

Abstract
Endometriosis is a significant risk factor for clear cell and endometrioid ovarian cancers and is often found contiguous with these cancers. Using whole‐genome shotgun sequencing of seven clear cell ovarian carcinomas (CCC) and targeted sequencing in synchronous endometriosis, we have investigated how this carcinoma may evolve from endometriosis. In every case we observed multiple tumour‐associated somatic mutations in at least one concurrent endometriotic lesion. ARID1A and PIK3CA mutations appeared consistently in concurrent endometriosis when present in the primary CCC. In several cases, one or more endometriotic lesions carried the near‐complete complement of somatic mutations present in the index CCC tumour. Ancestral mutations were detected in both tumour‐adjacent and ‐distant endometriotic lesions, regardless of any cytological atypia. These findings provide objective evidence that multifocal benign endometriotic lesions are clonally related and that CCCs arising in these patients progress from endometriotic lesions that may already carry sufficient cancer‐associated mutations to be considered neoplasms themselves, albeit with low malignant potential. We speculate that genomically distinct classes of endometriosis exist and that ovarian endometriosis with high mutational burden represents one class at high risk for malignant transformation. © 2015 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: endometriosis, ovarian cancer, clear cell carcinoma, cancer precursor, sequencing
Introduction
Endometriosis affects one in ten reproductive‐aged women and is a common cause of pelvic pain and infertility. It is defined as the ectopic presence of endometrium (epithelium and stroma) outside of the uterus, typically elsewhere in the pelvis, such as on the ovaries. Its aetiology remains somewhat controversial, although the dominant theories suggest either a uterine origin, dispersing via retrograde menstruation or lymphatics, or an extra‐uterine origin, wherein ectopic tissue would come about through coelomic metaplasia or abnormal differentiation 1, 2, 3. Treatments for endometriosis typically focus on alleviation of symptoms and hormonal manipulation. A common concern of patients with endometriosis is whether the condition may elevate their risk of cancer.
Despite being considered a benign condition, endometriosis has been shown to be a significant risk factor for developing ovarian cancer. In several recent epidemiological studies, the presentation of endometriosis elevated the risk for clear cell ovarian carcinoma (CCC) and endometrioid ovarian carcinoma (ENOCa) by ∼ three‐ and ∼ two‐fold, respectively. These ovarian cancer histotypes represent the second and third most common forms and account for > 20% of all epithelial ovarian carcinomas 4. Aside from a slight increased risk for low‐grade serous carcinomas in a single study, there is no significant increase in risk for other ovarian cancer histotypes 5, 6, 7. Endometriosis is frequently noted as comorbidity with ovarian cancer 8, 9. Numerous retrospective analyses report concurrent endometriosis in 30–40% of CCCs, while it is reported in < 10% of high‐grade serous ovarian carcinomas (HGSCs) 7. Atypical endometriosis, with marked cytological atypia of the epithelial cell component, has been noted to be present much more frequently in both ENOCa and CCC compared to other histological types 8. It is not uncommon to observe a morphological continuum wherein normal‐appearing endometriosis occurs directly adjacent to, or contiguous with, endometriosis having cytological atypia and finally with frank carcinoma. This circumstantial evidence alone suggests a strong link between endometriosis and cancer; however, only recently have molecular features proven a relationship to CCC and ENOCa. Initially, patterns of loss of heterozygosity (LOH) from CCC and ENOCa tumours mirrored in adjacent endometriosis supported a model wherein the carcinoma derived directly from endometriosis 10, 11, 12. More conclusive evidence of a clonal relationship between CCC and atypical endometriosis came with the discovery of loss‐of‐function mutations in ARID1A, a member of the SWI/SNF chromatin‐remodelling complex, in roughly half of CCCs and one‐third of ENOCa cases 13, 14; identical ARID1A mutations were shown to be present in concurrent endometriosis from these cases 13. More recently, amplification of the MET proto‐oncogene in atypical endometriosis and adjacent CCC has also been noted 15, 16. All studies so far have focused on single genes, and a broader view of this cancer precursor lesion has not been described. Distant foci of endometriosis without cytological atypia have failed to show clonal ARID1A mutations 13; however, cellularity and low‐resolution (Sanger) sequencing employed for validation may have impeded detection. A clonal relationship has been established in at least a subset atypical endometriosis specimens and adjacent carcinoma; however, questions remain about co‐occurring distant endometriotic lesions – can these share a clonal relationship with the cancer, and what is the risk for malignant transformation?
Herein we used whole‐genome sequencing of seven clear cell carcinomas, along with targeted sequencing across multiple adjacent and metastatic tumour sites, concurrent endometriosis and other benign gynaecological lesions from each patient. Our goals were to examine the overall pattern of somatic mutations within the full CCC genome, investigate whether a clonal relationship exists between cancer and adjacent or distant endometriotic lesions and, finally, to assess whether specific genes or gene families are mutated in progression from endometriosis to CCC.
Materials and methods
Cohort description
Collection and use of specimens was reviewed by the local research ethics board and approved under protocols H05‐60119, H08‐01411 and H09‐02153. Samples used for whole‐genome shotgun sequencing (WGSS) were selected from the OVCARE gynaecological tissue bank. Specimens underwent expert pathological review (author CBG) with immunohistochemical (IHC) staining of tumours to ensure proper diagnosis. All CCCs were WT1‐negative and HNF1B‐positive by IHC (see supplementary material, Figure S2). Archival blocks and slides were obtained from Vancouver General Hospital Anatomical Pathology and used for identification and sampling of additional tumours, including metastases, endometriosis and other benign lesions (reviewed by CBG, HLC and ANK; see supplementary material, Table S1).
DNA extraction
For WGSS, genomic DNA from frozen tumour sections or buffy coat from blood (germline reference) was extracted using the Gentra Pure Gene DNA Kit and standard techniques, as previously described. This DNA was whole‐genome amplified (WGA), using the Repli‐G kit (Qiagen), prior to use in the Haloplex amplicon kit (Agilent) for deep sequencing. DNA from additional tumours, endometriosis and benign lesions were isolated from formalin‐fixed, paraffin‐embedded (FFPE) tissues by coring, macrodissection from haematoxylin and eosin (H&E)‐stained sections, or laser‐capture microdissection (LCM) from stained sections, depending on their size and location (see supplementary material, Table S1). DNA from cored or macrodissected FFPE tissue was extracted using the QIAamp DNA FFPE kit (Qiagen). DNA from LCM FFPE tissues was extracted using the Picopure DNA Kit (Life Technologies). Samples with < 150 ng yield were subject to whole‐genome amplification, using the Ovation FFPE Whole Genome Amplification Kit (Nugen) and Ovation dsDNA module (Nugen), as marked in Table S1 (see supplementary material).
WGSS sequencing
SOLiD whole‐genome shotgun sequencing (WGSS) libraries for seven tumour/normal pairs were generated as previously described 17. One sample had data from both SOLiD and Illumina HiSeq available and this was incorporated into the analysis, as previously described 18 (for analysis details, see Supplementary materials and methods). Exon 1 of ARID1A was also sequenced using Sanger methodology, due to WGSS coverage deficiency 13.
Orthogonal validation of predicted somatic alterations using targeted deep sequencing
A custom Haloplex (Agilent) amplicon library was designed using Agilent SureDesign with small amplicon design (200 bp range) and FFPE optimization. Prior to preparation of Haloplex libraries, the quality and input of DNA was evaluated and adjusted, according to the manufacturer's recommendations. Haloplex custom libraries were then produced for all available specimens (see supplementary material, Table S1), according to the manufacturer's recommendations. Libraries were indexed, pooled and sequenced on an Illumina MiSeq, using 150 bp end reads (300 cycle v. 2 chemistry; Illumina) sequencing kits to a median depth of > 1300×; plots of coverage are shown in Figure S5 (see supplementary material).
Results
Whole‐genome and targeted deep sequencing of CCC
WGSS to a median depth > ×30 in both index tumours and matched germline DNA was performed to established a baseline of somatic mutations, DNA copy‐number aberrations (CNAs) and genomic architecture. With respect to CNAs, our data were consistent with previous reports on CCC CNAs 19, 20, 21, 22, 23; however, our cohort was too limited for a detailed comparison. CNA profiles were variable across specimens (Figure 1A), with the proportions of genomes altered ranging from 14% (case 3) to 65% (case 7). Gains of the 8q arm were the most obvious large‐scale and common events, with some level of copy number gain on 8q being observed in all cases. We observed DNA copy number gains in a number of previously documented CCC‐associated genes, including regions of 17q: HNF1B (5/7), PPM1D (5/7), ERBB2 (4/7), STAT3 (4/7); 3q PIK3CA (4/7); 7q: MET (2/7) and 20q: ZNF217 (6/7), including one high‐level gain of PIK3CA observed in case 7. Despite other reports showing common amplification of MET in CCC 15, 16, we did not observe any such events in our cohort, neither did we observe classic high‐level amplification of ERBB2, as has been previously reported in breast and mucinous ovarian cancer 24, 25, 26.
Figure 1.
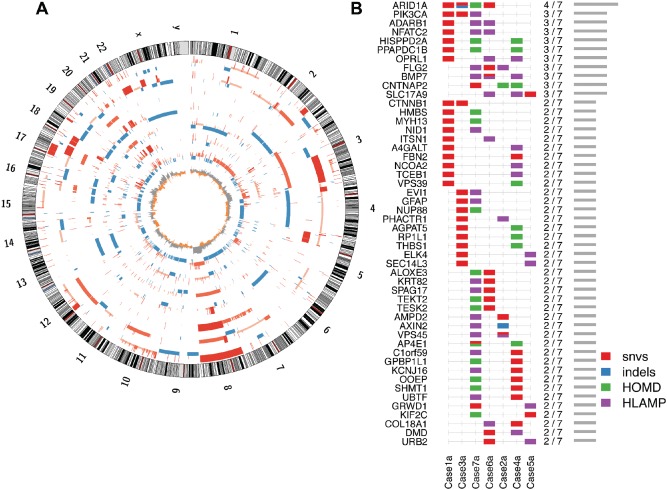
Overview of somatic genomic architecture and mutational pattern across sampled CCC tumours. (A) Circos plot, showing somatic DNA copy number gains (red) and losses (blue) in each CCC primary tumour, in order from the outermost ring (cases 1–7): bar heights indicate relative gains or losses, eg higher red bars indicate high‐level amplification, short blue bars indicate heterozygous deletions and the innermost ring shows the frequency of the gains (grey) and losses (orange) among the seven cases. (B) Genes most frequently affected by a somatic INDEL, SNV or extreme copy number event [homozygous deletion (HOMD) or high‐level amplification (HLAMP)]: at least one known somatic truncating variant in ARID1A is not illustrated here, as it was not found in whole‐genome data, neither was it able to be validated in our deep‐sequencing assay (case 4; see supplementary material, Figure S1), due to low coverage, likely related to high GC content
We predicted 39–163 coding SNVs and indels/case, with an overall somatic mutation load of 0.23–0.86 SNVs/Mb (see supplementary material, Table S1). A total of 847 predicted somatic mutations were selected for validation, using targeted deep sequencing (Haloplex; see supplementary material, Supplementary materials and methods). Targets included all predicted somatic coding variants, as well as a number of non‐coding positions, so as to sample at least 110 positions from each case (see supplementary material, Table S1 and Supplementary materials and methods). Following validation (median coverage × 1316), ARID1A mutations were confirmed in 4/7 cases, only one of which appeared to carry a bi‐allelic loss‐of‐function alterations (Figure 1B). Sanger‐based sequencing of ARID1A, exon 1, revealed one additional complex deletion and insertion predicted to result in early termination, in a case without a previously identified alteration (case 4; see supplementary material, Figure S1). Homozygous deletions were not observed at the ARID1A locus; however, hemizygous loss was observed over the entire ARID1A locus in case 7, which did not have another ARID1A mutation or loss of ARID1A protein expression (see supplementary material, Figure S2). PIK3CA‐activating mutations were seen in two cases, giving a total of three notable alterations, two SNVs and one high‐level amplification. This is similar to the expected frequency across both CCC and ENOCa, where alterations are observed in up to 45% of cases 4, 27, 28, 29. Somatic SNVs or indel alterations affecting other SWI/SNF members were not detected.
Our cohort was too small to make any claims of enrichment or mutations within any pathway; however, connections can be drawn between more frequently observed somatic alterations (Figure 1) and ARID1A/PIK3CA‐related pathways. For example, NCOA2 is known to interact with the SWI/SNF complex via BRG1 and BAF57 and has been implicated in potentiation of ER‐response genes 30. NCOA2 may also positively modulate AKT/mTOR signalling, as has been noted for other p160‐family members (NCOA3) 31. Both CTNNB1, particularly when affected by 'hotspot' mutations, as observed in our cohort, and NFATC2 positively regulate WNT signalling 32, 33. TCEB1 (Elongin C) 'hotspot mutants', described in clear‐cell renal carcinoma (ccRC), promote HIF1a stability by preventing interaction with VHL and the assembly of the VHL‐linked E3–ligase complex that degrades HIF1 34. Like its renal counterpart, ovarian CCC expresses high levels of both HIF1A and EPAS1(HIF2A) 23. Finally, TCEB1 is also known to form an E3–ligase complex with BAF250b (ARID1B; a paralogue of ARID1A and alternative SWI/SNF component) to mono‐ubiquitinate histone H2B 35. In this case it is important to note mono‐ubiquitination, as this is less likely to be linked directly to protein degradation than modification of function 36.
CCC mutations across lesions and precursors
To investigate the distribution of mutations beyond the index tumour specimens, we applied Haloplex deep sequencing to 33 additional sites across the seven cases, including carcinoma and putative precursor lesions, as well as benign tissues. Four cases had at least one other focus of carcinoma (including metastases) aside from the fully sequenced specimen; five cases had at least one focus of endometriosis with atypia; three had endometriosis (without atypia); three had other benign müllerian conditions (including endosalpingiosis, adenomyosis, endometrial polyps and leiomyomata) and six had normal endometrium available to sample (Table 1; see also supplementary material, Table S1). Atypical endometriosis was distinguished through morphological examination: the presence of endometrial‐like stroma adjacent to glands lined by endometrial‐type epithelial cells showing cytological atypia, but less atypia than was present within the carcinoma and less stratification and proliferation of the epithelium (again, compared to the carcinoma from the same case). When directly contiguous with a cancer, the different levels of atypia observed between the so‐called 'atypical endometriosis' and carcinoma may be subjective; however, atypical endometriosis, in the context of clear cell ovarian carcinoma, has been shown to be distinct from the cancer through maintenance of endometrial‐type stroma, retained ER‐expression and reduced HNF1B expression compared to the carcinoma 37. In the case of endometriosis with cytological atypia that is not contiguous with the tumour, metastatic spread of the carcinoma can be ruled out by the presence of endometrial‐type stroma alone.
Table 1.
Summary of specimen types and number collected for each patient
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | |
|---|---|---|---|---|---|---|---|
| Primary CCC | 1 | 1 | 1 | 1 | 3 | 1 | 2 |
| Metastasis | 2 | 1 | 2 | ||||
| Non‐CCC neoplasm | 2 | ||||||
| Atypical endometriosis | 1 | 1 | 2 | 1 | 1 | ||
| Endometriosis | 2 | 1 | 3 | ||||
| Other benign lesions | 1 | 1 | 2 | ||||
| Normal endometrium | 1 | 1 | 1 | 1 | 1 | 1 |
Most mutations in the primary index carcinomas were conserved across all other cancer/borderline tumour specimens from the same patient, with variable degrees of conservation ranging from 46% (case 1b, contralateral ovary ENOCa) to 88% (case 5c, ipsilateral fallopian tube intraluminal mass; Figure 2; see also supplementary material, Figure S3). Likewise, variability in conservation was also observed between neoplastic sites within the same case: eg 88% of somatic variants were shared between specimens 5c and 5f, while 5b and 5e shared only 73%. In case 1 we observed conservation of ancestral mutations across foci of distinctly different histology from the index CCC specimen (borderline endometrioid tumour and ENOCa), with 46% of variants conserved in the contralateral ovarian ENOCa specimen.
Figure 2.
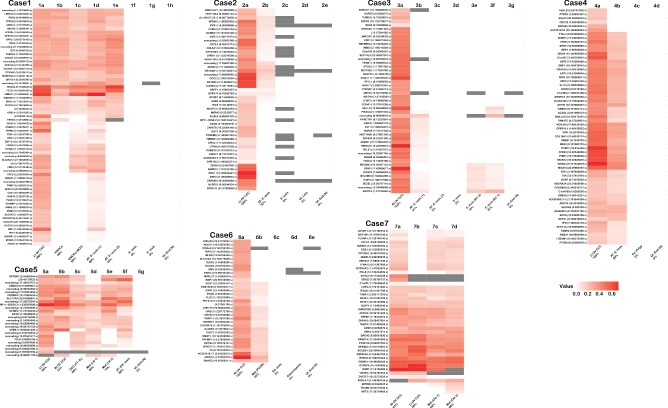
Heat map summary of verified somatic SNVs (s) and INDELs (i) in primary CCC tumours and other related or benign specimens from each case. Whenever possible, multiple samples of tumour, metastatic disease, putative precursor (including endometriosis) and/or benign lesion were examined from each case (see also supplementary material, Table S1, Figure S4). In all cases the left‐most specimen is the primary tumour used in WGSS and samples are ordered using their presumed relationship to the primary tumour, from other ovarian sites, metastatic sites, endometriosis (with and without atypia) to benign lesions and normal uterine endometrium. Patient‐matched germline DNA was used as the reference in both WGSS and deep sequencing confirmation experiments; intensity of red increases with allelic frequency; grey bars denote positions with insufficient coverage. Sub‐specimens are labelled under each panel; the percentage listed in the label refers to the conservation of somatic mutations compared to the index tumour specimen (see also supplementary material, Figure S3). Gene symbol, genomic position and mutation type is noted at the left of each panel; additional detail can be found in Tables S1 and S2 (see supplementary material): Lt OV, left ovary; Rt OV, right ovary; E‐osis, endometriosis; AT‐E‐osis, atypical endometriosis; ES‐osis, endosalpingiosis; Met, metastasis; EN‐Polyp, endometrial polyp; Ut‐End(N), uterine endometrium (normal); FT‐IL, Fallopian tube intraluminal fragment; LN, lymph node; BOT, borderline ovarian tumour; Ut‐Leio, uterine leiomyoma; RC, rectosigmoid colon; Om, omentum; PCDS, posterior cul‐de‐sac
Unlike in the endometriotic lesions, we saw no evidence of somatic mutations in any normal endometrium or other benign lesion. When considering both CCC and endometriosis samples within each case, there appeared to be a cluster of mutations that was present across specimens, including at least one specimen of atypical or non‐atypical endometriosis in all cases (Figure 2). The fraction of detectable somatic mutations that was shared between endometriosis and patient‐matched carcinomas ranged from 15% (case 3, distant endometriosis without atypia) to 98% (case 1, carcinoma‐adjacent atypical endometriosis; see also supplementary material, Figure S3), suggesting that, in some cases, atypical endometriosis shared the near‐complete signature of somatic coding changes detected in the primary index sample. With the exception of PIK3CA and ARID1A, the constituents of the conserved mutations were generally unique to a case. In cases with somatic ARID1A and/or PIK3CA mutations, we consistently found these mutations to be present across all cancer specimens, as well as any ancestrally linked atypical endometriosis or non‐atypical endometriosis specimens from those cases (Figure 2; see also supplementary material, Figure S4).
As noted above, ancestral events, defined as (clonal) somatic mutation of moderate‐high allelic frequency found across multiple specimens from the same patient, were seen in variable proportions, ranging from 15% (case 3) to 98% (case 1) of total genetic abnormalities in different cases. Case 3 provided a crucial example of ancestral mutations (15% of the total number of events), including ARID1A in endometriosis without atypia in two separate specimens from distant, extra‐ovarian foci (Figure 3). It should also be noted that in all three cases with multiple sampled foci of endometriosis (three of five cases with endometriosis), at least one endometriotic lesion did not share any somatic mutation with the index primary carcinoma.
Figure 3.
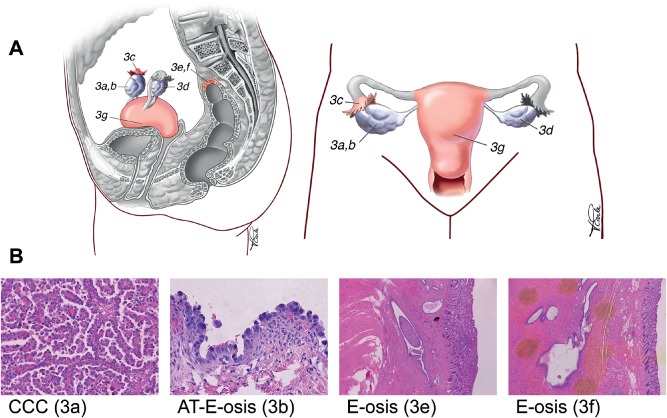
Specimens collected from case 3. (A) Anatomical diagram of the genital tract, indicating the positions of the specimens collected. The main tumour mass was on the right ovary (3a), with adjacent atypical endometriosis present around the ovary (3b) and tube (3c). Additional foci of endometriosis without atypia were also sampled from around the left ovary (3d) and the rectosigmoid colon (3e, 3f). Normal endometrium was also sampled (3 g). (B) H&E‐stained sections corresponding to the index tumour, atypical endometriosis (3b) and distant endometriosis without atypia (3e). The specimens shown appear to be clonally related, based on discovery of identical somatic mutations (see also Figure 2)
Discussion
In patients with clear cell ovarian carcinoma (CCC), we found that some concurrent endometriosis lesions shared a high proportion of CCC‐associated somatic mutations. We also observed several endometriosis lesions that shared the full complement, or near‐full complement, of coding somatic mutations seen in the tumour. However, there were also endometriotic lesions with none of the mutations seen in the CCC.
Some level of intratumoural heterogeneity was also observed in the cancer samples from an individual case; however, the overall architecture of ancestral mutations remained relatively stable from endometriosis to cancer. No one mutation suddenly became dominant as a function of allelic frequency. Further, no single gene (or gene family) was identified that was consistently gained in tumours compared to their matched endometriotic lesions. Mutations in so‐called 'cancer genes' 38 were also uncommon as later genetic events or descendent mutations. Two exceptions were a 'second' ARID1A mutation in case 3, appearing only in the index carcinoma, and CTNNB1 hotspot region mutations. These latter CTNNB1 alterations are more common to the ENOCa type 27, 39; nonetheless, case 1 has a CTNNB1 mutation present in sampled CCC, co‐existing ENOCa and endometrioid borderline tumour, as well as atypical endometriosis. In contrast, case 3 seems to have acquired CTNNB1 mutations after the transition from atypical endometriosis to CCC; no co‐existent endometrioid differentiation was observed in case 3.
The relationship between endometriosis and CCC appears similar to what has been observed in invasive breast cancers compared to ductal carcinoma in situ (DCIS) 40. Specifically, PIK3CA mutations appear early, with no detectable differences in allelic frequency from precursor to frank carcinoma. In the case of CCC, disruption of pathways tied to ARID1A and PIK3CA are likely to be obligatory at early steps in CCC tumour development, possibly even in the formation of endometriosis at high risk for malignant transformation. In fact, in a recently described mouse model, loss of Arid1a and activating mutation of Pik3ca were sufficient to generate ovarian clear cell tumours that phenotypically and molecularly resembled the human disease 41. Findings of somatic mutations in a cancer precursor are not surprising; at least one recent review suggested that a significant number of somatic mutations may in fact 'originate prior to tumour initiation′ 42; however, this is usually confined to so‐called 'passenger events'. Given the high degree of conservation observed in some endometriosis lesions compared to their corresponding carcinomas, and especially the occurrence of presumed driver mutations, our data suggest that the somatic genetic changes in these endometriosis lesions are sufficient to allow tumourigenic transformation, and may support a model wherein the critical final carcinoma‐transforming changes are not mediated by somatic mutation (eg epigenetic modifiers).
In all relevant cases, at least one endometriotic lesion and all carcinoma specimens from a given patient had a clear ancestral relationship to the index tumour sample, demonstrating that these foci originated from a single event in that individual. In cases with multiple foci of endometriosis, we also observed lesions that did not share any somatic mutations with the primary carcinoma. It is reasonable to conclude that these 'unrelated' endometriotic lesions are probably not of the same genetic lineage as their patient‐matched carcinomas. Given the frequency of endometriosis in women of reproductive age, it is likely that at least two genetically defined subgroups of endometriosis exist with respect to subsequent risk of cancer development, high and low risk of progression, and we postulate that endometriosis sharing mutations with a synchronous carcinoma are representative of 'high‐risk', while those lesions not sharing any mutations with the carcinoma may represent 'low‐risk' lesions.
No CCC‐associated somatic mutations were observed in normal (uterine) endometrium specimens from any patients; however, we feel that these data are not sufficient to favour either a uterine or a non‐uterine origin for endometriosis. The limited sampling available from surgical specimens, along with the general biology of the uterus, where epithelial cells are regularly shed (especially prior to menopause when endometriosis is more common), could make the discovery of CCC‐associated mutations in archival samples of endometrium extremely challenging. The implication of the findings in case 3, with clonality demonstrated for bland endometriosis of the rectosigmoid serosa and the ovarian CCC and atypical endometriosis, indicates that these anatomically separate lesions originated from a single site, rather than initiating through metaplasia at multiple different sites.
Previous studies have identified shared patterns of LOH 10, 11 as well as single gene abnormalities 13, 15 clonally linking endometriosis adjacent to CCC and ENOCa. We extend these observations, demonstrating that some endometriosis lesions have an extensive subset of the mutations present in the associated CCC, and have thus progressed further towards cancer than was previously appreciated. This is apparent in case 3; the index CCC had 37 coding somatic mutations, 15 (41%) of which were present in the adjacent atypical endometriotic lesion. Two specimens of bland, distant endometriosis on the rectosigmoid colon, non‐contiguous with the cancer, carried ∼20% (cases 3e and 3f) of these coding mutations, including the ARID1A and PIK3CA mutations. These lesions exhibited no atypia, and the risk of transformation to malignancy at that site, given that endometriosis‐associated cancers are most commonly found on the ovaries, is likely very low. Nonetheless, these are all definitively related to a common initiating lesion. This critical finding provides conclusive evidence that an ancestral lesion must have proliferated, disseminated and recolonized multiple foreign environments and, in general, suggests that endometriosis could, in some circumstances, be considered a neoplasm. We hypothesize that the risk for malignant transformation is most likely determined by a combination of high mutational burden, epigenetic modifications potentially influenced by SWI/SNF dysfunction 43 (eg ARID1A mutations) and, finally, the ovarian micro‐environment. These data contrast sharply with the generally accepted view of endometriosis as a benign, non‐neoplastic condition.
