

Abstract
Reported herein is the one‐pot synthesis of trifluoromethylated amines at room temperature using the bench‐stable (Me4N)SCF3 reagent and AgF. The method is rapid, operationally simple and highly selective. It proceeds via a formal umpolung reaction of the SCF3 with the amine, giving quantitative formation of thiocarbamoyl fluoride intermediates within minutes that can readily be transformed to N‐CF3. The mildness and high functional group tolerance render the method highly attractive for the late‐stage introduction of trifluoromethyl groups on amines, as demonstrated herein for a range of pharmaceutically relevant drug molecules.
Keywords: amines, synthetic methods, thiocarbamoyl fluoride, trifluoromethylation
Fluorination confers molecules with improved properties and function, affecting lipophilicity, solubility, conformational and metabolic stability features that impact numerous branches of chemistry, including the agrochemical and pharmaceutical arenas.1 While up to 40 % of all active compounds feature at least one fluorine atom, nitrogen makes up for an even greater abundance of 80 %.2 In view of the widespread dissemination of both elements in pharmaceuticals, it is somewhat puzzling that their combined functionality, that is, trifluoromethyl amines, are scarcely investigated to date. This might be a consequence of the lack of safe, general and high‐yielding methodology to access these compounds as well as encountered difficulties in their purifications (see Figure 1).3 The latest advances in the methodological repertoire include the direct N‐trifluoromethylation of certain amines, nitriles and azoles with electrophilic4 or radical‐based reagents,5 posing continued challenges in terms of generality and late‐stage synthetic applications to compounds with incompatible additional functionality. Alternatively, N‐CF3 compounds can be made in a two‐step sequence from free amines via oxidative transformations of dithiocarbamates.6 Access to the latter can be non‐trivial however with respect to functional group compatibility, as their syntheses generally require strong base, alkylating or toxic reagents.7
Figure 1.
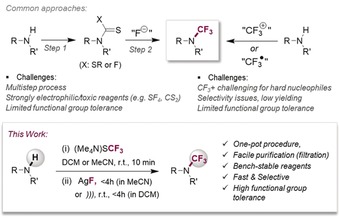
Overview of methods to access N‐CF3 compounds and this work.
A mild, safe and operational simple synthetic route to N‐CF3 compounds would hence be desirable and may enable numerous avenues in chemical and biomedical research. For example, the frequently encountered facile oxidation of amines in metabolic processes should be less pronounced under N‐CF3 modification.8 In addition, our calculations of selected drug molecules indicate similar conformational preferences but increased logP values for N‐CF3 vs. N‐Me.9 Moreover, a promising isolated study found the N‐CF3 analogue of floxacin to retain similar antibacterial activities as its methylated counterpart.10
We herein demonstrate the rapid and selective formation of a wide range of trifluoromethylated amines from secondary amines. The high‐yielding, one‐pot strategy is based on a polarity inversion and in situ formation of a thiocarbamoyl fluoride intermediate using the bench‐stable (Me4N)SCF3 reagent.
As part of our ongoing mechanistic and methodological program11 that also focusses on the generation of fluorine‐containing compounds,12 we recently developed metal‐catalyzed C−SCF3 bond formation protocols of aryl (pseudo)halides, using the bench‐stable “SCF3 −” source (Me4N)SCF3.13, 14 During these investigations, we encountered the unexpected formation of a challenging class of compounds, that is, a thiocarbamoyl fluoride. The subjection of one equivalent of (Me4N)SCF3 to N‐methylaniline in toluene at room temperature remarkably gave rise to facile and quantitative formation of N‐methyl‐N‐phenylthiocarbamoyl fluoride 1 a within minutes without any need for additional reagents or catalysts (see Scheme 1). Although thiocarbamoyl chlorides are well known and widely reported, interestingly, the fluoride equivalent has seen much less precedence.15 The few reported syntheses relied on the use of highly toxic and strongly electrophilic reagents.16 As such, our discovery constitutes a considerable advance, being safe, convenient, rapid and utilizing a bench‐stable solid as the sole reaction partner for quantitative transformation within minutes. To test the generality, we explored a variety of additional secondary amines for their reactivities with (Me4N)SCF3. Scheme 1 shows two additional examples of isolated thiocarbamoyl fluorides (and Tables 1 and 2 report their follow‐up usage). Notably, through the addition of hexane after 10 min reaction time, all side‐products are precipitated, allowing facile isolation of the thiocarbamoyl fluoride upon filtration, if desired.
