

Abstract
Lignin is an abundant biopolymer with a high carbon content and high aromaticity. Despite its potential as a raw material for the fuel and chemical industries, lignin remains the most poorly utilised of the lignocellulosic biopolymers. Effective valorisation of lignin requires careful fine‐tuning of multiple “upstream” (i.e., lignin bioengineering, lignin isolation and “early‐stage catalytic conversion of lignin”) and “downstream” (i.e., lignin depolymerisation and upgrading) process stages, demanding input and understanding from a broad array of scientific disciplines. This review provides a “beginning‐to‐end” analysis of the recent advances reported in lignin valorisation. Particular emphasis is placed on the improved understanding of lignin's biosynthesis and structure, differences in structure and chemical bonding between native and technical lignins, emerging catalytic valorisation strategies, and the relationships between lignin structure and catalyst performance.
Keywords: bioengineering, biorefining, catalysis, lignin, lignocellulose
1. Introduction
Lignocellulosic biomass, an intricate and complex architecture of three classes of biopolymer—cellulose, hemicelluloses and lignin—is an abundant and renewable resource. The separation, isolation, and subsequent chemical transformation of the three constituent polymer groups can afford a broad and multifunctional array of bio‐derived value‐added fuels, chemicals and materials. If these products are obtained by an integrated system of (catalytic) reaction pathways, i.e., in a so‐called biorefinery operation, the optimal potential of each component and thus the maximum value of the biomass feed as a whole can be achieved.
Lignin, a complex and water‐insoluble aromatic polymer, is derived primarily from methoxylated hydroxycinnamyl alcohol building blocks, the prototypical monolignols. Unlike cellulose, with a well‐defined sequence of monomeric units that are linked by regular β‐1,4‐glycosidic bonds, lignin is characterised by a variety of distinct and chemically different bonding motifs, each demanding different conditions for cleavage when selective depolymerisation is targeted. Although structurally more complex, the higher carbon‐content and lower oxygen content of lignin, relative to the polysaccharide or holocellulose fraction, render it an attractive feedstock for the production of biofuels and chemicals. Notably, the highly‐functionalised and aromatic nature of lignin presents the potential for the direct preparation of aromatic specialty and fine chemicals, circumventing the requirement for full defunctionalisation to “BTX” (benzene, toluene and xylenes) and subsequent refunctionalisation to desired platform chemicals. Nonetheless, owing to challenges associated with effective separation of oxygenated aromatics via distillation or other means, full defunctionalisation to arenes and alkanes will also be of importance for the production of chemicals and fuel components from lignin and its products. Such opportunities for valorisation together with its abundance have, amongst other reasons, motivated significant research activity into the catalytic valorisation of lignin.
An understanding of (and control over) the coupled and interdependent processing steps, for conversion of the initial biomass feedstock to the intended lignin‐derived product, requires a collaborative approach encompassing a variety of scientific disciplines (e.g., genetic engineering to increase the homogeneity of the polymer or the proportion of easily‐cleavable linkages, the development of effective analytical techniques for lignin structural determination, reactor engineering and improved catalytic systems for the depolymerisation of lignin and downstream processing, and related processes for product separation), as outlined in several review articles (Table 1).
Table 1.
Selected lignin review articles published since 2010, highlighting different focal topics, ordered from primarily concerned with upstream (e.g., lignin biosynthesis, structure) to downstream (e.g., catalytic transformations) process steps.
| Focal topic(s) | Ref. |
|---|---|
| Lignin biosynthesis & structure | 1, 2, 3, 4, 5, 6, 7, 8, 9 |
| Bioengineering of lignins | 8, 9, 10, 11, 12, 13 |
| Biotic/abiotic stress and effects on lignification | 6, 7, 8, 9, 10, 11 |
| Lignin depolymerisation | 14 |
| Catalytic (deoxygenative) valorisation to fuel and chemicals | 15, 16, 17, 18 |
| Pyrolysis, hydrodeoxygenation, catalytic upgrading | 19, 20, 21 |
| Oxidative valorisation of lignin | 22, 23, 24 |
| Lignin for polymers and composites | 25, 26, 27, 28, 29, 30 |
| Lignin analytics | 31, 32, 33, 34, 35, 36, 37 |
| Biodegradation of lignin | 38, 39, 40 |
The available review papers of lignin typically cover limited or specific aspects, or focus only on one or few stages of the lignin valorisation pathway. Here we aim to offer a “start‐to‐finish” analysis of the progress achieved in lignin valorisation, with a particular focus on the past five years, considering all of the interconnected stages of catalytic lignin biorefinering. On this basis, biosynthesis/genetic engineering of the lignin phenylpropanoid pathway, early‐stage catalytic conversion of lignin (ECCL) beginning with lignocellulosic biomass, and the catalytic valorisation of isolated technical lignins to products are each discussed in sequence, where possible drawing connections between separate stages of processing. Particular emphasis is placed on the characteristics of the interunit bonding in the lignins, and the profound impact of various “upstream” biorefining processes on the abundance of different labile (e.g., C−O) and recalcitrant (e.g., C−C) linkages (and thus also on the downstream processing of technical lignins). This review also aims to demystify or update several of the general statements and some of the misapprehensions often encountered in the current literature, thereby fostering a better understanding of the relationships between lignin molecular properties and the performance of catalytic systems.
This review is broadly subdivided into “upstream” and “downstream” sections—defined as the processes leading up to the separation and isolation of lignin (from the polysaccharide fraction), and subsequent depolymerisation and chemical modification of the isolated lignins to yield valorised products, respectively. Whilst this arbitrary division is imperfect (e.g., ECCL can be regarded as a combination of upstream and downstream elements), it is useful from a conceptual point of view as it mirrors the activities of the petrochemical industry.
The upstream section introduces the biosynthetic “phenylpropanoid pathway” with the genes and enzymes involved in the biosynthesis of lignins’ monomeric units. Phenylpropanoid genetic engineering strategies to give altered lignins are compared and contrasted. Subsequently, the chemical structure and bonding properties of native or protolignin (i.e., the lignins occurring in the plant cell wall) in addition to those of some technical isolated lignins (e.g., Kraft and Organosolv lignins) are reviewed, showing the relationship between the severity of a depolymerisation method and the prevalence of specific bonding motifs in the isolated technical lignins. Lastly, another emerging frontier of research, the catalytic upstream refining methods based on early‐stage conversion of lignin, is presented and discussed.
The downstream processing section covers the catalytic valorisation of isolated lignin streams into commodity chemicals, fuels and, discussed to a far lesser extent in this review, materials. Firstly, some relevant market and economic considerations that underpin lignin valorisation are detailed. Subsequently, an array of “mild” (oxidative, reductive, redox‐neutral) and “harsh” lignin depolymerisation strategies are described and compared.
The final section summarises the progress made in this field of research and proposes future directions for catalytic lignin valorisation research. An exhaustive coverage of all topics or contributions relevant to catalytic lignin valorisation is, indeed, impossible owing to the breadth of lignin research. Therefore, analytical methods for the characterisation of lignins and lignin products are discussed only succinctly. Furthermore, a detailed discussion of enzymatic and biological downstream processes is largely omitted.
2. Upstream Processing
2.1. Bioengineering of Lignins
To improve the economic feasibility of a biorefinery, biomass must be comprehensively converted into value‐added products; this includes the lignin stream. The intricate connectivity (not only by physical arrangement but also via actual covalent bonding) between cellulose, hemicelluloses, and lignin poses a challenge for the direct enzymatic saccharification of cellulose into glucose or, for example, to improve the digestibility of forage crops for animal feed.41 This difficulty has motivated plant biologists to modify the phenylpropanoid biosynthesis—a multi‐step, multi‐enzyme biosynthetic pathway for the preparation of nine‐carbon propenylated p‐hydroxyphenyl derivatives originating from the amino acid phenylalanine (or tyrosine)—in order to modify the molecular structure of lignin and/or the proportion incorporated into the plant cell wall.11 Modifications to the phenylpropanoid pathway may exert changes to plant fitness because the branch‐points of the pathway feed into a variety of other metabolic systems essential to plant growth and development. For a comprehensive discussion, the reader is referred to a recent review article.11
The majority of phenylpropanoid genetic strategies have been directed towards a decrease in lignin content across plant species, with research relating to biomass conversion being targeted at hardwoods,42, 43, 44 softwoods,45 monocots (grasses),46, 47, 48, 49, 50, 51, 52, 53 and dicots (including Arabidopsis and alfalfa/truncatula).43, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74 However, an increase in saccharification yield will not boost the economics of a biorefinery operation if the lignin fraction, at 15–30 wt % of dry biomass, becomes a more recalcitrant material.
In this section, recent and relevant genetic modifications of the lignin biosynthetic pathway are discussed, providing an overview of the impact of up‐ and down‐regulation of an array of genes for enzymes involved in the biosynthesis of lignin building blocks on the lignin structure. A survey of current literature related to structural modifications of lignin demonstrates an almost limitless potential for improved utilisation of both the carbohydrate and lignin fractions of lignocellulosic biomass, via catalytic processing.2 An ideal combination of genetic modifications would yield a plant with identical or improved growth compared to the wild‐type. The lignin fraction does not necessarily have to be increased or reduced in quantity, but should be constructed in such a way as to facilitate chemical (or enzymatic) deconstruction under milder conditions than currently required, e.g., by incorporating a restricted subset of linkages or precursor units.1 Bioengineering of lignin, therefore, may aim at enhancing the saccharification yield from biomass whilst simultaneously allowing for the improved valorisation of lignin via subsequent catalytic treatment.1 Optimal conditions for any bioengineering strategy will be determined, at least in part, by the targeted products, and whether these target species evolve from the carbohydrate stream or the lignin stream.
2.2. The Phenylpropanoid Pathway
Scheme 1 summarises the complete phenylpropanoid pathway, outlining enzymes directly involved in the biosynthesis of the prototypical lignin monomers. These monolignols, p‐coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol, are involved in lignification, the polymerisation process that creates the lignin polymer, affording the so‐called “H”, “G” and “S” units, respectively. The phenylpropanoid pathway starts with phenylalanine (Phe), but tyrosine (Tyr) may also be consumed in monocots.75 Phenylalanine is first deaminated to cinnamate (by the enzyme phenylalanine ammonia‐lyase, PAL), and cinnamate is then hydroxylated to p‐coumarate (by cinnamate 4‐hydroxylase, C4H). If tyrosine is used instead as the starting point, this two‐stage enzymatic process is circumvented and Tyr is directly converted into p‐coumarate via deamination (by tyrosine ammonia‐lyase, TAL, but also by PAL which is not absolutely specific for Phe).76
Scheme 1.

Summary of the phenylpropanoid pathway focusing on the formation of native lignin monomer precursors.11, 66 Asterisks (*) represent points at which the intermediate itself may be introduced as a monomer or used to create a new monomer that is used by the plant in its lignification following genetic modification (see Scheme 2). Enzymes are abbreviated as follows: phenylalanine ammonia‐lyase (PAL); cinnamate 4‐hydroxylase (C4H); tyrosine ammonia‐lyase (TAL); 4‐coumarate: CoA ligase (4CL); p‐coumarate 3‐hydroxylase (C3H); shikimate/quinate hydroxycinnamoyltransferase (HCT); cinnamoyl‐CoA reductase (CCR); cinnamyl alcohol dehydrogenase (CAD); caffeoyl shikimate esterase (CSE); caffeic acid O‐methyltransferase (COMT); caffeoyl‐CoA O‐methyltransferase (CCoAOMT); ferulate 5‐hydroxylase (F5H); hydroxycinnamaldehyde dehydrogenase (HCALDH). These enzymes were often named according to the assumed substrates at the time of discovery. They therefore do not necessarily reflect the true preferred substrate. For example, the preferred substrate for F5H is actually coniferaldehyde;95, 96 some have suggested renaming it CAld5H, but the name has not been well adopted. Similarly, the preferred substrates for COMT are 5‐hydroxyconiferaldehyde or 5‐hydroxyconiferyl alcohol;95, 97 AldOMT has been suggested as an alternative name,98 although COMT persists as the preferred name among lignin researchers.
At p‐coumarate, the sequence of enzymatic reactions may diverge to afford either p‐coumaroyl‐CoA (by 4‐coumarate: CoA ligase, 4CL) or caffeate via a second hydroxylation of the aromatic ring (C3H or C4H). In the normal monolignol biosynthetic pathway, the p‐coumaroyl‐CoA is then converted into p‐coumaroyl shikimic/quinic acid (by hydroxycinnamoyltransferase, HCT), or via reduction (cinnamoyl‐CoA reductase, CCR) to p‐coumaraldehyde, which may subsequently be reduced to p‐coumaryl alcohol (cinnamyl alcohol dehydrogenase, CAD); p‐coumaryl alcohol is incorporated into lignin to produce H units, which are usually found in low abundance.
Caffeate can also be converted into ferulate by methylation of the 3‐hydroxy group of the ring (caffeic acid O‐methyltransferase, COMT) to produce ferulic acid and then feruloyl‐CoA (by 4CL), which is more normally considered to arise directly from methylation of caffeoyl‐CoA by CCoAOMT (Scheme 1). Feruloyl‐CoA is then subsequently reduced to coniferaldehyde (by CCR). Coniferaldehyde represents the branching point between the formation of the predominant G (coniferyl‐alcohol‐derived) and S (sinapyl‐alcohol‐derived) units. The hydroxylation of coniferaldehyde (by F5H) and subsequent methylation of the product (by COMT) constitutes the major pathway toward sinapaldehyde. The final stage for the formation of both G and S units is the CAD‐catalysed reduction of the aldehyde moiety to yield the corresponding primary alcohol. Coniferaldehyde may be recycled back into the phenylpropanoid pathway via oxidation of the aldehyde to yield ferulic acid (by hydroxycinnamaldehyde dehydrogenase, HCALDH).
Although, in non‐specialised literature, it is generalised that just the three monolignols are the building blocks of lignins, a number of alternative monomers may be naturally introduced into the lignin structure in normal wild‐type plants or through their genetic modification—the latter, in which products of incomplete monolignol biosynthesis are incorporated into the lignin (and can often also be found at low levels in wild‐type plants), is discussed in Section 2.3.
Natural alternative monomers can also include a number of structures that remain underappreciated for their contributions to lignins in some plants. Of particular note are the acylated monolignols, monolignol acetates, p‐hydroxybenzoates, and p‐coumarates.1 Confusion has arisen with these last two products that are often incorrectly quantified as H‐lignin components. H‐lignin units, by definition, result from the monolignol p‐coumaryl alcohol. The monolignol conjugates, in contrast, are monolignol (usually coniferyl and sinapyl alcohol) ester conjugates of p‐hydroxybenzoic and p‐coumaric acids. Although p‐coumarate esters, in particular, derive from p‐coumaroyl‐CoA on the pathway, p‐coumarate cannot be considered to be a monolignol, nor is it a lignin monomer. In all cases, the monolignol moiety of the conjugate has been found to couple into lignin in the usual manner, whereas the p‐hydroxybenzoate and p‐coumarate moieties do not, as they remain as free‐phenolic pendant “decorations” on the γ‐OH groups of the C3‐sidechain of lignin's various units. The reason has been deduced to be the more facile radical transfer (to more stable G and S radicals) than radical coupling.1, 77, 78 Notably, as pendant esters can occur at significant levels, and indeed may be some of the easiest and more valuable products to “clip off” from various lignin streams, they must be seen as, and are, part of the lignin. In spite of this, they must not be confused with the monomers that enter the radical coupling reactions that typify lignification and create the polymer backbone itself.
The radical coupling products from monolignol conjugates are structurally analogous to those from the monolignols themselves except in the case of their β‐β coupling or their cross‐coupling with a monolignol. In each case, novel tetrahydrofuran (THF) structures result in the lignin instead of the normal resinols; it is these structures that provide the evidence that acylated lignins derive from pre‐acylated monolignols.79, 80, 81 THF structures are present to the almost complete exclusion of the resinol moieties in maize, where essentially all of the β‐β coupling appears to be from sinapyl p‐coumarate dimerisation. Similarly, THF structures are dominant in particularly highly naturally acetylated lignins (such as in curauá) where the monomers are almost exclusively monolignol acetates.80, 82 A gene required for monolignol p‐coumaroylation has been characterised,83, 84, 85, 86 but the genes for analogous p‐hydroxybenzoylation and acetylation remain unknown.
Among other important natural monomers, as reviewed,11, 87, 88, 89 are: dihydroconiferyl alcohol in softwoods that, under the oxidative conditions of lignification, can also produce guaiacylpropan‐1,3‐diols;90, 91, 92 tricin, a flavone from another pathway entirely, only recently identified in grasses.80, 93 In addition, as reviewed,1, 94 ferulates on arabinoxylans, and the dehydrodimers (and higher oligomers) that result from them, must also be considered lignin monomers in the broadest sense, as should the tyramine ferulates that are found incorporated into various of the Solanaceae species (e.g., tobacco, tomato).
Although a wealth of research has contributed to the collective knowledge of biochemical pathways of lignin and phenol derivatives in plants, the interplay between multiple genes (general and plant‐specific) as well as the effects upon lignin biogenesis have only been partially elucidated.8, 11 In this regard, it is perhaps surprising for a pathway that was long ago considered to be fully described, that new enzymes and new pathway steps continue to emerge, for example, the CSE‐catalysed conversion of caffeoyl shikimic or quinic acid to caffeic acid.66
2.3. Bioengineered Lignins
Following biosynthesis of the lignin building blocks, the lignin monomers are transported to the secondary plant cell walls, whereupon they are incorporated into the lignin macromolecular structure via laccase‐/peroxidase‐induced radical polymerisation reactions, affording several structural motifs (Scheme 1).41 The precise roles of laccases/peroxidases in the final polymerisation of lignin monomers are at present only partially understood.41, 61, 64, 70, 124, 125, 126, 127, 128 Laccases, though capable of inducing the radical reactions for the polymerisation of lignin subunits, are many and varied. They perform several functions in plant development, rendering the identification of lignin‐specific laccases difficult.125 Nevertheless, downregulation of various specific laccases has resulted in significantly altered lignification, implying that they certainly play a role in lignification.61, 129, 130 Further investigation is also required to identify individual (or groups of) peroxidases involved in lignin biosynthesis.8 For example, Peroxidase 4 was recently discovered to be involved in syringyl lignin formation in Arabidopsis thaliana,70 but in a mutant, a decrease in the proportion of syringyl units was seen only under optimum lighting conditions, and was also dependent on the age of the plant. This finding highlights the complexity of the interplay of biotic or abiotic stress, and how genetically engineered plants sometimes do not develop the change expected because of factors not related to genetic improvement.131 In this context, some attempts at determining holistic effects (i.e., encompassing bioengineering and an examination of the effects on other pathways, if not on external, “ambient” factors) have been reported.54, 107, 132
Table 2 summarises contemporary research relating to genetic modifications of the phenylpropanoid pathway and effects on plant structure and saccharification yield.
Table 2.
Summary of reported genetic modifications within the phenylpropanoid pathway and their effects on saccharification yield, total lignin content, lignin composition/structure (and/or the effect on metabolites) and plant phenotype; TG (transgenic), M (mutation), ND (not determined).
| Entry | Species | Gene alteration | Type | Compositional/structural change in lignin or metabolites | Effect on lignin content | Saccharif‐ ication yield[a] | Phenotypic effect | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | Arabidopsis thaliana | PAL deficient | M | Phe overaccumulation; S/G↑ flavonol glycosides↓ | ↓ | ND | No change | 54 |
| 2 | Brachypodium distachyon | PAL downregulation | TG | S/G↑; ferulate↓ | ↓ | 2×↑ | Delayed development; root growth↓ | 99 |
| 3 | Capsicum annum, C. chinense and Solanum tuberosum | C4H upregulation | TG | S/G↑ | ↓ | ND | Curled leaves, dwarfism, or no change | 55 |
| 4 | Arabidopsis ref3 | C4H deficient | M | S/G↑; New cinnamoylmalate | ↓ | ND | Dwarfism, male sterility, collapsed vasculature | 56 |
| 5 | Populus alba x grandidentata | C3H downregulation | TG | H ≈100×↑; S/G↓ | 50 %↓ | ND | No change | 100 |
| 6 | Lolium perenne | CCR downregulation | TG | No change | ↓ | ↑ | No change | 101 |
| 7 | Arabidopsis thaliana | CCR deficient | M | Ferulic acid‐coniferyl alcohol ether dimers in metabolites; sinapoyl malate ≈4×↓ | 25‐35 %↓ | ND | Dwarfism, delayed senescence | 102 |
| 8 | Pinus radiata | CCR downregulation | TG | Increase of p‐coumaroyl hexose, caffeic acid hexoside and ferulic acid hexoside; H↓ and G↓ | ca. 50 %↓ | ↑ | ND | 67 |
| 9 | Maize | CCR deficient | M | H‐units strongly decreased; S/G slightly↑ | Slight↓ | ↑ | No change | 103 |
| 10 | Nicotiana tabacum | CCR downregulation | TG | S/G↑; β‐O‐4 units↓; introduction of ferulic acid and sinapic acid | ↓ | ↑ | Orange xylem; Less severe: no change; more severe: dwarfism, collapsed vessels | 104 |
| 11 | Populus tremula x Populus alba | CCR downregulation | TG | Ferulic acid incorporated into lignin | 5‐24 %↓ | 15 %↑ | Orange xylem; Less severe: no change; More severe: dwarfism | 105 |
| 12 | Populus tremula x Populus alba | CCR downregulation | TG | Oligolignols↓ | ↓ | ≈20 %↑ | Orange‐coloured wood | 105, 106, 107 |
| 13 | Medicago truncatula | CAD deficient | M | ≈95 % Sinapaldehyde‐ and conifer‐ aldehyde‐derived | Increase in wall‐bound lignin moieties | ND | None at 22 °C; Dwarfed at 30 °C | 43 |
| 14 | Maize | CAD downregulation | TG | S/G↓ | No change | ↑ | No change | 108 |
| 15 | Triticum sinskajae | CAD downregulation | TG | H↓; S/G↑ | No change | ND | Slight dwarfism | 109 |
| 16 | Brachypodium distachyon | CAD deficient | M | Increase in β‐O‐4‐ and 4‐O‐5‐coupled sinapaldehyde units; increase in free phenolic groups | ↓ | ↑ | No change | 110 |
| 17 | Pinus taeda | CAD deficient | M | Incorporation of coniferaldehyde and dihydroconiferyl alcohol; increase in vanillin, ferulic acid, p‐coumaric acid, coniferaldehyde and p‐hydroxybenz‐ aldehyde | ↑ | ↑ | Dark‐brown wood | 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111 |
| 18 | Panicum virgatum | CAD downregulation | TG | Hydroxycinnamaldehydes↑ | Lignin↓ and cutin↓ | ↑ | No change | 112 |
| 19 | Koshihikari x Chugoku 117 rice | CAD deficient | TG, M | ND | ↓ | ↑ | No change | 113 |
| 20 | Arabidopsis thaliana | HCT deficient | M | H↑; S≈0; G≈0 | ↓ | ND | Severe dwarfism | 57 |
| 21 | Populus nigra | HCT downregulation | TG | H 17×↑ | No change | ND | Dwarfism | 114 |
| 22 | Medicago sativa | CCoAOMT deficient | TG | Incorporation of 5OH‐CA producing novel 5OH‐G units as benzodioxanes | ≈20 % decrease | ≈10 % increase in cellulose | No change | 42 |
| 23 | Brown midrib‐3 Maize | COMT deficient | M | S/G↓; incorporation of 5OH‐CA producing benzodioxanes | ↓ | Improved | No change | 115 |
| 24 | Arabidopsis thaliana | COMT deficient | M | S/G↓; trimeric moiety 5OHG‐5OHG‐G; benzodioxane linkages | No change | ND | No change | 58 |
| 25 | Saccharum officinarum cv. CP88‐1762 | COMT deficient | M | S↓; p‐coumarate (on lignin)↓ | 6‐12 %↓ | 28‐32 %↑ | No change | 116 |
| 26 | Arabidopsis thaliana | F5H deficient | M | S 70‐75 %↓; G↑ | No change | ND | Lack of 2° wall structure in inter‐fascicular and xylem fibres | 65 |
| 27 | Arabidopsis thaliana | F5H overexpression | TG | ≈100 % S (i.e., G↓, S↑) | ↓ | No change | Decreased plant stiffness | 117, 118 |
| 28 | Arabidopsis thaliana | F5H deficient | M | ≈100 % G (i.e., G↑, S↓) | No change | Decrease | Decreased plant stiffness | 117 |
| 29 | Brassica napus | COMT, C4H, C3H, F5H | TG | No change | 26‐40 % decrease in the seeds | ND | No change | 102 |
| 30 | Arabidopsis thaliana | F5H upregulation; COMT downregulation | TG | Lignin>70 % 5OH‐CA‐derived; ≈90 % benzodioxane units | ↓ | ND | Dwarfism; male sterility | 60, 119 |
| 31 | Arabidopsis thaliana | CCR and CAD deficient | TG | Increase in interunit bonding; incorporation of coniferaldehyde, sinap‐ aldehyde, ferulic acid | 50 %↓ | ↓ | Dwarfism; male sterility | 62 |
| 32 | Arabidopsis thaliana | CSE deficient | M | H↑ | ↓ | 4×↑ | No change | 66 |
| 33 | Populus alba x grandidentata | FMT introduced | TG | Ester linkages introduced into lignin backbone; S/G↑ | Little change | 78 %↑ | No change | 44 |
| 34 | M. sativa | C3H downregulation | TG | H 65×↑ | 50 %↓ | ND | Dwarfism | 120 |
| 35 | Populus tremula x alba | F5H upregulation | TG | 97.4 % S; (i.e., G↓, S↑) | Little change | Pulp yield higher, pulp brighter | No change | 121, 122 |
[a] Measured at low conversion extent; yields at full conversion will not differ.123
In general, downregulation of the genes associated with enzymes responsible for the early steps of phenylpropanoid biosynthesis, i.e., for enzymes PAL, C4H and 4CL (Scheme 1), results in decreased flux through the pathway, and consequently lowers lignin yields (entries 1, 2 and 4). Although researchers long favoured this approach of producing less of the problematic component, plants require lignin, so adverse agronomic effects can result if the lignin reduction is too severe.133
PAL downregulation results in an over‐accumulation of Phe. However, for Arabidopsis thaliana (entry 1), PAL downregulation had no observable impact on the plant phenotype, despite a significant decrease in lignin content. Incorporation of Tyr, as an alternative starting point into the phenylpropanoid pathway, may explain this result.75
3‐Hydroxylation of p‐coumarate by C3H was originally thought to occur at either the acid or the CoA level, until researchers showed the presence of a new enzyme, HCT, in various plants that produce p‐coumaroyl shikimic or quinic acid conjugates that are the preferred substrates of C3H. Following the hydroxylation, HCT was conjectured to return the product to the CoA level as caffeoyl‐CoA. However, again the pathway appears to be more complex as another enzyme, CSE, is now firmly established in some plants as returning the product of the C3H reaction back down to caffeic acid, upon which 4CL must again act to produce the CoA derivative.
The advantage of discovering all of the genes associated with the expression of the enzymes in the biosynthesis of lignin building blocks is that there are now more ways to perturb the system. In general, however, the main result of downregulating HCT, C3H, or CSE is a relative increase in the H‐unit level. Downregulation or deficiency of HCT (Scheme 1, Table 2, entries 20 and 21) leads to a significant decrease in growth, a reduction in the lignin quantity, over‐accumulation of flavonoids, and a predictable rise in the relative level of H‐units. The c3h mutant of Arabidopsis, called ref8, is a particularly stunted plant that does not produce seed but has H‐only lignin. Intriguingly, however, the agronomic issues do not arise from the change to H‐lignin per se—co‐downregulating a pair of mediator genes results in recovery of seed production as well as much of the dwarfing yet retains the novel high‐H lignin characteristics.134
To proceed toward the monolignols, p‐coumaroyl‐CoA, feruloyl‐CoA, and/or sinapoyl‐CoA are first reduced to the corresponding aldehydes (via CCR). CCR‐downregulation has attracted considerable interest (entries 6–12), particularly in Europe, as Arabidopsis and then poplar plants proved to be significantly more readily saccharifiable, and strikingly so even in the absence of a pretreatment.105 Plants show a small growth penalty and have lower lignin levels, but the most intriguing characteristic was that ferulic acid, as a monomer, was shown to be incorporated (at low levels) into the lignins.106, 107 As the incorporation produced novel acetal branch‐points in the polymer, and as acetals are readily cleaved with acid, the analysis of CCR‐deficient plants have produced another strategy for engineering lignins that are easier to chemically degrade.
The enzymatic conversion of coniferaldehyde is an important branching point in the lignin biosynthetic pathway, leading to G‐units (CAD), to S‐units (F5H, COMT and CAD), or back to ferulic acid (HCALDH). The overexpression of F5H with a powerful lignin promoter (entry 27) leads to a lignin almost exclusively composed of S‐units, whereas downregulation or deficiencies in F5H (entry 28) results in a predominance of G‐units.117 However, in both cases in Arabidopsis, a decrease in plant stiffness, caused by the lack of secondary plant wall structure in the interfascicular and xylem fibres, was observed.65 For lignins composed primarily of G‐units, no change in overall lignin content was observed, yet the saccharification yield of the biomass decreased, indicating the formation of a more recalcitrant biomass.117
Two classes of O‐methyltransferases, the so‐called CCoAOMT and COMT enzymes (Scheme 1), are involved in producing the 3‐ and 5‐methoxyl groups on G and S monomers. 5‐Hydroxyconiferaldehyde, when 5‐O‐methylation is deficient, reduces to 5‐hydroxyconiferyl alcohol that is then integrally incorporated into lignins in COMT‐deficient plants (entries 23 and 24). The resulting 5‐hydroxyguaiacyl units react by typical 4‐O‐β‐coupling with any of the hydroxycinnamyl alcohol monomers (the prototypical monolignols or further 5‐hydroxyconiferyl alcohol), but the internal trapping of the intermediate quinone methide product by the novel 5‐OH results in the formation of benzodioxane structures in the polymer.11, 92, 135, 136, 137, 138, 139 Benzodioxane levels can be amplified through a combination of F5H upregulation and COMT downregulation. In Arabidopsis, the resulting plants may incorporate up to ca. 70 % 5‐hydroxyconiferyl alcohol monomer and produce benzodioxane levels of as high as 90 % in the polymer (entry 30). However, the phenotypic outcome at this extreme level was abnormal plant growth.60, 119 CCoAOMT, invoked earlier in the pathway, has a similar effect on guaiacyl units and results in the incorporation of caffeyl alcohol into lignin, but only in softwoods (that do not make S‐lignin);140 attempts to even downregulate both OMTs (and various genes) in hardwoods, or dicots in general, have not produced any authentic evidence for caffeyl alcohol incorporation. Furthermore, other OMTs (or other genes besides those currently targeted) appear to be implicated as even strong downregulation of CCoAOMT and/or COMT results in the production of the normal G and S monolignols, albeit at a lower level.141
The downregulation of CAD, producing the CAD enzyme that is involved in the final step of production of all conventional lignin monomers, often results in the introduction of hydroxycinnamaldehydes into lignification, resulting in various types of atypical unit in the lignin structure (entries 17 and 18). Both coniferaldehyde and sinapaldehyde may be introduced into the lignin structure as monomers in their own right.42, 58, 60, 115 Particularly intriguing was the observation that coniferaldehyde will not, in vitro or in vivo, β‐O‐4‐cross‐couple with G units and is therefore poorly integrated into gymnosperm (G‐only) lignins. In contrast, coniferaldehyde readily cross‐couples with S units so it, and sinapaldehyde that readily cross‐couples with both G and S units, are well incorporated into dicot lignins. Recent CAD misregulation examples show just how high the level of non‐canonical monomers can be tolerated in plants that grow more or less normally, at least under some conditions. A CAD‐deficient mutant of Medicago truncatula has reportedly some 95 % of its lignin derived from hydroxycinnamaldehydes.43 In Arabidopsis, manipulation of the G and S monomer synthesis coupled with CAD deficiency has been examined, again producing plants that have essentially none of their lignin derived from the conventional monolignols, and plants that are derived almost solely from either coniferaldehyde or sinapaldehyde.142
Entirely new or non‐traditional monomers can be utilised in lignification, resulting in novel structures in the lignin polymer. To be completely accurate, some of these are also found in normal wild‐type plants at low levels (by sensitive analytical methods). For example, downregulation/suppression of C4H, C3H, CCR, COMT or CCoAOMT resulted in the incorporation of monomers other than the three H, G, or S monolignols into the lignin structure, regardless of the plant species. The nature of the monomers that are alternatively incorporated in the lignin structure vary between species, but include: ferulic acid;67, 102, 104, 105 coniferaldehyde;43, 62, 111 sinapaldehyde;43, 110, 111 5‐hydroxyconiferyl alcohol (5OH‐CA),42, 44, 58, 115 caffeyl alcohol,140, 143 and monolignol ferulates.44 In addition, various new products, or enhanced levels, arise in the extractable low molecular weight metabolites from actively lignifying tissues. Examples include sinapic acid,104 p‐coumaroyl hexose,67 caffeic acid hexoside,67 and various hydroxycinnamate esters.56 The alternative monomers that have been found incorporated into the lignin structure are summarised in Table 3; the monomers and metabolites are summarised in Scheme 2.
Table 3.
Summary of various alternative monomers incorporated into lignin, and the mutation(s) responsible for their incorporation (Scheme 2).
| Entry | Alternative monomer | Effect on lignin structure | Gene responsible |
|---|---|---|---|
| 1 | Ferulic acid | New acetal branch‐points in lignin | CCR 67, 102, 104, 105 |
| 2 | Sinapaldehyde | Sinapaldehyde integrally incorporated into polymer, enhanced unsaturation, and increased free‐phenolics | CAD 43, 110, 111 |
| 3 | Coniferaldehyde | Coniferaldehyde integrated into polymer chain in G/S lignins only, enhanced unsaturation, increased free‐phenolics | CAD/CAD‐CCR 43, 62, 91, 111 |
| 4 | 5‐Hydroxyconiferyl alcohol | Benzodioxane units in polymer; linear polymer if 100 % 5OHCA | COMT, F5H‐COMT 42, 58, 60, 115 |
| 5 | Caffeyl alcohol | Benzodioxane units in polymer; linear polymer if 100 % caffeyl alcohol | CCoAOMT 140, 143 |
| 6 | Monolignol ferulates | Incorporation of ester linkages into the polymer backbone | FMT (Ferulate monolignol transferase) 1, 44 |
Scheme 2.
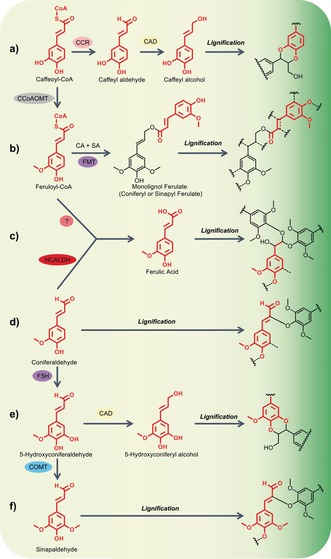
Incorporation of non‐native monomers in the lignification process, via genetic modification to the phenylpropanoid biosynthetic pathway (Scheme 1, see asterisked intermediates that are either the new monomers themselves, or give rise to a new monomer via the subsequent transformations shown here). The incorporation of new monomers (highlighted in red) by the same radical coupling modes gives rise to new structures in the lignin polymer (Table 3), mainly as a result of new opportunities for rearomatisation of the intermediate quinone methides: a) In CCoAOMT‐deficient softwoods, caffeyl alcohol is incorporated into the lignins producing benzodioxane structures. Some plants, such as vanilla, make lignin in their seed coats entirely from caffeyl alcohol, producing a polymer that is almost entirely composed of long chains of benzodioxane units. b) Employing a novel monolignol transferase enzyme, FMT, Feruloyl‐CoA can be conjugated to a monolignol, coniferyl alcohol (CA) or sinapyl alcohol (SA), to produce monolignol ferulate conjugates. Formation of labile ester linkages in the lignin polymer backbone can be achieved via lignification with a proportion of its monomer pool as these monolignol ferulate conjugates. Conjugates of this type are incorporated into the lignin structure in the same manner as conventional monomers.1 c) Ferulic acid itself can be formed either via the action of the enzyme HCALDH or by downregulation of CCR (Scheme 1). During lignification, double‐β‐O‐4‐coupling is undergone, to yield acid‐labile acetal bonds in the lignin. d) The intermediate, coniferaldehyde, accumulates in CAD‐deficient plants and can be incorporated into lignins via β‐O‐4‐coupling (shown) as well as other modes (not shown); the β‐O‐4‐coupling product is unsaturated due to the acidity of the β‐proton in the quinone methide intermediate that allows rearomatisation via H‐abstraction. e) In COMT‐deficient angiosperms, the intermediate 5‐hydroxyconiferaldehyde that accumulates can still be reduced by CAD, producing 5‐hydroxyconiferyl alcohol that, like caffeyl alcohol, produces benzodioxane analogues in the lignin. f) Sinapaldehyde also incorporates into lignins in CAD‐deficient angiosperms.
Inspired by three independent findings regarding lignin structure, namely that 1) ferulates were excellent lignin monomers and were implicated in monocot lignification, 2) monolignol conjugates were being used in lignification in certain natural plants, and 3) monolignol substitution was becoming increasingly evident with the study of transgenics, an attempt was made to “redesign lignin for processing”.1, 44
In preliminary model studies, it was estimated that with ca. 25 % incorporation of monolignol ferulate conjugates, the alkaline pulping temperature could be decreased from 160–170 °C to 100 °C for the same degree of delignification.1 At an incorporation level of ca. 65 % (that is likely unattainable), the pulping process on the model system could operate at just 30 °C whilst maintaining equivalent lignin removal from the biomass.1 The insertion of ester linkages into the lignin backbone may feasibly retain the lignin quantity whilst also maintaining the natural function. In a recent investigation,44 it was estimated that cell wall lignin could be augmented with perhaps 7–20 % of its units derived from monolignol ferulates via a specific transferase enzyme, FMT. The genetically modified biomass exhibited increased delignification and higher fibre yields after mild alkaline pretreatments compared to the control. Coniferyl ferulate and sinapyl ferulate monomer conjugates may be incorporated into the lignin macrostructure analogous to that of a dimer of G, S or H units (Scheme 2 b).
In certain instances, similar phenotypic characteristics may be exhibited by the same genetic mutation for different plant species; with Nicotiana tabacum (Table 2, entry 10)104 and Populus tremula x Populus alba (entry 11)105 severe downregulation of CCR results in dwarfed plants, for example. By contrast, the same genetic mutation can also have a profoundly different result depending on the selected plant type: for CAD‐deficient Pinus taeda (entry 17),111 a slightly higher quantity of lignin is observed compared to the wild‐type, yet for CAD‐deficient Koshihikari x Chugoku 117 rice (entry 19),113 a decrease in lignin content occurred. In some cases in which the plant phenotype was not affected, the saccharification yield increased regardless of the lignin content.
2.4. Practical Challenges of Lignin Bioengineering
Currently, the majority of bioengineering strategies have been directed towards decreasing the lignin content in order to achieve higher saccharification yields/improved fermentability. However, to ensure economic feasibility of a biorefinery, the recalcitrance of the lignin fraction must also be considered.1 In light of this consideration, preparation of lignin with a more chemically labile structure (e.g., by incorporating some ester bonds into the backbone of the polymer rather than dealing with the more recalcitrant ether linkages) is an interesting prospect (Scheme 2 b).
Research on plant improvement towards better lignin processing is a non‐trivial task. One major challenge is the time required for production and growth of the biomass. For most softwoods and hardwoods, this is between 5 and 30 years.131 To alleviate this limitation, the bulk of research on lignin bioengineering has been directed towards angiosperms with a shorter life cycle (e.g., the model dicot, Arabidopsis thaliana and, more recently, the model monocot, Brachypodium distachyon). Arabidopsis and Brachypodium are useful models of longer life‐cycle analogues such as Poplar/Aspen and the commercially produced monocots (grasses), exhibiting many similar biochemical and genetic traits. Model plants are valuable for indicating possible directions for genetic alterations. Nevertheless, the strategies still need to be applied to the actual softwood or hardwood trees, or grasses, at some stage of the development, and it is crucial to not assume trait portability between gymnosperms, dicots, and monocots. Differences in the phenotypic behaviour of any one plant species may even vary according to whether the plants are grown in a greenhouse or in the field.131 Application into readily transformable, moderately rapid growing and commercially important plants, is often via Poplar, Aspen, or Eucalypts (for hardwoods), and corn, barley, and switchgrass (for monocots); Loblolly and Radiata pine (Pinus taeda, Pinus radiata) are among the best for applications to softwoods, but the development remains difficult and slow.
Phenotypic alterations (i.e., changes in the morphology, in development, or behaviour) of a plant are common upon perturbing the phenylpropanoid pathway (although specific explanations for individual phenotypic changes are difficult to pinpoint). Dwarfism is typically accompanied by collapsed xylem vessels and, therefore, decreased water transport.43, 56, 57, 60, 62, 65, 102, 104, 105, 109, 114 However, contrary to prior belief, over‐accumulation of flavonoids is not necessarily associated with a decrease in plant growth.13 The precise cause of dwarfism remains elusive.
The challenges associated with predicting plant physiology and long‐term growth performance upon slight genetic modification has led to the proposal of using high‐throughput multi‐trait genetic modification. In this strategy, large numbers of genetically modified plants are screened for favourable growth traits at multiple stages during their life‐cycle.131 Plants are exposed to both biotic and abiotic stresses. Tolerant plants are carried on into further studies (assessed on the basis of growth, saccharifiability, and/or lignin composition) whilst non‐tolerant plants are discarded. This method of high‐throughput screening allows rapid identification of a plant with a lignin that can be chemically deconstructed. After this plant has been discovered, work may then be undertaken to try to identify the genetic changes responsible for the plant improvement. One high‐throughput strategy for multi‐trait genetic engineering, modified to include lignin screening, is illustrated in Scheme 3.131
Scheme 3.

