cells exhibited distorted interactions between E2 and AR (wt) signaling (Fig. 7B, C). Nonetheless, R5020 had no effect (Fig. 7B, C). Therefore, the behavior of the extrinsic AR (wt) in response to E2 or R5020 was highly similar among the Rv22, MCF‐7, and LNCaP cells (Figs. 6A and 7B, C). On the other hand, when the AR (Thr‐Ala877) was co‐transfected, E2 and R5020 in addition to DHT in the MCF‐7 cells (Fig. 7C), but only R5020 in the Rv22 cells (Fig. 7B), gained the ability to stimulate promoter activity. These results might imply that the partial loss of the AR's ligand specificity due to the AR (Thr‐Ala877) mutation occurred in a non‐cell‐specific manner. Next, we found that the Rv22 cells had the known AR mutation of His‐Tyr874 in their LBD (data not shown), which was previously reported to cross‐react with E2 and progesterone (Taplin et al., 1995; Duff and McEwan, 2005), but we confirmed that the AR in the MCF‐7 cells had no mutation in either allele (data not shown).
Taken together, the cross‐reactive recognition of the AR (wt) by both DHT and E2 was rather widely conserved among several cell lines (Fig. 8).
Figure 8.
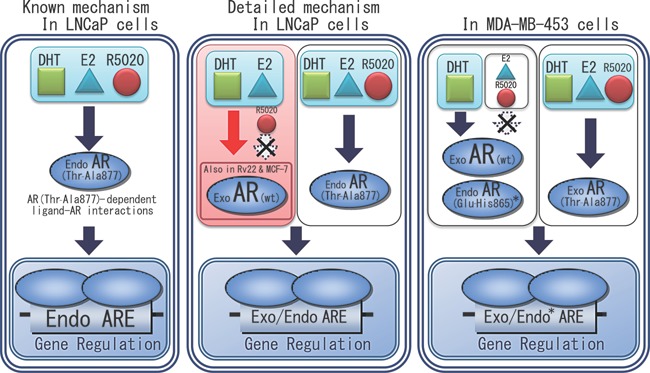
Diverse interactions of the AR with the steroidal ligands in several cell lines. (Left) Previous reports by others postulated that the endogenous AR (Thr‐Ala877) upregulated or downregulated the AR target genes mediated not only by DHT, but also by E2 and R5020 in LNCaP cells. (Middle) In addition, we found that the extrinsic wild‐type AR overexpressed in the LNCaP cells responded to E2. Even in the Rv22 and MCF‐7 cells, the exogenous AR (wt) exhibited a skewed crosstalk with E2 in ARE‐containing reporter gene activity. (Right) In MDA‐MB‐453 cells, the endogenous AR (Glu‐His865), as well as the exogenous AR (wt), responded only to DHT, while the exogenous AR (Thr‐Ala877) maintained its responses to E2 and R5020. *Endo AR (Glu‐His865) regulates the endogenous FKBP51 gene. Exo AR (wt) and Exo AR (Thr‐Ala877) regulate the exogenous ARE‐containing reporter gene. Throughout the figure, “Exo” denotes “exogenously introduced” and “Endo” denotes “endogenously expressed”.
Discussion
To further extend our previous finding of ligand‐dependent repression of PTHrP via the aberrant DHT‐ERα axis in MCF‐7 cells (Kajitani et al., 2011), we performed a series of experiments to unravel whether such a distorted ligand‐NR interaction also occurs in steroid hormone‐dependent prostate LNCaP cells. In this study, we confirmed the physiological signaling pathway between wt AR and E2 in LNCaP cells as well as other sex hormone‐sensitive cells such as Rv22, and MCF‐7 cells.
To analyze the interaction between hormones and either AR (Thr‐Ala877) or AR (wt) in the LNCaP cells, we employed intracellular localization analysis. As expected, E2 along with DHT clearly brought about nuclear retention of both the extrinsic and intrinsic ARs (Thr‐Ala877) (Figs. 3 and 6C). Moreover, we found that E2 markedly promoted nuclear translocation of the EGFP‐AR (wt) when exogenously added or re‐introduced after knockdown of the endogenous AR (Thr‐Ala877) (Fig. 6D, E). To our knowledge, this is the first report visualizing wt‐AR nuclear translocation after E2 treatment in hormone‐sensitive cultured cells expressing no functional ERα. Taken together with the subsequent reporter assay, these results demonstrated that the AR (Thr‐Ala877) did not necessarily contribute to the E2 crosstalk and that the AR (wt) alone conferred skewing of the E2 response (Fig. 6A).
In this scenario, we assumed that the contribution of the interaction between the extrinsic AR (wt) and the endogenous AR (Thr‐Ala877) on the effect of each hormone is negligible. As shown in the AR immunoblot (Supplemental Fig. 6), the total amount of each AR reached 2‐fold that of the endogenous AR (Thr‐Ala877) (Supplemental Fig. 6, pcDNA3.1 lane) under an estimated transfectional efficiency of 5% (Fig. 6B–E). This indicates that each transfectant expressing the introduced gene was under the influence of either type of the co‐introduced AR of approximately 20‐fold higher abundance compared with the endogenous AR (Thr‐Ala877). Last but not least, MCF‐7 cells with endogenous expression of only the wt AR exhibited similarly aberrant ARE‐reporter activation by E2 when extrinsic AR (wt) was introduced (Fig. 7C). These observations strongly underpin our hypothesis that the aberrant crosstalk driven by E2 was exclusively elicited through activation of the bona fide AR (wt).
In contrast, unlike E2, R5020 did not affect the ARE‐mediated reporter activity when the AR (wt) was exogenously overexpressed (Fig. 6A, red line on R5020 graph). Notwithstanding, R5020, like E2, shifted the introduced EGFP‐AR (wt) from the cytoplasm to the nucleus (Fig. 6D). However, when EGFP‐AR (wt) was re‐introduced after silencing the endogenous AR (Thr‐Ala877) expression (Fig. 6E), R5020 selectively conferred both cytoplasmic and nuclear re‐distribution to the EGFP‐AR (wt), while its nuclear distribution in the E2‐treated cells was unaltered. It is tempting to speculate from these observations that the intranuclear AR retention was necessary but not sufficient for the subsequent hormonal responses. Our results showing that the AR (Thr‐Ala877) was constitutively present within the nucleus even in the absence of added hormones (Fig. 6C) support this contention.
Taken together, it can be considered that the aberrant E2‐AR (wt) signaling observed in the LNCaP cells mirrors our previous finding that the endogenous ERα, but not the AR which was otherwise functional in MCF‐7 cells, mediated DHT‐induced PTHrP gene repression (Kajitani et al., 2011). However, the underlying mechanism in such an intricate mutual crosstalk remains unknown.
Furthermore, our experiments using TSA revealed that DHT, E2 and R5020 commonly recruited HDAC‐corepressor complexes and coactivators to PTHrP and PSA genes, respectively, in LNCaP cells (Fig. 5). Of note, the comprehensive expression profile of the genes located downstream of the AR (Thr‐Ala877) and regulated by either of the three steroid hormones, DHT, E2, or R5020, revealed that more than half but not all genes overlapped in the LNCaP cells (Fig. 4). Such non‐overlapping regulation suggests a substitute for the AR (Thr‐Ala877) that is capable of mediating E2‐ or R5020‐specific gene regulation. The ERβ was reported to be expressed in LNCaP cells (Lau et al., 2000; Bektic et al., 2004) or a membrane form(s) of the unknown receptors might be involved in this process. On the other hand, we realized that even the PTHrP or PSA genes were not selected as overlapping candidates under our stringent criteria (not shown), implying that the actual numbers of the target genes commonly regulated by these hormones might be considerably underestimated. In this sense, we consider it reasonable to use the canonical, but not gene‐specific ARE as a consensus AR target sequence in our reporter assays.
The recent development of chromatin immunoprecipitation analysis by genome‐wide techniques revealed unforeseen illegitimate interactions between NRs and versatile chromatinized DNA sequences, but such concepts were invariably based on the presumption that the corresponding cognate NR is faithfully expressed for its authentic ligand. For instance, inhibition of the E2‐ERα‐dependent gene regulatory axis by dexamethasone (Dex) has been rigorously studied in MCF‐7 cells (Karmakar et al., 2013). In other murine mammary cell lines, Dex‐GR (glucocorticoid receptor) changed the DNA accessibility of the E2‐ERα complex, resulting in its binding to novel specific sites (Miranda et al., 2013). In the MDA‐MB‐453 cells, which are ERα‐negative but AR‐positive, DHT‐AR signaling hijacked the E2‐ERα cistrome (Robinson et al., 2011). As such, it is possible that reciprocal interactions between the liganded NRs resulted in occasional intricate crosstalk.
In addition, our observation that not only the LNCaP cells, but also the AR (wt)‐expressing MCF‐7 and Rv22 cells, exhibited E2‐mediated regulation through the ARE‐containing luciferase reporter (Figs. 6A, 7B and C) strongly suggests that the AR (wt) interacted directly or indirectly with E2 in certain sex hormone‐responsive cancer cells. Regardless, this straightforward hypothesis is very hard to prove in living cells. Reports on classical cloning of NRs, especially homodimer‐requiring species, have often described the ambiguous ligand selectivity of these NRs (Lubahn et al., 1988). Nonetheless, unlike the present study, such “cross‐reactivity” was usually found when ligand concentrations were at supra‐physiological levels.
For the distortions observed in this study to occur at least in some cancer cells but not in others, the following predispositions might be required: a security switch for steroid hormones such as 11β‐hydroxysteroid dehydrogenase 2, which inactivates cortisol in aldosterone target cells (Arriza et al., 1987; Funder et al., 1988; Fuller et al., 2012); or an alteration of the enzymatic activity involving the intratumoral conversion of steroidal substances (Mohler et al., 2011). Recently, germline polymorphisms in the CYP1B1, SULT2B1, COMT, NQO1, and NQO2 genes, which play roles in multiple estrogen metabolism pathways, were reported to be closely associated with prostate cancer progression (Levesque et al., 2014). Further, a history of anti‐hormonal therapies might affect the repertoire of NR binding (Mitsiades, 2013).
In summary, we identified a novel regulatory mechanism(s) by which the AR and steroid hormones closely interact with each other in several hormone‐sensitive cancer cell lines. In versatile cells, the AR (wt) accepts E2 as an aberrant ligand and regulates PTHrP and PSA gene expression regardless of the mutation in its LBD. We successfully demonstrated by fluorescence analysis a probable in vivo interaction between E2 and both the AR (wt) and AR (Thr‐Ala877). We speculate the existence of unknown regulatory mechanistic links between the AR signaling axis and E2 in LNCaP cells and other sex hormone‐responsive cancer cells (Fig. 8), and revealing these will be a novel finding. It is desirable to develop drugs targeting such cell‐type‐specific crosstalk between sex hormones with the ability to overcome anti‐hormone resistance in certain types of prostate as well as breast cancers.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Figure S4.
Supplementary Figure S5.
Supplementary Figure S6.
Supplementary Figure S7.
Supporting Information.
Acknowledgments
We thank Drs. Shigeo Horie and Shunji Takahashi for providing the cell lines and Dr. Sigeaki Kato for providing the human AR expression vector.
Conflicts of interest: None.
Literature Cited
- Ahlstrom M, Pekkinen M, Lamberg‐Allardt C. 2009. Dexamethasone downregulates the expression of parathyroid hormone‐related protein (PTHrP) in mesenchymal stem cells. Steroids 74:277–282. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. 1987. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science 237:268–275. [DOI] [PubMed] [Google Scholar]
- Asadi F, Kukreja S. 2005. Parathyroid hormone‐related protein in prostate cancer. Crit Rev Eukaryot Gene Expr 15:15–28. [DOI] [PubMed] [Google Scholar]
- Bektic J, Berger AP, Pfeil K, Dobler G, Bartsch G, Klocker H. 2004. Androgen receptor regulation by physiological concentrations of the isoflavonoid genistein in androgen‐dependent LNCaP cells is mediated by estrogen receptor beta. Eur Urol 45:245–251; discussion251. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Greenberg NM, Scher HI, Harris JM, Marshall VR, Tilley WD. 2001. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res 7:1273–1281. [PubMed] [Google Scholar]
- Ceraline J, Cruchant MD, Erdmann E, Erbs P, Kurtz JE, Duclos B, Jacqmin D, Chopin D, Bergerat JP. 2004. Constitutive activation of the androgen receptor by a point mutation in the hinge region: A new mechanism for androgen‐independent growth in prostate cancer. Int J Cancer 108:152–157. [DOI] [PubMed] [Google Scholar]
