

Abstract
Hydrogen/deuterium exchange coupled with mass spectrometry (HDX MS) has become a powerful method to characterize protein conformational dynamics. Workflows typically utilize pepsin digestion prior to MS analysis to yield peptide level structural resolution. Tandem mass spectrometry (MS/MS) can potentially facilitate determination of site-specific deuteration to single-residue resolution. However; to be effective, MS/MS activation must minimize the occurrence of gas-phase intramolecular randomization of solution-generated deuterium labels. While significant work has focused on understanding this process in positive-ion mode, little is known about hydrogen/deuterium (H/D) scrambling processes in negative-ion mode. Here, we utilize selectively deuterated model peptides to investigate the extent of intramolecular H/D scrambling upon several negative ion mode MS/MS techniques, including negative-ion collision induced dissociation (nCID), electron detachment dissociation (EDD), negative-ion free radical initiated peptide sequencing (nFRIPS), and negative ion electron capture dissociation (niECD). H/D scrambling was extensive in deprotonated peptides upon nCID and nFRIPS. In fact, the energetics required to induce dissociation in nCID are sufficient to allow histidine C-2 and Cβ hydrogen atoms to participate in the scrambling process. EDD and niECD demonstrated moderate H/D scrambling with niECD being superior in terms of minimizing hydrogen migration, achieving ~30% scrambling levels for small c-type fragment ions. We believe the observed scrambling is likely due to activation during ionization and ion transport rather than during the niECD event itself.
Keywords: Hydrogen/deuterium exchange, Hydrogen/deuterium scrambling, Negative-ion mode tandem mass spectrometry, EDD, niECD, FRIPS, CID
Graphical Abstract
INTRODUCTION
Hydrogen/deuterium exchange (HDX) coupled with mass spectrometry (MS) is a powerful method for examining protein conformation and dynamics [1–5]. In HDX, protein amide hydrogen atoms are exchanged for deuterium (or vice versa). The location and rate of exchange depend on solvent accessibility and protection derived from hydrogen bonding and are thus indicative of protein three-dimensional structure and unfolding/refolding rate. When such deuterated samples are analyzed by a mass spectrometer, a mass shift of õne Dalton is observed for each exchanged hydrogen atom. The structural resolution of this technique, however, is typically limited to the peptide level with pepsin being the proteolytic enzyme of choice due to quenching at low pH (where most enzymes are inactive) to minimize deuterium back exchange prior to and during MS analysis. Peptic peptides with overlapping sequences are required to measure H/D exchange rates at individual amino acid residues [6–9].
Alternatively, dissociation via tandem mass spectrometry (MS/MS) could potentially be used to improve structural resolution, either by fragmenting proteolytic peptide ions, or to enable top-down (intact) analysis of deuterated proteins. However, the most common MS/MS activation technique, positive-ion mode collision-induced dissociation (CID), has been reported to cause intramolecular hydrogen/deuterium (H/D) scrambling due to the increased vibrational energy of protonated peptide/protein ions prior to dissociation and the up to millisecond time scale of CID [10–14]. Gas-phase H/D scrambling rearranges the deuteration pattern indicative of solution-phase protein structure/dynamics and, thus, erases the desired information.
Electron-based activation methods, e.g., electron-capture/transfer dissociation (ECD/ETD), have demonstrated the ability to induce peptide backbone dissociation with minimum levels of H/D scrambling at carefully tuned instrument settings [15, 16]. In addition to these electron-based techniques, ultraviolet photodissociation was recently reported to proceed with minimum H/D scrambling [17, 18]. ECD/ETD have seen increased utilization in HDX workflows [19–27], but are either incompatible or ineffective for low charge-state precursor ions such as the short peptides frequently generated upon pepsin digestion [28]. Furthermore, intramolecular hydrogen/deuterium migration upon ETD has recently been proposed to follow regio-selective behavior rather than a simple randomization process [29], thus further complicating spectral interpretation.
Negative-ion mode MS/MS is gaining interest for proteomic analysis due to the importance of acidic post-translational modifications [30–37]. While the use of HDX in this polarity has been limited, applications towards carbohydrates and MALDI-generated peptide anions have been reported [38, 39] with prevalent H/D scrambling observed in MALDI tandem time-of-flight CID of peptide anions [39]. However, intramolecular H/D scrambling behavior in electrospray-generated peptide anions has, to our knowledge, not been examined.
Here, we measure the extent of H/D scrambling in several negative-ion mode MS/MS techniques, including negative-ion mode CID (nCID), electron detachment dissociation (EDD) [40, 41], negative-ion electron capture dissociation (niECD) [34], and negative-ion mode free radical induced peptide sequencing (nFRIPS) [35, 42, 43]. Observed scrambling behaviors lend insight into the energetics of these processes.
EXPERIMENTAL
Materials
The model peptides P1 (HHHHHHIIKIIK) [44], P3 (DDDDDDIIEIIE), and P4 (DDDDDIIEII) were synthesized by GenicBio Limited (Shanghai, China). D2O (99.9 atom % D) was purchased from Sigma-Aldrich (St. Louis, MO). o-TEMPO-Bz-NHS was synthesized as described previously [35]. All other chemicals were obtained at HPLC grade from Thermo Fisher Scientific (Waltham, MA) and used without further purification.
Peptide Derivatization
Acetylation of P1 (here referred to as peptide P2) was achieved by mixing a 110 mM P1 solution in 30 μL methanol with a 20 μL 25% solution of acetic anhydride in methanol. Following three hour incubation at room temperature solvents were removed under vacuum.
o-TEMPO-Bz-NHS was dissolved in acetonitrile at a concentration of 22 mM. The P1 conjugation reaction was buffered with 100 mM triethylamine acetate (pH 8) and performed using a four-fold excess of label overnight at 37°C. These selected conditions enabled targeted labeling of the N-terminus. The reaction solution was 60% acetonitrile which, after completion, was evaporated using a vacuum centrifuge followed by redissolution in water. The poor water solubility of o-TEMPO-Bz-NHS enabled crude extraction of the labeled peptide. For control experiments this purified stock solution was diluted to 1–5 µM and directly infused into the instrument.