In general, a precursor and risk‐elevating relationship between endometriosis and ovarian cancer is well accepted. As has been noted previously, the growth factor‐rich environment provided in the ovary might be critical in supporting the full malignant transformation of endometriosis into these endometriosis‐associated cancers. As histotypes of ovarian cancer are now clearly acknowledged as distinct diseases 44, endometriosis is well recognized as a risk factor specific to clear cell and endometrioid types 5, 6. Despite this, endometriosis, without concurrent cancer, is still generally regarded as a benign condition. Given the frequency of endometriosis compared to CCC and ENOCa, this view is warranted and remains true for the vast majority of affected women. Herein we demonstrate that some endometriosis, occurring synchronously with clear cell carcinoma, shows both a mutational pattern that is highly conserved between the so‐called precursor and frank carcinoma, as well as neoplastic behaviour, supported by clonally related yet distant endometriotic lesions in a paradigm similar to borderline tumours. Further investigation may allow for early identification of lesions at 'high risk' for malignant transformation and could provide opportunities for screening, monitoring or treatment. Specifically, questions need to be asked around the presence of somatic alterations in non‐cancer‐associated endometriosis. If targetable mutations, such as PIK3CA‐activating changes, are found, should targeted 'cancer' treatments be explored, and is the presence of such mutations sufficient to define endometriosis at risk of malignant transformation?
Author contributions
MSA, AB, YKW, CBG, SPS and DGH conceived, designed and provided oversight of all project components and experiments; MSA, JS, WY, LMP and PJY collected samples and conducted experiments; HLC, ANK and CBG reviewed all specimens for pathology and diagnosis; MAM and MH oversaw whole‐genome sequencing; AB, YKW, GH, MRA, HF and SPS analysed all sequencing data and provided bioinformatics support; MSA, AB and YKW interpreted data from all experiments and produced the final dataset; MSA wrote the first manuscript draft; and all authors contributed to the final version.
Supplementary material on the internet.
The following supplementary material may be found in the online version of this article:
Supplementary materials and methods
Figure S1. Complex deletion/inversion in ARID1A exon 1, detected in case 7 using Sanger sequencing; a NGS‐based strategy showed poor performance of this GC‐rich region, forcing the more traditional Sanger‐sequencing method for exon 1 in all index tumour specimens
Figure S2. Immunohistochemical (IHC) staining of each primary tumour, to support diagnosis of CCC and provide protein expression data on ARID1A
Figure S3. Overview of determining levels of conserved variants within and between specimens
Figure S4. Sequence alignment (BAM) files illustrating the somatic variant in PIK3CA carried in case 3
Figure S5. Boxplots illustrating median coverage/specimen across our Haloplex deep‐sequencing assay
Table S1. Haloplex deep sequencing specimen manifest and coverage detail
Table S2. List of somatic variants detected in Haloplex deep sequencing
Supporting information
AppendixS1. Supplementary materials and methods
FigureS1. Complex deletion/inversion in ARID1A exon 1 was detected in case 7, using Sanger sequencing. Next‐generation sequencing (NGS)‐based strategy showed poor performance of this GC‐rich region, forcing the more traditional Sanger sequencing method for exon 1 in all index tumour specimens. The allelic frequency of this variant was not quantifiable and is therefore omitted from clonal analysis of deep sequencing
FigureS2. Immunohistochemical (IHC) staining of each primary tumour was done in order to support the diagnosis of CCC, as well as to provide protein expression data on ARID1A; all markers were also noted to be either positive (+) or negative (−). Typical CCC tumours show morphological features, including clear cytoplasm, hobnailing and papillary structures. Characteristic CCC IHC profiles are virtually always WT1‐negative and HNF1B‐positive, as were all samples in our cohort. IHC for ARID1A shows distinct nuclear staining and is visible in the nuclei of stromal cells, even when absent in the tumour, as a core‐specific control. All cases were evaluated in TMA format with triplicate cores; heterogeneity was not observed in any of the markers. Somatic alterations of ARID1A, detected through whole‐genome sequencing, targeted deep sequencing and Sanger sequencing, are denoted below the IHC. Only case 3 was determined to have two somatic truncating changes to ARID1A
FigureS3. Overview of determining levels of conserved variants within and between specimens: Blue box, minimum (solid) and maximum (dashed) conservation levels of variants across neoplastic specimens from case 1; yellow box, minimum (solid) and maximum (dashed) conservation levels of variants across endometriosis specimens from case 1
FigureS4. Sequence alignment (BAM) files illustrating the somatic variant in PIK3CA carried in case 3. The variant is clearly visible in the index tumour specimen (3a) and can be predicted as somatic in cases 3b and 3 f. In case 3e, one of two distant endometriosis specimens, low read count due to inherent low cellularity of small endometriotic lesions and moderate sample quality from FFPE source material hindered analysis. Our standard protocol, applying the binomial exact test to infer the somatic variants (see Supplementary materials and methods), was not sufficient to support a conclusive somatic variant call (adjusted p value = 0.005944391 > p value threshold = 0.001); nonetheless, manual examination of the data suggested that the PIK3CA variant was present
FigureS5. Box plots illustrating median coverage per specimen across our Haloplex deep‐sequencing assay
TableS1. Haloplex deep sequencing specimen manifest and coverage detail
TableS2. List of somatic variants detected in Haloplex deep sequencing
Acknowledgements
We thank all the women who donated samples used in this study. In addition, we would like to thank the Genetic Pathology Evaluation Centre (GPEC), the Centre for Translational and Applied Genomics (CTAG) and the Department of Anatomical Pathology at Vancouver General Hospital for technical assistance. We further thank the Cheryl Brown Ovarian Cancer Outcomes unit for providing clinical data, and the laboratories of Dr Aikou Okamoto and Dr Anne‐Marie Mes‐Mason for their collaborative assistance to the OVCARE team. The authors graciously thank the Gray Family Ovarian Clear Cell Carcinoma Research Resource, which provided start‐up and continued funding critical to this project. In addition, this work was funded by research grants from the Canadian Cancer Society (Grant No. 701603), the BC Cancer Foundation and the Vancouver General Hospital and University of British Columbia Hospital Foundation (to OVCARE; http://www.ovcare.ca). The Dr Chew Wei Memorial Professorship in Gynecologic Oncology provided support to DGH. The Michael Smith Foundation for Health Research Scholars and the Canada Research Chair in Computational Cancer Genomics provided support to SPS. Computational infrastructure support was provided by Genome British Columbia and Genome Canada. The Nelly Auersperg Grant from the Women's Health Research Institute and the Canadian Foundation for Women's Health General Research Grant provided support to PJY.