- Chirgwin JM, Guise TA. 2000. Molecular mechanisms of tumor‐bone interactions in osteolytic metastases. Crit Rev Eukaryot Gene Expr 10:159–178. [PubMed] [Google Scholar]
- Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. 1996. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate‐specific antigen promoter. J Biol Chem 271:6379–6388. [DOI] [PubMed] [Google Scholar]
- Dougherty KM, Blomme EA, Koh AJ, Henderson JE, Pienta KJ, Rosol TJ, McCauley LK. 1999. Parathyroid hormone‐related protein as a growth regulator of prostate carcinoma. Cancer Res 59:6015–6022. [PubMed] [Google Scholar]
- Duff J, McEwan IJ. 2005. Mutation of histidine 874 in the androgen receptor ligand‐binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Mol Endocrinol 19:2943–2954. [DOI] [PubMed] [Google Scholar]
- Endo K, Ichikawa F, Uchiyama Y, Katsumata K, Ohkawa H, Kumaki K, Ogata E, Ikeda K. 1994. Evidence for the uptake of a vitamin D analogue (OCT) by a human carcinoma and its effect of suppressing the transcription of parathyroid hormone‐related peptide gene in vivo. J Biol Chem 269:32693–32699. [PubMed] [Google Scholar]
- Falzon M. 1996. DNA sequences in the rat parathyroid hormone‐related peptide gene responsible for 1,25‐dihydroxyvitamin D3‐mediated transcriptional repression. Mol Endocrinol 10:672–681. [DOI] [PubMed] [Google Scholar]
- Fuller PJ, Yao Y, Yang J, Young MJ. 2012. Mechanisms of ligand specificity of the mineralocorticoid receptor. J Endocrinol 213:15–24. [DOI] [PubMed] [Google Scholar]
- Funder JW, Pearce PT, Smith R, Smith AI. 1988. Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science 242:583–585. [DOI] [PubMed] [Google Scholar]
- Gaddipati JP, McLeod DG, Heidenberg HB, Sesterhenn IA, Finger MJ, Moul JW, Srivastava S. 1994. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res 54:2861–2864. [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore JL, Scott JA, Bouizar Z, Robling A, Pitfield SE, Riese DJ, 2nd , Foley J. 2008. Amphiregulin‐EGFR signaling regulates PTHrP gene expression in breast cancer cells. Breast Cancer Res Treat 110:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatz JA, Heath JK, Southby J, O'Keeffe LM, Kiriyama T, Moseley JM, Martin TJ, Gillespie MT. 1994. Dexamethasone regulation of parathyroid hormone‐related protein (PTHrP) expression in a squamous cancer cell line. Mol Cell Endocrinol 101:295–306. [DOI] [PubMed] [Google Scholar]
- Grigoryev DN, Long BJ, Njar VC, Brodie AH. 2000. Pregnenolone stimulates LNCaP prostate cancer cell growth via the mutated androgen receptor. J Steroid Biochem Mol Biol 75:1–10. [DOI] [PubMed] [Google Scholar]
- Ha YS, Goodin S, DiPaola RS, Kim IY. 2013. Enzalutamide for the treatment of castration‐resistant prostate cancer. Drugs Today (Barc) 49:7–13. [DOI] [PubMed] [Google Scholar]
- Hickey RC, Samaan NA, Jackson GL. 1981. Hypercalcemia in patients with breast cancer. Osseous metastases, hyperplastic parathyroid tissue, or pseudohyperparathyroidism? Arch Surg 116:545–552. [DOI] [PubMed] [Google Scholar]
- Horoszewicz JS, Leong SS, Chu TM, Wajsman ZL, Friedman M, Papsidero L, Kim U, Chai LS, Kakati S, Arya SK, Sandberg AA. 1980. The LNCaP cell line–A new model for studies on human prostatic carcinoma. Prog Clin Biol Res 37:115–132. [PubMed] [Google Scholar]
- Ikeda K, Lu C, Weir EC, Mangin M, Broadus AE. 1989. Transcriptional regulation of the parathyroid hormone‐related peptide gene by glucocorticoids and vitamin D in a human C‐cell line. J Biol Chem 264:15743–15746. [PubMed] [Google Scholar]
- Inoue D, Matsumoto T, Ogata E, Ikeda K. 1993. 22‐Oxacalcitriol, a noncalcemic analogue of calcitriol, suppresses both cell proliferation and parathyroid hormone‐related peptide gene expression in human T cell lymphotrophic virus, type I‐infected T cells. J Biol Chem 268:16730–16736. [PubMed] [Google Scholar]
- Kajitani T, Tamamori‐Adachi M, Okinaga H, Chikamori M, Iizuka M, Okazaki T. 2011. Negative regulation of parathyroid hormone‐related protein expression by steroid hormones. Biochem Biophys Res Commun 407:472–478. [DOI] [PubMed] [Google Scholar]