High‐throughput multi‐gene engineering scheme.131 Final field trial stages are of critical importance to identify potential environmental impacts, (e.g., toxicity to insects, impact on soil chemistry). For a thorough review of environmental risk assessments for genetically‐engineered trees, the reader is directed to other literature.154 Adapted from Plant Sci. 2013, 212, 72–101.
Immunostimulatory activities of different lignins depend on their structure, neutral sugar content, molecular weight, and degree of polymerisation.144 Although research has been conducted on the response of lignin to abiotic stress (e.g., drought, salinity, wounding, low temperature, and UV‐B radiation), these studies typically focused on how lignin quantity was affected and not on structural analysis of lignin, therefore there are several pending questions.41 Abiotic stress directly impacts the formation of lignin via the phenylpropanoid and lignin biosynthetic pathways.5, 41, 99, 145, 146, 147, 148, 149, 150, 151 The response of these pathways to abiotic stresses is crucial for understanding the full biological role of lignin in the plant.5, 145 Effects of abiotic stress can be analysed either by individually introducing stresses, e.g., growing the plant in a medium with high salinity),150, 152 or by monitoring how the plants perform in a greenhouse setting vs. in field trials.153 Recent review literature provides a comparison of the impacts of biotic and abiotic stress on plant fitness when transferring from a controlled greenhouse environment to field trials.131 However, even for non‐transgenic tree varieties, it is hard to predict the behaviour and performance of the mature tree from greenhouse analyses.
To conclude, the rational incorporation of chemically‐labile linkages into the chemical structure of lignin represents a promising area of future investigation. Furthermore, developments in “high‐throughput multi‐gene engineering” will aid in identifying specific effective strategies to reduce the recalcitrance of the native lignin polymer. Notably, the wealth of variables associated with each stage of the valorisation stream (and difficulties associated with assigning the observed phenotypic changes to specific biotic or abiotic influences), in conjunction with the highly multidisciplinary nature of the research (from the initial introduction of the genetic mutation through to eventual chemical processing), render any bioengineering strategy challenging. In order to genetically engineer a more chemically deconstructible lignin whilst maintaining full functionality of the plant species, a combination of different types of expertise is required. To achieve this, long‐term research objectives, funding security, and commitment from multiple complementary research teams are mandatory.
2.5. Structural Features of Native Lignins
Over the past three decades, plant biochemists have made significant progress in the understanding and manipulation of genes associated with the expression of enzymes involved in the phenylpropanoid biosynthetic pathway, enabling modification of the prevalence and nature of lignin's building blocks, thus altering the distribution of structural motifs or linkages as well as the content of lignin in the plant biomass. However, manipulation of the biosynthesis of lignin monomers is just one of many key variables of lignification (i.e., the polymerisation of phenylpropanoid units rendering native protolignins). In fact, the concentrations of monolignols are governed not only by their relative prevalence, following their biosyntheses, but also by the transport and diffusion of monomers to the secondary plant cell wall.31 Moreover, other physical stress conditions (e.g., temperature, pressure, salinity and light) may dramatically affect lignification.131 These variables may partially offset the anticipated benefits from genetic manipulation of the phenylpropanoid biosynthetic pathway.
The lignification process is induced by the action of peroxidase and laccase enzymes producing the corresponding phenolic radicals that undergo cross‐coupling reactions to incorporate the monomer units into the growing lignin polymer. Certain peroxidases and laccases are believed to oxidise lignin oligomers directly.155 In these instances, the reaction was suggested to involve a one‐electron transport chain from the active site to the enzyme surface.155, 156 Despite the key roles of peroxidases and laccases in initiating the polymerisation, lignification per se is not an enzyme/protein‐controlled process. In fact, lignification is a “solution‐like” chemical process, as evidenced by the lack of optical activity in native lignins.155, 157 This fact implies that lignification neither occurs in the proximity of an enantioselective enzyme cavity nor is affected by the chiral environment of the surrounding polysaccharides.
Although coupling of lignin monomers into the growing polymer is evidenced to take place in a combinatorial and non‐stereospecific manner, i.e., lacking a specific sequence of monomers,158 there is ample evidence for the cell's control of the composition and structure by altering monomer supply. It was long ago revealed that, in dicots, lignification proceeded from H to G to S (in overlapping regimes) with cell maturity.159 More recently, under optimised conditions (pH 9, 488 nm excitation wavelength), fluorescence microscopy images were able to identify lignins at specific regions in Pine and Poplar cell structures that were likely enriched with G or S units, (Figure 1).160 An investigation of 25 Chinese hardwood biomass species demonstrated similar topochemical patterns of lignin whereby, e.g., in diffuse‐porous hardwoods the vessel cell walls incorporated predominantly guaiacyl (G) lignin yet the fibres were primarily composed of syringyl (S) lignin.161 Softwood lignins are predominantly G, although high‐compression‐wood zones are particularly H‐rich.162, 163 There is little question that harsh and unselective delignification processes will dismantle any such arrangement of enriched domains in hardwoods, affording a more uniform distribution of units in the isolated technical lignins through the recombination of lignin fragments dissolved in the extraction liquor. However, the development of soft delignification methods may represent a novel but challenging strategy to explore for the selective extraction of (H‐), G‐ or S‐type units, with potential for reducing the costs associated with product separation in the catalytic downstream processing of lignin streams.
Figure 1.

Topochemical distribution of likely G (magenta) and S (green) enriched lignin domains of lignocellulose feedstocks: monochrome fluorescence images of Pine (A) and Poplar (B) woods, and; the corresponding images coloured according to the fluorescence emissions (C and D, respectively); spectral image of Salix chilensis demonstrating the difference in guaiacyl and syringyl distributions in vessels and middle lamella from fibre walls (E); uniform distribution of lignin units in Acacia melanoxylon (F) and Eucalyptus nitens (G). The scale bars represent 60 μm. Reproduced with permission from IAWA Journal 2013, 34, 3–19, copyright 2013 Brill.160
The advent of multidimensional NMR methods in the 1980s enabled easier characterisation of complex molecules, including lignins. Short‐range 1H‐13C correlation experiments, such as the now popular Heteronuclear Single‐Quantum Coherence (HSQC) experiment, have taken over from the traditional 1D 1H and 13C experiments that suffered from insufficient resolution to distinguish subtle structural details, and are now widely employed for the investigation of lignin composition and structure. The interested reader is referred to thorough recent review articles on the methodology, potential and limitations of NMR spectroscopy for the characterisation of lignin, even without requiring its isolation from the cell wall.33, 34 HSQC experiments have been instrumental in the identification and (approximate) estimation of the relative abundance of bonding motifs of the types A, B, C, D and F (Scheme 5) and other structural elements that may occur both in untreated native lignins (e.g., spirodienone moieties, derived from β‐1 coupling of a monolignol with a preformed β‐ether unit),164 and residual linkages in the depolymerised material.165 Expansive literature, and two book chapters,31, 166 describe its application to deduce the changes in normal units and to elucidate and validate new products in the many transgenics.
Scheme 5.

Diagram highlighting the bonding motifs and potential linkages as targets for depolymerisation (in green), % occurrence values from the literature, and bond dissociation energies for a range of commonly encountered linkages/bonding motifs in native lignins.177, 178, 179, 180, 186, 187 It is important to point out that many of these % occurrence values, although reported in the literature, are unlikely or even untenable: β‐1 moieties likely do not exceed 1–2 % of all structures (substantially lower than the 9 % reported); it is impossible to encounter 19–27 % of 5‐5‐units in any lignin (it is probably restricted to a maximum of approximately 9 % in softwoods), and; the abundance of 4‐O‐5‐linkages in softwoods is almost certainly much lower than the 4–7 % claimed.
Despite the obvious value of 1H‐13C HSQC NMR spectroscopy for lignin characterisation, the information provided should not be over‐interpreted. The difficulty of performing quantitative analysis is indeed an important limitation of HSQC NMR in general. Semi‐quantitative determination of integral ratios is, however, possible when the 1H‐13C pairs are located in similar chemical environments, e.g., Cα‐Hα signals for lignin sidechains or C2‐H2/C6‐H6 aromatic signals, because 1 J CH assumes similar values under this condition.165, 167, 168 The 1 J CH dependence of polarisation transfer was previously an issue, with cross‐peaks having different response factors. However, various adiabatic variants in particular minimise this problem. Indeed, progress has been achieved for quantitative HSQC NMR, using so‐called QQ‐HSQC and HSQC0, with pulse sequences allowing better quantification of the identified functionalities.169, 170, 171 However, these methods still fail for rapidly and differentially relaxing samples. For example, in lignins, correlation peaks from the more mobile endgroups, including the p‐coumarates and p‐hydroxybenzoates that adorn some lignin sidechains, relax much more slowly than those from units in the polymer backbone and are consequently overestimated by often large factors. Methods for overcoming such issues are still actively sought. Regardless, regular HSQC NMR experiments still offer highly valuable semi‐quantitative, relative information on the linkage abundance, allowing for comparison of lignin structures and whole plant cell compositions.167, 168, 172 It should also be noted that 31P NMR provides quantitative data on the nature of the various OH groups in lignin, and is also capable of quantifying various interunit linkage types in lignins.173
The most common structural element in native lignins is the β‐ether (usually indicated by the letter “A” in HSQC NMR studies of lignins, Scheme 5), accounting for 50–80 % or more of the measurable interunit linkage types. The threo (syn) and erythro (anti) forms of the arylglycerol‐β‐aryl ethers are present in amounts that reflect the kinetics of the proton‐assisted rearomatisation of the quinone methide (by water); β‐guaiacyl ethers form in approximately equal proportions, whereas β‐syringyl ethers form with erythro‐isomers predominating by ca. 3:1; in both cases the thermodynamic ratio is close to 50:50, indicating that lignification is not under thermodynamic control.174, 175 As for all the units in lignins, β‐ether units do not possess any optical activity, implying that both the radical coupling itself and the addition of water to the quinone methide intermediate produce fully racemic products. Lignification is therefore concluded, as originally theorised,176 to be simply a chemical reaction, independent of proteinaceous control.155, 158, 168
DFT calculations performed on molecular models have predicted values of bond dissociation energy (BDE) for the β‐O‐4 bond between 54 and 72 kcal mol−1.177, 178, 179, 180 Notably, substituent effects can have a significant impact on the BDE of a β‐O‐4 bond. For example, oxidation of the α‐hydroxy group to a ketone was found to lower the BDE by 15 kcal mol−1.178
A phenylcoumaran unit B has a five‐membered ring that results from internal trapping of the intermediate quinone methide by the phenolic‐OH following the β‐5 coupling (Scheme 5). Again it is racemic, but the ring‐closure is trans‐selective such that there is only a single isomer of the dimeric unit. DFT calculations predict the α‐O‐4 bond of phenylcoumaran molecular models to have a low value of BDE (50–56 kcal mol−1), suggesting that these structural motifs can easily undergo radical cleavage under conditions of high severity.
Resinol structures C that are formed via β‐β coupling can only occur at the monomer stage, i.e., in dimerisation or crossed dimerisation reactions. In fact, sinapyl alcohol produces syringaresinol as its only authenticated dimer in peroxidase‐H2O2‐catalyzed reactions. Structures A and B can also form directly from the coupling of coniferyl alcohol with another monolignol, but most of these structures in lignin arise from the more common monomer‐oligomer cross‐coupling reactions that extend the polymer chain.155 Biphenyl linkages D from 5‐5‐coupling, that almost always result in dibenzodioxocin structures D2 after addition of the next monolignol to the chain, are only obtained from the coupling of two preformed oligomers.155
In 2001, spirodienone bonding motifs (F) were discovered in the structure of native lignins, shedding light on the divergence within the wood chemistry community regarding the occurrence and frequency of the β‐1 linkage.164 These spirodienone structures are particularly prone to undergo ring opening under mildly acidic conditions, leading to the formation of structures containing solely the β‐1 linkage (Scheme 4). The discovery of spirodienone structures through modern 2D NMR methods illustrates how challenging the structural elucidation of native lignins may be, as the native polymer is modified even by mild isolation methods.
Scheme 4.

Degradation of (native) spirodienone (F) structures under mildly acidic conditions.164
It was recently suggested that the thermodynamics of the radical reactions leading to lignification may govern the selectivity to the formation of different bonding motifs in the native lignins (Figure 2). Dimerisation of lignols was explored by DFT calculations at the M06‐2X/6‐311++G(d,p) level of theory.181 The formation of the β‐O‐4 linkage was predicted to be the most exothermic coupling reaction (for an H‐H or G‐G dimer, approximately 15 kcal mol−1 more favourable than the corresponding 4‐O‐5 linkage). This thermodynamic data concurs with the β‐O‐4 linkage being more abundant than 4‐O‐5 motifs. However, this prediction should not be taken as evidence that lignification is a thermodynamically controlled process. It is also important to bear in mind that 4‐O‐5 units are formed by coupling of oligomers, whereas lignification is mostly the result of sequential addition of monomers to the chain; the formation of β‐ether units would therefore prevail over 4‐O‐5 counterparts, regardless of the thermodynamics. Most importantly, there is convincing evidence that the processes leading to the formation of lignin linkages are instead kinetically controlled, as previously noted.
Figure 2.

Calculated ΔH r ranges for homo‐coupling and cross‐coupling reactions, yielding a variety of lignin linkages and bonding motifs (M06‐2X/6‐311++G(d,p) level of theory). Adapted with permission from J. Phys. Chem. B 2012, 116, 4760–4768.181 Copyright 2012 American Chemical Society.
From the DFT predictions, the enthalpy values for self‐/cross‐coupling reactions of lignols become gradually less exothermic values in the order: β‐O‐4> β‐β> β‐5≫ 5‐5> β‐1> 4‐O‐5.181 Not surprisingly, this ranking contradicts that originated from BDE values (Scheme 5), giving the false impression that β‐O‐4 linkages would be the most stable against homolytic cleavage of the C−O bond. Notably, one should consider that the initial and final states for the studies on BDE and heat of dimerisation of lignols are not identical. Accordingly, these sets of theoretical predictions cannot directly be compared.
The absence, or at least extremely low abundance, of non‐cyclic α‐O‐4 linkages, in addition to the discovery of eight‐membered ring structures D2, constitutes an important update to the classical models of lignin proposed by Nimz (1974)182 and Adler (1977).31, 183 These dibenzodioxocin motifs became recognised as crucial branching points in native lignins.184 However, various researchers have so far found evidence only for structures of type D2 in which the added monolignol unit remains free‐phenolic (to form a “U” type branch).185 Although D2 units could also implicate the joining of three chains, there is currently no evidence that D2 units forms a true “Y” branch. The same can be said for 5‐O‐4‐units in lignins. If this is the case, then native lignins must be thought of as essentially linear, and not branched to any significant degree. Indeed, although lignins are referred to as “polymer networks”, the quantity of condensed structures (i.e., aromatic units linked to others via their 3 or 5 ring positions in ways that form Y‐type structures) occurring in native lignins is now understood to be very low,184, 185 which at least in part explains the ease of depolymerising native lignins. This observation is in strong contrast with technical lignins, for which prior upstream treatment often results in a highly condensed structure (as discussed in further detail later in this review).
The progress made in the identification of native substructures of lignin, elucidated largely by NMR methods and validated using synthetic model compound data, allows for accurate identification of specific linkages/bonding motifs occurring in native lignins. It has been noted that 2D NMR is capable of pinpointing only a fraction of all the linkages because detection by HSQC is limited to C−H fragments a priori, with the technique being blind to other linkage patterns containing non‐protonated tertiary or quaternary carbons, such as in 5‐5 (biaryl) and 4‐O‐5 (biaryl ether) structures.168 However, this is not actually true. Essentially all of the 5‐5‐linked units in native lignins are in the form of dibenzodioxocins D2 that are easily seen, well dispersed and, in principle, quantified. Even the 4‐O‐5 structures leave signatures, because their C2–H2 and C6–H6 correlations are unique (at least in the G‐only softwoods).
Finally, it is important to note that as polymer growth occurs, the likelihood of observing two identical lignin macromolecules becomes vanishingly small.88, 158 On this basis, the sequencing of lignin building blocks in a similar manner as performed for amino acid residues (in “proteomics”) is impossible and, in any case, of limited value for the design of catalytic lignin conversion processes. Strategies for lignin conversion ought to be designed bearing in mind the occurrence of linkage/bonding motifs rather than a specific macromolecular structure. Due to lignin's racemic nature, its complexity, and its largely unknown associations with other cell wall polymers, many macromolecular aspects of the structure remain elusive.
2.6. Structural Features of Technical Lignins
Native lignins undergo extensive chemical transformation as a result of pulping or pretreatment processes. The extent of structural modification hinges upon the process “severity” (i.e., temperature and duration of cooking and concentration of pulping.188, 189, 190, 191, 192, 193 The abundances of different C−O and C−C linkages in technical lignins, therefore, are likely to differ substantially from those determined for the native lignin,190, 194 a realisation that greatly impacts any choices made for further depolymerisation. An example of the impact of pretreatment severity on the nature of the lignin isolated is shown in Figure 3, in which mild acetolysis produces a lignin high in β‐ethers, whereas almost no native lignin identity remains following the harshest pretreatment.
Figure 3.

Influence of pretreatment severity on the nature of a processed lignin as revealed by HSQC NMR spectroscopy. Knowing the method by which a lignin sample is prepared does not yield sufficient information about structural properties—it must be characterised. Here, a comparison is drawn between A) a maize Enzyme Lignin (EL), isolated in the lab, with B–D) lignins that are precipitated from an acetosolv process,195 in which acetic acid is the organic solvent. As can be seen by examining especially the β‐ether A correlations (cyan) but also the general nature of the aromatics, the mild process in (B) produces a rather native‐like lignin, with β‐ethers largely intact, and only a little “distortion” of the aromatics; the “lignin” does however contain significant levels of polysaccharide‐derived material (as seen by the additional grey peaks). With the medium‐severity treatment (C), which uses added mineral acid and a lower level of AcOH, β‐syringyl ethers have disappeared, the β‐ether level in general is lower, more tricin has detached from the polymer, and the aromatics are decidedly more complex. Under the highest severity conditions, in (D), no recognizable structural features (other than methoxyl and general aromatic signals) are evident—it has no β‐ethers. Any or all of these could be marketed as “acetosolv” lignin yet, clearly, those processes that rely on β‐ether cleavage may be wholly effective with the low‐severity material but would be completely worthless against the high‐severity material; material D is not valueless, nevertheless the method for valorisation will be highly different from material B. At some point, when effective processes for lignin utilisation evolve, techno‐economic analysis must be used to optimise the production of not just the sugars or pulp, but also the best lignin component, to fully optimise the profitability of the biorefinery. L:W=liquor to wood ratio. Contours in the NMR spectra, where they are sufficiently well resolved, are color‐coded to match the structures below; overlapping peaks are simply colored gray along with peaks from polysaccharides or other unidentified materials.
Structural characterisation (via 2D NMR and/or chemical degradation methods, e.g., acidolysis, thioacidolysis, etc.) of the technical lignins before catalytic treatment, and any remaining technical lignin after catalytic treatment, must become common practice in this field. The effectiveness of a catalyst either for performing a particular chemical transformation or for the cleavage of a specific linkage can then be better assessed.194 Such a strategy circumvents the difficulties associated with the limitations of simple model compounds. Indeed, these often offer a poor representation of the actual lignin structure (reactions on model compounds are discussed in greater detail later in this review), and such results can thus typically only with difficulty be extrapolated to “real” lignins. Model compounds of high structural fidelity can nonetheless provide valuable insight and aid in the elucidation of changes in the lignin structure upon catalytic processing. For instance, in the oxidation of Kraft lignin with DDQ, the characteristic resinol (from β‐β‐coupling) signal was found to disappear. The reaction performed on a model compound of appropriate complexity allowed for the identification of an unexpected pyron‐4‐one product.196 It is also worth noting that model compounds can be particularly useful for screening for the best reactions and conditions. Thus, although there is no guarantee that a reaction that works well on a model compound will also perform well on an actual lignin sample, the converse is almost always upheld, i.e., it is almost universally true that little is to be expected of a reaction on lignin if that reaction does not perform well on a lignin model compound of sufficient fidelity.
The distinct bonding features of technical lignins are described in the following subsections, describing the predominant structural modifications brought about by Kraft and Organosolv processing and their implications for catalysis. Herein, emphasis is placed on these two specific pulping processes for two main reasons. Firstly, Kraft pulping is operated on a large scale commercially, producing the largest volumes of lignin‐containing streams.197 In addition, the chemistry of Organosolv pulping underpins the novel class of valorisation processes referred to here as “Early‐stage Catalytic Conversion of Lignins” (ECCL)198 or the “Lignin‐first” strategy.277
2.6.1. Kraft Pulping Process
Globally, the Kraft process is the dominant technology of the pulp and paper industry. Approximately 130 million tons of Kraft pulp are generated annually.197 Surprisingly, this technology is one of the few persistent examples of a chemical organic process, performed at a million‐ton‐scale, that is stoichiometric and not catalytic. The Kraft process originated in 1879 in Danzig, Prussia (nowadays Gdańsk in Poland). It is so‐named after the German word for “strength”, because of the superior resilience of the pulps vs. those obtained from the earlier soda and sulfite processes. The long‐term success of the Kraft process lies in the recyclability of the inorganic pulping agents (Na2S/NaOH) and, more recently, in the efficient generation of electricity by the Kraft recovery boiler.197 In the boiler, the black liquor obtained from the pulping step (which contains the lignin fraction) is incinerated and Na2S is regenerated (Na2SO4+2 C→Na2S+2 CO2), i.e., the lignin's carbon is employed as a reducing agent. The boiler produces high‐pressure steam that powers turbo‐generators.197 Modern Kraft mills generate a considerable electricity surplus, which is often sold back to the local electrical grid.199 Considering the global scale of the process (incineration of lignin generates ca. 700 million tons of high‐pressure steam per year),200 Kraft lignin actually constitutes one of the world's most important biofuels.199 Notably, Kraft lignin black liquor represents the largest share of renewable biofuel in the Finnish and Swedish energy matrices.199
Although Kraft lignins currently constitute the largest lignin stream by volume, they are not available commercially in isolated form in the same abundance. This is because the black lignin liquor plays a key role as an internal energy supply and to recover the inorganic chemicals used in the pulping process. Nonetheless, the diversion of a fraction of Kraft lignin away from fuel use and towards the production of bulk, specialty or fine chemicals may be economically viable if the price of a lignin‐derived product exceeds the price of electricity, once all further downstream costs (i.e., for lignin isolation from the alkaline liquor, neutralisation, chemical transformation, product separation and purification) are accounted for. Despite recent advances, there are currently no widespread catalytic processes for the valorisation of Kraft lignins into bulk or fine chemicals. This fact can at least partly be attributed to the highly complex and condensed nature of the Kraft lignin, with a prevalence of highly recalcitrant linkages/bonding motifs in addition to a considerable sulfur content, an established catalyst poison. These properties of Kraft lignins render them challenging feedstocks for downstream catalytic valorisation.
Delignification of wood fibres can be regarded as a heterogeneous process in which lignin is “peeled” away from the residual lignocellulosic matrix via lignin depolymerisation.201 In the Kraft process, the wood fibres are treated with “white liquor” (a 1 mol l −1 NaOH and 0.25–0.70 mol l −1 Na2S aqueous solution) at temperatures of 165–175 °C. The process is maintained at this maximum temperature for 1–2 h, depending on the type of wood feedstock, the desired extent of delignification, and the exact digestion temperature.202 Throughout the pulping process, it is essential to ensure a liquor pH value >10, to avoid re‐deposition of lignin residues onto the remaining cellulosic fibres.203 The degradation and dissolution of lignin fragments from Spruce wood into the cooking liquor, as a function of time and programmed temperature, is displayed in Figure 4 a.188 The quantification of β‐ether units (as inferred from analytical acidolysis) of Kraft lignin from Pinewood, both isolated from the liquor and residual in the pulp (functions of time and programmed temperature), are displayed in Figure 4 b.190
Figure 4.

Reaction profiles for Kraft delignification of two native softwood biomass feedstocks: a) evolution of Spruce wood lignin into the liquor (green) as a function of cooking time and temperature, and; b) the quantification of β‐ethers in Pine wood lignin (quantified via analytical acidolysis), isolated from the liquor (light red) and residual lignin in the pulp (dark red), as a function of cooking time and temperature. For each graph, the programmed temperature (blue) increases steadily up to a fixed maximum of 170 °C.188, 190
The evolution of lignin into the liquor can be categorised into three approximate stages: initial (0–15 %), bulk (15–60 %) and final (60–90 %) delignification (shown for Spruce Kraft lignin in Figure 4 a).188 Analysis of the evolution of β‐ether content in Kraft lignins provides valuable insight into the design of future catalytic valorisation technologies. During initial delignification, the lignin dissolved in the liquor still has significant quantities of β‐ether units (approximately half of that estimated for the residual lignin in the pulp).193 On this basis, it is a reasonable assumption that the lignins following initial treatment will still exhibit good reactivity under mild conditions, as the subunits are linked largely via these relatively weak ether linkages. However, during the bulk delignification stage, the content of β‐ethers drastically decreases for both the liquor‐phase (Kraft) and solid‐residue lignins.190, 203, 204 At the final delignification point, both lignins exhibit approximately one seventh the content of β‐ethers present in the initial native lignin, as inferred from analytical acidolysis data.190 Recent HSQC NMR characterisation has confirmed that some β‐O‐4 and other bonding motifs (e.g., phenylcoumaran (β‐5) and resinol (β‐β) structures) are still present in a Kraft lignin, albeit indeed with very low abundance.196
β‐Ether units predominate in native lignins. Therefore, a substantial level of research has been devoted to understanding the fundamental aspects of β‐ether cleavage under the Kraft processing conditions. Detailed information regarding the mechanisms involved is contained in several recent book chapters.203, 204 Represented in Scheme 6 is a network of reactions underpinning the formation of recalcitrant, highly‐condensed and cross‐linked (C‐C) Kraft lignin from native feedstocks rich in β‐ether (C‐O) linkages.
Scheme 6.
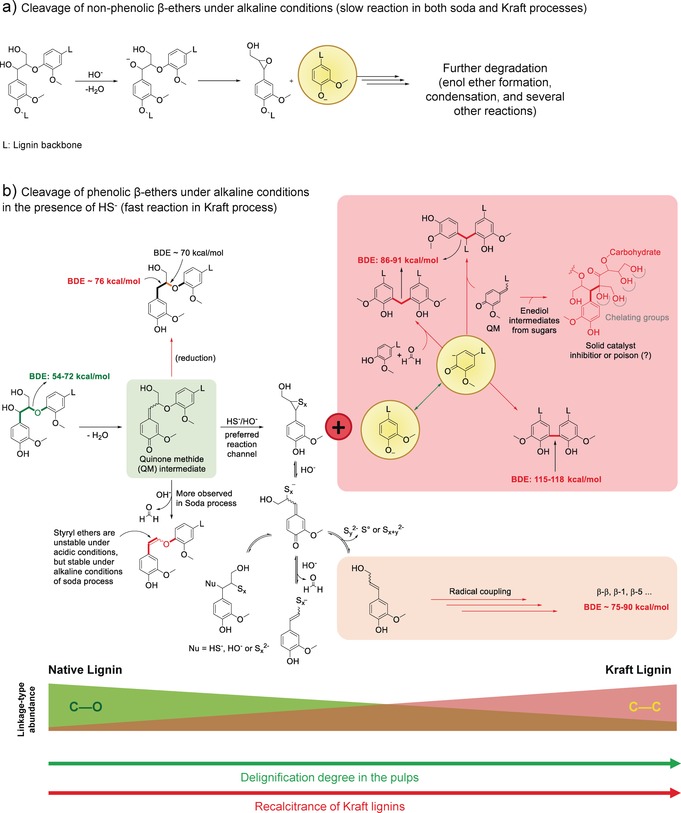
Reaction pathways for the conversion of β‐O‐4‐rich native lignins to recalcitrant and highly‐condensed/cross‐linked Kraft lignins via a quinone methide (QM) intermediate (shaded green). The Kraft lignins are characterised by C‐C linkages with high bond dissociation energies (86–118 kcal mol−1, shaded red).203 For clarity, the Scheme depicts only G‐units.
Kraft pulping shares one of its major depolymerising reactions with that of soda pulping, the cleavage of internal, non‐phenolic β‐ethers occurring in native lignins via an epoxide mechanism (Scheme 6 a). The second major depolymerising reaction, which is exclusive to the Kraft process, involves the trapping of quinone methide (QM) intermediates in an essential step in the cleavage of free‐phenolic β‐aryl ethers, including those that get produced by these non‐phenolic β‐ether cleavage reactions (Scheme 6 b).203, 205 Under the conditions of the Kraft process (HS−/OH−), the QM undergoes addition of nucleophilic HS−, followed by the elimination of a phenolate species via anchimeric assistance and formation of a thioepoxide.203 This is the particular reaction that explains the improvement seen upon introduction of sulfide (and the HS− produced during the process) to the earlier soda (NaOH‐only) process. In soda pulping, the retro‐aldol elimination of γ‐CH2OH as formaldehyde from the QM occurs more frequently. Such a transformation leads to styryl ether (sometimes called by the less specific term, vinyl ether) structures that are quite stable under alkaline conditions (but are prone to hydrolysis under acidic conditions).203 More importantly, the formaldehyde released in the soda process can react with any free‐phenolic guaiacyl unit (at its 5‐position) where subsequent o‐QM formation and condensation at C5 with another phenolic unit can result in additional condensation producing diphenylmethane structures (Scheme 6 b, shaded in red).206
The actual reactive species in Kraft pulping is not the sulfide (di)anion, but rather the hydrosulfide anion, HS−, as noted above. Partial oxidation of HS− generates polysulfide species (Sx 2−) that are assumed to promote one‐electron transfer reactions.204 However, the redox sulfur chemistry involved remains poorly understood. The sulfur‐containing lignin species may undergo a variety of subsequent reactions, leading to deoxygenation of the alkyl side chain. Subsequent radical coupling with lignin‐derived monomers partially regenerates oligomeric species (Scheme 6 b, shaded in beige). Under the harsh conditions of the Kraft process, the radical couplings are under thermodynamical control. Consequently, multiple alternative and highly stable C‐C cross‐linked structures (for example, β‐β, β‐1 and β‐5) are formed, replacing the C−O bonds found in native lignins.203, 207
Several studies have demonstrated the kinetic resolution of β‐ether diastereoisomers under Kraft (or soda) processing conditions. As can be readily predicted from the anti‐elimination mechanism whereby the nucleophile, ‐S−, must attack Cα from the opposite side of the O‐aryl leaving group, the erythro isomers were found to be cleaved faster than threo isomers by studies performed on both model compounds and true lignocellulose.188 As a result, threo isomers of the β‐ether units will predominate in the residual Kraft lignin.188
The reduced β‐ether structures, containing the deoxygenated α‐CH2 groups (Scheme 6 b), are relatively stable under the Kraft pulping conditions.203 These structures are thus a valid target for catalytic processing via hydrogenolysis, as they are more refractory than the original β‐ether units towards acid‐catalysed hydrolysis. The high BDE values (76 kcal mol−1) for such deoxygenated bonding motifs (approximately 20 kcal mol−1 higher than the most labile β‐O‐4 linkages) suggest that they will be far more resistant to (homolytic) cleavage.178
Under the harsh conditions of the Kraft process, the free‐phenolic guaiacyl units are prone to undergo multifarious repolymerisation processes (Scheme 6 b, region shaded in red).203, 205 Accordingly, there is a noticeable increase in molecular weight of lignin fragments in the liquor (i.e., those peeled from the lignocellulosic matrix) measured at the three stages (initial, bulk and final) of delignification (Figures 4 and 5).191 As noted above, formaldehyde, liberated by the elimination of γ‐CH2OH groups, plays a critical role in the repolymerisation process (see Section 3.4.4) but this formaldehyde elimination is markedly less problematic in Kraft pulping than in the soda process.203 In addition, lignin condensation reactions (some involving QM intermediates), the cross‐condensation of lignin fragments with reducing sugar end groups on polysaccharide polymers, oxidative coupling of phenolic guaiacyl units forming biphenyl (5‐5‐linked) structures, and radical cross‐coupling processes are also viable reaction channels creating refractory structural motifs.203, 207 Regardless of the exact mechanism, the repolymerised oligomeric and polymeric lignin fragments are characterised by very strong, highly recalcitrant C−C linkages (BDE: 70‐118 kcal mol−1).178
Figure 5.
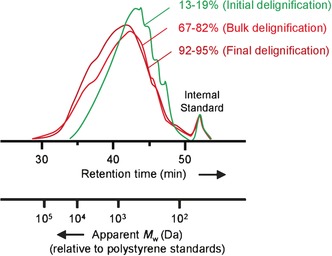
Superimposed gel permeation chromatography (GPC) traces of the black liquor obtained following varying extents of delignification from Pine wood.190
Repolymerisation of lignin fragments during the Kraft process, therefore, poses not only kinetic challenges associated with catalysis, but also impedes the overall thermodynamic efficiency of downstream processing. In fact, the thermodynamic costs associated with breaking stable C−C bonds will invariably be 30–60 kcal mol−1 higher (Scheme 6 b) than those required for the cleavage of β‐ether linkages in native lignins. Thereby, it is highly desirable to “tune” the Kraft process in order to avoid recondensation of the lignin fragments. Unfortunately, this is not a trivial task, due to the multitude and chemical variety of repolymerisation channels.205 Alternatively, continuous extraction of monomeric and oligomeric species from the black liquor could be an effective strategy, and would alleviate problems associated both with the self‐condensation of lignin and the condensation of lignin with hemicellulosic sugars.208 The removal of soluble carbohydrates is also desirable so as to avoid problems in subsequent downstream processing. As a case in point, it was recently shown that the presence of carbohydrates in the feed exerts a negative impact on the HDO reaction of guaiacol, taken as a model compound for lignin‐derived phenolics over Ru/C catalysts.209
In conclusion, although modification of the already highly optimised Kraft process is conceivable, it is important to consider that, at least at present, high‐quality cellulose fibres remain as the primary target because of their value (Kraft pulp global prices at 600–800 US$/ton as of January 2016).210 Accordingly, a productive line of investigation would be to devise an optimised system for maximising both the cellulose pulp quality and the quality of lignin (i.e., improving its susceptibility towards mild downstream valorisation), rather than exclusively focusing on the development of improved catalysts for treatment of highly‐condensed Kraft lignins. In fact, most technologies for (reactive) lignocellulose fragmentation have been developed with maximisation of the potential of the cellulosic fibres in mind, with lignin considered mainly as a by‐product, or worse, as a waste. However, there is a growing recognition that if future pulp and paper industry operations are to be commercially successful, valorisation also of the lignin fraction is mandatory.
2.6.2. Pulping with Organic Solvents—Organosolv Lignins
One of the serious drawbacks of Kraft pulping is the emission of malodourous organosulfur compounds.197, 204 This problem has long motivated the search for more environmentally‐benign alternatives. In this context, pulping with organic solvents was first reported in 1931 as an alternative to processes based on sulfurous chemicals.211 At temperatures of ca. 180 °C, the treatment of wood chips in aqueous ethanol (1:1 vol/vol) was demonstrated to be effective at releasing a major fraction of lignin and hemicelluloses into the solution, enabling the isolation of high‐purity cellulosic fibres.211 Despite this valuable finding, pulping in organic solvents remained a dormant field until the late 1960s, after which research activity intensified.212 At that time, the umbrella term “Organosolv” was established, in reference to the many variants of delignification processes performed in organic solvents.213 In this Section, properties of Organosolv lignins derived from treatment with and without added acid are discussed, owing to their importance for cellulosic biorefineries.214, 215, 216, 217 Organosolv processes performed under alkaline conditions have also been the focus of some investigations.212 However, their chemical features are similar to those of aqueous alkaline processes (e.g., the soda process, Scheme 6 a).
Organosolv processes have been commercially evaluated at pilot‐scale. One notable example is the Alcell process developed by Repap Enterprises Inc. (acquired by UPM‐Kymmene Corporation in 2000). The demonstration plant yielded over 5 000 tons of pulp from various northern hardwood feedstocks, generating consistent data, and the process was considered competitive with established Kraft pulping.218 Despite the great potential of the more environmentally benign Organosolv technology for pulping of lignocellulose, no process has yet survived longer than five years of operation at demonstration scale. Notably, as the installation of a Kraft mill represents a multi‐billion € investment, the replacement of well‐established technology is difficult and requires regulation to change the global industry. Moreover, unlike the Kraft process in which Kraft liquor is incinerated to recover the inorganics, in the Organosolv processes the incineration of the Organosolv liquor is prohibitive as it destroys the organic solvents used in the pulping process. This fact makes solvent recovery (and therefore lignin isolation) mandatory. Recent optimisation of the Alcell process has centred on the pretreament of plant biomass for enzymatic saccharification of cellulose.216, 219, 220, 221 In this context, Lignol Innovations Corporation was the proprietor of an integrated process involving solvent pretreatment of lignocellulose, saccharification, fermentation, and product recovery.222 Once again, the process was not brought to commercialisation. Tentatively, one primary cause may be the relative lack of high‐value applications to absorb the high‐quality, sulfur‐free isolated lignins, as suggested by techno‐economic analyses recently performed on Organosolv processes.223, 224
A variety of viable Organosolv solvent/water mixtures has emerged over the past 50 years. Typically, the organic solvent is a low‐weight primary alcohol (e.g., methanol, ethanol), a cyclic ether (e.g., 1,4‐dioxane, tetrahydrofurfuryl alcohol), a ketone (e.g., acetone), or a diol (e.g., ethylene glycol), in order to effectively dissolve the liberated lignin and hemicelluloses.212 Low‐molecular‐weight alcohols are favoured for their high volatility, and their consequent ease of removal after cooking. For improved delignification of the pulps, the organic solvent/water mixture must exhibit a Hildebrand parameter (δ) of approximately 23±2 MPa1/2, which corresponds to the solvent parameter of lignin.225 The organic solvent/water mixture plays at least two crucial roles in an Organosolv process:226, 227 1) the impregnation of the plant tissue (transferring the catalyst or reagent to the lignin through the polysaccharide matrix), and 2) the transport of the soluble lignin fragments from the matrix to the bulk solution.
Typically, Organosolv processes may operate at cooking temperatures of 180–195 °C, for a cooking duration of 30–90 min, an ethanol concentration of 35–70 % (wt/vol), and a liquor‐to‐solid ratio ranging from 4:1 to 10:1.212 Organosolv treatment typically results in extensive removal of lignin (>70 %) with minimum loss of cellulose (<2 %).212 Variants of the Organosolv process, performed with an added acid catalyst (e.g., HCl, H2SO4, oxalic acid, acetic acid, and formic acid) have been exploited for pretreatment of lignocellulose, in order to aid subsequent enzymatic saccharification of the cellulosic fraction.212, 228 Here, the pH of the Organosolv liquor is typically between 2 and 4, and at temperatures of 140–190 °C the lignocellulosic matrix undergoes a solvolytic reaction leading to partial or full “peeling” of hemicelluloses and extensive delignification. Again, cleavage of a fraction of the β‐ether linkages is essential to the delignification process.192 A proposed reaction network for degradation of lignin under acidic conditions is shown in Scheme 7.
Scheme 7.

a) Reaction channels (based on the acidolysis method) for the acid‐catalysed depolymerisation/degradation of lignin under Organosolv conditions, via “Hibbert's ketone” intermediates.205, 228 b) Inset highlighting measured concentrations of sum and individual species along the pathway of acidolysis of milled wood lignin (Pine), as a function of reaction time. For clarity, the scheme depicts only G‐units.
The reaction network is derived from analytical acidolysis data of lignins and model compounds. Acidolysis enables the abundance of β‐ether linkages to be semiquantified according to the known hydrolysis of arylglycerol‐β‐aryl ethers in the presence of HCl in 1,4‐dioxane/water (9:1 vol/vol).229 Such conditions resemble those used in some lignocellulose solvent pulping or acid treatment processes.212 The sum of compounds 3, 5, 6, 6′ and 10 (Scheme 7 b) is often used as the descriptor for β‐ether abundance.
Recently, acidolysis of lignin β‐ethers was revisited, in order to examine certain observed discrepancies in the rates of phenolic/non‐phenolic β‐ether hydrolyses (Figure 6).230 Hydrolysis of non‐phenolic model compounds (e.g., 1‐phenyl‐2‐phenoxyethanol highlighted in black in Figure 6) was approximately two orders of magnitude slower than for phenolic counterparts (shown in green or red in Figure 6), with 0.2 m aqueous H2SO4 at 150 °C. This observation similarly demonstrates the importance of selecting an appropriate lignin model compound for investigations regarding β‐ether cleavage. Clearly, the lack of the phenol moiety renders β‐ether species more recalcitrant and far less reactive towards hydrolysis. Therefore, it appears necessary for the model compound to incorporate a phenol or etherified phenol group para to the β‐ether alkyl chain (representing a lignin monomer at the end of or inside a chain, respectively) in order for the model to accurately reflect the reactivity of the β‐ether unit in a native or technical lignin.
Figure 6.

Graph highlighting relative rates of β‐O‐4 ether cleavage for phenolic and non‐phenolic model compounds, under 0.2 m aqueous H2SO4/150 °C conditions. Adapted with permission from ACS Sustain. Chem. Eng. 2014, 2, 472–485.230 Copyright 2014 American Chemical Society.
The reaction pathways of phenolic/non‐phenolic β‐ether hydrolyses were similarly examined by using DFT calculations.230 The predictions showed that both the cleavage of the ether linkage and elimination of the γ‐CH2OH group (as formaldehyde) are energetically feasible in the acidolysis of phenolic β‐O‐4 alkyl aryl ethers. With increasing acidity of the liquor, elimination of the γ‐CH2OH moiety (forming intermediate 2′ Scheme 7 a) begins to become the predominant mechanism; equimolar quantities of competing 2′ and intermediates 3–8 were formed by subjecting phenolic β‐ether model compounds to 0.2 m aqueous H2SO4/150 °C conditions for 2 h.230
The in situ evolution of formaldehyde may facilitate repolymerisation (condensation) of lignin fragments, affording diphenylmethane structures (see also Section 3.4.4). Critically, this side‐reaction is pH dependent.231 Repolymerisation occurs at fast rates outside the pH window of 2–7. Therefore, for relatively mild acid‐catalysed Organosolv processes, formaldehyde‐induced repolymerisation of phenolic fragments may play a less significant role as a repolymerisation channel, compared to Kraft, soda or base‐catalysed Organosolv processes. However, experimental evidence supporting this assumption is still pending.
Importantly, compound 3 in Scheme 7 belongs to a family of compounds known as “Hibbert's ketones”. Intermediates 4–8 are formed via tautomerisation and hydride transfer. Tautomerisation of 3 may also convert the keto group into a γ‐aldehyde group. The co‐existence of all these species at varying equilibrium concentrations substantially increases the complexity of the system, with respect to elucidating either further depolymerisation or repolymerisation processes.232
Examining the acid‐catalysed Organosolv depolymerisation reaction network presented in Scheme 7, it is apparent that the sum of products derived from the cleavage of β‐ether structural motifs (Scheme 7 b) reaches a maximum shortly after one hour, whereupon it begins to steadily decrease in concentration. The disappearance of specific phenolic monomers represents a primary and recurring problem for cross‐laboratory reproducibility of acidolysis experiments.32, 229 The duration of the acidolysis procedure should be optimised on a sample‐by‐sample basis, owing to the variability of lignin composition in terms of H, G, and S‐units. Consumption of these intermediates occurs as a result of condensation reactions involving the Hibbert's ketones. In wood pulping, the condensation of lignin fragments can occur either with lignin still immobilised on the lignocellulose matrix, or in solution involving the lignin fragments detached from the plant tissue. Therefore, condensation processes of this type occurring in lignocellulosic feedstocks will inhibit delignification.
Recently, the early‐stage conversion of Hibbert's ketones through a Raney‐Ni‐catalysed hydrogen‐transfer reaction (using 2‐propanol as a hydrogen‐donor and solvent) was shown to substantially suppress the repolymerisation processes.198, 233 As a result, the lignin stream was obtained as a viscous oil rather than a solid. Such hydrogen‐transfer reactions are discussed in Section 2.7. More recently, the protection of the aldehyde intermediates as acetals (using a 1,2‐diol, e.g., ethylene glycol as a protective group) confirmed the importance of stabilising these intermediates for improved mono‐aromatics yields (from acidolysis of Walnut dioxosolv lignin using triflic acid as the catalyst, see Section 3.3.3 for further details).234 These two pieces of evidence indicate that the fractionation of lignocellulosic biomass and valorisation of lignin can mutually benefit from a firmer understanding of the complex chemistry underpinning Organosolv processes, and from trapping/passivating reactive intermediates. In addition to the reactivity of the lignin fraction, hemicelluloses undergo varying extents of hydrolysis during acid‐catalysed Organosolv processes. Ordinarily, removal of the organic solvent by distillation under reduced‐pressure suffices to cause precipitation of water‐insoluble lignin fragments, whilst hemicellulosic sugars and oligomers remain in the aqueous solution.223 Nevertheless, distillation of the organic solvent is invariably an energy‐intensive step.224, 235 The “Organocat” process seems to overcome this constraint, whereby fractionation of lignocellulose occurs via the initial oxalic/formic acid‐catalysed shell‐peeling of lignin and hemicelluloses, in a biphasic 2‐methyltetrahydrofuran/water system.236, 237 Lignin and hemicellulosic fragments are thus immediately partitioned into organic and aqueous phases, respectively, upon liberation from the lignocellulose matrix. In this manner, the cellulose could be isolated as a pale yellow solid.236, 237
Organosolv processes may also be performed in the absence of an added acid, with good delignification results.[212, 238. 239] For such processes, the deacetylation of hemicelluloses suffices to bring about a pH decrease from 7 to ca. 4, initiating acid‐catalysed solvolysis of the most labile β‐ether linkages.198 The proportion of hemicellulosic hydroxy residues occurring as acetyl groups is approximately 1 % in softwoods, and between 3 and 6 % in hardwoods and perennial grasses. Due to the in situ evolution of acetic acid, the process may be regarded as autocatalytic.239 Importantly, under close to pH‐neutral conditions, lignin may undergo solvolysis via radical‐type reactions, in addition to acid‐catalysed transformations.240
Organosolv processing without added acid is often assumed to afford technical lignins retaining the majority of native β‐ether linkages.241 In this context, in the chemical literature dating from the 1960s to the mid‐1990s, it had been assumed that cleavage of non‐cyclic α‐O‐4 linkages was the primary explanation for solvolytic release of lignin fragments.212, 228, 238, 242 However, it is important to revisit these claims in light of the current understanding that α‐O‐4‐type bonding motifs are cyclic and mostly associated with phenylcoumaran and dibenzodioxocin bonding motifs.31, 155 Therefore, the cleavage of uncommon or even non‐existent non‐cyclic α‐O‐4 linkages should make a minimal contribution to lignin depolymerisation. Notably, full scission of true α‐O‐4‐containing bonding motifs should involve concomitant breaking of several other even stronger structural elements to result in depolymerisation. For instance, depolymerisation at phenylcoumaran motifs is only effective when both the α‐O‐4 (BDE=50‐56 kcal mol−1)180 and Cα−Cβ (BDE=54‐63 kcal mol−1)180 bonds are cleaved. Fragmentation of lignin at dibenzodioxocin linkages, as a result of cleavage of α‐O‐4 and β‐O‐4 bonds, will bring about a reduction in lignin branching (in the absence of recondensation). Furthermore, comprehensive lignin depolymerisation at dibenzodioxocin structural motifs also demands the breaking of strong and recalcitrant biphenyl 5–5 linkages (Scheme 5) that are likely to persist following the relatively mild treatment conditions of an Organosolv process with or without added acid catalysts.
Typically, the content of β‐O‐4 linkages can be assessed by analytical protocols based on chemical degradation (e.g., acidolysis, thioacidolysis, DFRC, and several others).32, 229 Unfortunately, there is little comparative data relating to the quantities of β‐O‐4 linkages remaining in lignin streams following various Organosolv processes. In a standalone report of β‐ether cleavage in an Organosolv process with added acid, the abundance of remnant β‐ethers in the isolated lignin was found to decrease dramatically in line with the delignification degree (cf. Figure 4 b for Kraft lignins). At 90 % delignification, only one quarter of the original β‐ether abundance remained in the isolated lignin—a value nearing those found for certain lignin streams from Kraft pulping (at the final stage of delignification), where just 10–15 % of bonding motifs can be inferred from the acidolysis products to be β‐ethers.190
As will be described in further detail in Section 3.2 “Catalytic downstream processing strategies”, the yield of monomers obtained from depolymerisation of lignin is often directly correlated with the abundance of β‐ether units in the lignin stream. Considering that retained β‐O‐4 linkages in the isolated technical lignins are (likely) the most reactive motifs towards depolymerisation, and that some Organosolv lignins show fractions of these linkages comparable to Kraft lignins, the blanket assertion that Organosolv lignins are always easier to depolymerise than lignin streams derived from Kraft process is, therefore, an erroneous generalisation. It is thus crucial to know the severity of the Organosolv process (see Figure 3). Ultimately, if lignin valorisation is tenable, techno‐economic modelling may be useful to determine whether the added value in keeping the lignin's β‐ethers can even offset a slightly lower sugar yield, i.e., whether the process can be better balanced for the value of all of the products. Similar caution should also be exercised for lignin‐enriched residues obtained from enzymatic saccharification of pretreated lignocellulosic feedstocks. The chemical nature of these lignins will depend strongly on the severity of the pretreatment method undertaken to enable the enzymatic saccharification (e.g., the presence of added acids or bases, temperature or process duration). Therefore, drawing general conclusions regarding the reactivity of such lignin streams is not possible without a detailed and thorough analysis employing HSQC NMR and chemical degradation protocols for each such lignin stream.
2.6.3. Other Fractionation Methods Based on Acid Catalysis
In addition to the more mainstream lignocellulose pulping and fractionation processes (e.g., Kraft and Organosolv) described in some detail above, in recent years research attention has been directed toward novel fractionation methods.201 Such methods typically target the isolation of pure cellulose, via controlled and mild delignification, for subsequent depolymerisation into glucose (saccharification), or perform the saccharification directly, obviating the need for enzymes. In contrast to the Kraft process (and some Organosolv processes) that expose the biomass to harsh, energy intensive conditions and generate waste streams, these novel approaches afford high yields of sugar monomers/dimers at low temperatures (100–180 °C) in the absence of costly and unrecyclable polysaccharidase enzymes.201
The isolation of pentose and hexose sugars from a packed bed of biomass (Corn stover, Maple wood, Loblolly Pine) has been recently demonstrated, by using the promoting effects of γ‐valerolactone (GVL) for mild aqueous acid hydrolysis.243, 244, 245, 246 High selectivity to different sugars obtained under varying conditions was attributed to the more facile hydrolysis of hemicelluloses relative to cellulose. By introducing a temperature gradient to the acid‐catalysed flow reaction, hemicelluloses were hydrolysed and isolated in an early fraction (150–180 °C), and cellulose in a latter fraction (180–220 °C), enabling separation of xylose (the main monomeric sugar from hemicelluloses) and glucose (from cellulose). Removal of the GVL (e.g., by phase separation upon adding liquid CO2 or NaCl), yielded a sugar stream concentrated at up to 127 g L−1 (i.e., 65 to 85 % of the highest concentrations obtained by enzymatic hydrolysis).247 The lignin fraction is also depolymerised in this process and can be separately isolated. As determined by 2D HSQC NMR, this lignin stream shows structural features similar to analytical lignins due to the low severity conditions employed for extraction (120 °C, 30 min, 80:20 wt % GVL:H2O). As an example of upgrading the lignin, a two‐stage hydrogenolysis process (first‐stage: 10 % lignin, 80 % THF, 8.5 % H3PO4 and 1.5 % H2O at 150 °C; second‐stage: solvent is replaced by heptane and temperature increased to 250 °C), over a Ru/C catalyst in an H2 atmosphere was reported. Up to 48 % of the carbon of the original lignin intake could be converted into mono‐aromatics that could then be extracted into a heptane solution, with methanol acting as a capping agent to form carboxylate esters.246
Solvent‐free, mechanocatalytic deep depolymerisation of polysaccharides has also been explored, beginning with either cellulose or crude lignocellulosic biomass.248, 249, 250, 251, 252, 253, 254 Here, the mechanocatalytic treatment affords a water‐soluble, depolymerised lignocellulose. The saccharification of the water‐soluble products renders high sugar yields (e.g., 88–92 % glucose, 3.5–8 % cellobiose, 93–98 % xylose relative to glucan and xylan fractions, respectively) and leads to precipitation of the lignin fraction as a sulfur‐free solid.248, 249, 250, 252, 253, 254 The lignin fractions isolated from mechanocatalysis of different biomass species (Pine, Beech and Sugarcane bagasse) closely resemble the lignins obtained via Organosolv processes, as determined by HSQC NMR analysis.249 Although mechanocatalytic processes are typically associated with the high energy costs for the ball‐milling operation, the energy requirement per kg of biomass diminishes drastically upon scale‐up from 1 g to 1 kg, demonstrating that this process might be suitable both energetically and economically.253
A further possibility is to employ protic ionic liquids (ILs) as a (“catalytic”) solvent, affording another variant of the Organosolv process denoted the “Ionosolv” process.255, 256, 257, 258, 259, 260, 261 As cellulose is insoluble in the new protic ILs, the process contrasts starkly with earlier methods for acid‐catalysed depolymerisation of cellulose in dialkylimidazolium ILs.262, 263, 264, 265, 266, 267 Accordingly, the acidic IL solvent acts specifically on lignin and hemicellulose. Delignification of lignocellulosic biomass (Miscanthus giganteus) is achieved at 120 °C by the cleavage of β‐ether units268 employing, e.g., 1‐butylimidazolium hydrogen sulfate or triethylammonium hydrogen sulfate, as a solvent.255, 256, 257, 258, 259, 260, 261 The lignin fraction dissolves in these ILs, and can be precipitated by the addition of water. Adopting this procedure, it is possible to recycle the IL for successive fractionation cycles. Effective application of ILs towards biomass valorisation has been previously hindered by (amongst other reasons) the high costs of IL precursors and synthesis, the derivatising nature of the IL, and difficulties regarding the separation and recycling of the IL.256, 269, 270, 271 However, triethylammonium hydrogen sulfate shows production costs close to those of conventional organic solvents and can be recycled in the process.259 Moreover, cellulosic fibres can be easily recovered by filtration as cellulose is insoluble in these ILs.
2.7. Early‐Stage Catalytic Conversion of Lignin as a Strategy for Biomass Fractionation
The Early‐stage Catalytic Conversion of Lignin (ECCL) or “Lignin‐first” strategy constitutes the backbone of emerging technologies for lignin valorisation. ECCL involves the concurrent extraction and catalytic conversion of the lignin fragments released from plant biomass in a one‐pot process. Employing heterogeneous catalysis in the fractionation of lignocellulose may fully alter the way that lignin is considered within current biorefinery schemes.201, 272 As illustrated in Scheme 8, current research into lignin utilisation is mostly devoted to depolymerisation of (repolymerised) lignin wastes, which are unavoidably generated by wood pulping (Kraft lignin and lignosulfonates) or cellulosic ethanol production, in addition to the catalytic upgrading of the low M w aromatics obtained. Strikingly, the emerging processes based on ECCL circumvent the inefficient sequence of depolymerisation (cleavage of weak C−O bonds), repolymerisation (formation of strong C−C bonds), and depolymerisation (through the cracking of C−C bonds formed in the previous step).
Scheme 8.

Process chains for valorisation of lignin isolated from conventional fractionation processes and from the emerging catalytic upstream biorefining processes based on Early‐stage Catalytic Conversion of Lignin (ECCL).
As described previously regarding the reactivity of lignins in both Kraft and Organosolv processes, β‐ethers are the primary target for depolymerisation of native lignins. Research and development in catalytic upstream biorefining processes based on ECCL have focused on chemical reduction of lignin fragments upon their removal from the lignocellulosic matrix. Such methods typically afford a highly‐aromatic lignin‐derived stream (whereby the most reactive functional groups have been deactivated through catalytic reduction, e.g., conversion of aldehydic intermediates into alcohols, and hydrodeoxygenation of ketones to methylene groups) and a holocellulose stream, as two distinct, stable and easily‐separable fractions.198 The two predominant approaches for such upstream catalytic processing of lignin under mild conditions are hydrogenation and deoxygenation reactions, using either a noble metal‐supported catalyst273, 274, 275, 276, 277, 278, 279, 280 or inexpensive Ni catalysts (notably, Raney‐Ni).198, 281, 282
ECCL performed on Birch wood sawdust (Ru/C catalyst, 3 MPa H2, 250 °C) has been investigated as a method to tune the alcohol functional group content of lignin oils. The carbohydrate fraction is retained as a pulp that is conducive to further upgrading, and the lignin fraction is collected separately as a highly aromatic oil containing up to 50 % of the carbon intake as mono‐aromatics.276 The synergistic use of Pd/C and ZnII (under an H2 pressure of 3.8 MPa at a temperature of 225 °C) has been also demonstrated as an effective method for the depolymerisation of genetically modified Poplar wood lignin (rich in S‐units), whilst retaining 95 % of the carbohydrate fraction.278 Increasing the proportion of S‐units in the lignin structure resulted in a higher yield of cleavable linkages under low severity conditions, and a correspondingly higher yield of monomer aromatic compounds, which was also observed in the Ru/C system.277, 278 Three reasons were proposed for this result. First, higher‐S lignins contain higher levels of β‐ethers. Second, the high concentration of S‐units minimises lignin re‐condensation, due to a lack of unsubstituted positions ortho to the phenol on the syringyl moiety. The third reason is the relative scarcity of G‐units, which reduces the complexity of the condensed units in lignin. There might also be reduced interconnectivity of the biopolymers within the lignocellulosic matrix, as G‐units have been suggested to have a higher propensity to crosslink with hemicelluloses.283 Catalytic delignification of high‐S wood is one prime example of the potential of the combination of genetic engineering and upstream biorefining based on ECCL.
Although molecular hydrogen is the cheapest source of hydrogen reducing equivalent, employing gaseous molecular hydrogen (H2) for hydrogenation places constraints on the process (e.g., reactor wall materials, solid feed systems, safety protocols). These limitations are circumvented by using a solvent capable of undergoing a hydrogen transfer reaction instead, for example secondary alcohols198, 281, 282 or formic acid.273, 274, 279 Hydrogen‐transfer strategies of this type have been demonstrated as effective methods for upstream processing of lignocellulosic materials under conditions of lower severity (180–200 °C, autogeneous pressure),233, 284, 285 when compared with aforementioned methods.
Importantly, to realise the full economic potential of a lignocellulose biorefinery that does not create unnecessary waste streams, the holocellulose fraction must also be valorised. Deconstruction of lignin may afford a solid carbohydrate pulp fraction, suitable either for undergoing full enzymatic hydrolysis into sugar monomers or for paper production (Figure 7).198, 275, 276, 277, 278, 282 Separation of the catalyst is important, so as to avoid contamination of the downstream products derived from the two streams. Furthermore, due to the strong possibility of poisoning by trace components in the lignocellulose feedstock, the catalyst must be relatively inexpensive. Recent investigations have centred on the use of Raney‐Ni, a magnetic catalyst.198, 282 This property allows for facile separation of the catalyst from both lignin oil and holocellulose streams. Holocellulosic fractions are obtained as predominantly catalyst‐free solids that are highly conducive to further downstream treatment (Figure 7 A).
Figure 7.

Holocellulosic fractions derived from two different Ni‐catalysed up‐stream processes: A) Raney‐Ni catalysed removal of lignin from Poplar wood chips (pulp conducive to further downstream processing198), and B) Ni/C catalysed removal of lignin from Birch wood saw dust (pulp not conducive to enzymatic hydrolysis281). Image (A) reproduced with permission from Angew. Chem. Int. Ed. 2014, 53, 8634–8639; Angew. Chem. 2014, 126, 8778–8783. Copyright 2014 John Wiley and Sons. Image (B) reproduced with permission from Energy Environ. Sci. 2013, 6, 994–1007. Copyright 2013 Royal Society of Chemistry.
The liquid‐phase extraction of lignin from Birch wood sawdust in alcoholic solutions has been reported, employing a Ni/C catalyst. Unfortunately, the recovered cellulose fraction could not be fully separated from the magnetic catalyst (Figure 7 B).281 Moreover, in the characterisation of the lignin fraction extracted from Birch wood, just three products were identified by GC characterisation.281 More recently, this process has been revisited by another research group,286 who obtained a product mixture that was more complex than previously reported. Moreover, the product spectrum and yield were demonstrated to depend heavily on both catalyst loading and biomass type and origin.286 As will be discussed in the next sections, high temperatures (300–450 °C) and elevated H2 pressures (MPa) are typically required for cracking the C−C bonds in technical lignins to produce low M w products in heterogeneously catalysed processes.287, 288 Conversely, strategies based on ECCL benefit from the intrinsically high reactivity of native lignins compared to condensed and therefore recalcitrant technical lignins. Indeed, from the solvolytically released lignin fragments, the ECCL directly produces monophenols and small oligomers (M w 100–400 Da).198, 277, 278 Notably, hemicellulose sugars released by solvolytic processes also play a key role in tuning the catalyst activity and selectivity, thus carrying major implications for the product distribution achieved by ECCL, as recently demonstrated for the process employing Raney Ni as a catalyst for H‐transfer reactions.[426] Moreover, the lignin oil products that are obtained can be upgraded under conditions of low‐severity similar to those employed in the conversion of phenolic model compounds of lignin and pyrolysis oil.289, 290, 291, 292 It is therefore clear that ECCL‐based strategies hold great promise for future lignin research.
3. Catalytic Downstream Processing of Isolated Lignin
3.1. Economic Considerations
When considering prospective applications of isolated lignin streams, the volume of lignin that such an application is able to absorb is of primary importance. Logically, where supply greatly exceeds demand, the large surplus will place a severe pressure on the market price, which may in turn put the profitability of the wider lignin valorisation process at risk. The glycerol market serves as an example of the disadvantages associated with dramatic oversupply. In fact, the prices of purified glycerol and crude glycerol were reduced by 50 % and 80 %, respectively, following the worldwide implementation of fatty‐acid transesterification for biodiesel production.293
It has long been recognised that there is a need for both “high‐volume and low‐value” and “low‐volume and high‐value” applications in order to achieve full economic use of isolated, technical lignins.294, 295 Seven categories of value‐added products from lignin, initially proposed by Glasser about 30 years ago, still remain relevant to this day: i) oil field chemicals; ii) agricultural chemicals; iii) asphalt extenders; iv) carbon black; v) adhesives; vi) engineering plastics and vii) specialty dispersants.294
Annually, more than 130 million tons of lignin are currently liberated in the paper‐and‐pulp industry, although at present only a very minor proportion is isolated and available.295 The figure for liberated lignin significantly increases when biorefining for transportation fuels is taken into account. For example, a biorefinery process producing ethanol from corn‐stover liberates approximately 0.5–1.5 kg of lignin per kg ethanol.296 Considering that about 40 % of the lignin‐rich residue would suffice to cover the heat and power demand for bioethanol production (including biomass pretreatment and ethanol distillation), it is clear that lignins released from the production of cellulosic ethanol will add to the already enormous pile of under‐utilised technical lignins that is produced (and in some cases isolated) from Kraft mills.
Vanillin, at present the only chemical commercially produced from lignin by oxidation of lignosulfonates, has a market volume of about 20 000 tons (Table 4).297, 298 With over 90 % of the synthetic vanillin used today being mineral oil‐derived, there is room for growth of lignin‐derived vanillin, considered closer in flavour/taste to natural vanilla extract than petrochemical guaiacol‐derived vanillin. However, the limited total market volume does not suffice to absorb the entire lignin output from biorefineries. By contrast, phenol is produced in annual quantities of approximately 8 million tons, whilst the mixture benzene, toluene and xylenes (BTX, precursors for most petrochemical aromatics, Table 4), is produced at 80 million tons per year.299 These markets hold the potential to employ a sufficiently large proportion of the lignin liberated from the paper‐and‐pulp and transportation fuel industries, so as to render the wider lignin valorisation process worthwhile.
Table 4.
Comparison of various potential products from lignin in terms of market volume, price and the maximum gravimetric yield from lignin, assuming a linear polymer of G monomer units at 196 g mol−1. Although market prices are in constant fluctuation, the data nevertheless serves to compare approximate sizes and yields of prospective lignin industries. Any residual carbon is assumed to be used for reforming to hydrogen.
| Compound | Volume (103 kg y−1) | Price ($ kg−1)[a] | Maximum theoretical yield from lignin (wt %) | H2 produced (kg per kg product) | CO2 produced (kg per kg product) |
|---|---|---|---|---|---|
| Benzene | 80 000 000 (combined) | 1.49 | 40 % | 0.181 | 2.254 |
| Toluene | 1.38 | 47 % | 0.088 | 1.433 | |
| Xylene | 1.36 | 54 % | 0.019 | 0.829 | |
| Phenol | 8 000 000 | 1.54 | 48 % | 0.172 | 1.871 |
| Vanillin | 20 000 | 10–15 | 78 % | 0.066 | 0.579 |
| 4‐Propylguaiacol | none | no (current) market | 85 % | −0.036 | 0.000[b] |
[a] Benzene, toluene, xylene and phenol prices are FOB U.S. Gulf as of 30th October 2012. For reference, the oil price (WTI future for delivery December) was $85.66 per barrel on that date. [b] Excluding carbon sources required to produce external hydrogen.
Potential approaches to producing value‐added products from lignin can be broadly divided into three sub‐categories: 1) direct use or as precursor for material applications; 2) as a feedstock for (drop‐in) transportation fuels; and 3) as a raw material for commodity or high‐value chemicals. Considering material use, the macroscopic or microscopic properties of the (technical) lignins must be acceptable without significant treatment, although some modifications may be exacted by physical or chemical treatment. In this manner, although the properties of technical lignin here constrain the number of possible applications, no further depolymerisation is required and processing is thus simpler.
The application of lignin in phenol‐formaldehyde resins is a typical example of this approach. Up to 50 % of the phenol content in this material can be replaced by lignosulfonate, Kraft, or Organosolv lignin without significantly compromising properties of the resin.300, 301, 302 Similarly, Kraft lignin may be oxypropylated and then used as a polyol component in the synthesis of rigid polyurethane foams.303 Lignin may also be employed as a feedstock for high‐quality carbon fibres, at present produced almost exclusively from poly(acrylonitrile).29 Although this application is beset by problems associated with the necessity to melt‐spin the precursor efficiently and rapidly convert it into carbon fibres at the carbonisation/graphitisation stage (requiring lignins with low polydispersity), there has nevertheless been considerable progress in this field.304 More unusual applications include the incorporation of lignin in water‐purification membranes,305 as a composite in battery cathodes,306 and as a starting material for the synthesis of highly porous carbon,307 which, for example, may find application as anode materials in lithium‐ion batteries.308 For more information on the materials application of lignins, the reader is directed to recent reviews.28, 309, 310
Fuel production from lignin requires both further depolymerisation of the technical lignin, and subsequent upgrading of the phenolic stream. Lignin has both the highest energy density and the lowest oxygen content of the three major components of lignocellulosic biomass, rendering it an attractive starting material for the production of transportation liquid fuels. The primary goal is, therefore, to generate a relatively volatile fraction with reduced oxygen content, whilst simultaneously retaining the energy content and limiting the quantity of hydrogen required for the catalytic upgrading. Interestingly, complete reduction towards alkanes may not be necessary. Research on the combustion properties of lignin‐derived small molecules suggests that cyclic alkyl alcohols and aromatic oxygenates could also find application as drop‐in fuel components, particularly in gasoline and diesel blends.311, 312, 313 However, this type of application is still in development, requiring further improvements to minimise soot and gas emissions.
Catalytic pyrolysis of biomass314, 315, 316, 317 and further catalytic upgrading of the pyrolysis oils19, 318, 319 have been extensively studied, typically employing an acidic zeolite (e.g., H‐USY and ZSM‐5) catalyst. A large quantity of catalyst is typically required, making catalyst stability of paramount importance. Deactivation occurs via extensive coking, leading to pore‐blockage and encapsulation of the active sites. Burning of the coke, followed by acid treatment, may partially restore activity.320, 321 Catalytic lignin pyrolysis efficiency will, in general, only be influenced to a limited extent by precise structural properties of the lignocellulosic feedstock. As the focus of this review is on relating lignin structure/bonding to susceptibility to valorisation through catalytic methods, catalytic pyrolysis strategies will not be covered in further detail here and the reader is referred to other reviews (see Table 1).
Catalytic depolymerisation in the liquid phase may provide more flexibility in terms of using lignin as a feedstock for both fuels and chemicals. Being the only primary component of lignocellulosic biomass containing aromatic subunits, production of aromatic (bulk) chemicals has, from the start, been an obvious and attractive route for lignin valorisation (Table 4). There are multiple potential aromatic targets, differing in the degree of chemical complexity and (correspondingly) production volume. The simplest products are BTX mixtures, which, as described previously, are an important feedstock for a wide array of petrochemical processes with a large market. Full hydrodeoxygenation of lignin streams to BTX may also facilitate the separation of products by fractional distillation as practiced in the oil refinery.284 Despite the potential to absorb large quantities of technical lignin, BTX may not be the best target for lignin valorisation as chemicals. In the interest of atom‐economy and energy efficiency,322 approaches that fully hydrodeoxygenate biogenic molecules, rendering (aromatic) hydrocarbons as products, which again need to be oxidised to afford the desired end‐products, should be considered as the preferred route in extreme cases only (i.e., to facilitate product separation,284 or for a lack of any other route for the production of a desired chemical).323 It has previously been recognised that redox‐neutral reactions performed on plant carbohydrates are desirable so as to preserve the intrinsically high functionality.322 A similar argument should be advanced for the valorisation of lignins.
For the BTX strategy, it even remains to be seen if attractive price premiums can be realised. For example, in October 2012 the benzene price was $1.49 kg−1 (FOB USG), and the price of high‐purity isolated lignin, though difficult to establish, is estimated at $0.25–0.50 kg−1.324 Nevertheless, it must be noted that, if lignin is idealised as a linear polymer of guaiacylglycerol (G units) with a monomer molecular weight of 196 g mol−1, the maximum gravimetric toluene yield is just 40 %. Although the remaining 60 % weight can, in principle, be employed towards the co‐generation of (at least) methanol, additional expenditure associated with sacrificial reagents (i.e., hydrogen and protective groups) and other operating costs serve to drive down potential profit margins.
Notably, both the atom efficiency, energy efficiency and cost competitiveness of obtaining chemicals from lignin can be improved when higher value bulk chemicals (e.g., phenol, cresols or adipic acid) can be directly obtained from lignin, circumventing the need to synthesise these compounds from a lignin‐derived BTX mixture. Occupying positions further up the value chain are synthetic precursors for pharmaceutical, agrochemical or other specialty applications. In fact, such high‐value precursors may be targeted in an initial mild lignin conversion strategy, prior to the application of harsher conditions in order to obtain higher‐volume, lower‐value bulk chemicals (e.g., BTX). In all discussions on chemicals production, it must be recognised that prices of bulk chemicals are strongly influenced by current oil prices and their price volatility, and therefore, the economic feasibility of any lignin‐to‐chemical pathway must be assessed with these factors in mind.
3.2. Catalytic Downstream Processing Strategies
As discussed in the previous sections, polymeric lignin streams vary widely in terms of their chemical nature. Some Organosolv lignins, mainly those extracted without added acid catalysts,192 may retain a considerable fraction of the original native β‐O‐4 linkages, as suggested by HSQC NMR analysis,249, 251 and evidenced by chemical degradation methods.194 By contrast, for lignins from chemical pulping processes (e.g., Kraft, soda lignins and lignosulfonates) β‐O‐4 linkages represent fewer than 10 % of the connections.190 In addition, extensive condensation of lignin fragments yields strong and recalcitrant carbon–carbon bonds. As a consequence of the highly varied and complex chemical/bonding properties of different technical lignins, there is, unfortunately, no “one‐size‐fits‐all” solution for catalytic downstream processing of these polymeric technical lignin streams. Nevertheless, contemporary methodologies for lignin utilisation and valorisation can be broadly divided into two primary categories: 1) convergent approaches, and 2) stepwise approaches (Scheme 9).
Scheme 9.

Two alternative approaches to depolymerisation of low‐value lignin streams: a) A funneling scheme for the stepwise, convergent generation of a limited number of end products, exemplified here for benzene, via depolymerisation‐dealkylation‐hydrodeoxygenation (HDO); b) A stepwise approach whereby lignin is first depolymerised under mild conditions affording high‐value fine chemicals, and residual technical lignin is then treated by more harsh depolymerisation conditions to afford fine/bulk chemicals and transportation fuels (and remnant material is used as fuel to heat/power the process). For the funneling scheme, downstream products are not necessarily of higher value, although costs associated with separation are reduced.
Prior to individual discussions of the various depolymerisation strategies, it is valuable to consider the statistics of depolymerisation in order to estimate the maximum yields of mono‐aromatics from lignins. Statistically, the release of a monomer from a finite polymeric chain containing cleavable and non‐cleavable bonds involves the cleavage of two bonds.325 Hence, the maximum yield of a monomer can be estimated by Equation (1), as reported in Ref. 326:
| (1) |
where: Y represents the sum of individual yields of mono‐aromatics, n is the number of monomers occurring in the polymer chain, and P corresponds to the fraction of cleavable/targeted bonds (e.g., for lignin, β‐O‐4 linkages). It has already been noted that upon increasing the polymeric chain towards a (hypothetical) infinite value of n, the yield of monomers quickly converges to Y≈P 2 already from n≈10.326
Figure 8 displays a graphical representation of the sum of individual yields of mono‐aromatic compounds, obtained from the depolymerisation of an infinite chain of lignin, for various values of P, for which typical β‐O‐4 contents of several classes of lignin streams have been taken.
Figure 8.

Graph representing Equation (1) for various P values. For lignin with a low percentage of cleavable bonds, it is close to impossible to obtain high individual yields of products under low severity conditions. Lignin varieties with a high proportion of reactive linkages (β‐O‐4, or esters in genetically modified plants) are required for high‐yield depolymerisation to be possible.
It is clear from Figure 8 that a high fraction of β‐O‐4 linkages (or other readily cleavable linkages) is essential to achieving high values of the sum of individual yields of mono‐aromatics from lignins. Considering native lignins with P values in the range of 0.35 to 0.85, the theoretical mono‐aromatics yields can be anticipated to be between 10 and 70 %. This estimate agrees well with the fact that the catalytic upstream biorefining based on ECCL is conducive to afford high yields of monomer products.198, 273, 278 Conversely, downstream treatment of technical lignins with P values lower than 0.2 (such as Kraft and certain Organosolv lignins) will lead to mono‐aromatic yields of less than 4 % if only the easily cleaved bonds are targeted.277 Notably, lignins derived from genetically engineered plants may hold the potential for even higher yields of lignin mono‐aromatics in catalytic upstream processes based on ECCL, as such native lignins may be tuned to P>0.85 (e.g., high‐syringyl lignins).
Despite the many advances seen in catalytic depolymerisation, Figure 8 clearly illustrates that, under low‐severity conditions, the transformation of technical lignins into a limited number of mono‐aromatics at high yields is very challenging, owing to the structural complexity and recalcitrance generated in the fractionation of lignocellulose. Therefore, successful valorisation of technical lignins will depend on addressing this challenge and making effective use of the structural diversity of the depolymerised products. Harsher methods will typically generate a complex mixture of compounds, adding the complication of product separation, which will take considerable effort, both in terms of energy as well as the need for large distillation columns, or other separation setups. In fact, structurally similar compounds with comparable boiling points, such as alkylphenols, may not be readily separated by distillation. Alternatively, if the application allows, targeting instead a mixture of compounds with a well‐defined specification in terms of macroscopic properties (e.g., analogous to petroleum‐derived surfactants, lubricants and fuels), might be a useful method to deal with this complexity and a means to valorise technical lignins.
An alternative strategy is to reduce the complexity of such a mixture of mono‐aromatics and oligomers in subsequent processes that convert a large number of the different constituent components into the same, single target compound (or a limited number of them). Such a convergent, funneling approach, schematically represented in Scheme 9 a, is also particularly well‐suited to biological systems because catabolic pathways within microorganisms typically converge to a small number of metabolites. The selected, recent examples discussed below highlight the potential for the consolidation of bio‐ and chemocatalytic transformation as part of a convergent approach for the valorisation of lignin streams. As a thorough discussion of such biological methods goes beyond the scope of this review, the interested reader is directed towards an extensive review on this topic327 and other key literature.328, 329, 330
The conversion of a mixture of lignin‐derived phenols into polyhydroxy acids of various chain length was demonstrated by using Pseudomonas putida.331 Employing a genetically‐engineered strain instead, the wide range of lignin‐derived aromatics present in the alkaline liquor obtained from a pretreatment of corn stover could be converted into cis,cis‐muconic acid in high yield (67 %). The cis,cis‐muconic acid could then be easily hydrogenated to adipic acid, one of the monomers for Nylon‐6,6.332 The biocatalytic oxidation of lignin‐derived phenols thus markedly contrasts with (non‐biological) lignin oxidation, e.g. over chalcopyrite, whereby a mixture of acids with different carbon chain lengths is invariably obtained.328
The ability of a microorganism to degrade the macromolecular lignin polymer itself into smaller phenolic intermediates would bring considerable advantages. Amycolatopsis sp. and Rhodococcus jostii strains were found capable of both secreting laccases and peroxides (thus enabling the oxidative depolymerisation of lignin) and of catabolising the released phenolics, for example.329 Employing these strains, a lignin conversion of up to 30 wt % could be achieved affording intracellularly‐stored polyhydroxy acids or fatty acids. As the catabolic pathways of the stored products may also be further genetically modified, these bacteria represent excellent starting platforms for improved strains capable of producing other high‐value products.
Alternatively, in a stepwise or cascade approach (Scheme 9 b), depolymerisation of technical lignins is conceived to occur gradually over several stages. Catalytic upstream biorefining methods based on ECCL may also be incorporated into such a cascade. The initial mild depolymerisation step is designed so as to be highly selective to specific bonding motifs, targeting the production of highly‐functionalised molecules (e.g., fine chemicals). The high value of the products may still ensure the economic feasibility of such an approach even when catalysts, reagents and separation of products obtained at low individual yields are accounted for. Nonetheless, the costs associated with costly reagents, (co)catalysts (e.g., TEMPO), molecular hydrogen and hydrogen donors, oxidants other than oxygen and (solvent‐derived) capping agents will certainly impact the overall economics of the downstream processes. Indeed, similar to the efforts that are required (for example) in upscaling the synthesis of fine chemicals from laboratory to commercial scale, after the proof‐of‐principle technology has been established for promising mild lignin depolymerisation routes, considerable development efforts are still required to allow larger scale production, paying particular attention to the viability of employing certain chemicals, solvents and catalysts on a commercial scale.
In this context, the manufacture of vanillin lends valuable insight into the challenges associated with any industrial‐scale lignin valorisation process.297 The oxidation of spent sulfite liquor to generate vanillin was operated intensively throughout the 1960s, 1970s and 1980s. Despite a high value (historically $10–15 kg−1) and an inexpensive catalyst, solvent and reagent (NaOH, water and oxygen, respectively), almost all liquor‐to‐vanillin plants had ceased production by the early 1990s. The low vanillin yield (2.5 % under optimum conditions) and large sodium hydroxide requirements resulted in the intolerably high generation of organic‐containing “caustic liquor” (160 kg per kg vanillin), rendering the overall procedure costly and uncompetitive against then emerging petroleum‐to‐vanillin technologies. Only Borregaard Industries in Norway still operates a similar process (with a copper catalyst), which presumably significantly increases the efficiency and decreases the amount of waste. This example clearly urges researchers to also consider the use of their “waste” streams when developing new lignin valorisation methods. Otherwise, novel technologies may share the same fate of most of the lignin‐to‐vanillin processes.
Critically, the residual lignin obtained from an initial mild depolymerisation step will invariably exhibit a larger fraction of resilient bonding motifs, compared to the starting material. Accordingly, in the cascade further downstream processing of the residual lignin stream under more severe conditions may yield bulk chemicals (e.g., organic acids, phenolics, BTX) or additives to transportation fuels. Finally, any heavy residues that remain even after severe treatment may potentially be subjected to catalytic cracking, or substitute for the asphaltene fraction of crude oil, or be incinerated for the generation of heat and power. This combination of strategies ensures that the lignin feedstock is used to maximum benefit. The individual processing steps of such an approach must be designed in order to accommodate an increasingly degraded and condensed lignin structure, and to tolerate or separate impurities introduced in previous steps.
In light of the convergent and stepwise approaches, there is a clear requirement for a “toolbox” of chemical lignin depolymerisation methods, operating under different severity conditions. In the following sections, multiple methods for the depolymerisation of lignin are discussed. Examples are subcategorised based on severity and selectivity of the procedures into so‐called “mild” procedures, that target specific bonding motifs in lignins using highly selective catalysts and reagents, and “harsher” procedures that use a regime in which concurrent thermal and catalytic reactions may take place. Selected examples are limited to those that specifically highlight the relationship between lignin structure and catalysis. Logically, this relationship is more difficult to assess under harsh depolymerisation conditions. In these instances, the discussion will also focus on other process variables (among others solvent effects and catalyst stability). Regardless of the somewhat artificial classification of the examples discussed based on process severity, the insights obtained from either category should be taken into account when devising new approaches to lignin depolymerisation.
3.3. Mild Depolymerisation Strategies
In recent years, an array of chemical depolymerisation methods has been developed that selectively target specific bonds within the lignin polymer,232 the majority of which target the β‐ethers. As already discussed, the generalisation that β‐O‐4 linkages constitute the predominant type in all varieties of isolated lignin is perhaps one of the major misconceptions that has unfortunately been propagated across the literature in this field. Logically, strategies that have proven effective for the cleavage of β‐O‐4 linkages in model compounds will not translate well into the processing of technical lignins that comprise little or none of this structural motif in their backbone. Nevertheless, conditions suitable for β‐O‐4 cleavage in model compounds are likely to translate well into the processing of some lignin varieties where a significant proportion of β‐ether linkages has been left intact after upstream treatment. Selected pathways for the selective cleavage of β‐ethers occurring in lignin (represented by a simple model compound) are summarised in Scheme 10. For the strategies outlined below, attention is devoted primarily to providing an overview of methods available for mild depolymerisation rather than a detailed mechanistic description of the chemistry involved. For further mechanistic details, the reader is directed to a recent review article.333
Scheme 10.

A variety of strategies for selective cleavage of the β‐O‐4 lignin linkages (represented by a simple dimer model compound), via C−O cleavage (green), C−C cleavage (red) and benzylic oxidation (blue).
3.3.1. Mild Oxidative Depolymerisation Pathways
Several oxidative routes targeting different end‐products from lignin have been reported, and are discussed in detail in a recent review article.334 Indeed, a large number of these oxidation methods target the cleavage of the β‐ethers, of which a number are discussed in this section. A chemoselective and organocatalytic method for the selective oxidation of the secondary (benzylic) alcohol of lignin model compounds and Aspen lignin was demonstrated. The system employing a (2,2,6,6‐tetramethylpiperidin‐1‐yl)oxyl (TEMPO) derivative, with HNO3 and HCl, performed best under mild conditions (Scheme 11 a).335 Moreover, models incorporating free phenolic groups (a functional group responsible for many of the difficulties encountered in oxidative lignin valorisation attempts) could be selectively oxidised. 2D HSQC NMR showed that the approach could be extended to Aspen lignin, with most of the S‐ and all of the G‐units in Aspen lignin selectively converted into the corresponding benzylic ketones. As mentioned in the discussion on lignin structure, the C−O ether bond of the oxidised β‐O‐4 substructures is substantially weakened. Subsequent redox‐neutral formic acid‐mediated cleavage of the oxidised β‐O‐4 substructures led, in the absence of a reducing metal, to the formation of simple ketone, diketone and phenol derivatives as primary products (Scheme 11 a).336 Extension of the strategy to an oxidised Aspen lignin (isolated via a mild cellulolytic enzyme protocol, and therefore, rich in β‐O‐4 linkages) also proved successful. A soluble fraction of low‐molecular‐weight aromatics was obtained, accounting for up to 61 wt % of the original lignin input, of which approximately 85 % (i.e., 51 wt % based on original lignin input) could be identified, with the diketone products expected from the model compound studies making up the major fraction. By contrast, a lignin in which the secondary alcohol was not oxidised prior to the cleavage reaction, afforded low monomer yields, highlighting the importance of generating the benzylic ketones to weaken the vicinal β‐O‐4 ether linkages and allow redox‐neutral cleavage. Having established the proof‐of‐principle of stepwise oxidative activation/redox‐neutral cleavage with this elegant approach, several practical aspects, including scalability, choice of solvent and chemicals used for the oxidation and cleavage steps and further reduction of the complexity of the product mixture obtained, now warrant further attention.
Scheme 11.
Mild oxidative pathways for cleavage of β‐ether lignin model compounds, via selective oxidation of the secondary alcohol functional group. The “Ar” groups represent simple aryl functionalities. For methods expanded to actual lignins, the results are indicated below the reaction scheme.
Along the same lines, a two‐step lignin degradation strategy has been applied to a series of lignin model compounds. Selective oxidation of the benzylic alcohol was achieved using “Bobbitt's salt” ([4‐AcNH‐TEMPO][BF4]) followed by a reductive, photocatalytic cleavage step, rendering the overall process redox‐neutral (Scheme 11 b).337 The reductive cleavage of the oxidised β‐O‐4 moiety by single electron transfer was accomplished using a common photoredox catalyst, [Ir(ppy)2(dtbby)][PF6], operating under visible light and employing three equivalents of formic acid and the base N,N‐diisopropylethylamine (DIPEA). As photocatalytic protocols can be expected to be less efficient for darkly coloured solutions, such as those expected for lignin streams (commonly varying from reddish brown to dark brown/black), reactions were also performed with the stepwise addition of a lignosulfonate material. In a batch reaction, the photocatalytic conversion suffered from ineffective radiation, but in a flow reactor the yields of mono‐aromatic products were restored. Unfortunately, no mention of the fate of the lignosulfonate was made.337 The viability of this photocatalytic strategy now needs to be demonstrated with actual lignin feeds. From an economic perspective it should be mentioned that while overall redox‐neutral, the coupled two‐step process still generates much stoichiometric waste. This, together with the costs associated with the expensive reagents and catalysts used remains to be addressed.
A third oxidation/cleavage strategy was applied to a Dioxasolv Birch lignin, employing a DDQ/tBuONO/O2 system for the selective oxidation of the β‐ether units at room temperature (Scheme 11 c).325 For the processing of Birch lignin, an alternative solvent to that used in the selective cleavage of model compounds was required, an important practical issue that is not to be underestimated. A mixture of 2‐methoxyethanol/1,2‐dimethoxyethane was found not only capable of dissolving the lignin feedstock, but also allowed for the catalytic DDQ reactions demonstrated on the model compounds. 2D HSQC NMR revealed a complete disappearance of the α‐C‐H cross‐peak of the β‐O‐4 linkages, with a concomitant appearance of a signal associated with the oxidised linkage. A subsequent stoichiometric reduction using metallic zinc performed in the same pot yielded a monomeric syringyl‐derived compound in 5 wt % isolated yield; it was also shown that the highly‐functionalised 3‐hydroxy‐1‐phenylpropan‐1‐one product provides many opportunities for further conversion into value‐added fine chemicals (Scheme 11 c).325 The production of this highly‐functionalised derivative, and therefore, the possibility of generating high‐value fine chemicals from this phenone product, may eventually justify the use of more expensive reagents and protocols, after which the remaining, lower‐quality and lower‐value condensed lignin could be further converted by harsher catalytic methods, i.e., by a cascading approach.
The same oxidation‐cleavage reaction was applied to a β‐ether polymer that included both G and S units, thus mimicking the structural complexity of lignin better than simple model compounds.325 Similar polymeric lignin mimics have been employed in additional studies.338, 339 Furthermore, polymers of greater sophistication have been investigated, incorporating a combination of β‐5, β‐β and β‐O‐4 linkages, and with control over the S:G ratio.340 It is anticipated that the use of such more advanced models, which incorporate more of the same bonding motifs as found in true lignin feedstocks, will offer further insight into the chemistry occurring in the actual lignin depolymerisation, and should be able to bridge the gap in terms of differences seen in reactivity between simple model compounds and actual lignin macromolecules. Indeed, the drop in cleavage activity seen in going from dimeric models, to the polymer, to the actual lignin shows the difficulties that are generally faced in translating chemistry developed on model compounds to real lignins. Importantly, though, as with the sodium formate/formic acid example, the main product obtained from lignin is the same as from the model compounds, which means that the chemistry on the lignin sample is well understood, allowing further optimisation.272 The latter would entail an assessment of the solvents used, the stoichiometric reagents, co‐catalysts and other additives, to improve the economic viability of such two‐step, activation‐before‐cleavage strategies.
Mild oxidative selective cleavage of a carbon–carbon (rather than the carbon–oxygen) bond in the β‐ether units represents an alternative route for lignin depolymerisation. A vanadium‐oxo complex incorporating a dipicolinate ligand and a copper(I) chloride/TEMPO system were compared alongside one another, with respect to the aerobic oxidation of a (not quite authentic) β‐ether model compound 1‐(3,5‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol (Scheme 12).341
Scheme 12.

Mild oxidative pathways catalysed by vanadium‐oxo complexes, CuCl/TEMPO or Cu(OTf)/TEMPO, for cleavage of β‐O‐4‐linked lignin model species: (upper) 1‐(3,5‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol, and: (lower) 1‐(4‐hydroxy‐3,5‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol.
In the reaction with copper(I) chloride/TEMPO and molecular oxygen, a 56 % combined yield of 3,5‐dimethoxybenzaldehyde (major) and 3,5‐dimethoxybenzoic acid (minor) was obtained, with minimal α‐ketone production. The vanadium system led not only to the corresponding α‐ketone (oxidation) in the presence of oxygen but also carbon‐carbon bond cleavage products were detected, albeit as minor products (<20 % yield). In a later investigation employing a vanadium‐oxo complex bearing 8‐quinolinate ligands, similar selectivity to the α‐ketone was observed (not shown in Scheme 12).342 Highlighting the importance of structural fidelity of model compounds, the use of a better analogue, a model compound with a phenolic hydroxy group [1‐(4‐hydroxy‐3,5‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol] led to carbon‐carbon bond cleavage with the quinolinato vanadium‐oxo catalytic system, generating 3,5‐dimethoxy‐p‐benzoquinone, an acrolein derivative, and a small quantity of the benzylic ketone compound (Scheme 12). Clearly, the oxidation selectivity is very sensitive to the choice of ligand structure, solvent and, most crucially, the model compound.24
Finally, the vanadium‐quinolinato complex was shown to also affect the oxidation of an ethanosolv lignin isolated from mixed hardwoods. The reduction of molecular weight seen by GPC was corroborated by the disappearance of characteristic cross‐signals for β‐O‐4, β‐5, β‐β and dibenzodioxocin bonding motifs, detected by HSQC NMR.343 However, the formation of volatile aromatics was not assessed. Therefore, it is unclear how the chemistry seen with the model compounds translates to actual lignin oxidation.
Similar to the vanadium‐catalysed example above, p‐benzoquinones could also be obtained by the oxidation of 1‐(4‐hydroxy‐3,5‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol with a cobalt‐Schiff base catalyst. Furthermore, partial loss of one of the ring methoxy substituent was also observed, suggesting both carbon‐carbon and carbon‐oxygen bond cleavage in the β‐O‐4 substructure.344 A copper‐catalysed oxidative process on the same model compound, by contrast, led primarily to an α‐ketone.345 These examples again outline the need of a careful choice of model compound and catalytic parameters, as the modification of a single substituent or metal complex can exert a profound influence on the predominant depolymerisation pathway(s).
Carbon‐carbon bond cleavage via metal‐free Baeyer–Villiger oxidation of 1‐(3,4‐dimethoxyphenyl)‐2‐(3‐methoxy‐5‐propylphenoxy)propan‐1‐one with aqueous H2O2 in HCO2H/CH2Cl2 was also demonstrated.346 The Cα−Cβ bond was successfully cleaved although only the 3,4‐dimethoxybenzoic acid product could be isolated. The corresponding aldehyde and phenol fragments were presumed to undergo oxidative polymerisation reactions.
Vanadium‐ and copper‐doped hydrotalcite materials were also found to depolymerise lignin under oxidative conditions,347 whereby the hydrotalcite is believed to act as a reservoir for the release of homogeneous copper and vanadium species. Catalysts doped both with vanadium and copper gave rise to synergistic properties, and the depolymerisation of an Organosolv lignin (extracted from Beech using mild water/ethanol conditions) led to a significant reduction in the apparent molecular weight (from 1100 to 300 Da, based on GPC analysis) in pyridine as solvent, under a 1.0 MPa pressure of O2. HSQC NMR spectra revealed the full conversion of β‐ether and resinol structures and p‐hydroxycinnamyl alcohols. The low molecular weight fraction was assumed to consist of dimers or trimers, which was further supported by analysis by MALDI‐TOF‐MS, but these were not further isolated or identified. The reaction was also performed on Kraft lignin, for which degradation of the β‐ether units was also observed.
An iron(III) DABCO complex was recently reported to oxidatively cleave lignin using hydrogen peroxide as the oxidant in a DMSO/water solvent mixture.348 A non‐phenolic β‐ether model compound was cleaved to afford guaiacol and veratraldehyde as the major products (in 47 % and 46 % maximum yield, respectively). The reaction is believed to proceed via a Fenton‐type radical initiation mechanism, with DMSO providing reactive methyl radicals. However, using a model compound with a free‐phenolic functionality the yield of guaiacol fell considerably (from 47 to 27 %) and no veratraldehyde was detected. The system was subsequently applied to an Organosolv lignin (isolated from Beech); HSQC NMR revealed cleavage of the β‐ether units, accompanied by the disappearance of characteristic peaks for resinol and phenylcoumaran structures. GPC analysis also revealed a significant reduction of the molecular weight, but no monomeric products were isolated.
Some other recent developments in mild oxidative depolymerisation of lignin include direct photochemical oxidation349 and radical oxidation performed in an ionic liquid solvent.350 The former strategy was conducted on a β‐ether model compound, which was cleaved by irradiation with visible light, in the presence of 10 mol % 1,4‐hydroquinone and 2 mol % Cu/AlO(OH). This reaction afforded the corresponding benzaldehyde and phenol derivatives (albeit at low yields of 10–20 %). The use of the ionic liquid 1‐benzyl‐3‐methylimidazolium bis(trifluoromethanesulfonyl)imide as the reaction solvent was found to promote the generation of the hydroperoxyl radical (HOO.) under an oxygen atmosphere in a metal‐free system. The cleavage of a β‐ether model rendered high yields (ca. 80 %) of the corresponding benzoic acid and phenol.350 However, these novel methods have yet to demonstrate their applicability in lignin depolymerisation.
3.3.2. Mild Reductive Depolymerisation Pathways
The tris(perfluorophenyl)borane‐catalysed reduction of an ether or alcohol with stoichiometric amounts of a hydrosilane is established organic synthetic methodology,351, 352 and has been recently applied to the reductive depolymerisation of lignin.353 At room temperature, β‐ether model compounds with increasing functionality could be effectively cleaved using excess triethylsilane (or other commercially‐relevant silanes) in CH2Cl2 (Scheme 13). The conversion of 2‐phenoxy‐1‐phenylethanol resulted in triethyl(phenoxy)silane and triethyl(phenethoxy)silane in yields exceeding 90 %, the latter compound being formed through the migration of the phenyl group. The corresponding alcohols could be obtained by acid‐catalysed hydrolysis. Alternatively, phenylalkanes can be produced by extending the reaction time or adding extra equivalents of reducing agent. Notably, methoxy substituents are also converted into silyl ethers under the applied conditions. The commercially relevant silanes poly(methylhydrosiloxane) and tetramethyldisiloxane were identified as equally effective reducing agents for the reaction performed on simple β‐ether model compounds.
Scheme 13.

Mild reductive cleavage of a simple β‐ether model compound, using excess triethylsilane and catalytic tris(perfluorophenyl)borane.326 For experiments with actual lignins, the results are indicated below the reaction scheme.
This mild reductive triethylsilane method was subsequently implemented on Formacell lignin derived from Black Poplar.326 Four mono‐aromatic compounds, representing both the silylated propanol and propane derivatives of guaiacol and syringol units, in net 20 wt % yield (i.e., accounting for weight added by silylation) relative to the input of lignin were isolated. Using lignin derived from the softwood Norway Spruce instead, a single silylated propylcatechol product could be obtained at a 21 wt % yield. Analysis of the residual lignin demonstrated that the β‐ether units were fully converted by the reduction procedure. In addition, Pinewood lignin extracted by Formacell, Ethanosolv, Methanosolv or Acetosolv methods afforded 25, 18, 12 and 4 wt % yields of silylated phenols, respectively, offering a clear example of the influence of different upstream methods on the availability of labile β‐ethers in the catalytic downstream processing of technical lignins.
Hydrosilane loading offered control over selectivity to either the propane or propanol derivatives. Hydrolysis of each obtained triethylsilylated product finally enabled isolation of the corresponding catechol. Although high yields may be obtained by this depolymerisation pathway, the greater‐than‐stoichiometric use of a silylating agent would need to be addressed (e.g., by regeneration and/or recycling) for the operation of such a process on the large scale required for lignin processing.
3.3.3. Mild Redox‐Neutral Depolymerisation Pathways
Redox‐neutral methods for the depolymerisation of lignin under mild conditions have also been developed. Typically, the redox‐neutral criterion can be achieved either by means of dehydrogenation of the lignin (or model) substrate itself, which provides the requisite H2 for cleavage of the C‐O ether (or other) bond, or through hydrolytic ether cleavage.
Redox‐neutral cleavage of lignin model compounds has been reported with the use of vanadium Schiff‐base complexes as catalysts.354 A complex containing a sterically bulky tridentate ligand led to high conversion of 1‐(4‐ethoxy‐3‐methoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol, with good selectivity to the α,β‐unsaturated ketone product 1‐(4‐ethoxy‐3‐methoxyphenyl)prop‐2‐en‐1‐one, the product of a formal elimination and dehydration reaction (Scheme 14 a). Mechanistic studies showed that for the reaction to proceed, a free α‐hydroxy group is required, whereas alkylation at the γ‐OH exerts no effect on the reaction outcome.
Scheme 14.

Select mild redox‐neutral cleavage pathways, a–g), for β‐ether lignin model substrates. For experiments performed using actual lignins, the results are indicated below the reaction scheme.
The same vanadium complex was applied to the depolymerisation of Organosolv lignins, extracted from Miscanthus giganteus using acetone, ethanol or 1,4‐dioxane.355 GPC results revealed a reduction in molecular weight of the lignins. The ethanosolv lignin underwent depolymerisation to a lesser extent compared to the other lignins studied. O‐ethylation of the benzylic alcohol moiety in lignin during ethanosolv pulping41 is responsible for this difference in reactivity, as a free α‐hydroxy group is required for the catalytic reaction, in line with the model compound studies. This outcome clearly shows that solvent choice in upstream fractionation of lignocellulose carries implications for the efficiency of the catalytic downstream processing of the isolated lignin. Finally, 2D NMR experiments confirmed the disappearance of characteristic β‐ether unit cross peaks in the treated lignins; units with other linkage types were largely unaffected by the depolymerisation procedure. However, the primary products observed were vanillin and syringaldehyde rather than the expected enone products. Further investigation is required in order to optimise the system and improve the yield of mono‐aromatics. Solvent selection and possible derivatising effects are clearly important points to be further studied.
A redox‐neutral, ruthenium‐Xantphos‐catalysed cleavage of β‐ether units has also been demonstrated (Scheme 14 b);338 2‐aryloxy‐1‐arylethanols were successfully cleaved to the corresponding phenols and acetophenones by employing 1 mol % RuH2(CO)(PPh3)3 and 1 mol % Xantphos in toluene. The reaction was expanded to the synthetic β‐ether polymer poly(4‐hydroxy‐1‐phenethanol), yielding 4‐hydroxyacetophenone in nearly quantitative yield. However, attempts to cleave models more similar to the actual lignin structural motifs afforded cleavage products only in low yield.356 Instead, a double dehydrogenated substrate was found to have chelated to the ruthenium metal centre, inhibiting further catalytic activity. An acetylated keto‐β‐ether model on the other hand did undergo cleavage, yielding both the acetophenone and propiophenone, but also a large amount of condensation products. Preliminary small‐scale experiments on an acetylated Kraft lignin did seem to suggest cleavage of the lignin, but the products could not be unambiguously identified.357
Ruthenium‐triphos complexes have also proved competent catalysts for the cleavage of 2‐aryloxy‐1‐arylethanols.358 Interestingly, when the triphos catalytic system was applied to the β‐ether model 1‐(3,4‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)‐propane‐1,3‐diol (Scheme 14 c), Cα−Cβ bond cleavage instead occurred, affording the corresponding benzaldehyde and 2‐guaiacylethanol species;359 a retro‐aldol mechanism with internal hydrogen transfer was proposed. In analogy to the oxidative vanadium‐catalysed cleavage, the examples of redox‐neutral ruthenium catalysis demonstrate that alternative cleaving mechanisms at the β‐O‐4 linkage may occur upon minor modification of the model compound, catalyst properties or solvent.
Mild cleavage of β‐ether units was also reported with a heterogeneous palladium on carbon catalyst, generating acetophenones and phenols.274 A broad array of 2‐aryloxy‐1‐arylethanol species could be cleaved using one equivalent of ammonium formate and 2.5 mol % of Pd/C at 80 °C, in a mixture of methyl tert‐butylether and water in air, affording the corresponding acetophenone in >90 % yield (Scheme 14 d). A model polymer, poly‐(4‐hydroxyphenyl)ethane‐1,2‐diol, could also be cleaved to obtain mono‐aromatics in excellent yield. In analogy to the ruthenium‐catalysed systems, the substituent at the γ‐position has a profound impact on the reactivity. The cleavage of aryl glyceryl ethers was challenging, requiring excess ammonium formate and a second reaction step incorporating formic acid, leading to the reduced arylpropane and arylpropanol products (Scheme 14 e). The reaction likely proceeds by initial dehydrogenation at the benzylic alcohol to form a ketone, which is the species susceptible to hydrogenolysis (with ammonium formate acting as a hydrogen donor). The catalytic procedure was also performed on a Dioxasolv lignin isolated from Pinus sylvestris; a modest reduction in molecular weight was observed by GPC, and HSQC NMR analyses demonstrated that 73 % of the β‐ether units (and certain ether resinol/coumaran units) were cleaved. Monomer products dihydroconiferyl alcohol and dihydro‐p‐coumaryl alcohol were detected by GC‐MS, but not quantified.
Recent investigations of the acid‐catalysed hydrolysis of a Dioxasolv walnut lignin have demonstrated the possibility of cleaving the aryl ether units, followed by reactive “trapping” of the generated aldehydes in the form of a cyclic acetal, preventing the products from subsequent recondensation (Scheme 14 f).234 Reaction of the simple β‐ether model compound 2‐(2‐methoxyphenoxy)‐1‐phenylethan‐1‐ol with 10 mol % triflic acid in 1,4‐dioxane at 140 °C in the absence of a trapping reagent yielded guaiacol in high yield, but 2‐phenylacetaldehyde was only detected in small quantities, presumably lost to secondary aldol condensation reactions. Incorporating 1.5 equivalents of ethylene glycol as a trapping agent, however, enabled isolation of the cyclic acetal in >90 % yield. Alternatively, the aldehyde intermediate may be trapped by in situ hydrogenation over Ru/C, although the product is a complex mixture of (semi‐) hydrogenated and hydrogenolysed species. Furthermore, the aldehyde intermediate could be trapped by decarbonylation with [IrCl(cod)]2 and PPh3, which afforded toluene in 73 % yield for the β‐ether model compound (2‐(2‐methoxyphenoxy)‐1‐phenylethan‐1‐ol. All three methods were further investigated for the depolymerisation of a Dioxasolv lignin. Trapping of lignin‐derived aldehyde products as acetals afforded a threefold increase in monomer yields relative to the control experiment, whereas hydrogenation improved the yield of monoaromatics by a factor of five. Decarbonylation demonstrated only a moderate increase in monoaromatics yield, however, which was attributed to the relatively slow rate of the iridium‐catalysed reaction.
Efficient depolymerisation of lignin by tandem hydrolysis‐decarbonylation has been demonstrated with water‐stable Lewis acids (such as scandium(III) or indium(III) triflate) rather than a Brønsted acid, to catalyse the first ether hydrolysis step.425 A homogeneous rhodium catalyst was selected for the decarbonylation step.425 The method (Scheme 14 g) was first validated with various model compounds to appropriately match the rate of the hydrolysis and decarbonylation steps, including the conversion of 1‐(3,4‐dimethoxyphenyl)‐2‐(2‐methoxyphenoxy)propane‐1,3‐diol to give guaiacol and 4‐methylveratrole in 88 % and 51 % yield, respectively. Cleavage occurs via initial α‐β dehydration, and subsequent hydrolysis of the resultant styryl ether, catalysed by the metal triflates. Decarbonylation of the reactive aldehyde intermediate then affords a methyl‐substituted aromatic compound. The dehydration/hydrolysis of α‐hydroxy‐β‐ethers has also previously been reported using a base360 or methyldioxorhenium as catalyst.361 Considering the tandem hydrolysis–decarbonylation, this method could also be translated to actual Dioxasolv lignins (from Poplar, Pine and brewer's spent grain) and Poplar sawdust, yielding up to 12 wt % mono‐aromatics. Interestingly, selectivity could be fine‐tuned by varying the strength and amount of the Lewis acid. To this effect, strong Lewis acids (e.g,. Ga(OTf)3) afforded primarily methyl‐substituted mono‐aromatics, whereas weak Lewis acids (e.g., Yb(OTf)3) led to 2‐propenyl‐substituted aromatics.
The aforementioned examples show that a broad array of mild oxidative, reductive, and redox‐neutral pathways for the depolymerisation of β‐ether lignin model compounds has emerged in the contemporary chemical literature. Moreover, several of these catalytic methods have been successfully applied to the treatment of lignin samples (typically Organosolv lignins isolated under relatively mild conditions, and thus assumed to still retain an appreciable fraction of the native β‐O‐4 linkages). Nevertheless, the examples also demonstrate that the modification of a single functional group (even when not in close proximity to the β‐O‐4 linkage), or modest changes to the catalytic system or process conditions, can result in pronounced changes in the chemistry observed and in the composition of the product mixture. Expectedly, neither (simple) model compounds nor synthetic β‐O‐4 polymers thus sufficiently represent the chemical complexity of the isolated technical lignins. Furthermore, economic constraints associated with the catalysts and reagents used have to be carefully considered with regard to large‐scale industrial implementation. Finally, the extraction of limited amounts of high‐value products should preferably be part of a cascade approach, in which the remnant lignin is also further valorised. To aid the latter, analytical data not only on the volatile or mono‐aromatic fraction, but also on the residual macromolecular components would provide valuable information on the action of homogeneous catalysts on the polymeric structure of lignin.
3.4. Harsh Depolymerisation Pathways
As discussed in the previous sections, the more reactive bonds (foremost, but not exclusively, β‐ethers) will have already been cleaved to a significant extent for technical lignins. The “toolbox” of mild and chemoselective catalytic cleavage methods (outlined in the previous sections) are, therefore, expected to prove largely ineffective for the depolymerisation and generation of value‐added products from such refractory, degraded lignins. For highly condensed, technical lignins, effective depolymerisation requires more severe conditions (i.e., higher operating temperatures or pressures) as they are cross‐linked by strong C−C bonds. The higher‐severity conditions reduce the ability of the catalyst to steer selectivity to specific target molecules; instead, the production of a lignin oil with more complex chemical composition is typical. Accordingly, the criteria against which successful depolymerisation is measured are altered. The degree of deoxygenation, boiling point range, degree of (ring) saturation, and molecular weight distribution often constitute key final properties of the product mixture, and the harsh depolymerisation process may be then tailored to produce a range of compounds that collectively exhibit the desired physical and chemical characteristics for the intended application.
It may be feasible for lignin oils to be further refined into mixtures of value‐added products, in analogy to the refining of crude oil, with eventual targets including BTX, phenols or cresols, which are employed as feedstocks to the chemical industry in sufficient volumes as to warrant large‐scale production. The price of the catalysts and recyclability, the intrinsic cost of the solvent and loss due to evaporation, decomposition and inhibition of substrate and catalyst, the infeasibility of using costly stoichiometric reagents (e.g., protective groups) and the environmental burden of any waste material are all factors that will determine both the economic and environmental feasibility of any prospective process based on harsh lignin depolymerisation.
A typical approach for the depolymerisation of lignin under harsh conditions employs either an acid or a base catalyst, a supported metal catalyst with hydrogen transfer capability, or a combination of both, at temperatures of up to around 400 °C. Under these operating conditions, it is sometimes difficult to distinguish the contributions of catalytic and thermochemical reactions to the process outcome. Indeed, significant conversion of lignin is often observed without added catalyst under “control” conditions. Solvents are typically water or lower alcohols, as well as phenols or even the products themselves in some cases. An external input of gaseous hydrogen may be employed in order to improve the catalytic activity for hydrogenolysis and carbonyl hydrogenation. Nonetheless, reforming of the alcohol solvent is often sufficient to supply the process with molecular hydrogen at low levels, minimising the extent of saturation of the aromatic rings. This aspect is of critical importance if the production of BTX from aromatic‐rich oils is desired. Alternatively, formic acid may be used as a hydrogen donor. Besides the lignin‐derived “bio‐oil”, an insoluble “char” is commonly obtained, and both water‐soluble organics and gaseous products may evolve.
In the next sections, analytical techniques commonly employed for characterisation of lignin oils and selected harsh depolymerisation methods are briefly described. Regarding the characterisation of lignin products, the discussion is neither exhaustive nor intended to be a strict instruction towards or against specific analysis methods. Instead, it aims to offer some constructive suggestions regarding key considerations for the effective characterisation of (gaseous, liquid or solid) lignin products. Finally, selected recent methods for depolymerisation are highlighted, ordered so as to clearly indicate ways in which important and recurring aspects of harsh depolymerisation procedures can be addressed. Crucially, although the following sections centre around harsh depolymerisation methods, the discussion is also highly relevant to the milder methodologies described in the previous sections.
3.4.1. Characterisation of the Lignin Products
Thorough characterisation of the often highly complex lignin‐derived products demands the use of a broad array of techniques for analysis of the liquid, solid and gaseous products obtained from lignin conversion. Regarding the liquid species, which are often the desired products obtained from mild or harsh depolymerisation methods, it should be noted that the terms “lignin oil”, “bio‐oil”, “product oil”, and “liquid product” are all equally ambiguous. Indeed, they may refer to a directly obtained liquid phase (after solids have been removed by filtration), via extraction into a solvent (commonly CH2Cl2 or EtOAc), or else through initial concentration by solvent removal. Although this seems to be merely a semantic problem, the lack of an unambiguous definition of the content of the liquid product (often quoted in wt % of the original lignin feedstock) hinders, to a certain extent, unbiased comparisons of the data. Residual solvent content in the oil, in particular water (3.3 wt % solubility in EtOAc at 20 °C), may lead to an overestimation of the yield, for example. Conversely, prolonged solvent removal under rotary evaporation conditions may lead to an underestimation of the yield of liquid products, as lighter (more volatile) fractions are likely to be removed alongside the solvent.
Concerning the chemical composition of the liquid products, characterisation is most often performed by using Gas Chromatography coupled with Mass Spectrometry or Flame Ionisation Detection (GC‐MS/GC‐FID). While the former can aid in identification of volatile products, quantification should be done with the latter. However, the frequently encountered practice of analysing lignin oil samples without the addition of an internal or external standard for GC makes thorough and quantitative evaluation difficult. This is further compounded by the fact that authentic samples for calibration are not available commercially for many of the volatile lignin products, which must then be (often laboriously) synthesised. Moreover, it must be recognised that the observed volatile compounds are often a poor representation of the net composition of the liquid products. In this context, Thermogravimetric Analysis (TGA) experiments of the liquid product, performed under an inert atmosphere (e.g., Ar, N2) and at a low heating rate (e.g., 5–10 °C min−1), are very useful for estimating the fraction of volatile compounds at the injector temperature.198
As conventional one‐dimensional GC techniques are actually not capable of fully resolving the individual components of the lignin oil, two‐dimensional GC methods (e.g., GC×GC‐MS/FID) offer several advantages over one‐dimensional GC techniques and are thus strongly recommended. In GC×GC images, each volatile component can be grouped together with analogous, structurally related species. Furthermore, this analytical technique markedly improves both the resolving power and sensitivity, enhancing the capability for cross‐checking against MS databases. Nonetheless, caution must be exercised when using the “National Institute of Standards and Technology” (NIST) and Wiley libraries to identify the lignin products detected by (GCx)GC‐MS. The libraries are a valuable yet non‐exhaustive resource of structural information; products anticipated from lignin depolymerisation are often absent from the database, potentially leading to misinterpretation of the observed compounds (for example, peaks assignments to unrealistic structural isomers).
As previously described, GC techniques are able to analyse only the volatile fraction of products in the lignin oil. However, the lignin oil will typically also contain non‐volatile and higher‐molecular‐weight components. Therefore, characterisation by GC must be complemented by techniques that analyse the whole oil. Specifically, Gel Permeation Chromatography (GPC) offers an approximate indication of molecular weight and size distribution,36 which may be compared against the determined quantities of volatile species. Noteworthy is the fact that, in GPC, the molecular weight is only indirectly inferred from the hydrodynamic volume of the analyte. Indeed, solvent effects and lack of suitable calibration standards seriously complicate molar mass determination. Cross‐linking can significantly decrease the apparent molecular weight observed by GPC, for instance, whereas branching might have an opposite effect, with both effects not being captured by the linear polymers used for calibration. Unsurprisingly, molar mass determination of an Alcell hardwood lignin gave widely different results by small angle neutron scattering (26 kDa), GPC (3 kDa), and electrospray ionisation (ESI) mass spectrometry (18–30 kDa).427 In addition, the neutron scattering results suggested that the material is much denser than expected for a linear polymer. It was proposed that lignin in solution has a rather compact structure reminiscent of a hyperbranched polymer or nanogel. As such a compact structure has a rather small hydrodynamic volume it thus appears to be of low molecular weight in GPC, explaining the observed discrepancy.
Mass spectrometry coupled with Electrospray Ionisation (ESI‐MS) is capable of resolving the complexity of the fraction of intermediate molecular weight (300–1500 Da), which is particularly difficult to analyse.318, 362, 363 Moreover, the use of multidimensional MS allows for structure elucidation from fragmentation patterns. Importantly, ESI‐MS/MS offers the possibility for advanced quantitative analysis not only of this intermediate fraction, but of complex mixtures of lignin products in general. Nevertheless, the intermediate molecular weight fraction requires more attention. As the analytical challenges here are in fact similar to those faced in the characterisation of the crude oil bitumen fraction,364, 365 analytical protocols used in this field may aid the analysis of lignin products in some instances.277, 366, 367, 368, 369
Some insight into the chemical reactivity and mechanisms underlying lignin depolymerisation may be gained from evaluation of the “carbon balance” of the transformation. In this context, determination of the elemental composition of residual fractions (not simply the volatile fraction) in addition to the liquid products is mandatory. Elemental analysis is also widely employed for the characterisation of the liquid product, with (atomic) C/H and C/O indices offering valuable information on oxygen content (reduction of oxygen content is a common target in the catalytic upgrading of lignin), hydrogen consumption, and heating value. A graphical representation of changes in C/H and C/O values in a Van Krevelen plot might furthermore provide insight into which reaction types dominate (e.g. dehydration, reduction, etc.). Gravimetric and elemental analyses of the residual solids are essential to aid in understanding any repolymerisation processes. Moreover, the severe conditions of “harsh depolymerisation” will likely generate a non‐negligible fraction of gaseous products; compositional analysis (e.g., Micro‐GC coupled with Mass Spectrometry) of the gas phase is, therefore, highly desired. Finally, the water‐soluble organics are clearly more difficult to isolate and quantify. Accordingly, determination of the total organic carbon content of the aqueous phase is important for a proper assessment of the carbon balance.370
Comprehensive lignin product analysis thus requires a considerable research effort, yet is rewarded with valuable information. This is illustrated by an investigation of the oligomeric fraction of depolymerised EMAL lignin after a Pd/C catalysed hydrogenolysis procedure.366 The obtained oil consisted of an approximately 1:1:2 mixture of mono‐, di‐aromatics and oligomers. After thorough extraction with diethyl ether, the higher molecular weight fraction was isolated and subjected to further analysis. With ESI‐MS, trimers and tetramers were detected and, based on their molecular weight, were found to be mostly 4‐propylguaiacol oligomers. HSQC and 31P NMR further revealed the inter‐unit linkages to be largely β‐5, 5‐5 and 4‐O‐5 linkages, with β‐1 and β‐β linkages present in small amounts. The dimeric and oligomeric fractions of the bio‐oils obtained from the reactive fractionation of birch sawdust over a Ru/C catalyst were also extensively characterised.277 Major dimeric products were found to have β‐1 and β‐5 linkages along with small amounts of 5‐5 and 4‐O‐5 linkages. Surprisingly, β‐β linkages, thought common in birch lignin, were not observed. Further HSQC analysis of the oligomer fraction also confirmed these bonds to be prevalent in the higher molecular weight residual lignin. Interestingly, similar to the lignin oil fractionation noted above, a method was recently reported for separating five different technical lignins into fractions of varying molecular weight. 31P NMR analysis on the separated fractions showed the lower molecular weight fractions to be less condensed.368 The development of preparative liquid chromatography procedures or organic solvent nanofiltration,367 followed by 2D NMR, would also be able to offer a further wealth of structural information. The insight that will be gained from these “advanced” fractionation and analysis techniques could well provide valuable information for the design of novel lignin (cascade) depolymerisation strategies.
Notably, there is a pressing need within the catalysis community working on lignin for standardisation of analytical protocols, to allow for a proper comparison of (emerging) processes. To meet this requirement, a collaborative and multi‐disciplinary approach between research groups is needed in order to translate the considerable number of analytical methods for lignin characterisation (developed by wood chemists) into standard protocols for the characterisation of lignin products obtained from catalytic reactions. Likewise, rational design and standarisation of “work‐up”/purification procedures is of paramount importance for effective comparison of processes for lignin depolymerisation.371 A similar desire for a best‐practices approach for the characterisation of bio‐oils obtained from catalytic pyrolysis was recently proposed in a review.369 Clearly, whilst the physical and chemical properties of the lignin products are influenced strongly by the choice of treatment, the methodologies used for evaluating their properties should be as consistent as possible across research laboratories.
3.4.2. Reactivity of Technical Lignins
Logically, the variety of structural motifs occurring in lignin streams, as a result of different sources and isolation methods, must be taken into account for the design of effective depolymerisation processes. Although it is tempting to directly compare different catalytic systems, this is often hampered by the fact that different lignin feedstocks have been employed. Indeed, for the mild methods, the relationship between available cleavable bonds and activity can more directly established. For harsher depolymerisation methods, it is much more difficult to recognise such a relationship, given that multiple bonds may be cleaved both under catalytic and thermal control. A systematic comparison of different lignin sources in the same catalytic process is therefore highly valuable, but unfortunately, such literature is very scarce.
In one of the few examples, the depolymerisation of four lignin samples (i.e., soda wheat straw, AFEX wheat straw, Organosolv Poplar, and ammonia Poplar), in the presence of Pt/Al2O3 catalysts at 300 °C in a methanol‐water solvent (Figure 9), was compared.194 Interestingly, there is a good correlation between the relative yields obtained from the catalytic depolymerisation and analytical chemical degradation by thioacidolysis, which is based on β‐ether bond cleavage. This example suggests that the thioacidolysis method constitutes a useful, correlative approach for ranking lignin streams according to their potential for production of mono‐aromatics products. Regarding the lignin residues, GPC traces after reaction were similar for each lignin, suggesting convergence to a highly condensed lignin that is resistant to further depolymerisation.
Figure 9.
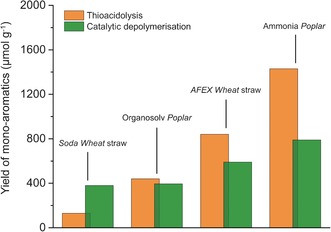
Comparison of lignin monomer yields via catalytic depolymerisation (green) vs. thioacidolysis (orange), of differing lignin samples, as a measure of degree of condensation.194
It is often asserted that Organosolv lignins are more amenable towards depolymerisation than those obtained from paper‐mill operations. Nevertheless, the fraction of readily cleavable linkages (primarily β‐O‐4) in several types of technical Organosolv lignins (mostly those obtained with added acid or base catalysts) has already been significantly reduced compared to the native lignin. An Organosolv lignin may therefore sometimes resemble Kraft lignin in recalcitrance. In some cases, Organosolv lignins may in fact be even more difficult to cleave; the structural profiles of Organolsolv lignins obtained from the different severities of the “same” organosolv process as shown in Figure 3 clearly illustrate this. Another example is provided by liquid‐phase reforming of lignin using a Pt/γ‐Al2O3 catalyst (with H2SO4 co‐catalyst),372 giving 18 wt % of mono‐aromatics from Kraft lignin compared to 9 wt % of mono‐aromatics with an Organosolv lignin. It remains to be verified whether the degree of condensation is responsible for the differences in yields obtained from the processing of the studied Kraft and Organosolv lignins. In any case, it is not enough to assume that a process (or a process name) defines a lignin—the lignin must be profiled first to allow any understanding of a process that is applied to it.
Another example highlighting the influence of structure on depolymerisation is that of candlenut‐derived lignin (Aleurites moluccana), catalysed by a copper‐containing porous metal oxide catalyst under relatively mild conditions in methanol (140 °C, 40 bar H2). This generated a series of 4‐propylcatechol‐type products in up to 64 % monomer yields.373 Compounds of this type have not been isolated from other lignins. Therefore, it is likely that the structure of the candlenut lignin differs from that in other feedstocks. As has been highlighted above in Section 2.3, homogeneous linear lignins derived solely from caffeyl alcohol or 5‐hydroxyconiferyl alcohol have been recently discovered, attesting to this possibility.143, 374
3.4.3. Solvents
The solubility of technical lignins will strongly influence the efficiency of liquid phase catalytic depolymerisation methods, including harsh processes. Among other reasons, it has been demonstrated that insoluble lignin fractions may cause increased char formation during depolymerisation.375 Lignin solubility is influenced, at least in part, by the structure, bonding properties and functional group density of the macromolecules. At room temperature, isolated lignins that closely resemble native lignins (e.g., cellulolytic enzyme lignins, certain Organosolv lignins) have low water solubility, and low solubility in pure polar organic solvents (e.g., ethanol, acetone, 1,4‐dioxane), but moderate solubility in alkaline solutions, and may be completely soluble in mixtures of polar organic solvents and water that have the right solvent factor—9:1 acetone:water and 96:4 dioxane water are established examples. By contrast, technical lignins (e.g., Kraft lignin) are often comparatively insoluble in the same organic solvents or their mixtures with water.372 Selected classes of imidazolium‐derived ionic liquids (ILs) have shown capability for dissolving both lignin and lignocellulosic biomass itself (in sawdust form),376, 377, 378, 379 in particular those with strongly hydrogen‐bonding basic anions to disrupt hydrogen bonding networks.380, 381, 382 However, the often‐associated high (aquatic) toxicities and other concerns associated with ILs still constitute barriers against their successful use in large‐scale processes. For a more thorough discussion of lignin in ILs, the reader is directed to two recent review articles.23, 379
Importantly, for any depolymerisation method, the actual solubility of lignin and solvent properties will markedly differ under process conditions from those determined at room temperature. At temperatures between 200 and 350 °C, under pressures higher than 10 MPa, many common solvents already experience near‐critical, critical, or even supercritical conditions, causing the reaction medium to possess very distinguished properties. For instance, even water, a highly polar solvent, shows a substantial decrease in polarity under near‐critical conditions. As a result, near‐critical water is completely miscible with toluene.383 Therefore, the assessment of the effects of lignin solubility upon the performance of catalytic depolymerisation must be performed under processing conditions, and not extrapolated from observations at room temperature.
Harsh depolymerisation methods are typically carried out in stainless steel pressure vessels. In this manner, the actual solubility of lignin (and the point at which solid residues begin to form) under such conditions is rarely known. However, the complete dissolution of an (initially insoluble) Organosolv lignin at 115–130 °C under neutral aqueous conditions was demonstrated in a pressure vessel equipped with a glass window (Figure 10 a).375 By contrast, dissolution of a Kraft lignin (Figure 10 b) was accompanied by agglomeration of solids onto the window. Gas bubbles were observed from the agglomerates, suggesting formation of gaseous products. In a 1:1 water:ethanol mixture, both Kraft and Organosolv lignin were fully dissolved at 115 °C and agglomeration was completely suppressed.372
Figure 10.

Images for dissolution of an a) Organosolv lignin and b) Kraft lignin (in water from 20 to 225 °C), taken in a high‐pressure autoclave equipped with an optical window. Adapted with permission from ChemSusChem 2011, 4, 369–378.375 Copyright 2011 John Wiley and Sons.
Apart from the fact that solvents serve as media for dissolution of lignin (fragments), they also often react with the lignin or with itself and/or interact with the catalyst.225 Such side reactions of the solvent may generate a complex array of products. For example, in the reaction of a Kraft lignin in the presence of a molybdenum carbide catalyst and ethanol solvent (280 °C), an oil yield of 160 wt % was achieved, which included a large amount of ethanol condensation products.384 In the absence of lignin, however, yields of these products were far lower, suggesting that lignin itself plays a role in promoting decomposition of the solvent. Solvent loss to side‐products may impact the economy of the process, depending on the value and difficulty of separation of these products and the value of, and ability to recycle, the solvent. However, reactivity of the solvent can also be advantageous. Decomposition of alcohols or formic acid may be employed for in situ hydrogen formation, facilitating hydrogenolysis.385 Furthermore, certain solvents may act as protective capping agents to prevent recondensation, or instead may even promote recondensation reactions, as described below.
Regarding the interactions between solvent and catalyst, in the hydrogenolysis of diphenyl ether (used as a model compound for recalcitrant ether structural motifs occurring in Kraft lignins) catalysed by Raney‐Ni, strong inhibition was observed when performing the reaction in methanol or 1,4‐dioxane, yet optimum reactivity was achieved in methylcyclohexane.233 Inhibition was attributed to strong adsorption of the solvents onto the catalyst surface, blocking the active sites. Regardless of the fact that lignin is insoluble in methylcyclohexane at room temperature, an 81 % conversion of Poplar Organosolv lignin into soluble products (cyclohexanols and cycloalkanes) was achieved in the presence of Raney‐Ni at 300 °C under 7 MPa H2 for 8 h. Clearly, this example demonstrates that thermolysis of technical lignins in the liquid phase constitutes a key step that brings aromatic fragments into solution, and therefore, enables the action of solid catalysts. Thermolysis of technical lignins usually starts to be relevant and contribute to heterogeneously catalysed processes at temperatures between 200 and 250 °C. As lignin thermally decomposes at these temperatures, releasing soluble fragments into the reaction medium, its catalytic conversion can be performed even in solvents in which lignin is insoluble at room temperature (e.g., methylcyclohexane). This observation was recently confirmed by two other studies on reductive depolymerisation of lignin to cycloalkanes performed in hydrocarbon solvents.386, 387
An interesting approach is to use the oil product from a biomass liquefaction process itself as the solvent, as reported for a biomass liquefaction process in guaiacol which afforded excellent bio‐oil (>93 % carbon) yields.388 Following the initial cycle, the produced oil was then used for subsequent biomass liquefaction and was demonstrated to be a still better solvent. Heavy fractions (>1 kDa) accumulated in the recycled oil, although this could be mitigated somewhat by selecting appropriate process parameters that balance the oil yield and the formation of heavy compounds.389 Although fairly common in industry, this latter approach of employing the product as a solvent has so far been little explored with respect to catalytic lignin depolymerisation. Considering all the above examples, it is clear that the choice of solvent for a (harsh) procedure of lignin depolymerisation is non‐trivial.
Performing catalytic lignin depolymerisation in the absence of any solvent constitutes another attractive prospect.390, 391, 392 The solvent‐free hydrogenolytic depolymerisation of a Kraft lignin was recently explored, employing supported sulfided CoMo and NiMo catalysts under 100 bar H2 at 350 °C.393 Yields of CH2Cl2‐soluble lignin oil increased when more basic catalyst supports were used. For instance, a NiMo catalyst supported on basic MgO‐La2O3 afforded the highest monomer yield (26 wt %), with 4‐alkylphenol compounds as the major constituents, as analysed by GC×GC‐MS.
Solvent‐free lignin depolymerisation may also be achieved by means of mechano‐catalytic ball milling using basic catalysts. The concept was initially demonstrated on β‐ether model compounds, employing 3.5 equivalents of NaOH (with Na2SO4 as a grinding auxiliary) and milling at 13.3 Hz to efficiently cleave the β‐ethers.394 Although guaiacol and syringol fragments could be isolated in good yields, the part of the model compound containing the propyl chain underwent several reactions, affording numerous unidentified products. Application of the procedure to an Organosolv lignin from Beech wood enabled a considerable reduction in the content of β‐ethers (analysed by HSQC NMR), although monomer identification or isolation was not reported.394
3.4.4. Prevention of Recondensation in Lignin Oils
It has long been recognised that the formation of reactive intermediates, particularly combinations of a phenol and aldehyde/ketone intermediate, leads to condensation and limits the efficacy of acid‐ or base‐catalysed lignin depolymerisation procedures.395 The repolymerisation channels are likely analogous to those previously described in the sections on isolation methods (Scheme 6), whereby disappearance of β‐O‐4‐containing fragments with the concomitant formation of a more recalcitrant lignin is observed. Correspondingly, there is a need to scavenge and “deactivate” such reactive phenol, carbonyl and/or alkene functionalities in order to increase the yield of lignin oil. In one such strategy, a base‐catalysed depolymerisation was performed with boric acid so as to convert the liberated phenols to the corresponding borate esters. The yield of bio‐oil more than doubled to 52 wt %, relative to the standard NaOH‐catalysed process.395 As previously noted, for hydrogenolytic processes with added transition metal catalysts, removal of reactive functional groups is implicit, typically by hydrogenation of exposed carbonyl functional groups to the corresponding alcohols or alkanes. This is demonstrated by the depolymerisation of cellulolytic enzyme lignin from bamboo over a physical mixture of an acidic catalyst (ultrastable zeolite Y) and a hydrogenolysis catalyst (Raney‐nickel), using methanol as both the solvent and hydrogen donor.396 Greatly improved bio‐oil yields were obtained when the two catalysts were concomitantly used (28 wt %, vs. 3/13 wt % with only the acid/Ni catalyst);396 the two catalysts thus appear to cleave different bonds in a complimentary manner.
The choice of solvent also has a profound influence on the propensity of depolymerised lignin oil to undergo recondensation. For example, the depolymerisation of lignin over copper‐containing (basic) porous metal oxides was initially reported in methanol.397 However, a significant beneficial effect of using ethanol as a solvent was later discovered.288, 371 Indeed, ethanol may function as a “capping agent” by ethoxylating reactive phenol moieties and preventing subsequent recondensation. HSQC NMR analysis of a Protobind‐derived lignin oil, depolymerised in ethanol over the same catalyst at 300 °C, demonstrated extensive alkylation, with phenol‐O alkylation preceeding ring C‐alkylation. Consequently, 17 wt % mono‐aromatics and 73 wt % THF‐soluble lignin oil were obtained under these conditions, whereas yields were limited to 6 wt % and 57 wt % in methanol.371
Although methanol similarly demonstrates the ability to alkylate lignin fragments,398 in situ formation of formaldehyde (from methanol) can occur, which undergoes condensation with phenols to form polymeric by‐products. This phenomenon was clearly demonstrated by model reactions with phenol in either methanol or ethanol, highlighting the formation of diphenylmethanes in methanol, yet the formation of alkyl and alkoxyphenols in ethanol (Figure 11). Due to increased steric bulk, the alkyl‐/alkoxyphenols are considerably less susceptible to recondensation. When employing a 1:1 methanol:ethanol solvent mixture, ethanol can furthermore “scavenge” any methanol‐derived formaldehyde and prevent condensation reactions with aromatic species. This effect is beneficial, as formaldehyde is also likely to form through elimination of the γ‐CH2OH groups on the propyl side‐chain of lignin units. Building on this insight, further optimisation of the reaction conditions with actual Protobind lignin allowed the formation of 60 wt % alkylated mono‐aromatics from lignin at 380 °C.288
Figure 11.

HSQC NMR spectra highlighting differences in reactivity of phenol in the presence of a) methanol or b) ethanol as solvent: with methanol, cross peaks corresponding to methylene bridges between phenol units are observed; with ethanol, C2‐alkylated phenols are instead visible. Adapted with permission from Green Chem. 2015, 17, 4941–4950.288 Copyright 2015 Royal Society of Chemistry.
3.4.5. Catalyst Stability and Lignin Impurities
Sufficient catalyst lifetime is essential for economically viable lignin downstream processing. Nevertheless, information on catalyst stability/deactivation is limited to a select number of depolymerisation systems. The general challenges associated with the stability of heterogeneous catalysts under hydrothermal conditions399 and specifically for the conversion of renewable feedstocks,400 have been reviewed.
One of the most common supports for metal catalysts, γ‐alumina, is known to undergo a phase transformation to boehmite in the presence of water.401, 402 Recently, catalyst stability was examined for liquid‐phase reforming of lignin over a Pt/γ‐Al2O3 catalyst.403 In this study, it was found that catalyst deactivation occurred by the formation of boehmite, which encapsulates the metal particles, dramatically reducing the quantity of catalytic sites. In the presence of lignin‐derived mono‐aromatics (e.g., guaiacol), the transformation of γ‐alumina into boehmite is inhibited by the coordination of phenolates onto the support surface. Lignin itself also has a great affinity for alumina, forming a coating and stabilising the textural properties of the catalyst support.
Impurities present in the lignin stream as a result of the lignin isolation procedure may also exert marked effects upon both reactivity and catalyst lifetime. No lignocellulose fractionation process is capable of yielding a lignin free from impurities (among others carbohydrates and ash). Despite the importance of thorough analysis of technical lignin impurities prior to subsequent catalytic downstream processing, such detailed characterisation is, unfortunately, often not presented in the literature. Residual sugars/carbohydrates are common impurities in lignin, particularly when milder fractionation procedures have been employed.404 The instability of sugars under acidic or basic conditions may lead to the formation of furans or humins via dehydration reactions, which can subsequently lead to catalyst deactivation by fouling/poisoning. Lignin and reactive carbohydrate fragments may also react, resulting in recondensation. Reforming of any sugars/carbohydrates present, on the other hand, may also reduce the demand for external hydrogen in hydrogenolysis/hydrogenation processes.
Recently, the influence of carbohydrate derivatives on catalyst performance was shown for the HDO of guaiacol catalysed by Ru/C,209 as furfural and 5‐hydroxymethylfurfural (5‐HMF) strongly inhibited activity. At 270 °C and under 40 bar pressure of H2, full conversion of guaiacol was observed after 1 h, reducing to just 30 % in the presence of equimolar furfural. Supporting DFT calculations of the Ru(0001) surface highlighted that furfural and 5‐HMF bind more strongly than guaiacol. In this manner, the reduced activity could be attributed to competitive adsorption at the active sites of the catalyst.
Residual (unsaturated) lipids may also be present in isolated lignins, and can influence catalyst performance through their olefinic and carboxylic acid functional groups. The former may lead to coking of a metal surface, the latter may coordinate to metal surfaces, potentially blocking active sites or promoting leaching of any active metal species.405, 406
Elements and functional groups not present in the native lignocellulose may be introduced into the lignin stream as a result of the upstream fractionation treatment, foremost sulfur under Kraft conditions (some sulfur impurities might also originate from amino acid residues) and alkali/alkali earth metals (from inorganics used in the chemical pulping of wood). Sulfur acts as a poison for many transition metals, in particular those belonging to the platinum group in common use as lignin hydrogenolysis catalysts.407 The precise mechanisms of sulfur poisoning in lignin valorisation processes, as well as the speciation and extent of incorporation of the sulfur in the lignin oil, are largely unknown. In general, predominantly sulfur‐free isolated lignin feedstocks are probably desirable for most catalysts. Lignin desulfurisation or the use of a sulfur‐tolerant catalyst408 are alternative strategies for the treatment of lignins with a high content of sulfur‐containing impurities. A Ni/C catalyst was, for example, demonstrated to be fairly robust against sulfur poisoning (using a sodium lignosulfonate feedstock in ethylene glycol at 200 °C and 50 bar of H2).408 In situ hydrodesulfurisation of the lignin feed was demonstrated by detection of H2S in the gas phase. Nevertheless, catalytically inactive NiS was identified by powder X‐ray diffraction after reaction. Interestingly, when typical cobalt‐molybdenum or nickel‐molybdenum sulfide catalysts are employed for lignin depolymerisation or the upgrading of a lignin oil by HDO, high sulfur content could actually be beneficial to retain the sulfided phase of the catalyst, preserving activity.
Where a solid acid (e.g., zeolite) catalyst is employed for downstream valorisation of lignin, the ion exchange of alkali (earth) metals with H+ may bring about a decrease in acidity and, therefore, activity. This hypothesis was verified by the deactivation of mixed silica‐alumina catalysts used for lignin depolymerisation in a solvent mixture of water:methanol (1:5) at 250 °C.409 Employing an Organosolv lignin, catalytic performance could be maintained for at least three successive cycles, leading to ca. 60 wt % of chloroform‐soluble oil. By contrast with a “dealkaline” lignin, significant deactivation was already observed in the second cycle, halving the oil yield, which was attributed to the presence of small amounts of sodium (ca. 29 mg per g lignin) able to ion exchange with active sites of the solid acid catalysts. Impurities in the lignin feedstock following upstream treatment, therefore, may have a profound inhibiting influence on downstream catalytic procedures.
3.5. Further Downstream Processing
The previous sections highlighted various mild and harsh depolymerisation routes for technical lignins. Nevertheless, the liquid products obtained from depolymerisation rarely already exhibit the desired properties, nor incorporate significant quantities of target species, for immediate utilisation in the intended applications. Therefore, catalytic upgrading processes are required for further chemical transformation to the desired products. Procedures typically focus on further reduction in the oxygen content via hydrodeoxygenation (HDO), and/or further cleavage within larger oligomeric fragments of the oil. HDO of liquid products from lignin and related model compounds is comprehensively described in recent review articles.19, 410, 411, 412, 413 Here, the discussion is limited to an overview of general principles and selected examples that highlight recent significant advances and specific challenges associated with the treatment of actual depolymerised lignin streams (i.e., lignin oils).
HDO approaches may be broadly categorised into direct and indirect strategies. Direct strategies employ a catalyst for selective hydrogenolytic cleavage of carbon‐oxygen bonds whilst avoiding ring‐hydrogenation reactions. Traditional catalysts are nickel‐ or cobalt‐doped molybdenum sulfides (which enjoy widespread use in the petrochemical industry for the removal of sulfur from crude oil streams410). Heteroatom removal (deoxygenation or desulfurisation) is achieved via coordination to a catalyst sulfur vacancy, followed by hydrogenolysis of the carbon‐heteroatom bond. Evolution of oxygen or sulfur as H2O/H2S then regenerates the active site. HDO typically proceeds more slowly than HDS owing to M−O bonds being stronger than the analogous M−S bonds. Acidic supports enhance the rate of HDO,414 although C‐alkylation of aromatic rings may also occur to some extent.415 Coke formation on the catalyst (similarly related to support acidity) also significantly limits the lifetime of the catalyst;412 alternative molybdenum/tungsten carbide416 and metal phosphide417, 418 catalysts, that follow different deoxygenation mechanisms, have partially overcome this difficulty.
By contrast, indirect deoxygenation strategies employ hydrogenation catalysts to saturate the aromatic ring, thereby weakening the C−O bonds, which are subsequently cleaved via acid‐catalysed dehydration. Typical catalysts contain noble metals (Pd, Pt, Ru, Rh) or Ni, supported on an acidic oxide or carbon.19 Both the acidic and metal functionalities of the catalyst are critical.419 Reaction of phenol in the presence of a Pd/C catalyst leads to the accumulation of cyclohexanone and subsequently cyclohexanol. However, in the presence of phosphoric acid, cyclohexanol rapidly dehydrates, rendering cyclohexene, which is also promptly hydrogenated to cyclohexane over the Pd/C catalyst. In several cases, the reaction of guaiacols proceeds via initial ring hydrogenation, followed by hydrolysis of the methoxy substituent to yield 1,2‐cyclohexanediols, which again undergo dehydration and hydrogenation rendering cyclohexane. A zeolite‐supported Ni catalyst demonstrated that both functions may be integrated into one material, and complex mixtures derived from biomass pyrolysis may be converted to alkanes.420 In this context, Ni/Al‐SBA‐15 was recently demonstrated to be capable of hydrodeoxygenating an Organosolv lignin with selectivity to cycloalkanes higher than 99 %.387 Owing to the similarities to hydrocarbons derived from petroleum, the lignin‐derived alkanes could well be refined into drop‐in fuels by subsequent conventional oil refinery processes. Moreover, in a broader perspective, this example highlights the importance of Al‐SBA‐15, as an acidic support alternative to zeolites or other acidic materials, for the HDO of phenolic streams.387
When arenes are desired, a supplementary dehydrogenation step is necessary following indirect deoxygenation. A mixture of Raney‐Ni and H‐BEA‐35 catalyst, with 2‐propanol as a hydrogen donor dissolved in an aliphatic hydrocarbon as a solvent, effectively converted phenol to benzene via an indirect pathway.284 By employing a liquid‐phase transfer‐hydrogen donor (2‐propanol) instead of gaseous H2, coverage of hydrogen on the nickel surface is significantly reduced, allowing for the dehydrogenation of cyclohexene intermediates to arenes as another potential route. The selectivity to arenes can therefore be fine‐tuned by modification of the available hydrogen in the form of 2‐propanol. This methodology is also applicable to complex pyrolytic bio‐oils and Organosolv lignins.
Examples of stepwise depolymerisation‐HDO processes are known, and serve to highlight some of the challenges that occur for real lignin streams. In one approach, lignin oils were prepared by liquid phase reforming over a 1 wt % Pt on γ‐Al2O3 catalyst, yielding 11 %, 9 % and 5 % mono‐aromatics for Organosolv, Kraft and sugarcane bagasse lignins, respectively.421 The product mixture incorporated mono‐, bis‐ and tris‐oxygenated aromatics, consistent with abundances of coumaryl, guaiacyl and syringyl functionalities in the feedstock. Subsequent HDO, using a Mo2C/C catalyst at 300 °C in dodecane, led to a monomer yield of 9 % (of the original lignin feedstock) for an Organosolv lignin, with a significant reduction in oxygen content. Interestingly, when beginning with bis‐oxygenated model compounds, mono‐oxygenated intermediates accumulate first before oxygen‐free products are formed, whereas mono‐oxygenates did not accumulate with the lignin oils.422 Complete deoxygenation of lignin‐derived bio‐oils is, however, still considerably more challenging than the conversion of oxygenated model compounds.
Birch wood sawdust was depolymerised via a hydrogenolysis procedure over a Pt/C catalyst, at 200 °C in a 1:1 water:1,4‐dioxane mixture with 1 % phosphoric acid.423 The product oil, extracted with cyclohexane, afforded a 46 wt % yield of propyl‐/propanol‐substituted mono‐aromatics, and 12 wt % of di‐aromatics. A second reaction step over Pd/C at 250 °C, using 5 wt % phosphoric acid in water, afforded alkanes in 94 % yield (relative to former monomer yield, divided between C8 (15 %) and C9 (85 %) alkanes). The dimer fraction similarly yielded 82 % of C14–C18 alkanes. Methanol had been generated from hydrolysis of methoxy groups.
Effective hydrotreating of 4‐(1‐propyl)guaiacol to 4‐(1‐propyl)cyclohexanol over a ceria‐supported nickel catalyst has been demonstrated.424 The system was subsequently applied to a lignin‐derived bio‐oil, obtained by ECCL of Pine sawdust using Ru/C in methanol at 250 °C and 30 bar H2. The oil incorporated 12 wt % (relative to the lignin feedstock) of 4‐(1‐propyl)guaiacol, and minor quantities of related products (e.g., 4‐ethylguaiacol, 4‐(1‐propyl)phenol). Subsequent hydrotreatment of the bio‐oil (3 wt % Ni/CeO2 catalyst, 40 bar H2, 300 °C, 200 min) resulted in full conversion, rendering a 73 % yield of the desired product, 4‐propylcyclohexanol. Notably, reaction times required for full conversion were five times longer with the actual bio‐oil, attributable to catalyst inhibition by some minor compounds. These results again illustrate that treatment of actual lignin or its oils is more challenging than the reaction of simple model compounds.
4. Concluding Remarks
This review article has presented a critical assessment comparing various strategies for “beginning‐to‐end” lignin valorisation, with a focus on the literature published since 2010. This survey of the recent lignin chemical literature clearly highlights several recurring phenomena. Importantly, at least two general widespread misconceptions regarding lignin molecular structures and bonding must be amended. Firstly, the assertion that “free” (acyclic) α‐O‐4 bonding motifs are present in the lignin macromolecules is now known to be incorrect, and it is understood that they are instead part of the D2 cyclic dibenzodioxocin moieties. The second misconception is the common practice of assuming that the β‐O‐4 linkage is necessarily the most abundant linkage in all lignins, including industrial lignins. The prevalence of this bonding motif in native lignin varies from 35–85 % depending on the plant type (hardwood, softwood, grass) and the exact species of lignocellulosic feedstock. Furthermore, it must be acknowledged that the abundance of the β‐O‐4 linkages is influenced by a multitude of processing variables, including lignin bioengineering (i.e., the up‐ and downregulation of specific enzymes of the phenylpropanoid pathway), any abiotic stress the plant encounters and, perhaps foremost, the severity of the conditions to which the lignin is exposed during any pretreatment or isolation process.
Clearly, the current lignin streams isolated via the Kraft process exhibit a dramatically reduced quantity of the labile β‐ether units. This principle will also hold true for certain Organosolv processes, particularly those performed with added acid or base catalysts and those run with long residence times. It is therefore not always correct to assume that the Kraft process must always generate a more recalcitrant and highly condensed lignin than Organosolv (or other) processes. In general, lignin recalcitrance and degree of condensation will, to a large extent, be determined by residence time of the lignin fragments (released upon treatment of wood) in both the pulp and liquor. As a case in point, solution‐phase lignins at the “initial delignification” stage of Kraft pulping are known to still retain a high proportion of the native β‐ether moieties. Therefore, the continuous extraction of this Kraft lignin stream may allow for improved ease of downstream valorisation.
To add to the chemical complexity of lignin, there is no single type of β‐ether unit, rather, a variety of such units exhibiting calculated values of BDE in the range of 54–72 kcal mol−1 depending on the chemical nature of the surrounding H, G, S (and other) lignin units. Therefore, some types of β‐O‐4 linkages may not necessarily be more easily cleaved than the β‐β or β‐5 linkages. Under typical harsh conditions, chemical changes to the latter types of linkages are also expected, and therefore the fate of such linkages must be evaluated in more detail.
The bioengineering of lignins to afford a higher abundance of such readily cleaved linkages (e.g., β‐O‐4, β‐β, or benzodioxane) may represent an effective strategy to improve catalytic lignin valorisation processes. It is possible to envision a “cascading” scheme in which these linkages are sequentially cleaved or modified. However, the implications of any upstream modification for all downstream processes must be carefully evaluated, a research area that has so far been scarcely explored. As the properties of isolated lignin depends strongly upon the fractionation process, there is real need for the development of catalytic upstream processes based on ECCL benefiting from the remarkable features of bioengineered lignins. Without such progress, lignin bioengineering may play a limited role in improving the efficiency of catalytic downstream processing, as the fractionation step may destroy the structural features generated by bioengineering.
Evaluation of the recent scientific literature shows that a wealth of oxidative, reductive and redox‐neutral methods for mild depolymerisation has been developed for dimeric or oligomeric lignin model compounds, and in selected cases also for lignin itself. It is noteworthy that, with several key exceptions, the optimum conditions for cleavage of model compounds do not translate well into those for real lignin depolymerisation reactions. Future investigations should therefore centre upon more complex models, or mixtures of models, that more accurately reflect the reactivity of lignin, and should apply the catalytic reactions to actual and well‐characterised lignin feedstocks themselves. Advanced synthetic lignin‐mimicking polymers are also particularly attractive in this sense, as they more accurately represent lignin's complexity than low‐molecular‐weight counterparts and may enable the bridging of this gap.
Catalytic lignin depolymerisation methods employing costly and stoichiometric reagents ought to be avoided, or are only warranted if high value components can be extracted and if the remainder of the lignin can still be valorised by other means. By contrast, convergent approaches (for example lignin depolymerisation‐hydrodeoxygenation) are likely to reduce the requirement for intermediate purification and separation stages.
The development of standardised analytical protocols for the characterisation of the starting feedstock and the generated bio‐oil(s) are of utmost importance. Particular attention must still be devoted to the development of methods for the analysis of the structure of residual intermediate‐molecular‐weight fractions. Indeed, characterisation of this component remains challenging. It is nevertheless evident that substantial progress has been made in developing depolymerisation methods that afford high yields of mono‐aromatic compounds and liquid products. These emerging approaches often achieve this by inhibiting recondensation channels by the action of capping agents, solvents, catalytic trapping pathways, and careful tuning of reaction parameters.
It is of great importance for future research to consider other factors, for example the influence of lignin pretreatment methods on downstream catalytic processes, not simply in terms of the resulting lignin structure, but also with regards to any impurities that are imparted to the lignin stream. Such impurities, which can greatly vary in nature, have been demonstrated to have a significant influence on the efficiency of depolymerisation. This influence is closely related to catalyst stability and the ability to recycle the catalyst during the long lifetimes that are required for actual commercial application.
Building upon the above conclusions and looking towards the future of lignin valorisation, effective and feasible catalytic valorisation strategies must consider every stage of the process stream, including contribution and expertise from a broad array of scientific disciplines. When designing lignin valorisation technologies, the volume of lignin that the envisaged application is able to absorb is of prime importance. Moreover, the size of the potential market and the price volatility of the targeted product are key considerations. In some circumstances, it will be appropriate to consider more than one possible target molecular and/or material. Advantages associated with a convergent approach—whereby lignin fragments are “funnelled” towards a limited, separable set of end products—or a stepwise, cascading approach—involving progressive production of high‐value and low‐value compounds—should be evaluated on a somewhat case‐by‐case basis. Integration of optimised catalytic lignin valorisation processes into existing cellulose‐centred technologies is important in order for the concept of an integrated “biorefinery” to be realised. As the majority of current biorefinery processes are primarily geared towards optimising sugar production, the (recondensed, and thus recalcitrant) technical lignin streams obtained are likely to require severe catalytic processing for their utilisation in the chemical and fuel industries. Future biorefineries ought to be designed with optimum valorisation of the whole biomass in mind, which potentially may lead to lignin streams more amenable to selective conversion (as with those obtained from the emerging catalytic treatments based on “Early‐stage Catalytic Conversion of Lignin”, ECCL or “lignin‐first” strategies).
In conclusion, the high natural abundance, high carbon content and highly‐functionalised nature of lignin render it an attractive feedstock for targeted valorisation to fuels, polymer composites, synthetic building blocks and valuable (e.g., pharmaceutical) precursors. At present, lignin is still heavily underutilised, being frequently employed simply as a low‐grade fuel. With the emergence of evermore selective and tailored lignin valorisation processes and the growing ability to alter lignin structure in the growing plant, lignin‐derived chemicals and materials may be expected to find increasingly widespread applications, opening up new avenues for the polymer, chemical and fuel industries.
Biographical Information
Roberto Rinaldi (Brazil, 1979) received his PhD from the State University of Campinas “UNICAMP” in São Paulo, Brazil (2006). He joined the Max‐Planck‐Institut für Kohlenforschung as a postdoctoral research fellow (2007), was promoted to Junior Group Leader in 2009, and received the “Sofja Kovalevskaja” award from the Alexander von Humboldt Foundation, thus becoming an Independent Group Leader (2009). Since 2015, he is a Senior Lecturer in the Department of Chemical Engineering, Imperial College London. His research focuses on the catalytic valorisation of lignin and cellulose.

Biographical Information
Robin Jastrzebski (The Netherlands, 1987) obtained his Bachelor's (2009) and Master's (2011) degrees from Utrecht University, The Netherlands. In 2016, he obtained his PhD degree, supervised by Dr. Pieter Bruijnincx and Prof. Bert Weckhuysen, at Utrecht University. His PhD research focused on development of novel catalytic technologies for the production of chemicals and fuels from biomass.

Biographical Information
Matthew T. Clough (United Kingdom, 1989) obtained his PhD from Imperial College London (2015), supervised by Prof. Tom Welton and Dr. Patricia Hunt, on a combined experimental/computational project investigating the thermal stabilities of ionic liquids and their mixtures with carbohydrates. Currently, he is a Postdoctoral Research Fellow at the Max‐Planck‐Institut für Kohlenforschung in Mülheim, Germany. His research interests include the catalytic valorisation of lignocellulose and ionic liquid chemistry.

Biographical Information
John Ralph (New Zealand, 1954) obtained his BSc (1976) from Canterbury University, New Zealand, and his PhD from the University of Wisconsin‐Madison (1982), supervised by Prof. Raymond A. Young. At present he is a Professor in the Department of Biochemistry, and is the Plants Area lead of the DOE's Great Lakes Bioenergy Research Center in Madison, Wisconsin; in 2015 he became Distinguished Professor of the Tokyo University of Agriculture and Technology.

Biographical Information
Marco Kennema (Canada, 1987) completed his Master's degree at the University of Guelph, Canada (2012). He is currently a PhD candidate in the group of Dr. Roberto Rinaldi at the Max‐Planck‐Institut für Kohlenforschung in Germany. His research interests include in‐line and in‐situ analytics, high‐throughput experiments, and valorisation of biomass.

Biographical Information
Pieter C. A. Bruijnincx (The Netherlands, 1979) obtained both his MSc (2002) and PhD (2007) degrees from Utrecht University. After a postdoctoral stay at the University of Warwick, UK, he returned to Utrecht to join the Weckhuysen group to work on the catalytic conversion of biomass for the production of bio‐based chemicals and fuels. He is currently a tenured Associate Professor and was recently elected as member of the Young Academy of the Royal Netherlands Academy of Arts and Sciences.

Biographical Information
Bert M. Weckhuysen (Belgium, 1968) obtained both his Master's (1991) and his PhD (1995) degrees from Leuven University, Belgium, after which he was a postdoctoral fellow with Prof. Israel Wachs and Prof. Jack Lunsford at Lehigh University and Texas A&M University, respectively. In 2000, he was appointed as Full Professor of Inorganic Chemistry and Catalysis at Utrecht University in The Netherlands. At present, he is Distinguished Professor of the Faculty of Science at Utrecht University.

Acknowledgements
R.R. acknowledges the financial support from the Alexander von Humboldt Foundation (Sofja Kovalevskaja Award 2010) and the Cluster of Excellence “Tailor‐Made Fuels from Biomass”. R.R. is grateful to Prof. Dr. Patricia Nunes da Silva (Institute of Mathematics and Statistics, State University of Rio de Janeiro, Brazil) for the valuable discussions on the statistics of depolymerisation processes. P.C.A.B. acknowledges the Netherlands Organisation for Scientific Research (NWO) for a Vernieuwingsimpuls Veni Grant. B.M.W. acknowledges the European Research Council (Advanced ERC grant no. 321140). P.C.A.B. and B.M.W. also gratefully acknowledge support from the European Commission (SuBiCat Initial Training Network, Call FP7‐PEOPLE‐2013, ITN, grant no. 607044) and from the CatchBio program, funded by the Smart Mix Program of The Netherlands Ministry of Economic Affairs and The Netherlands Ministry of Education, Culture, and Science. J.R. was funded in part by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE‐FC02‐07ER64494), and in part by Stanford University's Global Climate and Energy Project.
R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx, B. M. Weckhuysen, Angew. Chem. Int. Ed. 2016, 55, 8164.
Contributor Information
Dr. Roberto Rinaldi, Email: rrinaldi@imperial.ac.uk.
Prof. Dr. John Ralph, Email: jralph@wisc.edu
Dr. Pieter C. A. Bruijnincx, Email: p.c.a.bruijnincx@uu.nl.
Prof. Dr. Bert M. Weckhuysen, Email: b.m.weckhuysen@uu.nl
References
- 1. Ralph J., Phytochem. Rev. 2010, 9, 65–83. [Google Scholar]
- 2. Vanholme R., Demedts B., Morreel K., Ralph J., Boerjan W., Plant Physiol. 2010, 153, 895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi R., Sun Y.-H., Li Q., Heber S., Sederoff R., Chiang V. L., Plant Cell Physiol. 2010, 51, 144–163. [DOI] [PubMed] [Google Scholar]
- 4. Weng J.-K., Chapple C., New Phytol. 2010, 187, 273–285. [DOI] [PubMed] [Google Scholar]
- 5. Magalhaes Silva Moura J. C., Valencise Bonine C. A., Fernandes Viana J. d. O., Dornelas M. C., Mazzafera P., J. Integr. Plant Biol. 2010, 52, 360–376. [DOI] [PubMed] [Google Scholar]
- 6. Zhao Q., Dixon R. A., Trends Plant Sci. 2011, 16, 227–233. [DOI] [PubMed] [Google Scholar]
- 7. Liu C.-J., Mol. Plant 2012, 5, 304–317. [DOI] [PubMed] [Google Scholar]
- 8. Bonawitz N. D., Chapple C., in Annual Review of Genetics, Vol. 44 (Eds.: A. Campbell, M. Lichten, G. Schupbach), 2010, pp. 337–363. [DOI] [PubMed] [Google Scholar]
- 9. Mottiar Y., Vanholme R., Boerjan W., Ralph J., Mansfield S. D., Curr. Opin. Biotechnol. 2016, 37, 190–200. [DOI] [PubMed] [Google Scholar]
- 10. Hatakeyama H., Hatakeyama T., in Biopolymers, Vol. 232 (Eds.: A. Abe, K. Dusek, S. Kobayashi), Springer, Berlin, Heidelberg, 2010, pp. 1–63. [Google Scholar]
- 11. Vanholme R., Morreel K., Darrah C., Oyarce P., Grabber J. H., Ralph J., Boerjan W., New Phytol. 2012, 196, 978–1000. [DOI] [PubMed] [Google Scholar]
- 12. Simmons B. A., Loqué D., Ralph J., Curr. Opin. Plant Biol. 2010, 13, 312–319. [DOI] [PubMed] [Google Scholar]
- 13. Bonawitz N. D., Chapple C., Curr. Opin. Biotechnol. 2013, 24, 336–343. [DOI] [PubMed] [Google Scholar]
- 14. Pandey M. P., Kim C. S., Chem. Eng. Technol. 2011, 34, 29–41. [Google Scholar]
- 15. Zakzeski J., Bruijnincx P. C. A., Jongerius A. L., Weckhuysen B. M., Chem. Rev. 2010, 110, 3552–3599. [DOI] [PubMed] [Google Scholar]
- 16. Azadi P., Inderwildi O. R., Farnood R., King D. A., Renewable Sustainable Energy Rev. 2013, 21, 506–523. [Google Scholar]
- 17. Kang S., Li X., Fan J., Chang J., Renewable Sustainable Energy Rev. 2013, 27, 546–558. [Google Scholar]
- 18. Li C., Zhao X., Wang A., Huber G. W., Zhang T., Chem. Rev. 2015, 115, 11559–11624. [DOI] [PubMed] [Google Scholar]
- 19. Saidi M., Samimi F., Karimipourfard D., Nimmanwudipong T., Gates B. C., Rahimpour M. R., Energy Environ. Sci. 2014, 7, 103–129. [Google Scholar]
- 20. Zeng Y., Zhao S., Yang S., Ding S.-Y., Curr. Opin. Biotechnol. 2014, 27, 38–45. [DOI] [PubMed] [Google Scholar]
- 21. Collard F.-X., Blin J., Renewable Sustainable Energy Rev. 2014, 38, 594–608. [Google Scholar]
- 22. Lange H., Decina S., Crestini C., Eur. Polym. J. 2013, 49, 1151–1173. [Google Scholar]
- 23. Chatel G., Rogers R. D., ACS Sustainable Chem. Eng. 2014, 2, 322–339. [Google Scholar]
- 24. Hanson S. K., Baker R. T., Acc. Chem. Res. 2015, 48, 2037–2048. [DOI] [PubMed] [Google Scholar]
- 25. Doherty W. O. S., Mousavioun P., Fellows C. M., Ind. Crops Prod. 2011, 33, 259–276. [Google Scholar]
- 26. Laurichesse S., Averous L., Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar]
- 27. Thakur V. K., Thakur M. K., Raghavan P., Kessler M. R., ACS Sustainable Chem. Eng. 2014, 2, 1072–1092. [Google Scholar]
- 28. Sen S., Patil S., Argyropoulos D. S., Green Chem. 2015, 17, 4862–4887. [Google Scholar]
- 29. Esposito D., Antonietti M., Chem. Soc. Rev. 2015, 44, 5821–5835. [DOI] [PubMed] [Google Scholar]
- 30. Ten E., Vermerris W., J. Appl. Polym. Sci. 2015, 132, 42069. [Google Scholar]
- 31. Ralph J., Landucci L. L., in Lignin and Lignans: Advances in Chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press, Boca Raton, 2010, pp. 137–244. [Google Scholar]
- 32. Lapierre C., in Lignin and Lignans: Advances in Chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press, Boca Raton, 2010, pp. 11–48. [Google Scholar]
- 33. Pu Y., Cao S., Ragauskas A. J., Energy Environ. Sci. 2011, 4, 3154–3166. [Google Scholar]
- 34. Wen J.-L., Sun S.-L., Xue B.-L., Sun R.-C., Materials 2013, 6, 359–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghaffar S. H., Fan M., Biomass Bioenergy 2013, 57, 264–279. [Google Scholar]
- 36. Tolbert A., Akinosho H., Khunsupat R., Naskar A. K., Ragauskas A. J., Biofuels, Bioprod. Biorefin. 2014, 8, 836–856. [Google Scholar]
- 37. Lupoi J. S., Singh S., Parthasarathi R., Simmons B. A., Henry R. J., Renewable Sustainable Energy Rev. 2015, 49, 871–906. [Google Scholar]
- 38. Thevenot M., Dignac M.-F., Rumpel C., Soil Biol. Biochem. 2010, 42, 1200–1211. [Google Scholar]
- 39. Bugg T. D. H., Ahmad M., Hardiman E. M., Rahmanpour R., Nat. Prod. Rep. 2011, 28, 1883–1896. [DOI] [PubMed] [Google Scholar]
- 40. Bugg T. D. H., Ahmad M., Hardiman E. M., Singh R., Curr. Opin. Biotechnol. 2011, 22, 394–400. [DOI] [PubMed] [Google Scholar]
- 41. Bauer S., Sorek H., Mitchell V. D., Ibáñez A. B., Wemmer D. E., J. Agric. Food Chem. 2012, 60, 8203–8212. [DOI] [PubMed] [Google Scholar]
- 42. Marita J. M., Ralph J., Hatfield R. D., Guo D. J., Chen F., Dixon R. A., Phytochemistry 2003, 62, 53–65. [DOI] [PubMed] [Google Scholar]
- 43. Zhao Q., Tobimatsu Y., Zhou R., Pattathil S., Gallego-Giraldo L., Fu C., Jackson L. A., Hahn M. G., Kim H., Chen F., Ralph J., Dixon R. A., Proc. Natl. Acad. Sci. USA 2013, 110, 13660–13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wilkerson C. G., Mansfield S. D., Lu F., Withers S., Park J. Y., Karlen S. D., Gonzales-Vigil E., Padmakshan D., Unda F., Rencoret J., Ralph J., Science 2014, 344, 90–93. [DOI] [PubMed] [Google Scholar]
- 45. Wagner A., Donaldson L., Ralph J., in Advances in Botanical Research. Lignins: biosynthesis, biodegradation and bioengineering, Vol. 61 (Eds.: L. Jouanin, C. Lapierre), Academic Press, Burlington, 2012, pp. 37–76. [Google Scholar]
- 46. Gallego-Giraldo L., Shadle G., Shen H., Barros-Rios J., Corrales S. Fresquet, Wang H., Dixon R. A., Plant Biotechnol. J. 2016, 14, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baxter H. L., Mazarei M., Labbe N., Kline L. M., Cheng Q., Windham M. T., Mann D. G., Fu C., Ziebell A., Sykes R. W., Plant Biotechnol. J. 2014, 12, 914–924. [DOI] [PubMed] [Google Scholar]
- 48. Shen H., Poovaiah C. R., Ziebell A., Tschaplinski T. J., Pattathil S., Gjersing E., Engle N. L., Katahira R., Pu Y., Sykes R., Biotechnol. Biofuels 2013, 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tschaplinski T. J., Standaert R. F., Engle N. L., Martin M. Z., Sangha A. K., Parks J. M., Smith J. C., Samuel R., Jiang N., Pu Y., Biotechnol. Biofuels 2012, 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu B., Escamilla-Treviño L. L., Sathitsuksanoh N., Shen Z., Shen H., Percival Zhang Y. H., Dixon R. A., Zhao B., New Phytol. 2011, 192, 611–625. [DOI] [PubMed] [Google Scholar]
- 51. Fu C., Mielenz J. R., Xiao X., Ge Y., Hamilton C. Y., Rodriguez M., Chen F., Foston M., Ragauskas A., Bouton J., Proc. Natl. Acad. Sci. USA 2011, 108, 3803–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Escamilla-Treviño L. L., Shen H., Uppalapati S. R., Ray T., Tang Y., Hernandez T., Yin Y., Xu Y., Dixon R. A., New Phytol. 2010, 185, 143–155. [DOI] [PubMed] [Google Scholar]
- 53. Shen H., Mazarei M., Hisano H., Escamilla-Trevino L., Fu C., Pu Y., Rudis M. R., Tang Y., Xiao X., Jackson L., Plant Cell 2013, 25, 4342–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rohde A., Morreel K., Ralph J., Goeminne G., Hostyn V., De Rycke R., Kushnir S., Van Doorsselaere J., Joseleau J. P., Vuylsteke M., Van Driessche G., Van Beeumen J., Messens E., Boerjan W., Plant Cell 2004, 16, 2749–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Millar D. J., Long M., Donovan G., Fraser P. D., Boudet A.-M., Danoun S., Bramley P. M., Bolwell G. P., Phytochemistry 2007, 68, 1497–1509. [DOI] [PubMed] [Google Scholar]
- 56. Schilmiller A. L., Stout J., Weng J.-K., Humphreys J., Ruegger M. O., Chapple C., Plant J. 2009, 60, 771–782. [DOI] [PubMed] [Google Scholar]
- 57. Li X., Bonawitz N. D., Weng J.-K., Chapple C., Plant Cell 2010, 22, 1620–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moinuddin S. G. A., Jourdes M., Laskar D. D., Ki C., Cardenas C. L., Kim K.-W., Zhang D., Davin L. B., Lewis N. G., Org. Biomol. Chem. 2010, 8, 3928–3946. [DOI] [PubMed] [Google Scholar]
- 59. Patten A. M., Jourdes M., Cardenas C. L., Laskar D. D., Nakazawa Y., Chung B.-Y., Franceschi V. R., Davin L. B., Lewis N. G., Mol. Biosyst. 2010, 6, 499–515. [DOI] [PubMed] [Google Scholar]
- 60. Vanholme R., Ralph J., Akiyama T., Lu F., Pazo J. R., Kim H., Christensen J. H., Van Reusel B., Storme V., De Rycke R., Rohde A., Morreel K., Boerjan W., Plant J. 2010, 64, 885–897. [DOI] [PubMed] [Google Scholar]
- 61. Berthet S., Demont-Caulet N., Pollet B., Bidzinski P., Cezard L., Le Bris P., Borrega N., Herve J., Blondet E., Balzergue S., Lapierre C., Jouanin L., Plant Cell 2011, 23, 1124–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thévenin J., Pollet B., Letarnec B., Saulnier L., Gissot L., Maia-Grondard A., Lapierre C., Jouanin L., Mol. Plant 2011, 4, 70–82. [DOI] [PubMed] [Google Scholar]
- 63. Sung C. B., Si-Yong K., Hyeun-Jong B., Hyoun-Sub L., Eui-Ho P., Hanhong B., Res. J. Biotechnol. 2012, 7, 23–33. [Google Scholar]
- 64. Herrero J., Esteban-Carrasco A., Miguel Zapata J., Plant Physiol. Biochem. 2013, 67, 77–86. [DOI] [PubMed] [Google Scholar]
- 65. Öhman D., Demedts B., Kumar M., Gerber L., Gorzsás A., Goeminne G., Hedenström M., Ellis B., Boerjan W., Sundberg B., Plant J. 2013, 73, 63–76. [DOI] [PubMed] [Google Scholar]
- 66. Vanholme R., Cesarino I., Rataj K., Xiao Y., Sundin L., Goeminne G., Kim H., Cross J., Morreel K., Araujo P., Welsh L., Haustraete J., McClellan C., Vanholme B., Ralph J., Simpson G. G., Halpin C., Boerjan W., Science 2013, 341, 1103–1106. [DOI] [PubMed] [Google Scholar]
- 67. Wagner A., Tobimatsu Y., Goeminne G., Phillips L., Flint H., Steward D., Torr K., Donaldson L., Boerjan W., Ralph J., Plant Mol. Biol. 2013, 81, 105–117. [DOI] [PubMed] [Google Scholar]
- 68. Ali M. B., D. H. McNear, Jr. , BMC Plant Biol. 2014, 14, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Herrero J., Esteban Carrasco A., Miguel Zapata J., Plant Physiol. Biochem. 2014, 80, 192–202. [DOI] [PubMed] [Google Scholar]
- 70. Fernández-Pérez F., Vivar T., Pomar F., Pedreño M. A., Novo-Uzal E., J. Plant Physiol. 2015, 175, 86–94. [DOI] [PubMed] [Google Scholar]
- 71. Wang H., Zhao Q., Chen F., Wang M., Dixon R. A., Plant J. 2011, 68, 1104–1114. [DOI] [PubMed] [Google Scholar]
- 72. Zhou R., Jackson L., Shadle G., Nakashima J., Temple S., Chen F., Dixon R. A., Proc. Natl. Acad. Sci. USA 2010, 107, 17803–17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhao Q., Gallego-Giraldo L., Wang H., Zeng Y., Ding S. Y., Chen F., Dixon R. A., Plant J. 2010, 63, 100–114. [DOI] [PubMed] [Google Scholar]
- 74. Naoumkina M. A., Zhao Q., Gallego-Giraldo L., Dai X., Zhao P. X., Dixon R. A., Mol. Plant Pathol. 2010, 11, 829–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jansen F., Gillessen B., Mueller F., Commandeur U., Fischer R., Kreuzaler F., Biotechnol. Appl. Biochem. 2014, 61, 646–654. [DOI] [PubMed] [Google Scholar]
- 76. Higuchi T., Ito Y., Kawamura I., Phytochemistry 1967, 6, 875–881. [Google Scholar]
- 77. Hatfield R., Ralph J., Grabber J. H., Planta 2008, 228, 919–928. [DOI] [PubMed] [Google Scholar]
- 78. Takahama U., Oniki T., Shimokawa H., Plant Cell Physiol. 1996, 37, 499–504. [Google Scholar]
- 79. Lu F., Karlen S. D., Regner M., Kim H., Ralph S. A., Sun R.-C., Kuroda K.-i., Augustin M. A., Mawson R., Sabarez H., Singh T., Jimenez-Monteon G., Zakaria S., Hill S., Harris P. J., Boerjan W., Wilkerson C. G., Mansfield S. D., Ralph J., BioEnergy Res. 2015, 8, 934–952. [Google Scholar]
- 80. Lan W., Lu F., Regner M., Zhu Y., Rencoret J., Ralph S. A., Zakai U. I., Morreel K., Boerjan W., Ralph J., Plant Physiol. 2015, 167, 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lu F., Ralph J., Org. Biomol. Chem. 2008, 6, 3681–3694. [DOI] [PubMed] [Google Scholar]
- 82. del Río J. C., Rencoret J., Marques G., Gutiérrez A., Ibarra D., Santos J. I., Jiménez-Barbero J. S., Zhang L., Martínez A. N. T., J. Agric. Food Chem. 2008, 56, 9525–9534. [DOI] [PubMed] [Google Scholar]
- 83. Smith R. A., Gonzales-Vigil E., Karlen S. D., Park J. Y., Lu F., Wilkerson C. G., Samuels L., Ralph J., Mansfield S. D., Plant Physiol. 2015, 169, 2992–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Petrik D. L., Karlen S. D., Cass C. L., Padmakshan D., Lu F., Liu S., Bris P., Antelme S., Santoro N., Wilkerson C. G., Plant J. 2014, 77, 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Withers S., Lu F., Kim H., Zhu Y., Ralph J., Wilkerson C. G., J. Biol. Chem. 2012, 287, 8347–8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Marita J. M., Hatfield R. D., Rancour D. M., Frost K. E., Plant J. 2014, 78, 850–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vanholme R., Morreel K., Ralph J., Boerjan W., Curr. Opin. Plant Biol. 2008, 11, 278–285. [DOI] [PubMed] [Google Scholar]
- 88. Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P. F., Marita J. M., Hatfield R. D., Ralph S. A., Christensen J. H., Phytochem. Rev. 2004, 3, 29–60. [Google Scholar]
- 89. Boerjan W., Ralph J., Baucher M., Annu. Rev. Plant Biol. 2003, 54, 519–546. [DOI] [PubMed] [Google Scholar]
- 90. Ralph J., Kim H., Peng J., Lu F., Org. Lett. 1999, 1, 323–326. [DOI] [PubMed] [Google Scholar]
- 91. Ralph J., MacKay J. J., Hatfield R. D., O'Malley D. M., Whetten R. W., Sederoff R. R., Science 1997, 277, 235–239. [DOI] [PubMed] [Google Scholar]
- 92. Ralph J., Lapierre C., Marita J. M., Kim H., Lu F., Hatfield R. D., Ralph S., Chapple C., Franke R., Hemm M. R., Phytochemistry 2001, 57, 993–1003. [DOI] [PubMed] [Google Scholar]
- 93. Del Río J. C., Rencoret J., Prinsen P., Martínez A. n. T., Ralph J., Gutiérrez A., J. Agric. Food Chem. 2012, 60, 5922–5935. [DOI] [PubMed] [Google Scholar]
- 94. Ralph J., Bunzel M., Marita J. M., Hatfield R. D., Lu F., Kim H., Schatz P. F., Grabber J. H., Steinhart H., Phytochem. Rev. 2004, 3, 79–96. [Google Scholar]
- 95. Osakabe K., Tsao C. C., Li L., Popko J. L., Umezawa T., Carraway D. T., Smeltzer R. H., Joshi C. P., Chiang V. L., Proc. Natl. Acad. Sci. USA 1999, 96, 8955–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Humphreys J., Hemm M., Chapple C., Proc. Natl. Acad. Sci. USA 1999, 96, 10045–10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Humphreys J. M., Chapple C., Curr. Opin. Plant Biol. 2002, 5, 224–229. [DOI] [PubMed] [Google Scholar]
- 98. Li L., Zhou Y., Cheng X., Sun J., Marita J. M., Ralph J., Chiang V. L., Proc. Natl. Acad. Sci. USA 2003, 100, 4939–4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cass C. L., Peraldi A., Dowd P. F., Mottiar Y., Santoro N., Karlen S. D., Bukhman Y. V., Foster C. E., Thrower N., Bruno L. C., Moskvin O. V., Johnson E. T., Willhoit M. E., Phutane M., Ralph J., Mansfield S. D., Nicholson P., Sedbrook J. C., J. Exp. Bot. 2015, 66, 4317–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ralph J., Akiyama T., Coleman H. D., Mansfield S. D., BioEnergy Res. 2012, 5, 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tu Y., Rochfort S., Liu Z., Ran Y., Griffith M., Badenhorst P., Louie G. V., Bowman M. E., Smith K. F., Noel J. P., Mouradov A., Spangenberg G., Plant Cell 2010, 22, 3357–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Derikvand M. M., Sierra J. B., Ruel K., Pollet B., Do C.-T., Thevenin J., Buffard D., Jouanin L., Lapierre C., Planta 2008, 227, 943–956. [DOI] [PubMed] [Google Scholar]
- 103. Tamasloukht B., Lam M. S.-J. W. Q., Martinez Y., Tozo K., Barbier O., Jourda C., Jauneau A., Borderies G., Balzergue S., Renou J.-P., Huguet S., Martinant J. P., Tatout C., Lapierre C., Barriere Y., Goffner D., Pichon M., J. Exp. Bot. 2011, 62, 3837–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Piquemal J., Lapierre C., Myton K., O'Connell A., Schuch W., Grima-Pettenati J., Boudet A. M., Plant J. 1998, 13, 71–83. [Google Scholar]
- 105. Van Acker R., Leple J.-C., Aerts D., Storme V., Goeminne G., Ivens B., Legee F., Lapierre C., Piens K., Van Montagu M. C. E., Santoro N., Foster C. E., Ralph J., Soetaert W., Pilate G., Boerjan W., Proc. Natl. Acad. Sci. USA 2014, 111, 845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ralph J., Kim H., Lu F., Grabber J. H., Leplé J. C., Berrio-Sierra J., Derikvand M. M., Jouanin L., Boerjan W., Lapierre C., Plant J. 2007, 53, 368–379. [DOI] [PubMed] [Google Scholar]
- 107. Leple J.-C., Dauwe R., Morreel K., Storme V., Lapierre C., Pollet B., Naumann A., Kang K.-Y., Kim H., Ruel K., Plant Cell 2007, 19, 3669–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fornalé S., Capellades M., Encina A., Wang K., Irar S., Lapierre C., Ruel K., Joseleau J.-P., Berenguer J., Puigdomenech P., Rigau J., Caparros-Ruiz D., Mol. Plant 2012, 5, 817–830. [DOI] [PubMed] [Google Scholar]
- 109. Konovalov A. A., Shundrina I. K., Karpova E. V., Nefedov A. A., Goncharov N. P., Russ. J. Genet. 2014, 50, 1161–1168. [Google Scholar]
- 110. d′Yvoire M. B., Bouchabke-Coussa O., Voorend W., Antelme S., Cezard L., Legee F., Lebris P., Legay S., Whitehead C., McQueen-Mason S. J., Gomez L. D., Jouanin L., Lapierre C., Sibout R., Plant J. 2013, 73, 496–508. [DOI] [PubMed] [Google Scholar]
- 111. MacKay J., Presnell T., Jameel H., Taneda H., O'Malley D., Sederoff R., Holzforschung 1999, 53, 403–410. [Google Scholar]
- 112. Saathoff A. J., Sarath G., Chow E. K., Dien B. S., Tobias C. M., PLoS ONE 2011, 6, e16416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ookawa T., Inoue K., Matsuoka M., Ebitani T., Takarada T., Yamamoto T., Ueda T., Yokoyama T., Sugiyama C., Nakaba S., Funada R., Kato H., Kanekatsu M., Toyota K., Motobayashi T., Vazirzanjani M., Tojo S., Hirasawa T., Sci. Rep. 2014, 4, 6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vanholme B., Cesarino I., Goeminne G., Kim H., Marroni F., Van Acker R., Vanholme R., Morreel K., Ivens B., Pinosio S., Morgante M., Ralph J., Bastien C., Boerjan W., New Phytol. 2013, 198, 765–776. [DOI] [PubMed] [Google Scholar]
- 115. Sattler S. E., Palmer N. A., Saballos A., Greene A. M., Xin Z., Sarath G., Vermerris W., Pedersen J. F., BioEnergy Res. 2012, 5, 855–865. [Google Scholar]
- 116. Jung J. H., Vermerris W., Gallo M., Fedenko J. R., Erickson J. E., Altpeter F., Plant Biotechnol. J. 2013, 11, 709–716. [DOI] [PubMed] [Google Scholar]
- 117. Ciesielski P. N., Resch M. G., Hewetson B., Killgore J. P., Curtin A., Anderson N., Chiaramonti A. N., Hurley D. C., Sanders A., Himmel M. E., Chapple C., Mosier N., Donohoe B. S., Green Chem. 2014, 16, 2627–2635. [Google Scholar]
- 118. Meyer K., Shirley A. M., Cusumano J. C., Bell-Lelong D. A., Chapple C., Proc. Natl. Acad. Sci. USA 1998, 95, 6619–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Weng J.-K., Mo H., Chapple C., Plant J. 2010, 64, 898–911. [DOI] [PubMed] [Google Scholar]
- 120. Ralph J., Akiyama T., Kim H., Lu F., Schatz P. F., Marita J. M., Ralph S. A., Reddy M. S., Chen F., Dixon R. A., J. Biol. Chem. 2006, 281, 8843–8853. [DOI] [PubMed] [Google Scholar]
- 121. Huntley S. K., Ellis D., Gilbert M., Chapple C., Mansfield S. D., J. Agric. Food Chem. 2003, 51, 6178–6183. [DOI] [PubMed] [Google Scholar]
- 122. Stewart J. J., Akiyama T., Chapple C., Ralph J., Mansfield S. D., Plant Physiol. 2009, 150, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Santoro N., Cantu S. L., Tornqvist C.-E., Falbel T. G., Bolivar J. L., Patterson S. E., Pauly M., Walton J. D., BioEnergy Res. 2010, 3, 93–102. [Google Scholar]
- 124. Ros Barceló A., Protoplasma 1995, 186, 41–44. [Google Scholar]
- 125. Berthet S., Thevenin J., Baratiny D., Demont-Caulet N., Debeaujon I., Bidzinski P., Leple J.-C., Huis R., Hawkins S., Gomez L.-D., Lapierre C., Jouanin L., in Lignins: Biosynthesis, Biodegradation and Bioengineering, Vol. 61 (Eds.: L. Jouann, C. Lapierre), 2012, pp. 145–172. [Google Scholar]
- 126. Zhao Q., Nakashima J., Chen F., Yin Y., Fu C., Yun J., Shao H., Wang X., Wang Z.-Y., Dixon R. A., Plant Cell 2013, 25, 3976–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lu S., Li Q., Wei H., Chang M.-J., Tunlaya-Anukit S., Kim H., Liu J., Song J., Sun Y.-H., Yuan L., Yeh T.-F., Peszlen I., Ralph J., Sederoff R. R., Chiang V. L., Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wang C.-Y., Zhang S., Yu Y., Luo Y.-C., Liu Q., Ju C., Zhang Y.-C., Qu L.-H., Lucas W. J., Wang X., Chen Y.-Q., Plant Biotechnol. J. 2014, 12, 1132–1142. [DOI] [PubMed] [Google Scholar]
- 129. Lu S., Li Q., Wei H., Chang M.-J., Tunlaya-Anukit S., Kim H., Liu J., Song J., Sun Y.-H., Yuan L., Proc. Natl. Acad. Sci. USA 2013, 110, 10848–10853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cesarino I., Araújo P., Mayer J. L. S., Vicentini R., Berthet S., Demedts B., Vanholme B., Boerjan W., Mazzafera P., J. Exp. Bot. 2013, 64, 1769–1781. [DOI] [PubMed] [Google Scholar]
- 131. Dubouzet J. G., Strabala T. J., Wagner A., Plant Sci. 2013, 212, 72–101. [DOI] [PubMed] [Google Scholar]
- 132. Dauwe R., Morreel K., Goeminne G., Gielen B., Rohde A., Van Beeumen J., Ralph J., Boudet A. M., Kopka J., Rochange S. F., Plant J. 2007, 52, 263–285. [DOI] [PubMed] [Google Scholar]
- 133. Voelker S. L., Lachenbruch B., Meinzer F. C., Kitin P., Strauss S. H., Plant Cell Environ. 2011, 34, 655–668. [DOI] [PubMed] [Google Scholar]
- 134. Bonawitz N. D., Im Kim J., Tobimatsu Y., Ciesielski P. N., Anderson N. A., Ximenes E., Maeda J., Ralph J., Donohoe B. S., Ladisch M., Nature 2014, 509, 376–380. [DOI] [PubMed] [Google Scholar]
- 135. Lu F., Marita J. M., Lapierre C., Jouanin L., Morreel K., Boerjan W., Ralph J., Plant Physiol. 2010, 153, 569–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Morreel K., Ralph J., Lu F., Goeminne G., Busson R., Herdewijn P., Goeman J. L., Van der Eycken J., Boerjan W., Messens E., Plant Physiol. 2004, 136, 4023–4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ralph J., Lapierre C., Lu F., Marita J. M., Pilate G., Van Doorsselaere J., Boerjan W., Jouanin L., J. Agric. Food Chem. 2001, 49, 86–91. [DOI] [PubMed] [Google Scholar]
- 138. Marita J. M., Ralph J., Lapierre C., Jouanin L., Boerjan W., J. Chem. Soc. Perkin Trans. 1 2001, 2939–2945. [Google Scholar]
- 139. Jouanin L., Gujon T., Sibout R., Pollet B., Mila I., Leplé J.-C., Pilate G., Petit-Conil M., Ralph J., Lapierre C., in Plantation Forest Biotechnology in the 21st Century (Eds.: C. Walter, M. Carson), Research Signpost, Kerala, 2004, pp. 219–229. [Google Scholar]
- 140. Wagner A., Tobimatsu Y., Phillips L., Flint H., Torr K., Donaldson L., Pears L., Ralph J., Plant J. 2011, 67, 119–129. [DOI] [PubMed] [Google Scholar]
- 141. Jouanin L., Goujon T., de Nadai V., Martin M.-T., Mila I., Vallet C., Pollet B., Yoshinaga A., Chabbert B., Petit-Conil M., Plant Physiol. 2000, 123, 1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Anderson N. A., Tobimatsu Y., Ciesielski P. N., Ximenes E., Ralph J., Donohoe B. S., Ladisch M., Chapple C., Plant Cell 2015, 27, 2195–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Chen F., Tobimatsu Y., Havkin-Frenkel D., Dixon R. A., Ralph J., Proc. Natl. Acad. Sci. USA 2012, 109, 1772–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Tsuji R., Koizumi H., Aoki D., Watanabe Y., Sugihara Y., Matsushita Y., Fukushima K., Fujiwara D., J. Biol. Chem. 2015, 290, 4410–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Cabane M., Afif D., Hawkins S., in Lignins: Biosynthesis, Biodegradation and Bioengineering, Vol. 61 (Eds.: L. Jouann, C. Lapierre), 2012, pp. 219–262. [Google Scholar]
- 146. Barros J., Serk H., Granlund I., Pesquet E., Ann. Bot. 2015, 115, 1053–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Liu J., Osbourn A., Ma P., Mol. Plant 2015, 8, 689–708. [DOI] [PubMed] [Google Scholar]
- 148. Rossi M., Trupiano D., Tamburro M., Ripabelli G., Montagnoli A., Chiatante D., Scippa G. S., Planta 2015, 242, 339–351. [DOI] [PubMed] [Google Scholar]
- 149. Shafi A., Chauhan R., Gill T., Swarnkar M. K., Sreenivasulu Y., Kumar S., Kumar N., Shankar R., Ahuja P. S., Singh A. K., Plant Mol. Biol. 2015, 87, 615–631. [DOI] [PubMed] [Google Scholar]
- 150. Srivastava S., Vishwakarma R. K., Arafat Y. A., Gupta S. K., Khan B. M., Physiol. Mol. Biol. Plants 2015, 21, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wang Y., Wang Q., Zhao Y., Han G., Zhu S., Gene 2015, 566, 95–108. [DOI] [PubMed] [Google Scholar]
- 152. Lawson S. S., Michler C. H., Transgenic Res. 2014, 23, 817–826. [DOI] [PubMed] [Google Scholar]
- 153. Voelker S. L., Lachenbruch B., Meinzer F. C., Strauss S. H., New Phytol. 2011, 189, 1096–1109. [DOI] [PubMed] [Google Scholar]
- 154. Häggman H., Raybould A., Borem A., Fox T., Handley L., Hertzberg M., Lu M.-Z., Macdonald P., Oguchi T., Pasquali G., Pearson L., Peter G., Quemada H., Séguin A., Tattersall K., Ulian E., Walter C., McLean M., Plant Biotechnol. J. 2013, 11, 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Barrière Y., Ralph J., Méchin V., Guillaumie S., Grabber J. H., Argillier O., Chabbert B., Lapierre C., C. R. Biol. 2004, 327, 847–860. [DOI] [PubMed] [Google Scholar]
- 156. Munk L., Sitarz A. K., Kalyani D. C., Mikkelsen J. D., Meyer A. S., Biotechnol. Adv. 2015, 33, 13–24. [DOI] [PubMed] [Google Scholar]
- 157. Akiyama T., Magara K., Meshitsuka G., Lundquist K., Matsumoto Y., J. Wood Chem. Technol. 2014, 35, 8–16. [Google Scholar]
- 158. Ralph J., Brunow G., Harris P. J., Dixon R. A., Schatz P. F., Boerjan W., in Recent Advances in Polyphenol Research, Vol. 1 (Eds.: F. Daayf, A. El Hadrami, L. Adam, G. M. Ballance), Wiley-Blackwell, Oxford, 2008, pp. 36–66. [Google Scholar]
- 159. Terashima N., Fukushima K., He L.-F., Takabe K., in Forage Cell Wall Structure and Digestibility (Eds.: H. G. Jung, D. R. Buxton, R. D. Hatfield, J. Ralph), American Society of Agronomy, Crop Science Society of America, Soil Science Society of America, Madison, 1993, pp. 247–270. [Google Scholar]
- 160. Donaldson L., IAWA J. 2013, 34, 3–19. [Google Scholar]
- 161. Wu J., Fukazawa K., Ohtani J., Holzforschung 1992, 46, 181–185. [Google Scholar]
- 162. Donaldson L., Singh A., in Cellular Aspects of Wood Formation (Ed.: J. Fromm), Springer, Berlin, 2013, pp. 225–256. [Google Scholar]
- 163. Timmel T. E., Compression Wood in Gymnosperms, Springer, Heidelberg, 1986. [Google Scholar]
- 164. Zhang L., Gellerstedt G., Chem. Commun. 2001, 2744–2745. [Google Scholar]
- 165. Sette M., Lange H., Crestini C., Comput. Struct. Biotechnol. J. 2013, 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Ralph J., Marita J. M., Ralph S. A., Hatfield R. D., Lu F., Ede R. M., Peng J., Quideau S., Helm R. F., Grabber J. H., Kim H., Jimenez-Monteon G., Zhang Y., Jung H.-J. G., Landucci L. L., MacKay J. J., Sederoff R. R., Chapple C., Boudet A. M., in Advances in Lignocellulosics Characterization (Ed.: D. S. Argyropoulos), TAPPI Press, Atlanta, 1999, pp. 55–108. [Google Scholar]
- 167. Mansfield S. D., Kim H., Lu F., Ralph J., Nat. Protoc. 2012, 7, 1579–1589. [DOI] [PubMed] [Google Scholar]
- 168. Ralph J., Landucci L. L., in Lignin and Lignans: Advances in Chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press, Boca Raton, FL, 2010, pp. 137–244. [Google Scholar]
- 169. Heikkinen S., Toikka M. M., Karhunen P. T., Kilpeläinen I. A., J. Am. Chem. Soc. 2003, 125, 4362–4367. [DOI] [PubMed] [Google Scholar]
- 170. Hu K., Westler W. M., Markley J. L., J. Am. Chem. Soc. 2011, 133, 1662–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Cheng K., Sorek H., Zimmermann H., Wemmer D. E., Pauly M., Anal. Chem. 2013, 85, 3213–3221. [DOI] [PubMed] [Google Scholar]
- 172. Samuel R., Foston M., Jaing N., Cao S., Allison L., Studer M., Wyman C., Ragauskas A. J., Fuel 2011, 90, 2836–2842. [Google Scholar]
- 173. Argyropoulos D. S., in Lignan and Lignans: Advances in Chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press/Taylor & Francis, New York, 2010, pp. 245–265. [Google Scholar]
- 174. Brunow G., Karlsson O., Lundquist K., Sipilä J., Wood Sci. Technol. 1993, 27, 281–286. [Google Scholar]
- 175. Ralph J., Schatz P. F., Lu F., Kim H., Akiyama T., Nelsen S. F., in Quinone Methides (Ed.: S. Rokita), Wiley-Blackwell, Hoboken, 2009, pp. 385–420. [Google Scholar]
- 176. Freudenberg K., Neish A. C., Constitution and Biosynthesis of Lignin, Springer, New York, 1968. [Google Scholar]
- 177. Kim S., Chmely S. C., Nimlos M. R., Bomble Y. J., Foust T. D., Paton R. S., Beckham G. T., J. Phys. Chem. Lett. 2011, 2, 2846–2852. [Google Scholar]
- 178. Parthasarathi R., Romero R. A., Redondo A., Gnanakaran S., J. Phys. Chem. Lett. 2011, 2, 2660–2666. [Google Scholar]
- 179. Younker J. M., Beste A., A. C. Buchanan, III , ChemPhysChem 2011, 12, 3556–3565. [DOI] [PubMed] [Google Scholar]
- 180. Younker J. M., Beste A., A. C. Buchanan, III , Chem. Phys. Lett. 2012, 545, 100–106. [Google Scholar]
- 181. Sangha A. K., Parks J. M., Standaert R. F., Ziebell A., Davis M., Smith J. C., J. Phys. Chem. B 2012, 116, 4760–4768. [DOI] [PubMed] [Google Scholar]
- 182. Nimz H., Angew. Chem. Int. Ed. Engl. 1974, 13, 313–321; [Google Scholar]; Angew. Chem. 1974, 86, 336–344. [Google Scholar]
- 183. Adler E., Wood Sci. Technol. 1977, 11, 169–218. [Google Scholar]
- 184. Lundquist K., Parkas J., Bioresources 2011, 6, 920–926. [Google Scholar]
- 185. Crestini C., Melone F., Sette M., Saladino R., Biomacromolecules 2011, 12, 3928–3935. [DOI] [PubMed] [Google Scholar]
- 186. Elder T., Energy Fuels 2013, 27, 4785–4790. [Google Scholar]
- 187. Elder T., Energy Fuels 2014, 28, 1175–1182. [Google Scholar]
- 188. Kringstad K. P., Morck R., Holzforschung 1983, 37, 237–244. [Google Scholar]
- 189. Gellerstedt G., Lindfors E., Svensk Papperstidning-Nordisk Cellulosa 1984, 87, R115–R118. [Google Scholar]
- 190. Gellerstedt G., Lindfors E. L., Lapierre C., Monties B., Svensk Papperstidning-Nordisk Cellulosa 1984, 87, R61–R67. [Google Scholar]
- 191. Robert D. R., Bardet M., Gellerstedt G., Lindfors E. L., J. Wood Chem. Technol. 1984, 4, 239–263. [Google Scholar]
- 192. Bose S. K., Francis R. C., J. Pulp Pap. Sci. 1999, 25, 425–430. [Google Scholar]
- 193. Gellerstedt G., Lindfors E.-L., Holzforschung 1984, 38, 151–158. [Google Scholar]
- 194. Bouxin F. P., McVeigh A., Tran F., Westwood N. J., Jarvis M. C., Jackson S. D., Green Chem. 2015, 17, 1235–1242. [Google Scholar]
- 195. Vázquez G., Antorrena G., González J., Freire S., J. Wood Chem. Technol. 1997, 17, 147–162. [Google Scholar]
- 196. Tran F., Lancefield C. S., Kamer P. C. J., Lebl T., Westwood N. J., Green Chem. 2015, 17, 244–249. [Google Scholar]
- 197. Ragnar M., Henriksson G., Lindström M. E., Wimby M., Blechschmidt J., Heinemann S., in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2014. [Google Scholar]
- 198. Ferrini P., Rinaldi R., Angew. Chem. Int. Ed. 2014, 53, 8634–8639; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8778–8783. [Google Scholar]
- 199. Jönsson J., Pettersson K., Berntsson T., Harvey S., Int. J. Energy Res. 2013, 37, 1017–1035. [Google Scholar]
- 200. Konnerth H., Zhang J., Ma D., Prechtl M. H. G., Yan N., Chem. Eng. Sci. 2015, 123, 155–163. [Google Scholar]
- 201. Rinaldi R., Angew. Chem. Int. Ed. 2014, 53, 8559–8560; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8699–8701. [Google Scholar]
- 202. Patt R., Kordsachia O., Süttinger R., Ohtani Y., Hoesch J. F., Ehrler P., Eichinger R., Holik H., Hamm U., Rohmann M. E., Mummenhoff P., Petermann E., Miller R. F., Frank D., Wilken R., Baumgarten H. L., Rentrop G.-H., in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000. [Google Scholar]
- 203. Dimmel D., Gellerstedt G., in Lignin and Lignans: Advances in Chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press, Boca Raton, 2010, pp. 349–391. [Google Scholar]
- 204. Ek M., Pulping Chemistry and Technology (Eds.: M. Ek, G. Gellerstedt, G. Henriksson), Vol. 2, Walter de Gruyter, Berlin, 2009. [Google Scholar]
- 205. Santos R. B., Hart P., Jameel H., Chang H., BioResources 2013, 8, 1456–1477. [Google Scholar]
- 206. Ahvazi B. C., Pageau G., Argyropoulos D. S., Can. J. Chem. 1998, 76, 506–512. [Google Scholar]
- 207. Chakar F. S., Ragauskas A. J., Ind. Crops Prod. 2004, 20, 131–141. [Google Scholar]
- 208. Alekhina M., Ershova O., Ebert A., Heikkinen S., Sixta H., Ind. Crops Prod. 2015, 66, 220–228. [Google Scholar]
- 209. Dwiatmoko A. A., Lee S., Ham H. C., Choi J.-W., Suh D. J., Ha J.-M., ACS Catal. 2015, 5, 433–437. [Google Scholar]
- 210.Data obtained from http://www.foex.fi/PIX/pulp-paper/, accessed on 21st January 2016.
- 211. Kleinert T., Tayenthal K. v., Angew. Chem. 1931, 44, 788–791. [Google Scholar]
- 212. Johansson A., Aaltonen O., Ylinen P., Biomass 1987, 13, 45–65. [Google Scholar]
- 213. Kleinert T. N., US3585104-A.
- 214. Pan X. J., Arato C., Gilkes N., Gregg D., Mabee W., Pye K., Xiao Z. Z., Zhang X., Saddler J., Biotechnol. Bioeng. 2005, 90, 473–481. [DOI] [PubMed] [Google Scholar]
- 215. Pan X., Gilkes N., Kadla J., Pye K., Saka S., Gregg D., Ehara K., Xie D., Lam D., Saddler J., Biotechnol. Bioeng. 2006, 94, 851–861. [DOI] [PubMed] [Google Scholar]
- 216. Pan X., Xie D., Kang K.-Y., Yoon S.-L., Saddler J. N., Appl. Biochem. Biotechnol. 2007, 137, 367–377. [DOI] [PubMed] [Google Scholar]
- 217. Zhao X., Cheng K., Liu D., Appl. Microbiol. Biotechnol. 2009, 82, 815–827. [DOI] [PubMed] [Google Scholar]
- 218. Pye E. K., Lora J. H., Tappi J. 1991, 74, 113–118. [Google Scholar]
- 219. Zhu J. Y., Pan X., R. S. Zalesny, Jr. , Appl. Microbiol. Biotechnol. 2010, 87, 847–857. [DOI] [PubMed] [Google Scholar]
- 220. Zhu J. Y., Pan X. J., Bioresour. Technol. 2010, 101, 4992–5002. [DOI] [PubMed] [Google Scholar]
- 221. Wildschut J., Smit A. T., Reith J. H., Huijgen W. J. J., Bioresour. Technol. 2013, 135, 58–66. [DOI] [PubMed] [Google Scholar]
- 222.Google Patents, search criterium: inassignee:“Lignol Innovations Ltd.”, accessed on Sept 14, 2015.
- 223. Michels J., Wagemann K., Biofuels Bioprod. Biorefin. 2010, 4, 263–267. [Google Scholar]
- 224. Viell J., Harwardt A., Seiler J., Marquardt W., Bioresour. Technol. 2013, 150, 89–97. [DOI] [PubMed] [Google Scholar]
- 225. Rinaldi R., in Catalytic Hydrogenation for Biomass Valorization, The Royal Society of Chemistry, London, 2014, pp. 74–98. [Google Scholar]
- 226. Chirkova Y. A., Kreitus A. E., Polym. Sci. U.S.S.R. 1989, 31, 2285–2291. [Google Scholar]
- 227. Balogh D. T., Curvelo A. A. S., Degroote R., Holzforschung 1992, 46, 343–348. [Google Scholar]
- 228. McDonough T. J., Tappi J. 1993, 76, 186–193. [Google Scholar]
- 229. Lundquist K., in Methods in Lignin Chemistry (Eds.: S. Lin, C. Dence), Springer, Berlin, Heidelberg, 1992, pp. 289–300. [Google Scholar]
- 230. Sturgeon M. R., Kim S., Lawrence K., Paton R. S., Chmely S. C., Nimlos M., Foust T. D., Beckham G. T., ACS Sustainable Chem. Eng. 2014, 2, 472–485. [Google Scholar]
- 231. Kumpinsky E., Ind. Eng. Chem. Res. 1995, 34, 3096–3101. [Google Scholar]
- 232. Deuss P. J., Barta K., Coord. Chem. Rev. 2016, 306, 510–532. [Google Scholar]
- 233. Wang X., Rinaldi R., Energy Environ. Sci. 2012, 5, 8244–8260. [Google Scholar]
- 234. Deuss P. J., Scott M., Tran F., Westwood N. J., de Vries J. G., Barta K., J. Am. Chem. Soc. 2015, 137, 7456–7467. [DOI] [PubMed] [Google Scholar]
- 235. Laure S., Leschinsky M., Frohling M., Schultmann F., Unkelbach G., Cellul. Chem. Technol. 2014, 48, 793–798. [Google Scholar]
- 236. vom Stein T., Grande P. M., Kayser H., Sibilla F., Leitner W., Dominguez de Maria P., Green Chem. 2011, 13, 1772–1777. [Google Scholar]
- 237. Grande P. M., Viell J., Theyssen N., Marquardt W., Dominguez de Maria P., Leitner W., Green Chem. 2015, 17, 3533–3539. [Google Scholar]
- 238. Aziz S., Sarkanen K. V., Tappi J. 1989, 72, 169–175. [Google Scholar]
- 239. Goyal G. C., Lora J. H., Pye E. K., Tappi J. 1992, 75, 110–116. [Google Scholar]
- 240. Lundquist K., Parkås J., J. Wood Chem. Technol. 2014, 35, 3–7. [Google Scholar]
- 241. Top Value-Added Chemicals from Biomass, Volume II-Results of Screening for Potential Candidates from Biorefinery Lignin (Eds.: J. E. Holladay, J. F. White, J. J. Bozell, D. Johnson), Pacific Northwest National Laboratory, 2007. [Google Scholar]
- 242. Sarkanen K. V., in Progress in Biomass Conversion, Vol. 2 (Eds.: S. Kyosti V, T. David A), Elsevier, Amsterdam, 1980, pp. 127–144. [Google Scholar]
- 243. Dumesic J. A., Martin A. D., Luterbacher J. S., Alonso D. M., US2015176090-A1; WO2015095399-A1.
- 244. Han J., Luterbacher J. S., Alonso D. M., Dumesic J. A., Maravelias C. T., Bioresour. Technol. 2015, 182, 258–266. [DOI] [PubMed] [Google Scholar]
- 245. Luterbacher J. S., Alonso D. M., Rand J. M., Questell-Santiago Y. M., Yeap J. H., Pfleger B. F., Dumesic J. A., ChemSusChem 2015, 8, 1317–1322. [DOI] [PubMed] [Google Scholar]
- 246. Luterbacher J. S., Azarpira A., Motagamwala A. H., Lu F. C., Ralph J., Dumesic J. A., Energy Environ. Sci. 2015, 8, 2657–2663. [Google Scholar]
- 247. Luterbacher J. S., Rand J. M., Alonso D. M., Han J., Youngquist J. T., Maravelias C. T., Pfleger B. F., Dumesic J. A., Science 2014, 343, 277–280. [DOI] [PubMed] [Google Scholar]
- 248. Meine N., Rinaldi R., Schüth F., ChemSusChem 2012, 5, 1449–1454. [DOI] [PubMed] [Google Scholar]
- 249. Käldström M., Meine N., Farès C., Rinaldi R., Schüth F., Green Chem. 2014, 16, 2454–2462. [Google Scholar]
- 250. Käldström M., Meine N., Farès C., Schüth F., Rinaldi R., Green Chem. 2014, 16, 3528–3538. [Google Scholar]
- 251. Käldström M., Meine N., Fares C., Schüth F., Rinaldi R., Green Chem. 2014, 16, 4994–4994. [Google Scholar]
- 252. Schüth F., Rinaldi R., Meine N., Käldström M., Hilgert J., Rechulski M. D. K., Catal. Today 2014, 234, 24–30. [Google Scholar]
- 253. Kaufman Rechulski M. D., Käldström M., Richter U., Schüth F., Rinaldi R., Ind. Eng. Chem. Res. 2015, 54, 4581–4592. [Google Scholar]
- 254. Käldström M., Rinaldi R., Schüth F., Meine N., DE102014102972-A1; WO2014139515-A2; WO2014139515-A3.
- 255. Brandt A., Ray M. J., To T. Q., Leak D. J., Murphy R. J., Welton T., Green Chem. 2011, 13, 2489–2499. [Google Scholar]
- 256. Schrems M., Brandt A., Welton T., Liebner F., Rosenau T., Potthast A., Holzforschung 2011, 65, 527–533. [Google Scholar]
- 257. Verdía P., Brandt A., Hallett J. P., Ray M. J., Welton T., Green Chem. 2014, 16, 1617–1627. [Google Scholar]
- 258. Chen L., Sharifzadeh M., MacDowell N., Welton T., Shah N., Hallett J. P., Green Chem. 2014, 16, 3098–3106. [Google Scholar]
- 259. George A., Brandt A., Tran K., Zahari S. M. S. N. S., Klein-Marcuschamer D., Sun N., Sathitsuksanoh N., Shi J., Stavila V., Parthasarathi R., Singh S., Holmes B. M., Welton T., Simmons B. A., Hallett J. P., Green Chem. 2015, 17, 1728–1734. [Google Scholar]
- 260. Brandt A., Murphy R. J., Leak D. J., Welton T., Hallett J., WO2012080702-A2; WO2012080702-A3; CA2821403-A1; EP2652193-A2; CN103370469-A; US2014073016-A1.
- 261. Hallett J. P., Welton T., Brandt A., WO2014140643-A1.
- 262. Binder J. B., Raines R. T., Proc. Natl. Acad. Sci. USA 2010, 107, 4516–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. de Oliveira H. F. N., Fares C., Rinaldi R., Chem. Sci. 2015, 6, 5215–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Li C., Wang Q., Zhao Z. K., Green Chem. 2008, 10, 177–182. [Google Scholar]
- 265. Li C., Zhao Z. K., Adv. Synth. Catal. 2007, 349, 1847–1850. [Google Scholar]
- 266. Rinaldi R., Meine N., vom Stein J., Palkovits R., Schüth F., ChemSusChem 2010, 3, 266–276. [DOI] [PubMed] [Google Scholar]
- 267. Rinaldi R., Palkovits R., Schüth F., Angew. Chem. Int. Ed. 2008, 47, 8047–8050; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8167–8170. [Google Scholar]
- 268.G. F. De Gregorio, T. Welton, J. P. Hallett, Elucidating the mechanism of lignin depolymerisation using acidic ionic liquids: A focus on ether cleavage, 248th National Meeting of the American Chemical Society 2014.
- 269. Ebner G., Schiehser S., Potthast A., Rosenau T., Tetrahedron Lett. 2008, 49, 7322–7324. [Google Scholar]
- 270. Wei X., Han Z., Zhang D., Carbohydr. Res. 2013, 374, 40–44. [DOI] [PubMed] [Google Scholar]
- 271. Clough M. T., Geyer K., Hunt P. A., Son S., Vagt U., Welton T., Green Chem. 2015, 17, 231–243. [Google Scholar]
- 272. Bruijnincx P. C. A., Weckhuysen B. M., Nat. Chem. 2014, 6, 1035–1036. [DOI] [PubMed] [Google Scholar]
- 273. Galkin M. V., Samec J. S. M., ChemSusChem 2014, 7, 2154–2158. [DOI] [PubMed] [Google Scholar]
- 274. Galkin M. V., Sawadjoon S., Rohde V., Dawange M., Samec J. S. M., ChemCatChem 2014, 6, 179–184. [Google Scholar]
- 275. Schutyser W., Van den Bosch S., Renders T., De Boe T., Koelewijn S. F., Dewaele A., Ennaert T., Verkinderen O., Goderis B., Courtin C. M., Sels B. F., Green Chem. 2015, 17, 5035–5045. [Google Scholar]
- 276. Van den Bosch S., Schutyser W., Koelewijn S. F., Renders T., Courtin C. M., Sels B. F., Chem. Commun. 2015, 51, 13158–13161. [DOI] [PubMed] [Google Scholar]
- 277. Van den Bosch S., Schutyser W., Vanholme R., Driessen T., Koelewijn S. F., Renders T., De Meester B., Huijgen W. J. J., Dehaen W., Courtin C. M., Lagrain B., Boerjan W., Sels B. F., Energy Environ. Sci. 2015, 8, 1748–1763. [Google Scholar]
- 278. Parsell T., Yohe S., Degenstein J., Jarrell T., Klein I., Gencer E., Hewetson B., Hurt M., Kim J. I., Choudhari H., Saha B., Meilan R., Mosier N., Ribeiro F., Delgass W. N., Chapple C., Kenttamaa H. I., Agrawal R., Abu-Omar M. M., Green Chem. 2015, 17, 1492–1499. [Google Scholar]
- 279. Dawange M., Galkin M. V., Samec J. S. M., ChemCatChem 2015, 7, 401–404. [Google Scholar]
- 280. Galkin M. V., Dahlstrand C., Samec J. S. M., ChemSusChem 2015, 8, 2187–2192. [DOI] [PubMed] [Google Scholar]
- 281. Song Q., Wang F., Cai J., Wang Y., Zhang J., Yu W., Xu J., Energy Environ. Sci. 2013, 6, 994–1007. [Google Scholar]
- 282. Ferrini P., Rinaldi R., EP2891748-A1; WO2015104262-A1.
- 283. You T.-T., Zhang L.-M., Zhou S.-K., Xu F., Ind. Crops Prod. 2015, 71, 65–74. [Google Scholar]
- 284. Wang X., Rinaldi R., Angew. Chem. Int. Ed. 2013, 52, 11499–11503; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11713–11717. [Google Scholar]
- 285. Geboers J., Wang X., De Carvalho A. B., Rinaldi R., J. Mol. Catal. A 2014, 388–389, 106–115. [Google Scholar]
- 286. Klein I., Saha B., Abu-Omar M. M., Catal. Sci. Technol. 2015, 5, 3242–3245. [Google Scholar]
- 287. Wang X., Rinaldi R., ChemSusChem 2012, 5, 1455–1466. [DOI] [PubMed] [Google Scholar]
- 288. Huang X., Koranyi T. I., Boot M. D., Hensen E. J. M., Green Chem. 2015, 17, 4941–4950. [Google Scholar]
- 289. Zhao C., Kou Y., Lemonidou A. A., Li X., Lercher J. A., Chem. Commun. 2010, 46, 412–414. [DOI] [PubMed] [Google Scholar]
- 290. Ohta H., Kobayashi H., Hara K., Fukuoka A., Chem. Commun. 2011, 47, 12209–12211. [DOI] [PubMed] [Google Scholar]
- 291. Rinaldi R., in Catalytic Hydrogenation for Biomass Valorization (Ed.: R. Rinaldi), The Royal Society of Chemistry, London, 2014, pp. 74–98. [Google Scholar]
- 292. Güvenatam B., Kurşun O., Heeres E. H. J., Pidko E. A., Hensen E. J. M., Catal. Today 2014, 233, 83–91. [Google Scholar]
- 293. Yang F., Hanna M. A., Sun R., Biotechnol. Biofuels 2012, 5, 13–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Chemical & Engineering News Archive 1984, 62, 19–20.
- 295. Strassberger Z., Tanase S., Rothenberg G., RSC Adv. 2014, 4, 25310–25318. [Google Scholar]
- 296. Humbird D., Davis R., Tao L., Kinchin C., Hsu D., Aden A., Schoen P., Lukas J., Olthof B., Worley M., Sexton D., Dudgeon D., Process Design and Economics for Biochemical Conversion of Lignocellulosic Biomass to Ethanol: Dilute-Acid Pretreatment and Enzymatic Hydrolysis of Corn Stover, Golden, Colorado, 2011. [Google Scholar]
- 297. Hocking M. B., J. Chem. Educ. 1997, 74, 1055–1059. [Google Scholar]
- 298. Bomgardner M. M., Chem. Eng. News 2014, 92, 10–14. [Google Scholar]
- 299. Cherubini F., Strømman A. H., in Biofuels (Eds.: A. Pandey, C. Larroche, S. C. Ricke, C.-G. Dussap, E. Gnansounou), Academic Press, Amsterdam, 2011, pp. 3–24. [Google Scholar]
- 300. Turunen M., Alvila L., Pakkanen T. T., Rainio J., J. Appl. Polym. Sci. 2003, 88, 582–588. [Google Scholar]
- 301. Wang M., Leitch M., Xu C., Eur. Polym. J. 2009, 45, 3380–3388. [Google Scholar]
- 302. Zhang W., Ma Y., Wang C., Li S., Zhang M., Chu F., Ind. Crops Prod. 2013, 43, 326–333. [Google Scholar]
- 303. Li Y., Ragauskas A. J., J. Wood Chem. Technol. 2012, 32, 210–224. [Google Scholar]
- 304. Baker D. A., Rials T. G., J. Appl. Polym. Sci. 2013, 130, 713–728. [Google Scholar]
- 305. Manjarrez Nevárez L., Ballinas Casarrubias L., Solis Canto O., Celzard A., Fierro V., Ibarra Gómez R., Gonzalez Sánchez G., Carbohydr. Polym. 2011, 86, 732–741. [Google Scholar]
- 306. Milczarek G., Inganäs O., Science 2012, 335, 1468–1471. [DOI] [PubMed] [Google Scholar]
- 307. Kijima M., Hirukawa T., Hanawa F., Hata T., Bioresour. Technol. 2011, 102, 6279–6285. [DOI] [PubMed] [Google Scholar]
- 308. Tenhaeff W. E., Rios O., More K., McGuire M. A., Adv. Funct. Mater. 2014, 24, 86–94. [Google Scholar]
- 309. Stewart D., Ind. Crops Prod. 2008, 27, 202–207. [Google Scholar]
- 310. Liu W.-J., Jiang H., Yu H.-Q., Green Chem. 2015, 17, 4888–4907. [Google Scholar]
- 311. Boot M., Frijters P., Luijten C., Somers B., Baert R., Donkerbroek A., Klein-Douwel R. J. H., Dam N., Energy Fuels 2009, 23, 1808–1817. [Google Scholar]
- 312. Zhou L., Boot M. D., Johansson B. H., Reijnders J. J. E., Fuel 2014, 115, 469–478. [Google Scholar]
- 313. McCormick R. L., Ratcliff M. A., Christensen E., Fouts L., Luecke J., Chupka G. M., Yanowitz J., Tian M., Boot M., Energy Fuels 2015, 29, 2453–2461. [Google Scholar]
- 314. Amen-Chen C., Pakdel H., Roy C., Bioresour. Technol. 2001, 79, 277–299. [DOI] [PubMed] [Google Scholar]
- 315. Mohan D., Pittman C. U., Steele P. H., Energy Fuels 2006, 20, 848–889. [Google Scholar]
- 316. Zhang Q., Chang J., Wang T., Xu Y., Energy Convers. Manage. 2007, 48, 87–92. [Google Scholar]
- 317. Mu W., Ben H., Ragauskas A., Deng Y., BioEnergy Res. 2013, 6, 1183–1204. [Google Scholar]
- 318. Owen B. C., Haupert L. J., Jarrell T. M., Marcum C. L., Parsell T. H., Abu-Omar M. M., Bozell J. J., Black S. K., Kenttaemaa H. I., Anal. Chem. 2012, 84, 6000–6007. [DOI] [PubMed] [Google Scholar]
- 319. Pham T. N., Shi D., Resasco D. E., Appl. Catal. B 2014, 145, 10–23. [Google Scholar]
- 320. Ma Z., van Bokhoven J. A., ChemCatChem 2012, 4, 2036–2044. [Google Scholar]
- 321. Iliopoulou E. F., Stefanidis S., Kalogiannis K., Psarras A. C., Delimitis A., Triantafyllidis K. S., Lappas A. A., Green Chem. 2014, 16, 662–674. [Google Scholar]
- 322. Dusselier M., Mascal M., Sels B., in Selective Catalysis for Renewable Feedstocks and Chemicals, Vol. 353 (Ed.: K. M. Nicholas), Springer International Publishing, 2014, pp. 1–40. [Google Scholar]
- 323. Williams C. L., Chang C.-C., Do P., Nikbin N., Caratzoulas S., Vlachos D. G., Lobo R. F., Fan W., Dauenhauer P. J., ACS Catal. 2012, 2, 935–939. [Google Scholar]
- 324.R. J. A. Gosselink, Lignin as a Renewable Aromatic Resource for the Chemical Industry, PhD Thesis, Wageningen University, The Netherlands, 2011.
- 325. Lancefield C. S., Ojo O. S., Tran F., Westwood N. J., Angew. Chem. Int. Ed. 2015, 54, 258–262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 260–264. [Google Scholar]
- 326. Feghali E., Carrot G., Thuery P., Genre C., Cantat T., Energy Environ. Sci. 2015, 8, 2734–2743. [Google Scholar]
- 327. Bugg T. D. H., Rahmanpour R., Curr. Opin. Chem. Biol. 2015, 29, 10–17. [DOI] [PubMed] [Google Scholar]
- 328. Ma R., Guo M., Zhang X., ChemSusChem 2014, 7, 412–415. [DOI] [PubMed] [Google Scholar]
- 329. Salvachúa D., Karp E. M., Nimlos C. T., Vardon D. R., Beckham G. T., Green Chem. 2015, 17, 4951–4967. [Google Scholar]
- 330. Mycroft Z., Gomis M., Mines P., Law P., Bugg T. D. H., Green Chem. 2015, 17, 4974–4979. [Google Scholar]
- 331. Linger J. G., Vardon D. R., Guarnieri M. T., Karp E. M., Hunsinger G. B., Franden M. A., Johnson C. W., Chupka G., Strathmann T. J., Pienkos P. T., Beckham G. T., Proc. Natl. Acad. Sci. USA 2014, 111, 12013–12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Vardon D. R., Franden M. A., Johnson C. W., Karp E. M., Guarnieri M. T., Linger J. G., Salm M. J., Strathmann T. J., Beckham G. T., Energy Environ. Sci. 2015, 8, 617–628. [Google Scholar]
- 333. Kärkäs M. D., Matsuura B. S., Monos T. M., Magallanes G., Stephenson C. R., Org. Biomol. Chem. 2016, 14, 1853–1914. [DOI] [PubMed] [Google Scholar]
- 334. Ma R., Xu Y., Zhang X., ChemSusChem 2015, 8, 24–51. [DOI] [PubMed] [Google Scholar]
- 335. Rahimi A., Azarpira A., Kim H., Ralph J., Stahl S. S., J. Am. Chem. Soc. 2013, 135, 6415–6418. [DOI] [PubMed] [Google Scholar]
- 336. Rahimi A., Ulbrich A., Coon J. J., Stahl S. S., Nature 2014, 515, 249–252. [DOI] [PubMed] [Google Scholar]
- 337. Nguyen J. D., Matsuura B. S., Stephenson C. R. J., J. Am. Chem. Soc. 2014, 136, 1218–1221. [DOI] [PubMed] [Google Scholar]
- 338. Nichols J. M., Bishop L. M., Bergman R. G., Ellman J. A., J. Am. Chem. Soc. 2010, 132, 12554–12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Parsell T. H., Owen B. C., Klein I., Jarrell T. M., Marcum C. L., Haupert L. J., Amundson L. M., Kenttamaa H. I., Ribeiro F., Miller J. T., Abu-Omar M. M., Chem. Sci. 2013, 4, 806–813. [Google Scholar]
- 340. Lancefield C. S., Westwood N. J., Green Chem. 2015, 17, 4980–4990. [Google Scholar]
- 341. Sedai B., Díaz-Urrutia C., Baker R. T., Wu R., Silks L. A. P., Hanson S. K., ACS Catal. 2011, 1, 794–804. [Google Scholar]
- 342. Hanson S. K., Wu R., Silks L. A. P., Angew. Chem. Int. Ed. 2012, 51, 3410–3413; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3466–3469. [Google Scholar]
- 343. Díaz-Urrutia C., Chen W.-C., Crites C.-O., Daccache J., Korobkov I., Baker R. T., RSC Adv. 2015, 5, 70502–70511. [Google Scholar]
- 344. Biannic B., Bozell J. J., Org. Lett. 2013, 15, 2730–2733. [DOI] [PubMed] [Google Scholar]
- 345. Sedai B., Baker R. T., Adv. Synth. Catal. 2014, 356, 3563–3574. [Google Scholar]
- 346. Patil N. D., Yao S. G., Meier M. S., Mobley J. K., Crocker M., Org. Biomol. Chem. 2015, 13, 3243–3254. [DOI] [PubMed] [Google Scholar]
- 347. Mottweiler J., Puche M., Räuber C., Schmidt T., Concepción P., Corma A., Bolm C., ChemSusChem 2015, 8, 2106–2113. [DOI] [PubMed] [Google Scholar]
- 348. Mottweiler J., Rinesch T., Besson C., Buendia J., Bolm C., Green Chem. 2015, 17, 5001–5008. [Google Scholar]
- 349. Mitchell L. J., Moody C. J., J. Org. Chem. 2014, 79, 11091–11100. [DOI] [PubMed] [Google Scholar]
- 350. Yang Y., Fan H., Song J., Meng Q., Zhou H., Wu L., Yang G., Han B., Chem. Commun. 2015, 51, 4028–4031. [DOI] [PubMed] [Google Scholar]
- 351. Piers W. E., in Adv. Organomet. Chem. Vol. 52 (Eds.: R. West, A. F. Hill), Elsevier/Academic Press, San Diego, 2005, pp. 1–76. [Google Scholar]
- 352. Fedorov A., Toutov A. A., Swisher N. A., Grubbs R. H., Chem. Sci. 2013, 4, 1640–1645. [Google Scholar]
- 353. Feghali E., Cantat T., Chem. Commun. 2014, 50, 862–865. [DOI] [PubMed] [Google Scholar]
- 354. Son S., Toste F. D., Angew. Chem. Int. Ed. 2010, 49, 3791–3794; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3879–3882. [Google Scholar]
- 355. Chan J. M., Bauer S., Sorek H., Sreekumar S., Wang K., Toste F. D., ACS Catal. 2013, 3, 1369–1377. [Google Scholar]
- 356. Wu A., Patrick B. O., Chung E., James B. R., Dalton Trans. 2012, 41, 11093–11106. [DOI] [PubMed] [Google Scholar]
- 357. Wu A., Lauzon J. M., James B. R., Catal. Lett. 2015, 145, 511–518. [Google Scholar]
- 358. vom Stein T., Weigand T., Merkens C., Klankermayer J., Leitner W., ChemCatChem 2013, 5, 439–441. [Google Scholar]
- 359. vom Stein T., den Hartog T., Buendia J., Stoychev S., Mottweiler J., Bolm C., Klankermayer J., Leitner W., Angew. Chem. Int. Ed. 2015, 54, 5859–5863; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5957–5961. [Google Scholar]
- 360. Dabral S., Mottweiler J., Rinesch T., Bolm C., Green Chem. 2015, 17, 4908–4912. [Google Scholar]
- 361. Harms R. G., Markovits I. I. E., Drees M., Herrmann h. c. m. W. A., Cokoja M., Kühn F. E., ChemSusChem 2014, 7, 429–434. [DOI] [PubMed] [Google Scholar]
- 362. Haupert L. J., Owen B. C., Marcum C. L., Jarrell T. M., Pulliam C. J., Amundson L. M., Narra P., Aqueel M. S., Parsell T. H., Abu-Omar M. M., Kenttaemaa H. I., Fuel 2012, 95, 634–641. [Google Scholar]
- 363. Jarrell T. M., Marcum C. L., Sheng H., Owen B. C., O'Lenick C. J., Maraun H., Bozell J. J., Kenttaemaa H. I., Green Chem. 2014, 16, 2713–2727. [Google Scholar]
- 364. Marshall A. G., Rodgers R. P., Proc. Natl. Acad. Sci. USA 2008, 105, 18090–18095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Herod A. A., Bartle K. D., Morgan T. J., Kandiyoti R., Chem. Rev. 2012, 112, 3892–3923. [DOI] [PubMed] [Google Scholar]
- 366. Torr K. M., van de Pas D. J., Cazeils E., Suckling I. D., Bioresour. Technol. 2011, 102, 7608–7611. [DOI] [PubMed] [Google Scholar]
- 367. Werhan H., Farshori A., Rudolf von Rohr P., J. Membr. Sci. 2012, 423–424, 404–412. [Google Scholar]
- 368. Boeriu C. G., Fiţigău F. I., Gosselink R. J. A., Frissen A. E., Stoutjesdijk J., Peter F., Ind. Crops Prod. 2014, 62, 481–490. [Google Scholar]
- 369. Venderbosch R. H., ChemSusChem 2015, 8, 1306–1316. [DOI] [PubMed] [Google Scholar]
- 370. Ardiyanti A. R., Venderbosch R. H., Yin W., Heeres H. J., in Catalytic Hydrogenation for Biomass Valorization, The Royal Society of Chemistry, London, 2014, pp. 151–173. [Google Scholar]
- 371. Huang X., Korányi T. I., Boot M. D., Hensen E. J. M., ChemSusChem 2014, 7, 2276–2288. [DOI] [PubMed] [Google Scholar]
- 372. Zakzeski J., Jongerius A. L., Bruijnincx P. C. A., Weckhuysen B. M., ChemSusChem 2012, 5, 1602–1609. [DOI] [PubMed] [Google Scholar]
- 373. Barta K., Warner G. R., Beach E. S., Anastas P. T., Green Chem. 2014, 16, 191–196. [Google Scholar]
- 374. Chen F., Tobimatsu Y., Jackson L., Nakashima J., Ralph J., Dixon R. A., Plant J. 2013, 73, 201–211. [DOI] [PubMed] [Google Scholar]
- 375. Zakzeski J., Weckhuysen B. M., ChemSusChem 2011, 4, 369–378. [DOI] [PubMed] [Google Scholar]
- 376. Pu Y., Jiang N., Ragauskas A. J., J. Wood Chem. Technol. 2007, 27, 23–33. [Google Scholar]
- 377. Kilpeläinen I., Xie H., King A., Granstrom M., Heikkinen S., Argyropoulos D. S., J. Agric. Food Chem. 2007, 55, 9142–9148. [DOI] [PubMed] [Google Scholar]
- 378. Mäki-Arvela P., Anugwom I., Virtanen P., Sjöholm R., Mikkola J. P., Ind. Crops Prod. 2010, 32, 175–201. [Google Scholar]
- 379. Brandt A., Grasvik J., Hallett J. P., Welton T., Green Chem. 2013, 15, 550–583. [Google Scholar]
- 380. Tan S. S. Y., MacFarlane D. R., Upfal J., Edye L. A., Doherty W. O. S., Patti A. F., Pringle J. M., Scott J. L., Green Chem. 2009, 11, 339–345. [Google Scholar]
- 381. Singh S., Simmons B. A., Vogel K. P., Biotechnol. Bioeng. 2009, 104, 68–75. [DOI] [PubMed] [Google Scholar]
- 382. Brandt A., Hallett J. P., Leak D. J., Murphy R. J., Welton T., Green Chem. 2010, 12, 672–679. [Google Scholar]
- 383. Kerton F., Marriott R., in Alternative Solvents for Green Chemistry (2), The Royal Society of Chemistry, London, 2013, pp. 82–114. [Google Scholar]
- 384. Ma R., Hao W., Ma X., Tian Y., Li Y., Angew. Chem. Int. Ed. 2014, 53, 7310–7315; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7438–7443. [Google Scholar]
- 385. Xu W., Miller S. J., Agrawal P. K., Jones C. W., ChemSusChem 2012, 5, 667–675. [DOI] [PubMed] [Google Scholar]
- 386. Kasakov S., Shi H., Camaioni D. M., Zhao C., Baráth E., Jentys A., Lercher J. A., Green Chem. 2015, 17, 5079–5090. [Google Scholar]
- 387. Wang X., Rinaldi R., Catal. Today 2016, 269, 48–55. [Google Scholar]
- 388. van Rossum G., Zhao W., Castellvi Barnes M., Lange J.-P., Kersten S. R. A., ChemSusChem 2014, 7, 253–259. [DOI] [PubMed] [Google Scholar]
- 389. Kumar S., Lange J.-P., Van Rossum G., Kersten S. R. A., Ind. Eng. Chem. Res. 2014, 53, 11668–11676. [Google Scholar]
- 390. Meier D., Ante R., Faix O., Bioresour. Technol. 1992, 40, 171–177. [Google Scholar]
- 391. Oasmaa A., Alén R., Meier D., Bioresour. Technol. 1993, 45, 189–194. [Google Scholar]
- 392. Kloekhorst A., Wildschut J., Heeres H. J., Catal. Sci. Technol. 2014, 4, 2367–2377. [Google Scholar]
- 393. Kumar C. R., Anand N., Kloekhorst A., Cannilla C., Bonura G., Frusteri F., Barta K., Heeres H. J., Green Chem. 2015, 17, 4921–4930. [Google Scholar]
- 394. Kleine T., Buendia J., Bolm C., Green Chem. 2013, 15, 160–166. [Google Scholar]
- 395. Roberts V. M., Stein V., Reiner T., Lemonidou A., Li X., Lercher J. A., Chem. Eur. J. 2011, 17, 5939–5948. [DOI] [PubMed] [Google Scholar]
- 396. Jiang Y., Li Z., Tang X., Sun Y., Zeng X., Liu S., Lin L., Energy Fuels 2015, 29, 1662–1668. [Google Scholar]
- 397. Barta K., Matson T. D., Fettig M. L., Scott S. L., Iretskii A. V., Ford P. C., Green Chem. 2010, 12, 1640–1647. [Google Scholar]
- 398. Voitl T., Rudolf von Rohr P., ChemSusChem 2008, 1, 763–769. [DOI] [PubMed] [Google Scholar]
- 399. Xiong H., Pham H. N., Datye A. K., Green Chem. 2014, 16, 4627–4643. [Google Scholar]
- 400. Lange J. P., Angew. Chem. Int. Ed. 2015, 54, 13186–13197; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13382–13394. [Google Scholar]
- 401. Rinaldi R., Fujiwara F. Y., Schuchardt U., Appl. Catal. A 2006, 315, 44–51. [Google Scholar]
- 402. Ravenelle R., Copeland J., Van Pelt A., Crittenden J., Sievers C., Top. Catal. 2012, 55, 162–174. [Google Scholar]
- 403. Jongerius A. L., Copeland J. R., Foo G. S., Hofmann J. P., Bruijnincx P. C. A., Sievers C., Weckhuysen B. M., ACS Catal. 2013, 3, 464–473. [Google Scholar]
- 404. Katahira R., Sluiter J. B., Schell D. J., Davis M. F., J. Agric. Food Chem. 2013, 61, 3286–3292. [DOI] [PubMed] [Google Scholar]
- 405. Hollak S. A. W., de Jong K. P., van Es D. S., ChemCatChem 2014, 6, 2648–2655. [Google Scholar]
- 406. Boga D. A., Liu F., Bruijnincx P. C. A., Weckhuysen B. M., Catal. Sci. Technol. 2016, 6, 134–143. [Google Scholar]
- 407. Bartholomew C. H., in Advances in Catalysis Vol. 31 (Ed.: D. D. Eley), New York, 1982. [Google Scholar]
- 408. Song Q., Wang F., Xu J., Chem. Commun. 2012, 48, 7019–7021. [DOI] [PubMed] [Google Scholar]
- 409. Deepa A. K., Dhepe P. L., ACS Catal. 2015, 5, 365–379. [Google Scholar]
- 410. Furimsky E., Appl. Catal. A 2000, 199, 147–190. [Google Scholar]
- 411. Huber G. W., Corma A., Angew. Chem. Int. Ed. 2007, 46, 7184–7201; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7320–7338. [Google Scholar]
- 412. Mortensen P. M., Grunwaldt J. D., Jensen P. A., Knudsen K. G., Jensen A. D., Appl. Catal. A 2011, 407, 1–19. [Google Scholar]
- 413. Zacher A. H., Olarte M. V., Santosa D. M., Elliott D. C., Jones S. B., Green Chem. 2014, 16, 491–515. [Google Scholar]
- 414. Shin E.-J., Keane M. A., Ind. Eng. Chem. Res. 2000, 39, 883–892. [Google Scholar]
- 415. Centeno A., Laurent E., Delmon B., J. Catal. 1995, 154, 288–298. [Google Scholar]
- 416. Jongerius A. L., Gosselink R. W., Dijkstra J., Bitter J. H., Bruijnincx P. C. A., Weckhuysen B. M., ChemCatChem 2013, 5, 2964–2972. [Google Scholar]
- 417. Li K., Wang R., Chen J., Energy Fuels 2011, 25, 854–863. [Google Scholar]
- 418. Zhao H., Li D., Bui P., Oyama S., Appl. Catal. A 2011, 391, 305–310. [Google Scholar]
- 419. Zhao C., Kou Y., Lemonidou A. A., Li X., Lercher J. A., Angew. Chem. Int. Ed. 2009, 48, 3987–3990; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4047–4050. [Google Scholar]
- 420. Zhao C., Lercher J. A., Angew. Chem. Int. Ed. 2012, 51, 5935–5940; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6037–6042. [Google Scholar]
- 421. Jongerius A. L., Bruijnincx P. C. A., Weckhuysen B. M., Green Chem. 2013, 15, 3049–3056. [Google Scholar]
- 422. Jongerius A. L., Jastrzebski R., Bruijnincx P. C. A., Weckhuysen B. M., J. Catal. 2012, 285, 315–323. [Google Scholar]
- 423. Yan N., Zhao C., Dyson P. J., Wang C., Liu L.-t., Kou Y., ChemSusChem 2008, 1, 626–629. [DOI] [PubMed] [Google Scholar]
- 424. Schutyser W., Van den Bosch S., Dijkmans J., Turner S., Meledina M., Van Tendeloo G., Debecker D. P., Sels B. F., ChemSusChem 2015, 8, 1805–1818. [DOI] [PubMed] [Google Scholar]
- 425.R. Jastrzebski, S. E. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen, P. C. A. Bruijnincx, ChemSusChem 2016, DOI: 10.1002/cssc.201600683. [DOI] [PMC free article] [PubMed]
- 426.C. Chesi, I. B. D. de Castro, M. T. Clough, P. Ferrini, R. Rinaldi, ChemCatChem 2016, DOI: 10.1002/cctc.201600235. [DOI]
- 427. Harton S. E., Pingali S. V., Nunnery G. A., Baker D. A., Walker S. H., Muddiman D. C., Koga T., Rials T. G., Urban V. S., Langan P., ACS Macro Lett. 2012, 1, 568–573. [DOI] [PubMed] [Google Scholar]