Scheme 1.

Generation of thiocarbamoyl fluorides via umpolung (top) and selectivity in reactivities with alternative nucleophiles (bottom).
Table 1.
Scope of trifluoromethylation of secondary amines.[a]


|
[a] Reaction conditions: Amine (0.2 mmol), (Me4N)SCF3 (46 mg, 0.26 mmol), AgF (76 mg, 0.6 mmol) solvent (1.5 mL). [b] Formation of thiocarbamoyl fluoride in 1 h. [c] Formation of thiocarbamoyl fluoride in 24 h. [d] Filtration and column chromatography performed.
Table 2.
N‐Trifluoromethylation of important drug molecules.[a]

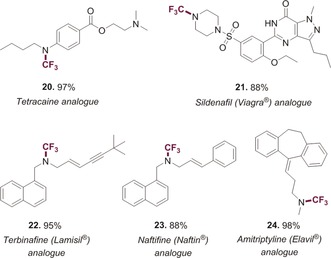
|
[a] Reaction conditions: Amine (0.2 mmol), (Me4N)SCF3 (46 mg, 0.26 mmol), AgF (76 mg, 0.6 mmol) solvent (1.5 mL).
Given this intriguing and non‐obvious reactivity of two nucleophiles (R2NH and CF3S−) to generate the highly electrophilic thiocarbamoyl fluoride quantitatively, we set out to explore this transformation in greater detail. On first sight, a reasonable scenario would appear to be the in situ generation of F2C=S upon fluoride elimination,17 which could then serve as an electrophile to the amine. However, dissolved (Me4N)SCF3 salt in MeCN neither generated F2C=S by itself nor under the addition of a small amount of water.18 Furthermore, if generated F2C=S were to trigger the observed reactivity, we would also expect to see transformation with nucleophiles other than secondary amines. However, subjection of the (Me4N)SCF3 reagent to tertiary amines or phosphines did not give rise to any reaction,19 indicating potential for selectivity which would otherwise not be given with standard electrophiles that would preferentially select for the most nucleophilic site (Scheme 1). In our case, selectivity for the “heteroatom‐H” site appears to be given.
We next investigated oxygen‐based nucleophiles.17b We observed that the reaction of phenol with (Me4N)SCF3 gives rapid formation of the thiocarbonate species ROC(S)OR within minutes. Similarly, also rather weakly nucleophilic alcohols, such as 2,2,2‐trifluoroethanol (N=1.11 according to Mayr's scale20) or hexafluoroisopropanol (N=−1.93) formed the corresponding thiocarbonates upon subjection to (Me4N)SCF3. On the other hand, the arguably more nucleophilic 4‐(trifluoromethyl)phenoxide did not markedly react with (Me4N)SCF3, showing only traces of product over the course of 15 h at room temperature (see Scheme 1, bottom).
These results clearly indicate that in contrast to established transformations to access these and related compound classes, the nucleophilicity of the to be functionalized heteroatom is not the decisive reactivity factor, therefore offering a platform for distinct selectivities and functional group tolerance in the context of late‐stage applications of this methodology.
That said, we next set out to further transform this valuable intermediate to trifluoromethyl amines. We anticipated that the distinct selectivity imposed in the first reaction step should ultimately translate to the final N‐CF3 compounds.
Building on Tyrra's report that described the AgF‐mediated conversion of a diethylcarbamoyl fluoride to N‐CF3,21 we subsequently investigated the feasibility of this protocol to convert a wider range of thiocarbamoyl fluorides, including those generated in situ under our conditions. Mixing N‐methylaniline in acetonitrile with (Me4N)SCF3 led to the quantitative formation of the corresponding thiocarbamoyl fluoride within 10 minutes at room temperature, as judged by quantitative 19F‐NMR analysis of the reaction mixture. Subsequent direct addition of AgF (3 equiv) to the same mixture gave complete conversion to the corresponding trifluoromethylated amine 1 in Table 1 within two hours. The reaction between AgF and the thiocarbamoyl fluoride was found to proceed smoothly at room temperature or higher temperature (50 °C).22 No side‐products, for example, arising from a Ritter‐type reaction as commonly observed under electrophilic trifluoromethylation of amines,4b were detected.
Notably, the only by‐products generated in this one‐pot two‐step sequence are salts [(Me4N)HF2 and Ag2S]. Those can readily be precipitated from the reaction mixture via the addition of low polarity solvents, such as hexane or pentane, leaving the desired N‐CF3 target compound as the only product dissolved in the organic phase (MeCN). Thus, only filtration and solvent removal are required to isolate the desired N‐CF3 compounds. For more volatile products, we identified that it is also possible to use the lower boiling solvent CH2Cl2 as reaction medium. Although AgF is not soluble in CH2Cl2, sonication of the reaction mixture mitigates this and the trifluoromethylamines are generated equally effectively.
This one‐pot protocol therefore presents a convenient, safe and high‐yielding route to trifluoromethylamines, and we subsequently set out to investigate its full potential. Table 1 presents an overview of the studied amines. We found our protocol to be compatible with aromatic as well as aliphatic secondary amines, generating the corresponding N‐CF3 products in excellent yields (98–81 %). Electron‐poor and rich amines were coupled with the same efficiency (see Table 1). Exclusive selectivity for the NH site and a high functional group tolerance were observed. Halogens (6, 11, 18), ester (2, 5, 7, 14, 17), nitrile (4), nitro (3, 15), amide (13), sulfonyl (19), methoxy (12, 16) as well as a heterocycle (18) were all tolerated. Two protected amino acids, that is, the glycine (7) and proline (2) derivatives, were also successfully synthesized. As such, the presented methodology constitutes a significant improvement to previous protocols in terms of operational simplicity and generality.
As a further test of our newly developed methodology for pharmaceutical and agrochemical applications, we next investigated nitrogen‐containing drug molecules of greater complexity. We successfully N‐trifluoromethylated tetracaine 20, a widely used anesthetic, in 97 % yield (Table 2).23 Similarly, there are numerous N‐Me containing drugs on the market, and we therefore also investigated the feasibility to synthesize their N‐CF3 bioisosteres.24 In this context, we primarily focused on pharmaceuticals that are listed on the “WHO Model List of Essential Medicines” (a list containing the most important medications required in any basic health system).25 We prepared the N‐CF3 analogue of Sildenafil 21 in high yield (88 %), the most widely used drug against erectile dysfunction (Viagra®),26 the antifungal agents terbinafine 22 (in 95 %),27 and naftifine 23 (88 %) as well as amitriptyline 24 (98 %)28 that finds use in the treatment of mental illnesses (see Table 2). We were pleased to obtain high yields in all cases, demonstrating the scope and applicability of this protocol in a pharmaceutical context, as heterocycles, tertiary amines, alkynes and alkenes were well tolerated.