- Karmakar S, Jin Y, Nagaich AK. 2013. Interaction of Glucocorticoid Receptor (GR) with Estrogen Receptor‐alpha (ERalpha) and Activator Protein 1 (AP1) in Dexamethasone‐Mediated Interference of ERalpha Activity. J Biol Chem 288:24020–24034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasono K, Isozaki O, Sato K, Sato Y, Shizume K, Ohsumi K, Demura H. 1991. Effects of glucocorticoids and calcitonin on parathyroid hormone‐related protein (PTHrP) gene expression and PTHrP release in human cancer cells causing humoral hypercalcemia. Jpn J Cancer Res 82:1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurebayashi J, Otsuki T, Tanaka K, Yamamoto Y, Moriya T, Sonoo H. 2003. Medroxyprogesterone acetate decreases secretion of interleukin‐6 and parathyroid hormone‐related protein in a new anaplastic thyroid cancer cell line, KTC‐2. Thyroid 13:249–258. [DOI] [PubMed] [Google Scholar]
- Lau KM, LaSpina M, Long J, Ho SM. 2000. Expression of estrogen receptor (ER)‐alpha and ER‐beta in normal and malignant prostatic epithelial cells: Regulation by methylation and involvement in growth regulation. Cancer Res 60:3175–3182. [PubMed] [Google Scholar]
- Levesque E, Laverdiere I, Audet‐Walsh E, Caron P, Rouleau M, Fradet Y, Lacombe L, Guillemette C. 2014. Steroidogenic germline polymorphism predictors of prostate cancer progression in the estradiol pathway. Clin Cancer Res 20:2971–2983. [DOI] [PubMed] [Google Scholar]
- Lindemann RK, Ballschmieter P, Nordheim A, Dittmer J. 2001. Transforming growth factor beta regulates parathyroid hormone‐related protein expression in MDA‐MB‐231 breast cancer cells through a novel Smad/Ets synergism. J Biol Chem 276:46661–46670. [DOI] [PubMed] [Google Scholar]
- Lindemann RK, Braig M, Ballschmieter P, Guise TA, Nordheim A, Dittmer J. 2003. Protein kinase Calpha regulates Ets1 transcriptional activity in invasive breast cancer cells. Int J Oncol 22:799–805. [PubMed] [Google Scholar]
- Liu B, Goltzman D, Rabbani SA. 1993. Regulation of parathyroid hormone‐related peptide production in vitro by the rat hypercalcemic Leydig cell tumor H‐500. Endocrinology 132:1658–1664. [DOI] [PubMed] [Google Scholar]
- Lu C, Ikeda K, Deftos LJ, Gazdar AF, Mangin M, Broadus AE. 1989. Glucocorticoid regulation of parathyroid hormone‐related peptide gene transcription in a human neuroendocrine cell line. Mol Endocrinol 3:2034–2040. [DOI] [PubMed] [Google Scholar]
- Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. 1988. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science 240:327–330. [DOI] [PubMed] [Google Scholar]
- Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, Grontved L, Schiltz RL, Hager GL. 2013. Reprogramming the chromatin landscape: Interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res 73:5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades N. 2013. A road map to comprehensive androgen receptor axis targeting for castration‐resistant prostate cancer. Cancer Res 73:4599–4605. [DOI] [PubMed] [Google Scholar]
- Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, Wilson EM. 2011. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res 71:1486–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore NL, Buchanan G, Harris JM, Selth LA, Bianco‐Miotto T, Hanson AR, Birrell SN, Butler LM, Hickey TE, Tilley WD. 2012. An androgen receptor mutation in the MDA‐MB‐453 cell line model of molecular apocrine breast cancer compromises receptor activity. Endocr Relat Cancer 19:599–613. [DOI] [PubMed] [Google Scholar]
- Mundy GR, Edwards JR. 2008. PTH‐related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol 19:672–675. [DOI] [PubMed] [Google Scholar]
- Ni L, Llewellyn R, Kesler CT, Kelley JB, Spencer A, Snow CJ, Shank L, Paschal BM. 2013. Androgen induces a switch in the androgen receptor from cytoplasmic retention to nuclear import. Mol Cell Biol 33:4766–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishishita T, Okazaki T, Ishikawa T, Igarashi T, Hata K, Ogata E, Fujita T. 1998. A negative vitamin D response DNA element in the human parathyroid hormone‐related peptide gene binds to vitamin D receptor along with Ku antigen to mediate negative gene regulation by vitamin D. J Biol Chem 273:10901–10907. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Nishimori S, Ogata E, Fujita T. 2003. Vitamin D‐dependent recruitment of DNA‐PK to the chromatinized negative vitamin D response element in the PTHrP gene is required for gene repression by vitamin D. Biochem Biophys Res Commun 304:632–637. [DOI] [PubMed] [Google Scholar]