No conflicts of interest were declared.
References
- 1. Rogers PA, D'Hooghe TM, Fazleabas A, et al. Defining future directions for endometriosis research: workshop report from the 2011 World Congress of Endometriosis in Montpellier, France. Reprod Sci 2013; 20: 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Eskenazi B, Warner ML. Epidemiology of endometriosis. Obstet Gynecol Clin N Am 1997; 24: 235–258. [DOI] [PubMed] [Google Scholar]
- 3. Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril 2012; 98: 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anglesio MS, Carey MS, Kobel M, et al. Clear cell carcinoma of the ovary: a report from the first Ovarian Clear Cell Symposium, June 24th, 2010. Gynecol Oncol 2011; 121: 407–415. [DOI] [PubMed] [Google Scholar]
- 5. Merritt MA, Green AC, Nagle CM, et al. Talcum powder, chronic pelvic inflammation and NSAIDs in relation to risk of epithelial ovarian cancer. Int J Cancer 2008; 122: 170–176. [DOI] [PubMed] [Google Scholar]
- 6. Pearce CL, Templeman C, Rossing MA, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case‐control studies. Lancet Oncol 2012; 13: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heidemann LN, Hartwell D, Heidemann CH, et al. The relation between endometriosis and ovarian cancer – a review. Acta Obstet Gynecol Scand 2014; 93: 20–31. [DOI] [PubMed] [Google Scholar]
- 8. LaGrenade A, Silverberg SG. Ovarian tumors associated with atypical endometriosis. Hum Pathol 1988; 19: 1080–1084. [DOI] [PubMed] [Google Scholar]
- 9. Vercellini P, Parazzini F, Bolis G, et al. Endometriosis and ovarian cancer. Am J Obstet Gynecol 1993; 169: 181–182. [DOI] [PubMed] [Google Scholar]
- 10. Jiang X, Morland SJ, Hitchcock A, et al. Allelotyping of endometriosis with adjacent ovarian carcinoma reveals evidence of a common lineage. Cancer Res 1998; 58: 1707–1712. [PubMed] [Google Scholar]
- 11. Prowse AH, Manek S, Varma R, et al. Molecular genetic evidence that endometriosis is a precursor of ovarian cancer. Int J Cancer 2006; 119: 556–562. [DOI] [PubMed] [Google Scholar]
- 12. Campbell IG, Thomas EJ. Endometriosis: candidate genes. Hum Reprod Update 2001; 7: 15–20. [DOI] [PubMed] [Google Scholar]
- 13. Wiegand KC, Shah SP, Al‐Agha OM, et al. ARID1A mutations in endometriosis‐associated ovarian carcinomas. N Engl J Med 2010; 363: 1532–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science (New York, NY) 2010; 330: 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamamoto S, Tsuda H, Miyai K, et al. Accumulative copy number increase of MET drives tumor development and histological progression in a subset of ovarian clear‐cell adenocarcinomas. Mod Pathol 2012; 25: 122–130. [DOI] [PubMed] [Google Scholar]
- 16. Yamamoto S, Tsuda H, Miyai K, et al. Gene amplification and protein overexpression of MET are common events in ovarian clear‐cell adenocarcinoma: their roles in tumor progression and prognostication of the patient. Mod Pathol 2011; 24: 1146–1155. [DOI] [PubMed] [Google Scholar]
- 17. McKernan KJ, Peckham HE, Costa GL, et al. Sequence and structural variation in a human genome uncovered by short‐read, massively parallel ligation sequencing using two‐base encoding. Genome Res 2009; 19: 1527–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eirew P, Steif A, Khattra J, et al. Dynamics of genomic clones in breast cancer patient xenografts at single cell resolution. Nature 2015; 518: 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sung CO, Choi CH, Ko YH, et al. Integrative analysis of copy number alteration and gene expression profiling in ovarian clear cell adenocarcinoma. Cancer Genet 2013; 206: 145–153. [DOI] [PubMed] [Google Scholar]
- 20. Kuo KT, Mao TL, Chen X, et al. DNA copy numbers profiles in affinity‐purified ovarian clear cell carcinoma. Clin Cancer Res 2010; 16: 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan DS, Lambros MB, Rayter S, et al. PPM1D is a potential therapeutic target in ovarian clear cell carcinomas. Clin Cancer Res 2009; 15: 2269–2280. [DOI] [PubMed] [Google Scholar]
- 22. Tan DS, Iravani M, McCluggage WG, et al. Genomic analysis reveals the molecular heterogeneity of ovarian clear cell carcinomas. Clin Cancer Res 2011; 17: 1521–1534. [DOI] [PubMed] [Google Scholar]
- 23. Anglesio MS, George J, Kulbe H, et al. IL6–STAT3–HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin Cancer Res 2011; 17: 2538–2548. [DOI] [PubMed] [Google Scholar]
- 24. Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER‐2/neu oncogene. Science (New York, NY) 1987; 235: 177–182. [DOI] [PubMed] [Google Scholar]
- 25. Anglesio MS, Kommoss S, Tolcher MC, et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J Pathol 2013; 229: 111–120. [DOI] [PubMed] [Google Scholar]
- 26. McAlpine JN, Wiegand KC, Vang R, et al. HER2 overexpression and amplification is present in a subset of ovarian mucinous carcinomas and can be targeted with trastuzumab therapy. BMC Cancer 2009; 9: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McConechy MK, Ding J, Senz J, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol 2014; 27: 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kuo KT, Mao TL, Jones S, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 2009; 174: 1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res 2004; 64: 7678–7681. [DOI] [PubMed] [Google Scholar]