Deuterium/hydrogen Exchange
Deuterated peptides were prepared by dissolving unmodified peptides or diluting (1:20) derivatized peptides in D2O for a minimum of 18 h at 4 °C. The peptide concentration was 1,000 μM, 500 μM , 50 μM, 100 μM, and 200 μM for nCID, nFRIPS, ECD, EDD, and niECD experiments, respectively. Deuterated peptides were diluted to 10–20 μM using cooled protiated ESI solvents: 49:49:2 methanol:water:formic acid for ETD and nFRIPS and 45:45:10:0.1 water:methanol:isopropanol:formic acid for nCID and positive ion mode ECD, both solvents at approximately pH 2.5. Peptides P3 and P4 were diluted into 100 mM NH4HCO3 in 50:50:0.3 H2O:isopropanol:formic acid (pH 4.0), or 45:45:10:0.05 H2O:methanol:isopropanol,ammonium hydroxide (pH 7.5) for niECD and EDD, respectively. These solutions were immediately transferred to a precooled syringe (Hamilton Co., Reno, NV) or to a Nanomate (Advion, Ithaca, NY) cooled to 0 °C. For syringe infusion, the syringe was mounted on a pump and cooled with ice bags or dry ice. The flow rate was 10 μL/h for the Nanomate and 2–5.0 μL/min for the syringe pump.
Mass Spectrometry
Negative-ion mode CID MS/MS spectra were recorded on an Orbitrap-XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) equipped with a Nanomate nanoESI source. Doubly deprotonated precursor ions were isolated with an 18 m/z window and fragmented at a normalized collision energy of 30%. Fragment ions were acquired in profile mode with four microscans and mass-analyzed in the Orbitrap. Maximum ion injection times were set to 200 ms for MS/MS. The automatic gain control targets were set to 1×105 for MS/MS in the Orbitrap. MS/MS data were gathered from m/z 210–1,600 in negative-ion mode.
ECD, niECD, and EDD MS/MS spectra were recorded on a 7 T SolariX Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (Bruker Daltonics, Billerica, MA) equipped with an Apollo II ESI source. For ECD experiments, an accumulation time of 0.4–0.5 s was used to optimize ion abundance. Mass spectra were acquired from m/z 200–2,000 with 32 scans for an overall acquisition time of less than one minute. Instrument parameters, including drying gas temperature, capillary exit voltage, deflector plate voltage, funnel 1 voltage, skimmer 1 voltage, and quadrupole isolation width, were carefully tuned in order to define regimes of low and high scrambling levels (Table S1). ECD of triply protonated P1 ions was performed with a bias voltage of - 1.0 V and an irradiation time of 0.15 to 0.40 s. A lens electrode placed in front of the cathode was set at + 9–10 V. For negative-ion mode experiments on the SolariX, the nebulizing gas, drying gas, and temperature were set to 1 bar, 4.0 L/min, and 50–200 °C, respectively. Ions were accumulated for 3 s prior to EDD experiments. Doubly deprotonated P3 ions were irradiated at a cathode bias voltage of - 19 V for 2 s while the lens electrode was held at - 10 V. Mass spectra were acquired from m/z 200–2,000 with 32 averaged scans, accumulated over 2–3 minutes. For niECD experiments, the singly deprotonated peptides P3 and P4 were irradiated at a cathode bias voltage of - 4.7 V for 2 s while the lens electrode was held 1.0–1.5 V lower than the cathode bias voltage to maximize the abundance of the charge-increased radical anion.
Negative-ion mode FRIPS and positive-ion mode ETD and CID spectra were acquired on a Thermo Scientific Orbitrap Fusion Lumos (Waltham, MA). To minimize scrambling, positive ion mode ETD and CID experiments were collected under the “softest” settings that allowed for stable electrospray. The spray voltage, sheath gas, auxiliary gas, and RF lens were set to 3,600 V, 2.5 (Arb), 1.5, and 10 %, respectively. The 3+ charge state was isolated with a 10 m/z window and either reacted for 17 ms or subjected to 26% CID. In both experiments, the generated product ions were measured in the orbitrap. For nFRIPS, spectra were acquired with 2,700 V spray voltage, 7 sheath gas, 2 Aux gas, 150 °C ion transfer tube temperature, and 30% RF lens. For nFRIPS fragmentation, an MS3 workflow was used: the precursor ion was first dissociated with 17% HCD and the homolytically cleaved product was then isolated with a 10 m/z window and further activated at 25% HCD. The resulting product ions were measured in the orbitrap.
Data Analysis
Raw precursor and product ion deuterium contents were calculated by subtracting the observed weighted average natural isotopic distribution from the observed weighted average of deuterated equivalents (to yield Δmexp). Observed product ion raw deuterium contents, were then converted to deuteration levels using the following equation:
| (1) |
where m100 corresponds to the weighted average isotopic distribution of the deuterated intact precursor ion and m0 corresponds to the weighted average of its natural isotopic distribution, respectively. D/H exchange rates and predicted deuterium content curves under no scrambling conditions were calculated with the HXPep software (Zhongqi Zhang, Amgen, Thousand Oaks, CA), see Table S2 for peptides P3 and P4, unique to this work. Theoretical 100% scrambling product ion curves were calculated by multiplying equation (1) with the following factor:
| (2) |
where HFrag is the number of exchangeable hydrogen atoms in a given fragment ion and HPre is the total number of exchangeable hydrogen atoms in the precursor ion. Note that precursor ion charge states and the mechanism by which fragment ions were generated were both taken into consideration when calculating theoretical deuterium contents under complete scrambling conditions [34, 41, 45–49]. All experimental data are shown as the average of triplicate measurements. In cases where incomplete product ion series were observed, the level of H/D scrambling was calculated according to the following equation:
| (3) |
where Δm100% and Δm0% are the theoretical mass increases of the corresponding species in the case of 100% scrambling and 0% scrambling, respectively.
RESULTS AND DISCUSSION
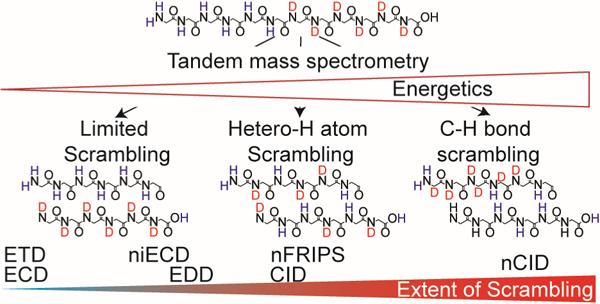
The exchange rate of backbone amide hydrogen atoms in unstructured peptides depends on pH, temperature, steric hindrance, and the inductive influence of neighboring side chains [15, 44, 50]. Utilizing these attributes, Rand et. al. designed the peptide HHHHHHIIKIIK (P1) to investigate H/D scrambling [15, 16, 44]. The difference in intrinsic exchange rates of histidine and isoleucine residues promotes preferential retention of deuterium on C-terminal residues. The degree by which deuteriums migrate to N-terminal residues prior to backbone dissociation determines the applicability of an MS/MS technique for accurately determining deuterium location within this peptide (Figure 1). Implementing a similar approach, we investigated the prevalence of H/D scrambling in a series of negative-ion mode MS/MS techniques.