- Perry JE, Tindall DJ. 1996. Androgens regulate the expression of proliferating cell nuclear antigen posttranscriptionally in the human prostate cancer cell line, LNCaP. Cancer Res 56:1539–1544. [PubMed] [Google Scholar]
- Pizzi H, Gladu J, Carpio L, Miao D, Goltzman D, Rabbani SA. 2003. Androgen regulation of parathyroid hormone‐related peptide production in human prostate cancer cells. Endocrinology 144:858–867. [DOI] [PubMed] [Google Scholar]
- Rabbani SA, Khalili P, Arakelian A, Pizzi H, Chen G, Goltzman D. 2005. Regulation of parathyroid hormone‐related peptide by estradiol: Effect on tumor growth and metastasis in vitro and in vivo. Endocrinology 146:2885–2894. [DOI] [PubMed] [Google Scholar]
- Rizzoli R, Feyen JH, Grau G, Wohlwend A, Sappino AP, Bonjour JP. 1994. Regulation of parathyroid hormone‐related protein production in a human lung squamous cell carcinoma line. J Endocrinol 143:333–341. [DOI] [PubMed] [Google Scholar]
- Robinson JL, Macarthur S, Ross‐Innes CS, Tilley WD, Neal DE, Mills IG, Carroll JS. 2011. Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J 30:3019–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche PJ, Hoare SA, Parker MG. 1992. A consensus DNA‐binding site for the androgen receptor. Mol Endocrinol 6:2229–2235. [DOI] [PubMed] [Google Scholar]
- Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL. 2010. Antitumour activity of MDV3100 in castration‐resistant prostate cancer: A phase 1–2 study. Lancet 375:1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B. 2007. Serum/glucocorticoid‐induced protein kinase‐1 facilitates androgen receptor‐dependent cell survival. Cell Death Differ 14:2085–2094. [DOI] [PubMed] [Google Scholar]
- Sterling JA, Oyajobi BO, Grubbs B, Padalecki SS, Munoz SA, Gupta A, Story B, Zhao M, Mundy GR. 2006. The hedgehog signaling molecule Gli2 induces parathyroid hormone‐related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res 66:7548–7553. [DOI] [PubMed] [Google Scholar]
- Sugimoto T, Shiba E, Watanabe T, Takai S. 1999. Suppression of parathyroid hormone‐related protein messenger RNA expression by medroxyprogesterone acetate in breast cancer tissues. Breast Cancer Res Treat 56:11–23. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. 1996. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: Relation to antiandrogen withdrawal syndrome. Prostate 29:153–158. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Sato N, Watabe Y, Masai M, Seino S, Shimazaki J. 1993. Androgen receptor gene mutations in human prostate cancer. J Steroid Biochem Mol Biol 46:759–765. [DOI] [PubMed] [Google Scholar]
- Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. 1995. Mutation of the androgen‐receptor gene in metastatic androgen‐independent prostate cancer. N Engl J Med 332:1393–1398. [DOI] [PubMed] [Google Scholar]
- Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, Stadler W, Hayes DF, Kantoff PW, Vogelzang NJ, Small EJ. 2003. Androgen receptor mutations in androgen‐independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol 21:2673–2678. [DOI] [PubMed] [Google Scholar]
- Veldscholte J, Berrevoets CA, Ris‐Stalpers C, Kuiper GG, Jenster G, Trapman J, Brinkmann AO, Mulder E. 1992. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol 41:665–669. [DOI] [PubMed] [Google Scholar]
- Walsh CA, Birch MA, Fraser WD, Lawton R, Dorgan J, Walsh S, Sansom D, Beresford JN, Gallagher JA. 1995. Expression and secretion of parathyroid hormone‐related protein by human bone‐derived cells in vitro: Effects of glucocorticoids. J Bone Miner Res 10:17–25. [DOI] [PubMed] [Google Scholar]
- Welsbie DS, Xu J, Chen Y, Borsu L, Scher HI, Rosen N, Sawyers CL. 2009. Histone deacetylases are required for androgen receptor function in hormone‐sensitive and castrate‐resistant prostate cancer. Cancer Res 69:958–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Supplementary Figure S1.
Supplementary Figure S2.
Supplementary Figure S3.
Supplementary Figure S4.
Supplementary Figure S5.
Supplementary Figure S6.
Supplementary Figure S7.
Supporting Information.