- 30. Belandia B, Orford RL, Hurst HC, et al. Targeting of SWI/SNF chromatin remodelling complexes to estrogen‐responsive genes. EMBO J 2002; 21: 4094–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou G, Hashimoto Y, Kwak I, et al. Role of the steroid receptor coactivator SRC‐3 in cell growth. Mol Cell Biol 2003; 23: 7742–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griesmann H, Ripka S, Pralle M, et al. WNT5A–NFAT signaling mediates resistance to apoptosis in pancreatic cancer. Neoplasia 2013; 15: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosenbluh J, Wang X, Hahn WC. Genomic insights into WNT/β‐catenin signaling. Trends Pharmacol Sci 2014; 35: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear‐cell renal cell carcinoma. Nat Genet 2013; 45: 860–867. [DOI] [PubMed] [Google Scholar]
- 35. Li XS, Trojer P, Matsumura T, et al. Mammalian SWI/SNF – a subunit BAF250/ARID1 is an E3 ubiquitin ligase that targets histone H2B. Mol Cell Biol 2010; 30: 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wright DE, Wang CY, Kao CF. Histone ubiquitylation and chromatin dynamics. Front Biosci 2012; 17: 1051–1078. [DOI] [PubMed] [Google Scholar]
- 37. Wiegand KC, Lee AF, Al‐Agha OM, et al. Loss of BAF250a (ARID1A) is frequent in high‐grade endometrial carcinomas. J Pathol 2011; 224: 328–333. [DOI] [PubMed] [Google Scholar]
- 38. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med 2004; 10: 789–799. [DOI] [PubMed] [Google Scholar]
- 39. Palacios J, Gamallo C. Mutations in the β‐catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res 1998; 58: 1344–1347. [PubMed] [Google Scholar]
- 40. Andrade VP, Ostrovnaya I, Seshan VE, et al. Clonal relatedness between lobular carcinoma in situ and synchronous malignant lesions. Breast Cancer Res 2012; 14: R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chandler R, Damrauer J, Raab J, et al. ARID1A–PIK3CA mutations promote ovarian clear‐cell tumorigenesis through pro‐tumorigenic inflammatory cytokine signaling. Nat Commun 2015; 6: 6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self‐renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA 2013; 110: 1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Skulte KA, Phan L, Clark SJ, et al. Chromatin remodeler mutations in human cancers: epigenetic implications. Epigenomics 2014; 6: 397–414. [DOI] [PubMed] [Google Scholar]
- 44. Kobel M, Kalloger SE, Boyd N, et al. Ovarian carcinoma subtypes are different diseases: implications for biomarker studies. PLoS Med 2008; 5: e232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
AppendixS1. Supplementary materials and methods
FigureS1. Complex deletion/inversion in ARID1A exon 1 was detected in case 7, using Sanger sequencing. Next‐generation sequencing (NGS)‐based strategy showed poor performance of this GC‐rich region, forcing the more traditional Sanger sequencing method for exon 1 in all index tumour specimens. The allelic frequency of this variant was not quantifiable and is therefore omitted from clonal analysis of deep sequencing
FigureS2. Immunohistochemical (IHC) staining of each primary tumour was done in order to support the diagnosis of CCC, as well as to provide protein expression data on ARID1A; all markers were also noted to be either positive (+) or negative (−). Typical CCC tumours show morphological features, including clear cytoplasm, hobnailing and papillary structures. Characteristic CCC IHC profiles are virtually always WT1‐negative and HNF1B‐positive, as were all samples in our cohort. IHC for ARID1A shows distinct nuclear staining and is visible in the nuclei of stromal cells, even when absent in the tumour, as a core‐specific control. All cases were evaluated in TMA format with triplicate cores; heterogeneity was not observed in any of the markers. Somatic alterations of ARID1A, detected through whole‐genome sequencing, targeted deep sequencing and Sanger sequencing, are denoted below the IHC. Only case 3 was determined to have two somatic truncating changes to ARID1A
FigureS3. Overview of determining levels of conserved variants within and between specimens: Blue box, minimum (solid) and maximum (dashed) conservation levels of variants across neoplastic specimens from case 1; yellow box, minimum (solid) and maximum (dashed) conservation levels of variants across endometriosis specimens from case 1
FigureS4. Sequence alignment (BAM) files illustrating the somatic variant in PIK3CA carried in case 3. The variant is clearly visible in the index tumour specimen (3a) and can be predicted as somatic in cases 3b and 3 f. In case 3e, one of two distant endometriosis specimens, low read count due to inherent low cellularity of small endometriotic lesions and moderate sample quality from FFPE source material hindered analysis. Our standard protocol, applying the binomial exact test to infer the somatic variants (see Supplementary materials and methods), was not sufficient to support a conclusive somatic variant call (adjusted p value = 0.005944391 > p value threshold = 0.001); nonetheless, manual examination of the data suggested that the PIK3CA variant was present
FigureS5. Box plots illustrating median coverage per specimen across our Haloplex deep‐sequencing assay
TableS1. Haloplex deep sequencing specimen manifest and coverage detail
TableS2. List of somatic variants detected in Haloplex deep sequencing