Figure 1.
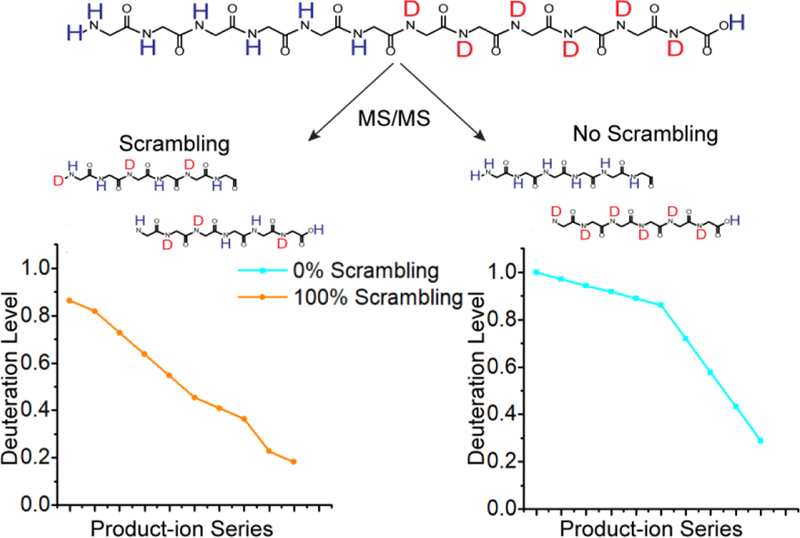
Illustration of a selectively deuterated peptide undergoing gas-phase dissociation with (left) and without (right) H/D scrambling. Blue and orange curves illustrate theoretical 0 and 100% scrambling levels, respectively, for a hypothetical series of y-type ions (y2-y11).
Previous positive-ion mode work demonstrated that the ionization process itself as well as the ion transport conditions prior to MS/MS both have the potential to induce H/D scrambling [15]. That work also characterized the variables that must be adjusted to minimize these scrambling processes. Little is known about the conditions needed to minimize these processes in negative-ion mode; thus, on the utilized Bruker SolariX and Thermo Fusion Lumos instruments, settings were initially chosen that minimized scrambling in positive-ion mode as confirmed by ECD and ETD, respectively (Figures S1 and S2). These positive-ion mode settings were then ported to negative-ion mode and modified as necessary to achieve signal-to-noise ratios sufficient for analysis (Tables S3 and S4). The Orbitrap XL is not equipped with activation techniques capable of examining the extent of scrambling; thus, the energetics of its ion source and ion transport were minimized according to literature values [15; Table S5].
H/D Scrambling in Collision Induced Dissociation of Electrosprayed Deprotonated Peptides
Upon nCID, the doubly deprotonated peptide P1 dissociated into a near complete series of y-type (y4-y11) and c-type fragment ions (c2-c10) (Figure S3). When deuterated P1 was dissociated, a corresponding set of isotopically enriched product ions was formed. Observed isotopic distributions for two representative fragment ions with and without deuterium incorporation are shown in Figures 2A (c3 fragment) and C (y9 fragment), respectively. The weighted average m/z values for the natural isotopic distributions are indicated with green solid lines and the weighted average m/z values for the deuterated species are indicated with grey solid lines. The dashed lines in Figures 2A and C represent the calculated weighted average m/z values under no scrambling (blue dashed line) and complete scrambling (orange dashed line) conditions, respectively. The N-terminal c3 fragment ion is expected to show a low deuteration level in the absence of scrambling whereas the C-terminal y9 fragment ion is expected to show a high deuteration level. Near complete scrambling is evident for the c3 fragment ion whereas the y9 ion shows a deuteration level even lower than that expected for 100% scrambling. Figure 2B illustrates the deuteration level of all observed nCID-derived c-type ions (black line) as well as their expected deuteration levels in the absence (blue line for 0%) and presence (orange line for 100%) of scrambling. Similar to c3, all c-type ions show deuterium contents indicative of high scrambling levels. Unlike the c-type ions but similar to the y9 ion, the deuteration level curve for the y-type ion series (Figure 2D, black line) does not follow the expected trend for 100% scrambling (Figure 2D, orange line). Unexpectedly, all these ions contain significantly less deuterium than previously observed for positive ion mode CID [51]. This apparent deuterium loss suggests that significantly more hydrogen atoms are participating in scrambling reactions in nCID compared with positive ion mode CID. Also, the slope differential between the experimental data and 100% scrambling curves in Figure 2D indicates deuterium enrichment onto the N-terminal side of the peptide rather than randomization of the deuterium content.
Figure 2.
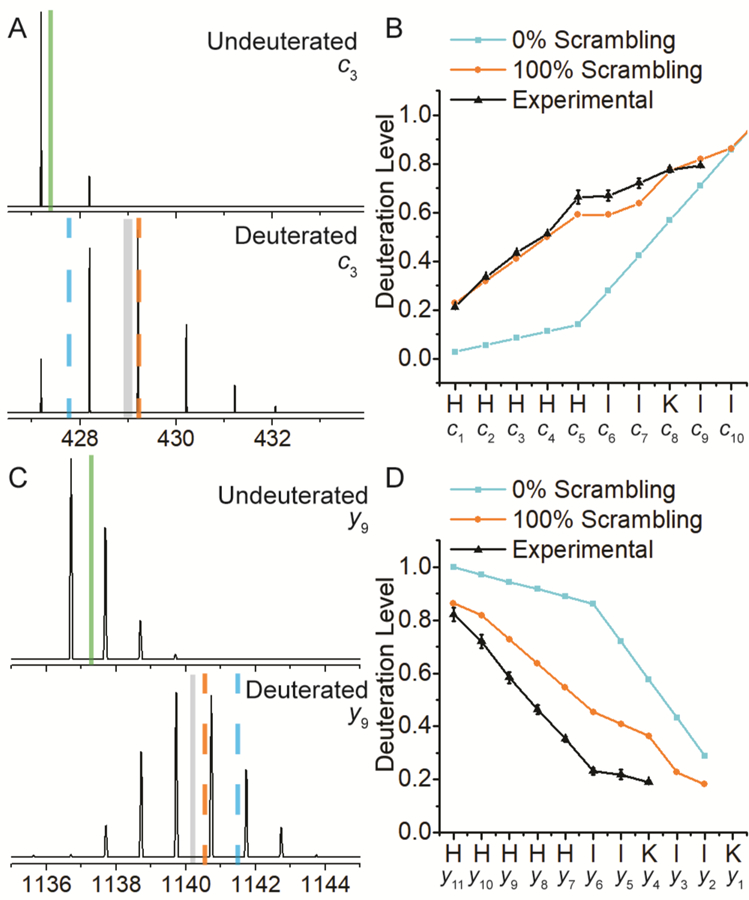
Observed isotopic distributions of c3 (A) and y9 (C) fragment ions from nCID of the natural and deuterated peptide P1 (HHHHHHIIKIIK). The weighted average m/z value for each species is illustrated by a solid green (natural) or grey (deuterated) line. Theoretical deuteration levels for 100 and 0% scrambling are denoted with dashed orange (100%) and blue (0%) lines, respectively. Deuteration levels for all measured P1 nCID-derived c- (B) and y-type (D) fragment ions. Blue and orange lines represent expected deuteration levels for 0 and 100% scrambling, respectively.
To examine whether this aberrant scrambling behavior is mediated by the lysine residues, P1 was acetylated, resulting in modification of the N-terminus and both lysine residues. Deuterated doubly deprotonated *HHHHHHIIK*IIK* (P2) was then subjected to nCID, yielding a near complete sequence of y-type fragment ions (y4-y11, data not shown). Similar to P1, the P2 y-ion series demonstrated deuterium enrichment onto the N-terminal amino acid residues (Figure S4), suggesting that primary amines do not mediate this unexpected H/D scrambling.
The histidine residues in P1/P2 constitute another possible source of excess hydrogen atoms. Thus, a new model peptide, DDDDDDIIEIIE (P3), was designed. Similar to P1, P3 relies on substantially different intrinsic exchange rates of isoleucine and aspartic acid to concentrate deuterium to the C-terminal residues upon D/H exchange. Following nCID, b- and y-type fragment ions were observed (Figure S5). H/D scrambling for the y-type fragment ion series falls within the theoretical limit (Figure 3). This result is in direct contrast to nCID of deuterated P1 and suggests that the additional hydrogen atoms participating in the scrambling reaction for P1 originate from the histidine residues.
Figure 3.
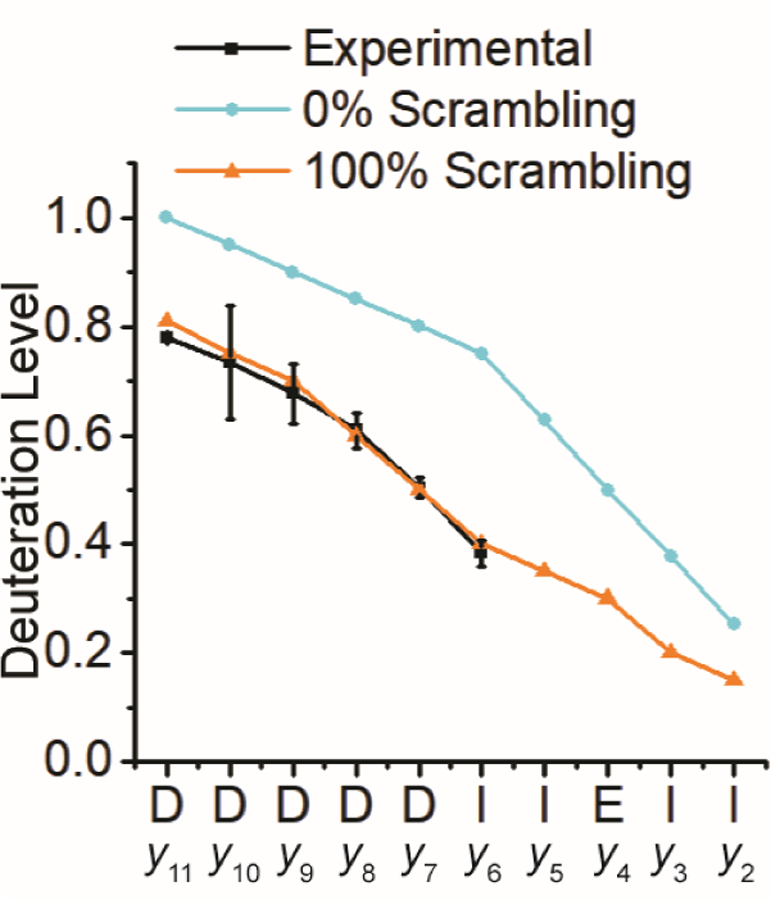
Deuteration levels observed for y-type ions from nCID of the peptide P3 (DDDDDDIIEIIE) as well as expected deuteration levels for 0% and 100% scrambling, respectively.
The histidine C-2 hydrogen atom is known to undergo exchange reactions and has been utilized to examine local histidine environments [52, 53]. In the gas phase, however, previous work in positive-ion mode reported that these hydrogen atoms do not participate in scrambling reactions [54]. On the other hand, deprotonation of histidine residues has been shown to promote this process [55] and critical energies for dissociating deprotonated peptides are known to be higher than those of protonated peptides [10, 56, 57]. Thus, histidine C-2 hydrogen atoms may undergo rearrangement in negative-ion mode. Accounting for these hydrogen atoms, the predicted 100% scrambling curve for P1 y-type ions (Figure 4A) is closer to the experimental nCID data (Figure 4A vs. 2D). However, the experimental curve (Figure 4A) is still beyond the theoretical deuterium migration limit and the slope differential between experimental data and predicted deuteration levels at 100% scrambling still suggests there are unaccounted hydrogen atoms participating in the scrambling process.
Figure 4.
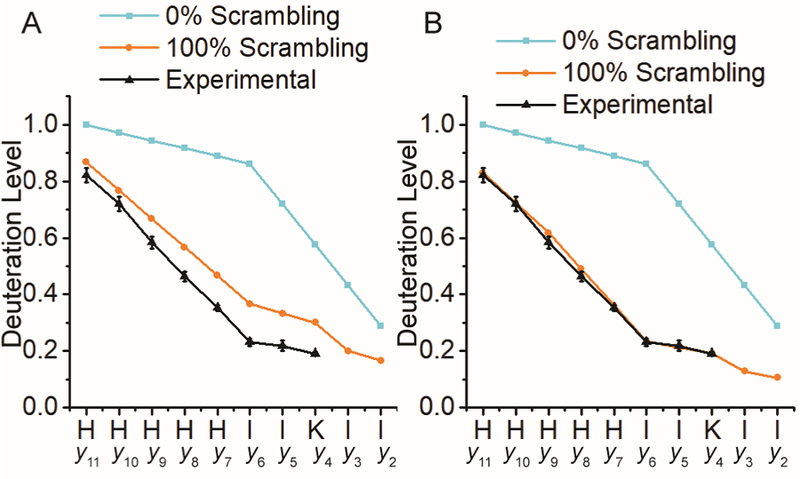
nCID-generated P1 deuteration levels overlaid with 0% and 100% scrambling curves with the 100% curve being adjusted to account for hydrogen exchange at histidine C-2 carbon atoms (A) and adjusted to account for both histidine C-2 and Cβ hydrogen atom exchange (B).
nCID-driven y-ion formation in the absence of deprotonated amino acid side chains has been proposed to involve hydrogen atom abstraction from α-carbon atoms [46]. The energy required to abstract Cα H atoms is in the same range as histidine Cβ-H bonds, which have the lowest bond dissociation energies (BDE) for Cβ-H bonds of any residue [58]. Additionally, when analyzing phenylalanine—another low Cβ-H BDE residue—containing peptides, Bowie et. al. observed nCID derived products that could only be explained via abstraction of Cβ-H atoms [58, 59]. Thus, at the energies necessary to induce nCID dissociation, these Cβ-H atoms may also need to be considered when calculating the 100% scrambling curve. When both the exchange of histidine C-2 and Cβ hydrogen atoms are considered, the experimental data follows the 100% scrambling limit closely (Figure 4B). nCID of P3 does not demonstrate this effect; likely because aspartic acid Cβ-H bonds have significantly higher BDEs and the y-type ions from this peptide are generated through a lower-energy aspartic acid-driven mechanism, analogous to the non-mobile proton positive-ion mode mechanism [48, 60]. The latter mechanism is supported by the extensive water loss observed for the b-type ion series (Figure S5), a hallmark of the aspartic acid-driven mechanism [48].
H/D Scrambling in Radical-Driven Dissociation of Peptide Anions
As with cations [10–14], the above data demonstrate that collisional activation of anions promotes extensive scrambling and is not amenable to improving the structural resolution of HDX experiments. Free radical initiated peptide sequencing (FRIPS) is a radical-driven technique that is achieved through derivatization of a peptide with a persistent radical-containing precursor [45, 61]. Upon low level collisional activation, this radical precursor undergoes homolytic cleavage, generating a radical that propagates throughout the peptide to yield a neutral loss, or an a- x-, c-, or z-type ion (Scheme S1). TEMPO-assisted FRIPS was recently applied to peptide anions [35, 42] and was shown to allow backbone cleavage without loss of highly labile posttranslational modifications, e.g., sulfation [35], suggesting potential applicability towards deuterated peptides. nFRIPS of P1 generated a near complete set of z-type ions (Figure S6). When deuterated P1 was dissociated with nFRIPS, the generated z-type ions demonstrated near complete hydrogen randomization (Figure 5A). However, in contrast to nCID of this peptide, no imidazole C-2 hydrogen migration was observed. We recently calculated that TEMPO-assisted nFRIPS requires approximately 50 kcal mol−1 to promote homolytic cleavage and subsequent backbone dissociation [35]. Our current result implies: 1) that heteroatom-hydrogen scrambling requires less than 50 kcal mol−1 to proceed, and 2) that histidine C-2 and Cβ hydrogen migration likely requires more than 50 kcal mol−1.
Figure 5.
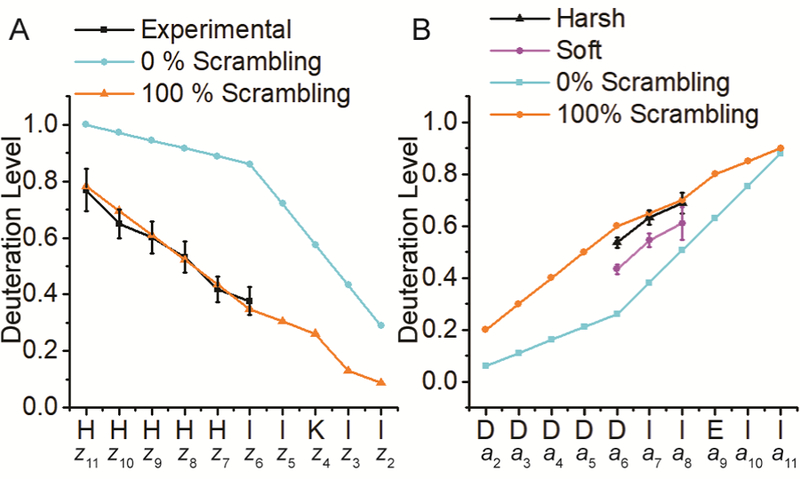
Deuteration levels of nFRIPS-generated z-type fragment ions (A) and EDD-generated a-type ions (B) from the peptide P3 as well as predicted 0% and 100% scrambling curves. EDD was performed after both “soft” and “harsh” ion source conditions.
EDD was the first anion-electron reaction technique reported for peptide MS/MS and generates primarily and x-type product ions [40, 41]. The seminal publication [41] demonstrated that EDD is a “soft” dissociation technique, potentially allowing application towards deuterated peptides. When applied to P1, which lacks acidic amino acid residues, EDD did not promote backbone fragmentation (data not shown). Thus, we focused on the acidic peptide P3. Upon electron irradiation, doubly deprotonated P3 underwent fragmentation, generating characteristic ions (Figure and x-type fragment S7). In particular, , , and were observed. However, upon deuterium incorporation, the low EDD fragmentation efficiency and resulting low fragment ion abundance, combined with signal dilution across broader isotopic envelopes, enabled the calculation of scrambling for only three product ions. Excitingly, these product ions demonstrated only moderate H/D scrambling (Figure 5B). The corresponding deuteration levels equate to a scrambling percentage of 51 for , 61 for , and 54 for .
To ensure that this reduction in scrambling (as compared to nCID and nFRIPS) was accurate and not due to an unforeseen mechanism, the instrument parameters—particularly the ion source—were modified to promote “harsh” conditions (Table S3). The harsher source once again promoted extensive hydrogen migration (Figure 5B), increasing the scrambling percentages to 81, 94, and 95 for , , , respectively. This result suggests that EDD is capable of limiting H/D scrambling for a-type ions. However, even under gentle ionization conditions scrambling levels are too high and the fragmentation efficiency is far too low to be feasible for HDX experiments. EDD utilizes ~20 eV electrons, which are significantly more energetic than the <1 eV electrons typically employed in ECD [41, 62]. It has been demonstrated that EDD involves both radical-driven fragmentation and vibrational excitation [63, 64]. Additionally, previous work demonstrated an uneven deuterium distribution in aspartic acid repeats following ETD [65]. This unexpected observation was attributed to the presence of nearby lysine residues. Although lysines are not present in the aspartic acid-rich model peptide studied here, it is possible that the observed H/D scrambling can also be attributed to this effect.
Negative-ion electron capture dissociation (niECD) is a radical-driven dissociation technique that is thought to occur through a similar mechanism as its positive-ion mode analogue. In niECD, electron energies are considerably lower (3.5–6.5 eV) than in EDD. However, niECD is most compatible with low charge-state anions [34] and, under conditions suitable for HDX, the singly deprotonated P3 ion did not show sufficient abundance for analysis (Figure S8A). To increase the abundance of the desired singly deprotonated precursor ion, a truncated version of P3 was synthesized. Following ESI, the singly deprotonated species of the peptide DDDDDIIEII (P4) demonstrated significantly higher abundance, rendering it amenable to niECD. Electron irradiation of undeuterated P4 resulted in the formation of primarily c-type ions (Figure S8B). Similar to EDD, upon deuteration, the niECD efficiency was not widely compatible with the broad isotopic distributions observed following HDX and only the , , , , , and product ions could be analyzed. Despite the utilization of low energy electrons, the z-type ion series demonstrated nearly 100% scrambling; 92, 84, and 100% for the , , and fragment ions respectively. niECD however, was able to minimize H/D scrambling to 32%±8% for the c2 fragment ion and 30%±4% for c3.
Interestingly, the degree of H/D scrambling (96%±30%) observed for fragment ion is much higher than that of the c2 and c3 ions. Similar inconsistencies were reported from ECD [15] and ETD [29], where the extent of scrambling appears to depend on the size of c-type ions with smaller c-ions demonstrating lower degrees of scrambling. This phenomenon may be related to the lifetime difference of the corresponding radical intermediates with their complementary -type fragments [37].
CONCLUSIONS
To investigate H/D scrambling in negative-ion mode, several MS/MS techniques were applied towards regio-selectively deuterated peptide anions. We demonstrated that nCID mediated extensive H/D scrambling within all peptides examined. Furthermore, nCID promotes aberrant scrambling profiles in histidine-containing peptides. This unexpected behavior is potentially due to the scrambling of histidine C-2 and Cβ-hydrogen atoms. Radical-driven fragmentation methods also promoted H/D scrambling: nFRIPS induced complete randomization of the deuterium profile whereas EDD and niECD proceeded with a moderate degree of H/D scrambling with niECD slightly outperforming EDD. Interestingly, the scrambling levels observed for different fragment ions from the same peptide upon EDD and niECD can be different, which may be attributed to the difference in chemical properties of exchangeable hydrogen sites and/or the conformation of the precursor peptide ions. Thus, more detailed analysis is needed to elucidate detailed mechanisms of intramolecular H/D migration. Overall, nCID, nFRIPS, EDD, and niECD are not amenable to HDX MS/MS experiments. For nCID, nFRIPS, and EDD, this incompatibility is likely due to the energetics associated with their mechanisms. For niECD, however, the energetics of the negative-ionization process itself may be too high. Despite limited practical applications, the observed H/D scrambling behavior provided insights into the energetics of these anion fragmentation techniques.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation CHE 1609840 and the University of Michigan. The Thermo Scientific Orbitrap Fusion Lumos was acquired via National Institutes of Health grant S10 OD021619. We would also like to thank Z. Zhang for kindly providing the HXPEP software.
REFERENCES
- 1.Pirrone GF, Iacob RE, Engen JR: Applications of Hydrogen/Deuterium Exchange MS from 2012 to 2014. Anal. Chem 87, 99–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wales TE, Engen JR: Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev 25, 158–170 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Hoofnagle AN, Resing KA, Ahn NG: Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct 32, 1–25 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Smith DL: Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci 2, 522–531 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konermann L, Pan J, Liu YH: Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev 40, 1224–1234 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Brier S, Maria G, Carginale V, Capasso A, Wu Y, Taylor RM, Borotto NB, Capasso C, Engen JR: Purification and characterization of pepsins A1 and A2 from the Antarctic rock cod Trematomus bernacchii. FEBS J 274, 6152–6166 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cravello L, Lascoux D, Forest E: Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom 17, 2387–2393 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Fajer PG, Bou-Assaf GM, Marshall AG: Improved Sequence Resolution by Global Analysis of Overlapped Peptides in Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom 23, 1202–1208 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Borotto NB, Zhang Z, Dong J, Burant B, Vachet RW: Increased β-Sheet Dynamics and D–E Loop Repositioning Are Necessary for Cu(II)-Induced Amyloid Formation by β−2-Microglobulin. Biochemistry 56, 1095–1104 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jørgensen TJD, Gårdsvoll H, Ploug M, Roepstorff P: Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation. J. Am. Chem. Soc 127, 2785–2793 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Hoerner JK, Xiao H, Dobo A, Kaltashov IA: Is there hydrogen scrambling in the gas phase? Energetic and structural determinants of proton mobility within protein ions. J. Am. Chem. Soc 126, 7709–7717 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Hagman C, Håkansson P, Buijs J, Håkansson K: Inter-molecular migration during collisional activation monitored by hydrogen/deuterium exchange FT-ICR tandem mass spectrometry. J. Am. Soc. Mass Spectrom 15, 639–646 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Ferguson PL, Pan J, Wilson DJ, Dempsey B, Lajoie G, Shilton B, Konermann L: Hydrogen/deuterium scrambling during quadrupole time-of-flight MS/MS analysis of a zinc-binding protein domain. Anal. Chem 79, 153–160 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Hamuro Y, Tomasso JC, Coales SJ: A Simple Test To Detect Hydrogen/Deuterium Scrambling during Gas-Phase Peptide Fragmentation. Anal. Chem 80, 6785–6790 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Rand KD, Adams CM, Zubarev RA, Jørgensen TJD: Electron Capture Dissociation Proceeds with a Low Degree of Intramolecular Migration of Peptide Amide Hydrogens. J. Am. Chem. Soc 130, 1341–1349 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Zehl M, Rand KD, Jensen ON, Jorgensen TJD: Electron Transfer Dissociation Facilitates the Measurement of Deuterium Incorporation into Selectively Labeled Peptides with Single Residue Resolution. J. Am. Chem. Soc 130, 17453–17459 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Brodie NI, Huguet R, Zhang T, Viner R, Zabrouskov V, Pan J, Petrotchenko EV, Borchers CH: Top-Down Hydrogen–Deuterium Exchange Analysis of Protein Structures Using Ultraviolet Photodissociation. Anal. Chem 90, 3079–3082 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Mistarz UH, Bellina B, Jensen PF, Brown JM, Barran PE, Rand KD: UV Photodissociation Mass Spectrometry Accurately Localize Sites of Backbone Deuteration in Peptides. Anal. Chem 90, 1077–1080 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Pan J, Zhang S, Parker CE, Borchers CH: Subzero Temperature Chromatography and Top-Down Mass Spectrometry for Protein Higher-Order Structure Characterization: Method Validation and Application to Therapeutic Antibodies. J. Am. Chem. Soc 136, 13065–13071 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Wang G, Kaltashov IA: Approach to Characterization of the Higher Order Structure of Disulfide-Containing Proteins Using Hydrogen/Deuterium Exchange and Top-Down Mass Spectrometry. Anal. Chem 86, 7293–7298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abzalimov RR, Bobst CE, Kaltashov IA: A New Approach to Measuring Protein Backbone Protection with High Spatial Resolution Using H/D Exchange and Electron Capture Dissociation. Anal. Chem 85, 9173–9180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan J, Heath BL, Jockusch RA, Konermann L: Structural Interrogation of Electrosprayed Peptide Ions by Gas-Phase H/D Exchange and Electron Capture Dissociation Mass Spectrometry. Anal. Chem 84, 373–378 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Bobst CE, Kaltashov IA: Enhancing the Quality of H/D Exchange Measurements with Mass Spectrometry Detection in Disulfide-Rich Proteins Using Electron Capture Dissociation. Anal. Chem 86, 5225–5231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rand KD: Pinpointing changes in higher-order protein structure by hydrogen/deuterium exchange coupled to electron transfer dissociation mass spectrometry. Int. J. Mass Spectrom 338, 2–10 (2013). [Google Scholar]
- 25.Landgraf RR, Chalmers MJ, Griffin PR: Automated Hydrogen/Deuterium Exchange Electron Transfer Dissociation High Resolution Mass Spectrometry Measured at Single-Amide Resolution. J. Am. Soc. Mass Spectrom 23, 301–309 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Going CC, Xia Z, Williams ER: Real-time HD Exchange Kinetics of Proteins from Buffered Aqueous Solution with Electrothermal Supercharging and Top-Down Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom 27, 1019–1027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masson GR, Maslen SL, Williams RL: Analysis of phosphoinositide 3-kinase inhibitors by bottom-up electron-transfer dissociation hydrogen/deuterium exchange mass spectrometry. Biochem. J 474, 1867–1877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rand KD, Zehl M, Jensen ON, Jørgensen TJD: Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal. Chem 81, 5577–5584 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Hamuro Y: Regio-Selective Intramolecular Hydrogen/Deuterium Exchange in Gas-Phase Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom 28, 971–977 (2017). [DOI] [PubMed] [Google Scholar]
- 30.McAlister GC, Russell JD, Rumachik NG, Hebert AS, Syka JEP, Geer LY, Westphall MS, Pagliarini DJ, Coon JJ: Analysis of the acidic proteome with negative electron-transfer dissociation mass spectrometry. Anal. Chem 84, 2875–2882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doerr A: Navigating the negative-mode proteome. Nat. Methods 12, 808–808 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Riley NM, Rush MJP, Rose CM, Richards AL, Kwiecien NW, Bailey DJ, Hebert AS, Westphall MS, Coon JJ: The Negative Mode Proteome with Activated Ion Negative Electron Transfer Dissociation. Mol. Cell. Proteomics 14, 2644–2660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson MR, Brodbelt JS: Integrating Weak Anion Exchange and Ultraviolet Photodissociation Mass Spectrometry with Strategic Modulation of Peptide Basicity for the Enrichment of Sulfopeptides. Anal. Chem 88, 11037–11045 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Yoo HJ, Wang N, Zhuang S, Song H, Håkansson K: Negative-ion electron capture dissociation: Radical-driven fragmentation of charge-increased gaseous peptide anions. J. Am. Chem. Soc 133, 16790–16793 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Borotto NB, Ileka KM, Tom CATMB, Martin BR, Håkansson K: Free Radical Initiated Peptide Sequencing for Direct Site Localization of Sulfation and Phosphorylation with Negative Ion Mode Mass Spectrometry. Anal. Chem 90, 9682–9686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huzarska M, Ugalde I, Kaplan DA, Hartmer R, Easterling ML, Polfer NC: Negative electron transfer dissociation of deprotonated phosphopeptide anions: choice of radical cation reagent and competition between electron and proton transfer. Anal. Chem 82, 2873–2878 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Hersberger KE, Håkansson K: Characterization of O -Sulfopeptides by Negative Ion Mode Tandem Mass Spectrometry: Superior Performance of Negative Ion Electron Capture Dissociation. Anal. Chem 84, 6370–6377 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Kostyukevich Y, Kononikhin A, Popov I, Nikolaev E: In-ESI Source Hydrogen/Deuterium Exchange of Carbohydrate Ions. Anal. Chem 86, 2595–2600 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Bache N, Rand KD, Roepstorff P, Ploug M, Jørgensen TJD: Hydrogen Atom Scrambling in Selectively Labeled Anionic Peptides Upon Collisional Activation by MALDI Tandem Time-of-Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom 19, 1719–1725 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Kjeldsen F, Silivra OA, Ivonin IA, Haselmann KF, Gorshkov M, Zubarev RA: Cα-C backbone fragmentation dominates in electron detachment dissociation of gas-phase polypeptide polyanions. Chem. Eur. J 11, 1803–1812 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Budnik BA, Haselmann KF, Zubarev RA: Electron detachment dissociation of peptide di-anions: An electron-hole recombination phenomenon. Chem. Phys. Lett 342, 299–302 (2001). [Google Scholar]
- 42.Lee J, Park H, Kwon H, Kwon G, Jeon A, Kim HI, Sung BJ, Moon B, Oh HB: One-step peptide backbone dissociations in negative-ion free radical initiated peptide sequencing mass spectrometry. Anal. Chem 85, 7044–7051 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Hage C, Ihling CH, Götze M, Schäfer M, Sinz A: Dissociation Behavior of a TEMPO-Active Ester Cross-Linker for Peptide Structure Analysis by Free Radical Initiated Peptide Sequencing (FRIPS) in Negative ESI-MS. J. Am. Soc. Mass Spectrom 28, 56–68 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Rand KD, Jørgensen TJD: Development of a Peptide Probe for the Occurrence of Hydrogen (1 H/ 2 H) Scrambling upon Gas-Phase Fragmentation. Anal. Chem 79, 8686–8693 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Lee M, Kang M, Moon B, Oh HB: Gas-phase peptide sequencing by TEMPO-mediated radical generation. Analyst 134, 1706–1712 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Bowie JH, Brinkworth CS, Dua S: Collision-induced fragmentations of the (M-H)- parent anions of underivatized peptides: An aid to structure determination and some unusual negative ion cleavages. Mass Spectrom. Rev 21, 87–107 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Brinkworth CS, Dua S, McAnoy AM, Bowie JH: Negative ion fragmentations of deprotonated peptides: Backbone cleavages directed through both Asp and Glu. Rapid Commun. Mass Spectrom 15, 1965–1973 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Wysocki VH, Tsaprailis G, Smith LL, Breci LA: Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom 35, 1399–1406 (2000). [DOI] [PubMed] [Google Scholar]
- 49.Syrstad EA, Tureček F: Toward a general mechanism of electron capture dissociation. J. Am. Soc. Mass Spectrom 16, 208–224 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Bai Y, Milne JS, Mayne L, Englander SW: Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Genet 17, 75–86 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jørgensen TJD, Gårdsvoll H, Ploug M, Roepstorff P: Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation. J. Am. Chem. Soc 127, 2785–2793 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Miyagi M, Wan Q, Ahmad MF, Gokulrangan G, Tomechko SE, Bennett B, Dealwis C: Histidine hydrogen-deuterium exchange mass spectrometry for probing the microenvironment of histidine residues in dihydrofolate reductase. PLoS One 6, e17055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dong J, Callahan, Katie L, Borotto NB, Vachet RW: Identifying Zn-bound histidine residues in metalloproteins using hydrogen-deuterium exchange mass spectrometry. Anal. Chem 86, 766–773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cebo M, Kielmas M, Adamczyk J, Cebrat M, Szewczuk Z, Stefanowicz P: Hydrogen–deuterium exchange in imidazole as a tool for studying histidine phosphorylation. Anal. Bioanal. Chem 406, 8013–8020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bradbury JH, Chapman BE, Pellegrino FA: Hydrogen-deuterium exchange kinetics of the C-2 protons of imidazole and histidine compounds. J. Am. Chem. Soc 95, 6139–6140 (1973). [DOI] [PubMed] [Google Scholar]
- 56.Harrison AG: Sequence-specific fragmentation of deprotonated peptides containing H or alkyl side chains. J. Am. Soc. Mass Spectrom 12, 1–13 (2001). [DOI] [PubMed] [Google Scholar]
- 57.Marzluff EM, Campbell S, Rodgers MT, Beauchamp JL: Low-Energy Dissociation Pathways of Small Deprotonated Peptides in the Gas Phase. J. Am. Chem. Soc 116, 7787–7796 (1994). [Google Scholar]
- 58.Moore BN, Julian RR: Dissociation energies of X-H bonds in amino acids. Phys. Chem. Chem. Phys 14, 3148–3154 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Eckersley M, Bowie JH, Hayes RN: Collision-induced dissociations of deprotonated peptides, dipeptides and tripeptides with hydrogen and alkyl α groups. An aid to structure determination. Org. Mass Spectrom 24, 597–602 (1989). [Google Scholar]
- 60.Burlet O, Yang CY, Gaskell SJ: Influence of cysteine to cysteic acid oxidation on the collision-activated decomposition of protonated peptides: Evidence for intraionic interactions. J. Am. Soc. Mass Spectrom 3, 337–344 (1992). [DOI] [PubMed] [Google Scholar]
- 61.Hodyss R, Cox HA, Beauchamp JL: Bioconjugates for Tunable Peptide Fragmentation: Free Radical Initiated Peptide Sequencing (FRIPS). J. Am. Chem. Soc 127, 12436–12437 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Axelsson J, Palmblad M, Håkansson K, Håkansson P: Electron capture dissociation of substance P using a commercially available Fourier transform ion cyclotron resonance mass spectrometer. Rapid Commun. Mass Spectrom 13, 474–477 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Wolff JJ, Laremore TN, Aslam H, Linhardt RJ, Amster IJ: Electron-Induced Dissociation of Glycosaminoglycan Tetrasaccharides. J. Am. Soc. Mass Spectrom 19, 1449–1458 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kinet C, Gabelica V, Balbeur D, De Pauw E: Electron detachment dissociation (EDD) pathways in oligonucleotides. Int. J. Mass Spectrom 283, 206–213 (2009). [Google Scholar]
- 65.Rand KD, Lund FW, Amon S, Jorgensen TJD: Investigation of amide hydrogen back-exchange in Asp and His repeats measured by hydrogen (1H/2H) exchange mass spectrometry. Int. J. Mass Spectrom 302, 110–115 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.