In summary, we reported a convenient, safe and operationally simple one‐pot protocol for the rapid and mild trifluoromethylation of secondary amines. The readily accessible bench‐stable (Me4N)SCF3 salt formally serves as CF3 source via initial reaction between the SCF3‐anion and the amine. Upon umpolung a rarely encountered and valuable thiocarbamoyl fluoride intermediate is generated quantitatively in situ that can subsequently be converted to the corresponding N‐CF3 compounds upon reaction with AgF. The purification consists solely of precipitation of the salt by‐products with non‐polar solvent, followed by filtration. The umpolung strategy allowed distinct selectivities in trifluoromethylation which is not governed by nucleophilicity, but instead by the availability of an “N‐H” unit. As such, the protocol displays a wide functional group tolerance, and was demonstrated to be compatible with carbonyl, alkene, alkyne, tertiary amine, nitrile, nitro, heterocycles as well as protected amino acids and relevant pharmaceuticals.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the RWTH Aachen, the MIWF NRW and the European Research Council (ERC‐637993) for funding. We are grateful to Julia Baums and Tomàs de Sousa Aguiar for their practical assistance.
T. Scattolin, K. Deckers, F. Schoenebeck, Angew. Chem. Int. Ed. 2017, 56, 221.
Contributor Information
Thomas Scattolin, http://www.schoenebeck.oc.rwth‐aachen.de/.
Prof. Dr. Franziska Schoenebeck, Email: franziska.schoenebeck@rwth-aachen.de
References
- 1.
- 1a. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]
- 1b. Purser S., Moore P. R., Swallow S., Gouverneur V., Chem. Soc. Rev. 2008, 37, 320–330; [DOI] [PubMed] [Google Scholar]
- 1c. Zimmer L. E., Sparr C., Gilmour R., Angew. Chem. Int. Ed. 2011, 50, 11860–11871; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12062–12074; [Google Scholar]
- 1d. Tomashenko O. A., Grushin V. V., Chem. Rev. 2011, 111, 4475–4521; [DOI] [PubMed] [Google Scholar]
- 1e. Liang T., Neumann C. N., Ritter T., Angew. Chem. Int. Ed. 2013, 52, 8214–8264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8372–8423; [Google Scholar]
- 1f. Champagne P. A., Desroches J., Hamel J.-D., Vandamme M., Paquin J.-F., Chem. Rev. 2015, 115, 9073; [DOI] [PubMed] [Google Scholar]
- 1g. Tlili A., Toulgoat F., Billard T., Angew. Chem. Int. Ed. 2016, 55, 11726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11900. [Google Scholar]
- 2.For a recent perspective, see: Vitaku E., Smith D. T., Njardarson J. T., J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Dmowski W., Kamiński M., J. Fluorine Chem. 1983, 23, 207–218; [Google Scholar]
- 3b. Harder R. J., Smith W. C., J. Am. Chem. Soc. 1961, 83, 3422–3424; [Google Scholar]
- 3c. Markovskij L. N., Pashinnik V. E., Kirsanov A. V., Synthesis 1973, 787–789; [Google Scholar]
- 3d. Klauke E., Angew. Chem. Int. Ed. Engl. 1966, 5, 848–848; [Google Scholar]; Angew. Chem. 1966, 78, 829–829; [Google Scholar]
- 3e. Pawelke G., J. Fluorine Chem. 1991, 52, 229–234; [Google Scholar]
- 3f. Abe T., Hayashi E., Baba H., Fukaya H., J. Fluorine Chem. 1990, 48, 257–279; [Google Scholar]
- 3g. Yagupolskii L. M., Kondratenko N. V., Timofeeva G. N., Dronkina M. I., Yagupolskii Y. L., Zh. Org. Khim. 1980, 16, 2508–2513. [Google Scholar]
- 4.
- 4a. Niedermann K., Früh N., Senn R., Czarniecki B., Verel R., Togni A., Angew. Chem. Int. Ed. 2012, 51, 6511–6515; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6617–6621; [Google Scholar]
- 4b. Niedermann K., Früh N., Vinogradova E., Wiehn M. S., Moreno A., Togni A., Angew. Chem. Int. Ed. 2011, 50, 1059–1063; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1091–1095; [Google Scholar]
- 4c. Umemoto T., Adachi K., Ishihara S., J. Org. Chem. 2007, 72, 6905–6917. [DOI] [PubMed] [Google Scholar]
- 5.For sulfoximine-derived N-CF3, see: Teng F., Cheng J., Bolm C., Org. Lett. 2015, 17, 3166–3169. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Kuroboshi M., Hiyama T., Tetrahedron Lett. 1992, 33, 4177–4178; [Google Scholar]
- 6b. Hagooly Y., Gatenyo J., Hagooly A., Rozen S., J. Org. Chem. 2009, 74, 8578–8582. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Azizi N., Aryanasab F., Saidi M. R., Org. Lett. 2006, 8, 5275–5277; [DOI] [PubMed] [Google Scholar]
- 7b. Walter W., Bode K. D., Angew. Chem. Int. Ed. Engl. 1967, 6, 281–293; [Google Scholar]; Angew. Chem. 1967, 79, 285–297; [Google Scholar]
- 7c. Chaturvedi D., Ray S., Tetrahedron Lett. 2006, 47, 1307–1309; [Google Scholar]
- 7d. Chen Z., Jin Y., Stang P. J., J. Org. Chem. 1987, 52, 4117–4118; [Google Scholar]
- 7e. Hori l., Hayashi T., Midorikawa H., Synthesis 1975, 727–729; [Google Scholar]
- 7f. Kiyoshi K., Katsuya M., Manabu K., Tamejiro H., Bull. Chem. Soc. Jpn. 1998, 71, 1973–1991. [Google Scholar]
- 8. Hlavica P., Gorrod J. W., Crit. Rev. Biochem. Mol. Biol. 1982, 12, 39–101. [DOI] [PubMed] [Google Scholar]
- 9.Our calculations of the logP values of N-CF3 versus N-CH3 drugs gave slightly higher values for N-CF3 LogP of Amitriptyline: 5.55 (wet octanol) and 5.56 (dry octanol). LogP of amitriptyline-CF3: 5.97 (wet octanol) and 6.06 (dry octanol). LogP of naftifine: 5.89 (wet octanol) and 5.94 (dry octanol). LogP of naftifine-CF3: 6.25 (wet octanol) and 6.35 (dry octanol). LogP of sildenafil: 1.16 (wet octanol) and 0.59 (dry octanol). LogP of sildenafil-CF3: 1.51 (wet octanol) and 1.02 (dry octanol); calculated with COSMOthermX (F. Eckert, A. Klamt, COSMOtherm, Version C30_1601, Release 12.15; COSMOlogic GmbH &Co. KG,Germany, 2010).
- 10. Asahina Y., Araya I., Iwase K., Iinuma F., Hosaka M., Ishizaki T., J. Med. Chem. 2005, 48, 3443–3446. [DOI] [PubMed] [Google Scholar]
- 11. Sperger T., Sanhueza I. A., Schoenebeck F., Acc. Chem. Res. 2016, 49, 1311. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Anstaett P., Schoenebeck F., Chem. Eur. J. 2011, 17, 12340–12346; [DOI] [PubMed] [Google Scholar]
- 12b. Sanhueza I. A., Nielsen M. C., Ottiger M., Schoenebeck F., Helv. Chim. Acta 2012, 95, 2231–2236; [Google Scholar]
- 12c. Sanhueza I. A., Bonney K. J., Nielsen M. C., Schoenebeck F., J. Org. Chem. 2013, 78, 7749–7753; [DOI] [PubMed] [Google Scholar]
- 12d. Nielsen M. C., Bonney K. J., Schoenebeck F., Angew. Chem. Int. Ed. 2014, 53, 5903–5906; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6013–6016; [Google Scholar]
- 12e. Aufiero M., Sperger T., Tsang A. S. K., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 10322–10326; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10462–10466. [Google Scholar]
- 13.
- 13a. Dürr A. B., Yin G., Kalvet I., Napoly F., Schoenebeck F., Chem. Sci. 2016, 7, 1076–1081; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Yin G., Kalvet I., Englert U., Schoenebeck F., J. Am. Chem. Soc. 2015, 137, 4164–4172; [DOI] [PubMed] [Google Scholar]
- 13c. Yin G., Kalvet I., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 6809–6813; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6913–6917. [Google Scholar]
- 14.For other applications of (Me4N)SCF3 in metal-catalyzed trifluoromethylthiolations, see:
- 14a. Zhang C.-P., Vicic D. A., J. Am. Chem. Soc. 2012, 134, 183; [DOI] [PubMed] [Google Scholar]
- 14b. Zhang C.-P., Vicic D. A., Chem. Asian J. 2012, 7, 1756. For a synthesis of reagent, see: [DOI] [PubMed] [Google Scholar]
- 14c. Naumann D., Hogea B., Yagupolskii Y. L., Tyrra W. E., J. Fluorine Chem. 2003, 119, 101. [Google Scholar]
- 15.Reaxys and SciFinder searches gave 6 and 4 entries, respectively, for thiocarbamoyl fluorides, date: 20.09.2016.
- 16.
- 16a. Harris J. F., J. Org. Chem. 1967, 32, 2063–2074; [Google Scholar]
- 16b. Yarovenko N. N., Vasil'eva A. S., Zh. Obshch. Khim. 1959, 29, 3754–3757. [Google Scholar]
- 17.The SCF3 anion has been proposed to be unstable to heat, see:
- 17a. Tavener S. J., Adams D. J., Clark J. H., J. Fluorine Chem. 1999, 95, 171. For recent use of this presumed side-reaction, see: [Google Scholar]
- 17b. Liu J.-B., Xu X.-H., Chen Z.-H., Qing F.-L., Angew. Chem. Int. Ed. 2015, 54, 897; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 911; [Google Scholar]
- 17c. Glenadel Q., Tlili A., Billard T., Eur. J. Org. Chem. 2016, 1955. [DOI] [PubMed] [Google Scholar]
- 18.With 10 % of water [relative to the amount of employed (Me4N)SCF3], up to 90 % of the reagent remained untouched over 96 h, indicating that small amounts of moisture present in the reaction mixture do not catalyze the decomposition of (Me4N)SCF3.
- 19.Following reference [17a], we generated SCF2 in situ upon reaction of SCCl2 with KF. Subsequent addition of (iPr)3P led to complete consumption of the phosphine, in contrast to our reaction. This indicates either SCF2 is not involved in our reaction, or the heteroatom-H is crucial for its selective formation.
- 20.Mayr's scale is based on the addition of nucleophiles to carbocations and Michael acceptors; see:
- 20a. Kempf B., Hampel N., Ofial A. R., Mayr H., Chem. Eur. J. 2003, 9, 2209–2218; [DOI] [PubMed] [Google Scholar]
- 20b. Richter D., Mayr H., Angew. Chem. Int. Ed. 2009, 48, 1958–1961; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1992–1995. [Google Scholar]
- 21. Tyrra W., J. Fluorine Chem. 2001, 109, 189–194. [Google Scholar]
- 22.We found AgNO3, AgBF4 and AgOTf are also effective in place of AgF, albeit less efficiently.
- 23.First synthesized in 1932. Otto E. (Winthrop Chem. Co Inc), US1889645, 1932.
- 24. Patani G. A., LaVoie E. J., Chem. Rev. 1996, 96, 3147–3176. [DOI] [PubMed] [Google Scholar]
- 25.For the complete list, see: http://www.who.int/medicines/publications/essentialmedicines/EML_2015_FINAL_amended_NOV2015.pdf, 20.09.2016.
- 26. Bell A. S., Brown D., Terrett N. K. (Pfizer Inc.), US5250534, 1993.
- 27. Richter F., Steiger M. (Novartis Ag Ltd.), US5681849, 1997.
- 28.(Merck and co., Inc.), FR1368709, 1964.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary