

Abstract
The non‐enzymatic acylative kinetic resolution of challenging aryl–alkenyl (sp2 vs. sp2) substituted secondary alcohols is described, with effective enantiodiscrimination achieved using the isothiourea organocatalyst HyperBTM (1 mol %) and isobutyric anhydride. The kinetic resolution of a wide range of aryl–alkenyl substituted alcohols has been evaluated, with either electron‐rich or naphthyl aryl substituents in combination with an unsubstituted vinyl substituent providing the highest selectivity (S=2–1980). The use of this protocol for the gram‐scale (2.5 g) kinetic resolution of a model aryl–vinyl (sp2 vs. sp2) substituted secondary alcohol is demonstrated, giving access to >1 g of each of the product enantiomers both in 99:1 e.r.
Keywords: acylation, isothiourea, kinetic resolution, organocatalysis, renewable resources
Introduction
Non‐enzymatic, acylative kinetic resolution (KR) is a powerful method for the preparation of enantiomerically enriched alcohols.1 In this regard, enantioselective Lewis base‐catalysed acylations are one of the most widely employed methodologies, and various catalyst structures and acyl transfer agents have been developed. In terms of substrate scope, non‐enzymatic acylative KRs are most commonly trialed on benzylic secondary alcohols for which the catalytic acylating agent must differentiate between the enantiomers of alcohols bearing a planar aryl (sp2) and a tetrahedral alkyl (sp3) substituent in order to obtain high selectivity (Figure 1 a).
Figure 1.

Lewis base‐catalysed KR of secondary alcohols.
Although less common, highly selective methods have also been developed for the KR of both alkynyl–alkyl (sp vs. sp3) and alkenyl–alkyl (sp2 vs. sp3) substituted secondary alcohols. In these systems the acylating agent must differentiate between the enantiomers of alcohols with a planar π‐system and a tetrahedral sp3 hybridized substituent. For example, a number of Lewis base organocatalysts have been utilized for the acylative KR of alkenyl–alkyl (sp2 vs. sp3) allylic alcohols (Figure 1 b).2, 3, 4, 5, 6, 7 Fu used planar‐chiral DMAP‐derived ferrocene catalyst 1 and acetic anhydride for the KR of a range of allylic alcohols, including two that had served as intermediates in natural product synthesis, with high selectivity factors, S (up to 80).2 Vedejs has also achieved high selectivity for the KR of allylic alcohols using chiral phosphine 2 and isobutyric anhydride (S up to 82).3 More recently, both Birman4 and Deng5 have used amidine catalysts 4–6 for the acylative KR of alkenyl–alkyl (sp2 vs. sp3) alcohols with moderate to good selectivity obtained across a range of substrates.
To date there are very few examples of the KR of secondary allylic alcohols bearing both planar alkenyl and planar aryl substituents (sp2 vs. sp2).8 This is likely to be due to the challenge of the catalytic acylating agent differentiating between enantiomeric alcohols with two planar sp2 hybridized substituents during the selectivity‐determining acylation step. To this end, Connon and co‐workers have studied the KR of a range of Morita–Baylis–Hillman (MBH) adducts 8 bearing aryl substituents, obtaining moderate selectivity (S up to 13) using chiral DMAP derivative 3 and isobutyric anhydride (Scheme 1 a).9 Mandai and Suga have also reported a single example of the KR of an aryl MBH adduct using a chiral phosphoric acid catalyst alongside acetyl chloride and DABCO (1,4‐diazabicyclo[2.2.2]octane).10 Deng and co‐workers have used amidine 7 as a catalyst for the acylative KR of aryl–alkenyl substituted alcohols 10, with moderate to good selectivity (S up to 24) obtained for a range of aryl substituents and simple 1,1‐disubstituted alkenes (Scheme 1 b).11
Scheme 1.

Lewis base‐catalysed acylative KR of aryl–alkenyl alcohols.
Herein, the challenge of resolving aryl–alkenyl (sp2 vs. sp2) substituted secondary alcohols is addressed using an isothiourea‐based organocatalyst (Scheme 1 c).12, 13 Isothioureas have previously been used as catalysts for the acylative KR of various secondary alcohols,14 as well as the desymmetrization of meso‐diols.15 In this report, we demonstrate that the isothiourea HyperBTM 12 can differentiate between the enantiomers of aryl–alkenyl (sp2 vs. sp2) substituted secondary alcohols. The selectivity of the KR has been assessed across a wide range of allylic alcohols, with good to excellent enantiodiscrimination observed for substrates bearing either electron‐rich or naphthyl substituents alongside an unsubstituted vinyl substituent.
Results and Discussion
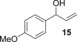
The reaction of (±)‐1‐(4‐methoxyphenyl)prop‐2‐en‐1‐ol 15 with propanoic anhydride (0.5 equiv) and i‐Pr2NEt (0.5 equiv) in CHCl3 was chosen as the starting point to identify suitable reaction conditions for the acylative KR of aryl–alkenyl (sp2 vs. sp2) substituted alcohols. The commercially available and readily prepared isothiourea HyperBTM 12 (1 mol %) was identified as the most promising in an initial screen of readily available catalysts, giving 44 % conversion into ester 16 with S=8,16, 17, 18 whereas both tetramisole 17 and BTM 18 gave poor conversion and lower selectivity (Table 1, entries 1–3). The absolute configuration of the major enantiomer of recovered alcohol (S)‐15 was confirmed by comparison of its specific rotation with literature values.19 Further optimization revealed that using isobutyric anhydride and lowering the reaction temperature to −40 °C gave improved selectivity (Table 1, entry 4). A solvent screen showed that both THF (S=16) and in particular toluene (S=21) gave improvements in selectivity (Table 1, entries 5 and 6). Further lowering the reaction temperature to −78 °C led to the efficient KR of (±)‐15 with excellent selectivity (S=29) considering the challenging aryl–alkenyl (sp2 vs. sp2) alcohol substitution (Table 1, entry 7). The catalyst loading could also be lowered to 0.25 mol % without an appreciable drop in either conversion or selectivity (Table 1, entry 8), although for practicality 1 mol % HyperBTM 12 was used to assess the reaction scope.
Table 1.
Reaction optimization.
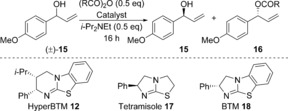
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Cat. (mol %) | R | Solvent | T [°C] | Conv. [%][a] | 15 e.r.[b] | 16 e.r.[b] | S [c] |
| 1 | 12 (1) | Et | CHCl3 | 0 | 44 | 76:24 | 83:17 | 8 |
| 2 | 17 (1) | Et | CHCl3 | 0 | 6 | 52:48 (ent) | 68:32 (ent) | 2 |
| 3 | 18 (1) | Et | CHCl3 | 0 | 21 | 52:48 | 58:42 | 2 |
| 4[d] | 12 (1) | i‐Pr | CHCl3 | −40 | 52 | 88:12 | 86:14 | 14 |
| 5[d] | 12 (1) | i‐Pr | THF | −40 | 51 | 90:10 | 87:13 | 16 |
| 6[d] | 12 (1) | i‐Pr | PhMe | −40 | 50 | 90:10 | 90:10 | 21 |
| 7 | 12 (1) | i‐Pr | PhMe | −78 | 50 | 92:8 | 92:8 | 29 |
| 8 | 12 (0.25) | i‐Pr | PhMe | −78 | 53 | 94:6 | 89:11 | 22 |
[a] Calculated by HPLC analysis. [b] e.r. determined by HPLC analysis. [c] Calculated using the equations developed by Kagan.17 [d] 0.6 equiv of anhydride used.
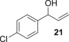
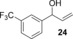
The optimized conditions for the KR of (±)‐15 were then tested for a range of vinyl alcohols bearing various aryl substituents (Tables 2, 3, and 4). Initial investigations probed the effect of varying the steric and electronic nature of the aryl group bearing a single substituent in either the para‐, meta‐, or ortho‐position (Table 2). Unsubstituted and aryl rings bearing electron‐donating methoxy substituents in either the para‐, meta‐, or ortho‐positions worked well, with excellent selectivity obtained in all cases (Table 2, entries 1, 2, 6 and 9, S=29–59). In contrast, the presence of an electron‐withdrawing CF3 substituent in any of the positions around the aryl ring led to a noticeable drop in selectivity (Table 2, entries 3, 7 and 10, S=7–11). For example, although 3‐methoxy substituted alcohol (±)‐23 gave S=59, the analogous 3‐CF3 substituted (±)‐24 gave S=11. Various halogen substituents were tolerated, allowing KR of alcohols 21, 22 and 25 with moderate levels of selectivity (Table 2, entries 4, 5 and 8, S=8–17). This observation is consistent with previous proposals for the acylative KR of aryl–alkyl (sp2 vs. sp3) substituted secondary alcohols using isothioureas, which typically give higher selectivity in the resolution of alcohols bearing electron‐rich aryl substitutents.14 In these processes, the aryl unit is thought to be the key recognition motif for enantiodiscrimination, being involved in π‐stacking with an electron‐deficient acyl ammonium intermediate during the acylation step.
Table 2.
KR of substituted aryl–vinyl (sp2 vs. sp2) secondary alcohols.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conv. [%][a] | Alcohol e.r.[b] (yield, %) | Ester e.r.[b] (yield, %) | S [c] |
| 1 |
|
50 | 92:8 (40) | 92:8 (34) | 29 |
| 2 |

|
41 | 82:18 (48) | 95:5 (35) | 35 |
| 3 |

|
52 | 83:17 (37) | 82:18 (41) | 8 |
| 4 |
|
48[d] | 87:13 (31) | 91:9 (30) | 17 |
| 5 |

|
35 | 68:32 (56) | 84:16 (28) | 8 |
| 6 |

|
43 | 86:14 (46) | 96:4 (40) | 59 |
| 7 |
|
50 | 86:14 (46) | 88:12 (50) | 15 |
| 8 |

|
54[d] | 89:11 (33) | 84:16 (34) | 12 |
| 9 |

|
52[d] | 95:5 (44) | N/D[e] (35) | 36 |
| 10 |

|
37 | 68:32 (59) | 82:18 (30) | 7 |
[a] Calculated by HPLC analysis. [b] e.r. determined by HPLC analysis. [c] Calculated using the equations developed by Kagan.17 [d] Conversion determined by 1H NMR analysis. [e] Enantiomers of ester inseparable by HPLC.
Table 3.
KR of poly‐substituted aryl–vinyl (sp2 vs. sp2) secondary alcohols.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conv. [%][a] | Alc. e.r.[b] (yield, %) | Ester e.r.[b] (yield, %) | S [c] |
| 1[d] |

|
37 | 78:22 (40) | 99:1 (26) | 110 |
| 2 |

|
60 | >99:1 (39) | 80:20 (50) | 44 |
| 3 |

|
51 | 94:6 (43) | 92:8 (47) | 33 |
| 4 |

|
22 | 61:39 (51) | 90:10 (17) | 11 |
| 5 |

|
49 | 97:3 (47) | >99:1 (45) | 1980[e] |
| 6 |

|
46 | 92:8 (41) | 98:2 (31) | 108 |
| 7 |

|
47 | 72:28 (31) | 75:25 (37) | 5 |
| 8 |

|
42 | 78:22 (50) | 88:12 (37) | 13 |
| 9 |

|
48 | 89:11 (34) | 92:8 (29) | 26 |
[a] Calculated by HPLC analysis. [b] e.r. determined by HPLC analysis. [c] Calculated using the equations developed by Kagan.17 [d] 48 h reaction time. [e] Determined by linear regression analysis (see text).
Table 4.
KR of heteroaryl–vinyl (sp2 vs. sp2) secondary alcohols.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conv. [%][a] | Alcohol e.r.[b] (yield, %) | Ester e.r.[b] (yield, %) | S[c] |
| 1 |

|
49 | 67:33 (42) | 68:32 (45) | 3 |
| 2 |

|
46 | 69:31 (46) | 73:27 (39) | 4 |
| 3 |

|
44 | 76:24 (49) | 84:16 (44) | 9 |
[a] Calculated by HPLC analysis. [b] e.r. determined by HPLC analysis. [c] Calculated using the equations developed by Kagan.17
Subsequent studies aimed to exploit this observation through testing the KR of aryl–vinyl alcohols bearing either poly‐substituted electron‐rich aryl‐substitutents or extended aromatic naphthyl units (Table 3). Excellent selectivity was observed with electron‐rich 2,6‐dimethoxy substituted aryl–alkenyl alcohol (±)‐28 (S=110), although the presence of two ortho‐substituents resulted in lower, but still acceptable, conversion over an extended 48 h reaction time due to the slower rate of acylation (Table 3, entry 1). The methodology was then applied to the KR of lignin‐derived alcohols (±)‐29 and (±)‐30 bearing methoxy‐substituted aryl rings (Table 3, entries 2 and 3). Pleasingly, the resolutions proceeded with excellent selectivity in both cases (S=44 and 33, respectively), allowing the recovered alcohols 29 and 30 to be isolated with high e.r. This demonstrates that the methodology can be used to access enantiomerically pure synthetic building blocks from renewable monomers derived from lignin, which is important for the continued drive for valorization of such feedstocks.18 Mesityl‐substituted allylic alcohol (±)‐31 also gave lower conversion into the corresponding ester, but the KR selectivity was reasonable (Table 3, entry 4, S=11). The KR of 2‐naphthyl substituted vinyl alcohol (±)‐32 gave exceptional selectivity, with the remaining alcohol 32 (97:3 e.r.) and the corresponding isobutyric ester (>99:1 e.r.) isolated with excellent e.r. at 50 % conversion (Table 3, entry 5). The presence of a 1‐naphthyl substituent also led to excellent selectivity (S=108) under the standard conditions (Table 3, entry 6). The selectivity observed with naphthyl substituents was surprisingly sensitive to further substitution on the naphthylene ring. For example, 6‐methoxy substituted naphthyl alcohol (±)‐34 gave dramatically lower selectivity (S=5) compared with the unsubstituted analogue (Table 3, entry 7). To probe the origin of the high selectivity using unsubstituted naphthyl alcohols, the KR protocol was tested on aryl substrates (±)‐35 and (±)‐36 containing 4‐phenyl and 3‐vinyl substituents, respectively (Table 3, entries 8 and 9). In both cases the KR gave good selectivity (S=13 and 26), although neither match the levels of enantiodiscrimination observed with the extended conjugation within the unsubstituted naphthyl examples.
For the resolution of (±)‐32, the exceptionally high selectivity, coupled with the accuracy of the HPLC analysis used to measure the e.r. values of both alcohol and ester, makes the calculation of an exact selectivity factor difficult. To validate the reported S value, repeat experiments were performed and product enantioselectivities measured at varying reaction conversions. The data obtained was plotted as shown in Figure 2, allowing the selectivity factor to be determined using linear regression.19 Good linear correlation of the data over a range of reaction conversions suggests that S=1980 for the KR of (±)‐32.
Figure 2.

Determination of the selectivity factor for the KR of (±)‐32 using linear regression.
Next, the use of heteroaryl–vinyl (sp2 vs. sp2) secondary alcohols in the KR was briefly assessed. Both 2‐ and 3‐pyridyl substituted alcohols (±)‐38 and (±)‐39 gave poor selectivity (Table 4, entries 2 and 3, S=3 and 4, respectively), whereas 2‐thiophenyl alcohol (±)‐40 gave better, but still moderate, results (Table 4, entry 3, S=9).
The effect of substitution on the alkene portion was then explored under the standard conditions (Table 5). The KR of 1,1‐disubstituted alkene (±)‐41 showed good reactivity and reasonable selectivity (Table 5, entry 1), although the selectivity was lower (S=10) than for the corresponding vinyl analogue (±)‐15 (S=29). The reaction with 1,2‐disubstituted alkene (±)‐42 did not proceed at −78 °C and gave a complex mixture of products when performed at 0 °C. However, the recovered alcohol and ester were both obtained in low e.r. so the selectivity is likely to be minimal (Table 5, entry 2). The use of 1,1,2‐trisubstituted alkene (±)‐43 also gave low levels of selectivity (Table 5, entry 3, S=3). As the 2‐naphthyl aryl substituent led to extremely high levels of enantiodiscrimination with unsubstituted allylic alcohol (±)‐32, the effect of alkene substitution within this series was also investigated. In this case, 1,1‐disubstituted alkene (±)‐44 gave higher selectivity (S=24, Table 5, entry 4) compared with (±)‐41, although again this was significantly lower than for vinyl substituted (±)‐32. The reactions of 1,2‐disubstituted (±)‐45 and 1,1,2‐trisubstituted (±)‐46 followed the same trend as previously and both gave relatively low selectivity (Table 5, entries 5 and 6, S=11 and 8). These results demonstrate that levels of enantiodiscrimination between the two enantiomers of aryl–alkenyl (sp2 vs. sp2) secondary alcohols decreases with increasing substitution on the alkenyl moiety.
Table 5.
Effect of alkene substitution.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conv. [%][a] | Alcohol e.r.[b] (yield, %) | Ester e.r.[b] (yield, %) | S[c] |
| 1 |

|
45 | 79:21 (48) | 84:16 (42) | 10 |
| 2[d] |

|
57 | 73:27 (42) | 65:35 (33) | N/D |
| 3 |

|
38 | 62:38 (64) | 70:30 (34) | 3 |
| 4 |

|
47 | 86:14 (45) | 92:8 (37) | 24 |
| 5 |

|
47 | 81:19 (51) | 85:15 (38) | 11 |
| 6 |

|
53 | 84:16 (45) | 80:20 (48) | 8 |
[a] Calculated by HPLC analysis. [b] e.r. determined by chiral HPLC analysis. [c] Calculated using the equations developed by Kagan.17 [d] Reaction performed at 0 °C.
Finally, as the catalytic system can effectively discriminate between the two planar sp2 hybridized substituents within aryl–alkenyl alcohols, the KR of some alternative classes of secondary alcohol were compared under the same reaction conditions (Table 6). Interestingly, the KR of aryl–vinyl substituted alcohol (±)‐32 (sp2 vs. sp2) gave higher levels of enantiodiscrimination than the analogous aryl–alkyl substituted alcohol (±)‐47 (sp2 vs. sp3), although in both cases the selectivity is excellent (Table 6, entries 1 and 2). However, the use of aryl–alkynyl alcohol (±)‐48 (sp2 vs. sp) gave poor selectivity (S=3) in the KR process (Table 6, entry 3). The catalytic system was also only poorly selective for the KR of vinyl‐alkyl alcohol (±)‐49 (sp2 vs. sp3) (Table 6, entry 4, S=3). This suggests that both aryl (sp2) and alkynyl (sp) groups are effective recognition motifs for enantiodiscrimination and may interact with the proposed acyl ammonium intermediate (vide infra) during the acylation step. Conversely, vinyl (sp2) and alkyl (sp3) substituents are poor recognition units and are unlikely to interact with the catalytic intermediate. Consequently, combining an effective recognition motif (such as aryl (sp2) and alkynyl (sp) groups) with a poor one (such as vinyl (sp2) and alkyl (sp3) units) leads to high enantiodiscrimination during KR, whereas alternative combinations result in low selectivity.
Table 6.
KR of different classes of secondary alcohols.

| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conv. [%][a] | Alcohol e.r.[b] (yield, %) | Ester e.r.[b] (yield, %) | S [c] |
| 1 |

|
52 | >99:1 (37) | 96:4 (39) | 152 |
| 2 |

|
49 | 97:3 (47) | >99:1 (45) | 1980[d] |
| 3 |

|
53 | 66:34 (35) | 64:36 (36) | 3 |
| 4 |

|
40 | 61:39 (35) | 68:32 (25) | 3 |
[a] Calculated by HPLC analysis. [b] e.r. determined by chiral HPLC analysis. [c] Calculated using the equations developed by Kagan.17 [d] Determined by linear regression analysis (see text).
To demonstrate the synthetic utility of this KR process to facilitate the separation of the two enantiomers of a racemic alcohol, the KR was performed on a preparative laboratory scale using 2.5 g (13.6 mmol) of (±)‐32 and 1 mol % of HyperBTM (Scheme 2). This highly selective reaction proceeded to 50 % conversion, allowing unreacted (S)‐32 to be recovered in 43 % yield (1.08 g) and 99:1 e.r. Isolated ester (R)‐37 was readily hydrolyzed under basic conditions to give (R)‐32 in 45 % yield (1.12 g) over the two steps and >99:1 e.r.
Scheme 2.

Preparative‐scale KR for the separation of (±)‐32.
The proposed catalytic cycle starts with a reversible acylation of HyperBTM 12 with isobutyric anhydride to form acyl ammonium intermediate 50 (Scheme 3 a). Turnover‐limiting acylation of the favoured enantiomer of the aryl–alkenyl alcohol is thought to occur with concomitant proton transfer to the carboxylate anion.20, 21 The i‐Pr2NEt may possibly act as a shuttle base to regenerate the catalyst and remove isobutyric acid. The sense of enantioselectivity observed can be rationalized by considering the interactions of the incoming alcohol with acyl ammonium 50 during the selectivity‐determining step (Scheme 3 b). Acyl ammonium 50 is thought to be conformationally locked due to a stabilizing non‐bonding O−S interaction (nO to σ*C−S),22 with the Re face blocked by the pseudo‐axial phenyl group. The fast‐reacting enantiomer of the aryl–alkenyl alcohol can adopt a conformation that has a potentially stabilizing aryl π‐cation interaction with the isothiourea (52), which is favoured over the potential alkenyl π‐cation interaction in the slow reacting enantiomer (53).23 This model is consistent with the higher selectivity observed for substrates bearing electron‐rich aryl rings due to the increased strength of the proposed cation‐π interaction in the favoured transition state in these cases.24 Conversely, increasing the substitution on the alkene makes this π‐system more electron rich, which decreases the difference in energy between the diastereomeric transition states and accounts for the lower selectivity obtained for these examples. A possible explanation for the enhanced selectivity with naphthyl substituents is the presence of an additional stacking interaction with the benzenoid ring of acyl ammonium 50 for the fast reacting enantiomer. Substitution of the naphthyl ring with electron‐donating substituents may destabilise these additional interactions,25 resulting in the observed loss in enantiodiscrimination.
Scheme 3.

a) Proposed mechanism. b) Stereochemical rationale.
Conclusion
The isothiourea HyperBTM 12 (1 mol %) can catalyze the acylative KR of a range of aryl–alkenyl (sp2 vs. sp2) substituted secondary alcohols with isobutyric anhydride. The catalytic system achieves effective enantiodiscrimination between the enantiomers of secondary alcohols bearing two planar sp2 hybridized substituents. The efficiency of the KR process has been assessed for a range of substituted aryl and heteroaryl moieties and various alkene substitution patterns. The highest selectivity is obtained when either electron‐rich or naphthyl aryl substituents are present in combination with a vinyl substituent. Conversely, the presence of either electron‐deficient aryl rings or substituted alkenes leads to lower levels of selectivity. The optimized KR process can be used to separate the two enantiomers of synthetically useful aryl–vinyl alcohols with high enantioselectivity (up to >99:1 e.r.) on a preparative scale at low catalyst loading (1 mol %). Ongoing work within this laboratory is focused upon the development of practical KR processes of challenging substrates and their applications in synthesis.
Experimental Section
General: For general experimental details, full characterisation data, 1H and 13C{1H} NMR spectra, and HPLC traces, see the Supporting Information.26
Representative procedure for the KR of aryl–alkenyl alcohols
The appropriate alcohol (1 equiv) was dissolved in PhMe (0.35 m) and the solution cooled to −78 °C. HyperBTM 12 (1 mol %), i‐Pr2NEt (0.6 equiv) and isobutyric anhydride (0.5 equiv) were added and the solution stirred at −78 °C for 16 h. The reaction was quenched with 1 m HCl, the solution diluted with EtOAc and washed successively with 1 m HCl (×2), NaHCO3 (×2) and brine. The organic layer was dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The alcohol and ester were purified by column chromatography and analysed by chiral HPLC.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council and CRITICAT Centre for Doctoral Training [Ph.D. studentship to S.F.M.; Grant code: EP/L016419/1 and EP/J018139/1] and The Leverhulme Trust [Early Career Fellowship to J.E.T.; ECF‐2014‐005] for financial support. A.D.S. thanks the Royal Society for a Wolfson Merit Award. We also thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.
S. F. Musolino, O. S. Ojo, N. J. Westwood, J. E. Taylor, A. D. Smith, Chem. Eur. J. 2016, 22, 18916.
Contributor Information
Dr. James E. Taylor, Email: jet20@st-andrews.ac.uk
Prof. Andrew D. Smith, Email: ads10@st-andrews.ac.uk
References
- 1.For reviews on non-enzymatic acylative KR, see:
- 1a. Spivey A. C., McDaid P., in Enantioselective Organocatalysis, (Ed.: P. I. Dalko), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2007, pp. 287–329; [Google Scholar]
- 1b. Spivey A. C., Arseniyadis S., in Asymmetric Organocatalysis (Ed.: B. List), Springer, Heidelberg, 2009, pp. 233–280; [Google Scholar]
- 1c. Müller C. E., Schreiner P. R., Angew. Chem. Int. Ed. 2011, 50, 6012–6042; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 6136–6167; [Google Scholar]
- 1d. Pellissier H., Adv. Synth. Catal. 2011, 353, 1613–1666. [Google Scholar]
- 2.
- 2a. Ruble J. C., Latham H. A., Fu G. C., J. Am. Chem. Soc. 1997, 119, 1492–1493; [Google Scholar]
- 2b. Bellemin-Laponnaz S., Tweddell J., Ruble J. C., Breitling F. M., Fu G. C., Chem. Commun. 2000, 1009–1010. [Google Scholar]
- 3. Vedejs E., MacKay J. A., Org. Lett. 2001, 3, 535–536. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Birman V. B., Jiang H., Org. Lett. 2005, 7, 3445–3447; [DOI] [PubMed] [Google Scholar]
- 4b. Li X., Jiang H., Uffman E. W., Guo L., Zhang Y., Yang X., Birman V. B., J. Org. Chem. 2012, 77, 1722–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Jiang S.-S., Gu B.-Q., Zhu M.-Y., Yu X., Deng W.-P., Tetrahedron 2015, 71, 1187–1191; [Google Scholar]
- 5b. Jiang S.-S., Xu Q.-C., Zhu M.-Y., Yu X., Deng W.-P., J. Org. Chem. 2015, 80, 3159–3169. [DOI] [PubMed] [Google Scholar]
- 6. Xu Q., Zhou H., Geng X., Chen P., Tetrahedron 2009, 65, 2232–2238. [Google Scholar]
- 7. Roy S., Chen K.-F., Chen K., J. Org. Chem. 2014, 79, 8955–8959. [DOI] [PubMed] [Google Scholar]
- 8.For a single example of an enzyme-catalysed KR of aryl–alkenyl alcohols, see: Marques F. A., Oliveira M. A., Frensch G., Sales Maia B. H. L. N., Barison A., Lenz C. A., P. G. Guerrero Jr. , Lett. Org. Chem. 2011, 8, 696–700. [Google Scholar]
- 9. Dálaigh C. Ó., Connon S. J., J. Org. Chem. 2007, 72, 7066–7069. [DOI] [PubMed] [Google Scholar]
- 10. Mandai H., Murota K., Mitsudo K., Suga S., Org. Lett. 2012, 14, 3486–3489. [DOI] [PubMed] [Google Scholar]
- 11. Hu B., Meng M., Jiang S., Deng W., Chin. J. Chem. 2012, 30, 1289–1294. [Google Scholar]
- 12.For a review on the use of isothioureas as organocatalysts, see: Taylor J. E., Bull S. D., Williams J. M. J., Chem. Soc. Rev. 2012, 41, 2109–2121. [DOI] [PubMed] [Google Scholar]
- 13.Birman and Okamoto independently reported the use of achiral isothioureas as an effective acylation catalysts, see:
- 13a. Kobayashi M., Okamoto S., Tetrahedron Lett. 2006, 47, 4347–4350; [Google Scholar]
- 13b. Birman V. B., Li X., Han Z., Org. Lett. 2007, 9, 37–40. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Birman V. B., Guo L., Org. Lett. 2006, 8, 4859–4861; [DOI] [PubMed] [Google Scholar]
- 14b. Birman V. B., Li X., Org. Lett. 2006, 8, 1351–1354; [DOI] [PubMed] [Google Scholar]
- 14c. Birman V. B., Li X., Org. Lett. 2008, 10, 1115–1118; [DOI] [PubMed] [Google Scholar]
- 14d. Zhou H., Xu Q., Chen P., Tetrahedron 2008, 64, 6494–6499; [Google Scholar]
- 14e. Zhang Y., Birman V. B., Adv. Synth. Catal. 2009, 351, 2525–2529; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14f. Nakata K., Shiina I., Heterocycles 2010, 80, 805; [Google Scholar]
- 14g. Shiina I., Nakata K., Ono K., Sugimoto M., Sekiguchi A., Chem. Eur. J. 2010, 16, 167–172; [DOI] [PubMed] [Google Scholar]
- 14h. Belmessieri D., Joannesse C., Woods P. A., MacGregor C., Jones C., Campbell C. D., Johnston C. P., Duguet N., Concellon C., Bragg R. A., Smith A. D., Org. Biomol. Chem. 2011, 9, 559–570; [DOI] [PubMed] [Google Scholar]
- 14i. Nakata K., Ono K., Shiina I., Heterocycles 2011, 82, 1171–1180; [Google Scholar]
- 14j. Shiina I., Ono K., Nakahara T., Chem. Commun. 2013, 49, 10700–10702; [DOI] [PubMed] [Google Scholar]
- 14k. Wang H.-Y., Yang K., Yin D., Liu C., Glazier D. A., Tang W., Org. Lett. 2015, 17, 5272–5275. [DOI] [PubMed] [Google Scholar]
- 15.Desymmetrization, see:
- 15a. Birman V. B., Jiang H., Li X., Org. Lett. 2007, 9, 3237–3240; [DOI] [PubMed] [Google Scholar]
- 15b. Merad J., Borkar P., BouyonYenda T., Roux C., Pons J.-M., Parrain J.-L., Chuzel O., Bressy C., Org. Lett. 2015, 17, 2118–2121. [DOI] [PubMed] [Google Scholar]
- 16. Kagan H. B., Fiaud J. C., in Topics in Stereochemistry, Vol 18, (Eds.: E. L. Eliel, S. H. Wilen), John-Wiley and Sons, New York, 1988; pp. 249–330. [Google Scholar]
- 17.Throughout this manuscript, reaction conversions and selectivity factors (S) were calculated using the equations outlined by Kagan in Ref. 16. Reaction conversions were mostly calculated from HPLC measurements according to the equation: c=(100×ee alcohol)/(ee ester+ee alcohol). Conversions could also be determined by 1H NMR spectroscopy, with the two methods providing consistent results. Selectivity factors were calculated using the reaction conversion (c) and the ee of the recovered alcohol according to the equation: S=ln{[(1−c)(1−ee alcohol)]/[(1−c)(1+ee alcohol)]}.
- 18.Authentic racemic samples of all isobutyric esters were synthesised using a DMAP-catalysed acylation, see the Supporting Information.
- 19.Throughout, the absolute configuration of the major enantiomer of the recovered alcohols were determined by comparison of specific rotations with literature values, see the Supporting Information. For alcohols without appropriate literature specific rotations the configurations were assigned by analogy.
- 20. Lancefield C. S., Ojo O. S., Tran F., Westwood N. J., Angew. Chem. Int. Ed. 2015, 54, 258–262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 260–264. [Google Scholar]
- 21.For a previous example of linear regression analysis to determine a high selectivity factorfor KR, see: Klare H. F. T., Oestreich M., Angew. Chem. Int. Ed. 2007, 46, 9335–9338; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 9496–9499. [Google Scholar]
- 22.For kinetic analysis on a related HBTM-catalyzed esterification of a cyclic secondary alcohol, see: Wagner A. J., Rychnovsky S. D., Org. Lett. 2013, 15, 5504–5507. [DOI] [PubMed] [Google Scholar]
- 23.For computational and kinetic studies on the DMAP-catalyzed acylation of alcohols, see:
- 23a. Xu S., Held I., Kempf B., Mayr H., Steglich W., Zipse H., Chem. Eur. J. 2005, 11, 4751–4757; [DOI] [PubMed] [Google Scholar]
- 23b. Larionov E., Mahesh M., Spivey A. C., Wei Y., Zipse H., J. Am. Chem. Soc. 2012, 134, 9390–9399. [DOI] [PubMed] [Google Scholar]
- 24.For a review on non-covalent sulfur interactions in medicinal chemistry, see:
- 24a. Beno B. R., Yeung K.-S., Bartberger M. D., Pennington L. D., Meanwell N. A., J. Med. Chem. 2015, 58, 4383–4438; For selected discussions of O-S interactions in isothiourea catalysis, see: [DOI] [PubMed] [Google Scholar]
- 24b. Liu P., Yang X., Birman V. B., Houk K. N., Org. Lett. 2012, 14, 3288–3291; [DOI] [PubMed] [Google Scholar]
- 24c. Abbasov M. E., Hudson B. M., Tantillo D. J., Romo D., J. Am. Chem. Soc. 2014, 136, 4492–4495; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24d. Robinson E. R. T., Walden D. M., Fallan C., Greenhalgh M. D., Cheong P. H.-Y., Smith A. D., Chem. Sci. 2016, 7, 6919–6927; For selected discussions of the origin of O-S interactions, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24e. Zhang X., Gong Z., Li J., Lu T., J. Chem. Inf. Model. 2015, 55, 2138–2153; [DOI] [PubMed] [Google Scholar]
- 24f. Ángyán J. G., Kucsman Á., Poirier R. A., Csizmadia I. G., J. Mol. Struct. 1985, 123, 189–201; [Google Scholar]
- 24g. Iwaoka M., Takemoto S., Tomoda S., J. Am. Chem. Soc. 2002, 124, 10613–10620; [DOI] [PubMed] [Google Scholar]
- 24h. Murray J. S., Lane P., Politzer P., Int. J. Quantum Chem. 2008, 108, 2770–2781; [Google Scholar]
- 24i. Brameld K. A., Kuhn B., Reuter D. C., Stahl M., J. Chem. Inf. Model. 2008, 48, 1–24. [DOI] [PubMed] [Google Scholar]
- 25.For a review on the importance of nitrogen cation–π interactions in asymmetric organocatalysis, see: Yamada S., Fossey J. S., Org. Biomol. Chem. 2011, 9, 7275–7281. [DOI] [PubMed] [Google Scholar]
- 26.The trends in selectivity observed upon variation of the aromatic substituent may be rationalised by considering the electrostatic component of the cation–π interation, see:
- 26a. Mecozzi S., West A. P., Dougherty D. A., Proc. Natl. Acad. Sci. USA 1996, 93, 10566–10571; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Ma J. C., Dougherty D. A., Chem. Rev. 1997, 97, 1303–1324; [DOI] [PubMed] [Google Scholar]
- 26c. Dougherty D. A., Acc. Chem. Res. 2013, 46, 885–893; Substituent effects in cation-π interactions have also been rationalised by considering local interactions between the aromatic substituent and the ion, see:23214924 [Google Scholar]
- 26d. Wheeler S. E., Houk K. N., J. Am. Chem. Soc. 2009, 131, 3126–3127; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26e. Wheeler S. E., Acc. Chem. Res. 2013, 46, 1029–1038. [DOI] [PubMed] [Google Scholar]
- 27.For reviews on aromatic interactions, see:
- 27a. Meyer E. A., Castellano R. K., Diederich F., Angew. Chem. Int. Ed. 2003, 42, 1210–1250; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 1244–1287; [Google Scholar]
- 27b. Martinez C. R., Iverson B. L., Chem. Sci. 2012, 3, 2191–2201; [Google Scholar]
- 27c. Krenske E. H., Houk K. N., Acc. Chem. Res. 2013, 46, 979–989; For an electrostatic model of aromatic-aromatic interactions, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27d. Hunter C. A., Sanders J. K. M., J. Am. Chem. Soc. 1990, 112, 5525–5534; For models considering local aromatic substituent interactions, see: [Google Scholar]
- 27e. Wheeler S. E., Houk K. N., J. Am. Chem. Soc. 2008, 130, 10854–10855; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27f. Wheeler S. E., J. Am. Chem. Soc. 2011, 133, 10262–10274. [DOI] [PubMed] [Google Scholar]
- 28.The data underpinning this research can be found at DOI: https://doi.org/10.17630/5b5c7f5a-7329-40b7-aaed-907c94dff692.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary