

Abstract
Multiplex immunoassays confer several advantages over widely adopted singleplex immunoassays including increased efficiency at a reduced expense, greater output per sample volume ratios and higher throughput predicating more resolute, detailed diagnostics and facilitating personalised medicine. Nonetheless, to date, relatively few protein multiplex immunoassays have been validated for in vitro diagnostics in clinical/point‐of‐care settings. This review article will outline the challenges, which must be ameliorated prior to the widespread integration of multiplex immunoassays in clinical settings: (i) biomarker validation; (ii) standardisation of immunoassay design and quality control (calibration and quantification); (iii) availability, stability, specificity and cross‐reactivity of reagents; (iv) assay automation and the use of validated algorithms for transformation of raw data into diagnostic results. A compendium of multiplex immunoassays applicable to in vitro diagnostics and a summary of the diagnostic products currently available commercially are included, along with an analysis of the relative states of development for each format (namely planar slide based, suspension and planar/microtitre plate based) with respect to the aforementioned issues.
Keywords: ELISA, In vitro diagnostics, Multiplex immunoassay
Abbreviations
- CLIA
The Clinical Laboratory Improvement Amendments
- FDA
Food and Drug Administration
- IVD
in vitro diagnostics
- MHRA
Medicines and Healthcare Products Regulatory Agency
- POC
point of care
- UKAS
United Kingdom Accreditation Service
1. Introduction
Over the past 20 years, the development of high‐throughput technologies for genomic and proteomic analysis has ushered in a novel era of biomarker discovery, which has thus far yielded numerous new markers, many of which maintain an undefined pathophysiological significance and limited practical clinical application 1, 2, 3, whereas others are already bearing a notable impact on disease diagnosis and therapy [4].
While contemporary singleplex ELISA formats are able to accurately diagnose diseases whereby characterisation of a single analyte is sufficient (i.e. HIV–1 p24); patient stratification and monitoring of more complex, multifactorial diseases such as cancers, graft‐versus‐host disease, autoimmune and neurodegenerative diseases require the analysis of multiple biomarkers in order to implement optimised therapeutic regimens 5. Accordingly, validation of novel biomarkers and their amalgamation into multiplex immunoassay panels confers the attractive prospect of simultaneous measurement of multiple analytes in a single patient sample, thereby minimising assay costs, time and sample volume while concurrently enabling progression monitoring and outcome prediction to anticipate potentially severe sequelae or adverse drug reactions. Moreover, the advent of multiplex technology complements an ongoing progressive shift towards personalised medicine with holistic, molecular fingerprinting of diseases via the identification and characterisation of biomarker profiles to accommodate greater diagnostic resolution between closely related disease phenotypes. For such reasons, it appears highly likely that multiplex immunoassays will continue to garner popularity and secure a mainstream role in research and eventually clinical spheres. The subject of this review will be an outline of the contemporary multiplex immunoassay formats in tandem with a perspective of the ongoing challenges that must be ameliorated prior to the widespread integration of the technology into clinical settings. Accordingly, the aim is towards clarifying the intricate and intertwined technological issues in the hope of promoting directives for future development.
2. Historical perspective on immunoassay development
Immunoassays such as singleplex/conventional ELISA formats have assumed a ‘workhorse’ role in the highly sensitive qualitative and quantitative detection of analytes within heterogeneous samples for over 50 years. Moreover, the advent of hybridoma technology in the 70s as a means of generating monoclonal antibodies 6 has facilitated the generation of highly robust, antibody‐based assays with a proclivity for standardisation and automation. While multiplex and singleplex ELISA adopt a common, ‘sandwich’ format (i.e. capture antibody, sample addition and detection antibody), the latter is typically developed upon addition of a colorimetric enzymatic substrate. Contrastingly, multiplex ELISA adopts chemiluminescent/fluorescent reporter systems, as enzymatic reporters are chemically incompatible for simultaneous analysis of multiple localised targets. On this basis, we adopt a terminology of ‘immunoassay’ in lieu of ‘ELISA’ throughout. The notion of immunoassay miniaturisation for diagnostics was initially conceived in 1963 when J.G. Feinberg and A.W. Wheeler developed a ‘microspot’ technique as a means of detecting autoimmune antibody and tissue antigens, whereby thyroglobulin immobilised in a microspot on cellulose acetate strips were incubated with serum from autoimmune thyroiditis patients 7 (Fig. 1). The presence of autoantibodies in the sera resulted in the spontaneous precipitation of thyroglobulin in the microspot. The ability of the technique to detect low levels of both the autoantibody and its cognate antigen led the authors to propose that such microspot assays ‘would have a particular advantage for routine use on clinical specimens because it is simple, sensitive, objective, quickly carried out and read, requires but minute quantities of serum and antigen, and provides a permanent record for the case files’. Subsequently, in 1989, R. Ekins devised the ambient analyte theory 8 postulating that miniaturisation of immunoassays (i.e. reduction of the capture antibody concentration) elicits an improved LOD. According to the law of mass action concerning the behaviour of solutions in dynamic equilibrium, only a diminutive proportion of the total concentration of analyte molecules would be captured on a spot composed of a reduced quantity of immobilised capture antibodies; thus, given ambient analyte conditions, the total concentration of analyte in a sample would not alter significantly despite the high affinity of the capture antibody in conjunction with the low total analyte concentration. Therefore, miniaturisation renders concentration dependency inasmuch that the quantity of captured analyte in the spot is representative of the total analyte concentration in the sample. A high overall sensitivity is achievable with miniaturisation as analyte measurement is always conducted while retaining the highest concentration per unit volume attainable for the given sample, with decreased reaction times due to curtailed diffusion distances. Taken together, Ekins outlined the fundamental microarray multiplex technology principles and envisioned their potential application in research and clinical diagnostics. Historically these findings have influenced the trajectory of multiplex immunoassay development. The necessity of protein microarray technology for the identification, quantification and functional analysis of proteins in basic and applied proteome research has been accentuated since the turn of the 21st century, in part due to a shift in genomics and proteomics towards the analysis of gene and cognate protein function from more holistic perspectives, but also by virtue of the fact that there is no absolute correlation between mRNA expression levels and corresponding protein expression. Furthermore, it is impossible to deduce the functional state of a protein solely from its expression level; thus, large‐scale screening operations implemented by the post‐genomic era have thus encompassed applications ranging from functional analysis of unknown genes to identification of disease‐related gene products, screening in drug discovery and clinical diagnostics. Notably, due to the proof of concept afforded by DNA microarrays and the ongoing necessity to develop high‐throughput technologies facilitating the analysis of proteome functionality, there has been a surge in the development and refinement of multiplex protein immunoassays (Fig. 2).
Figure 1.
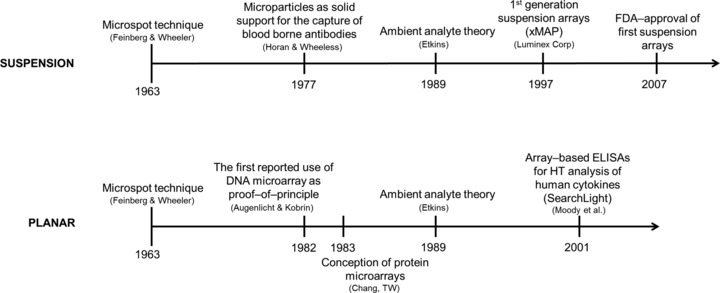
Timelines charting the theoretical and practical milestones in the development of multiplex technology for suspension and planar immunoassay formats. The first commercial suspension array was available before the first planar‐based array 94, 95, 96.
Figure 2.
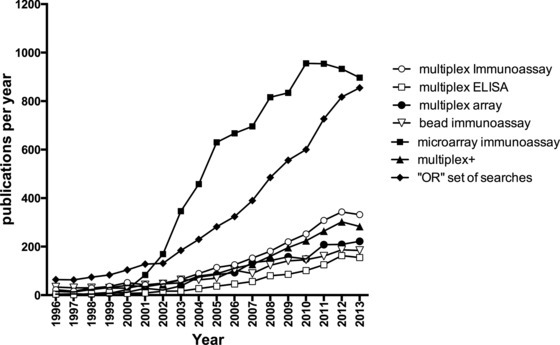
Number of publications published per year, as found within the PubMed database (sampling 1996–2013) using the following keywords search parameters. Multiplex ELISA, multiplex immunoassay, multiplex array, microarray immunoassay, ‘or’ set of searches is derived from accumulating the search terms above using a logical or to combine: (((((multiplex immunoassay) or multiplex ELISA) or multiplex bead) or multiplex cytokine) or multiplex array) or bead immunoassay multiplex+ = ((bead) or (cytokine) or (ELISA) or (array) or (protein) or (antibody) or (antigen) and immunoassay) and multiplex. Numbers were obtained by the PubMed build‐in year count facility, by downloads of CSV files for each search performed, after reviewing the returned papers. The year 2013 was chosen as a cutoff as current year PubMed papers are still increasing each month at the time of writing.
3. Commercially available multiplex immunoassays
Several high‐content protein microarrays for comparative profiling are available commercially (Tables 1 and 2) 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, however, of these, relatively few have been validated for in vitro diagnostics (IVD) applications; moreover, despite the theoretical multiplexing capacities of the platforms, commercially available panels appear to only harness a fraction of this capacity, seldom exceeding 20 individual analytes 46, 60, 61, 62. Contemporary multiplex immunoassay systems may be divided into the two described formats: planar assays and microbead‐based suspension assays (Fig. 3). The planar format includes platforms such as the Mesoscale Discovery Technology Platform (MSD®) and the Q–Plex™ array (Quansys Biosciences) whereby high‐affinity capture ligands are immobilised discretely on a solid phase, 2D support, usually in a microtitre plate format, although multiplexed assays on functionalised glass slides are also available. The immobilised ligands are subsequently exposed to treatment with the sample and probing with detection antibodies labelled with a reporter system. The suspension format includes platforms such as Luminex™, Cytometric Bead Arrays and Bio–PlexPro™ whereby high‐affinity capture ligands are immobilised discretely on fluorescently activated plastic microbeads and mixed with the sample in liquid phase. Subsequent addition of detection antibodies labelled with a reporter dye enables high‐resolution analysis of specific fluorescent signal via flow cytometric methods. Notably, for both formats a reciprocal approach known as RP protein array may also be adopted, whereby sample material representing the proteomic repertoire of the isolate source is bound to the array surface and subsequently probed with antibodies specific to the target components. However, in contrast to conventional sandwich/capture‐based assays, the heterogeneity of the immobilised sample necessitates an imperative for validated, highly specific antibodies to negate false‐positive readouts 45.
Table 1.
Commercially available multiplex arrays suitable for a variety of clinical indications
| Indication(s) | Target | Company | Product | Platform/format | FDA approved |
|---|---|---|---|---|---|
| Allergies | Antibodies | Phadia | ImmunoCAP® ISAC | Planar array | – |
| Autoimmune | Antibodies | INOVA Diagnostics | QUANTA Plex® | Luminex | + |
| Allergies | Antibodies | ImmuneTech | MyAllergyTest | Luminex | + |
| Allergies (domestic allergens) | Antibodies | INDOOR Biotechnologies | IgE–QBA™ | Luminex | – |
| Autoimmune | Antibodies | Immucor | BeadChip™ | Bead array | + |
| Autoimmune | Antibodies | Theradiag | FIDIS™ System | Luminex | + |
| Autoimmune, Infectious Disease | Antibodies | Zeus Scientific | AtheNA Multi–Lyte® Test System | Luminex | + |
| Autoimmune, Infectious Disease | Antibodies | Bio–Rad Laboratories | Bio–Plex® 2200 Autoimmune and Infectious Disease Panels | Luminex | + |
| Autoimmune | Antibodies | Whatman | FAST Quant® | Planar array | – |
| Multiple | Proteins | Myriad RBM | Human MAPs | Luminex | – |
| Cardiovascular, Infectious Disease | Antibodies + Proteins | Biomérieux | VIDAS® range | VIDAS | + |
| Cardiovascular | Proteins | Alere | Triage® Cardiac Panel | Triage | – |
| Infectious Disease | Antibodies | Arrayit Corporation | Pathogen Antigen Microarrays | Planar array | – |
| Infectious Disease | Antibodies | DiaMex | Optiplex Borrelia | Luminex | – |
| Infectious Disease | Antibodies | Focus Diagnostics | Plexus™ HerpeSelect® | Luminex | + |
| Multiple | Proteins | Randox | Biochip Array | Planar | + |
| Neurodegenerative disorders | Proteins | Innogenetics NV | INNO–BIA AlzBio3 | Luminex | + |
| HLA typing | Antibodies | Gen–Probe | LIFECODES range | Luminex | + |
| Acute Phase Protein Profiling | Proteins | R&D Systems | Human Cytokine Array Panel A | Planar array | – |
| Multiple | Antibodies + Proteins | RayBiotech | Quantibody® | Planar array | – |
| Acute Phase Protein Profiling | Proteins | Quansys | Q–Plex™ array | Planar array | – |
| Cytokine Profiling | Proteins | Aushon | Ciraplex® | Planar array | – |
| Multiple | Proteins | Meso Scale Discovery | MULTI–ARRAY microplate | Planar array | – |
| Cytokine Profiling | Proteins | QuantiScientifics, LLC | A2® Multiplex ELISA Human Cytokine Panel | Planar array | – |
| Multiple | Proteins | eBioscience | FlowCytomix™ Multiplex | Flow cytometry | – |
| Cytokine Profiling | Proteins | BD Biosciences | CBA Human Th1/Th2/Th17 Kits | Flow cytometry | – |
| Multiple | Antibodies + Proteins | Illumina | VeraCode™ | Luminex | – |
Assays that have progressed past pre‐clinical validation studies and attained regulatory body approval for their clinical application are designated with a ‘+’ in this table. Those that have demonstrated clinical viability and applicability in pre‐clinical validation studies but not demonstrated compliance with regulatory parameters are designated with a ‘–’ in this table but a ‘+’ in Table 2.
Table 2.
Commercially available multiplex arrays suitable for IVD, which have demonstrated clinical viability and applicability in pre‐clinical validation studies but not demonstrated complete compliance with regulatory parameters (at time of writing) are designated with a ‘–’ in Table 1, but with a ‘+’ in this table
| Company | Product | Validation | Reference |
|---|---|---|---|
| Phadia | ImmunoCAP® ISAC | + | 9, 10, 11, 12, 13 |
| INDOOR Biotechnologies | IgE–QBA™ | + | 14, 15, 16 |
| Bio–Rad Laboratories | Bio–Plex® 2200 Autoimmune and Infectious Disease Panels | + | 17, 18, 19, 20, 21 |
| Whatman | FAST Quant® | + | 17, 22, 23, 45 |
| Myriad RBM | Human MAPs | + | 24, 25, 26, 27, 28 |
| Alere | Triage® Cardiac Panel | + | 29, 30 |
| Arrayit Corporation | Pathogen Antigen Microarrays | – a | |
| DiaMex | Optiplex Borrelia | – | |
| R&D Systems | Human Cytokine Array Panel A | + | 31, 32, 33, 34 |
| RayBiotech | Quantibody® | + | 35, 36, 37, 38, 39, 40, 41 |
| Quansys | Q–Plex™ array | + | 42, 43, 44, 45 |
| Aushon | Ciraplex® | – a | |
| Meso Scale Discovery | MULTI–ARRAY microplate | + | 17, 45, 46, 47, 48, 49, 50 |
| QuantiScientifics, LLC | A2® Multiplex ELISA Human Cytokine Panel | – | |
| eBioscience | FlowCytomix™ Multiplex | + | 51, 52, 53, 54, 55 |
| BD Biosciences | CBA Human Th1/Th2/Th17 Kits | + | 56, 57, 58 |
| Illumina | VeraCode™ | + | 59 |
Commercially available multiplex arrays that have demonstrated neither clinical viability/applicability nor compliance with regulatory body approval parameters but nonetheless may theoretically be applicable for clinical applications are designated with a ‘–’ in this table and Table 1.
Manufacturer claims validation, although no supportive published data are provided.
Figure 3.
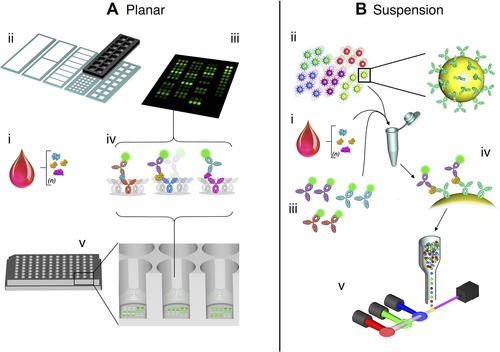
Multiplex formats in common use include planar‐based assays or suspension‐based assays. (A) Planar arrays can be produced in two formats, either slide based or microtitre based. A common starting point for such assays, as with ELISA, would be a serum sample extracted from blood (i). Unlike microtitre plate based formats, slide‐based formats support numerous layouts whereby repeated or individual assays composed of specific sets of antibodies are printed robotically upon the activated slide surface (ii). The sample matrix is applied and discrete assays are spatially separated by a frame and gasket, whereby they may be subsequently treated as individual microtitre wells, subject to blocking, washing, etc. Internal standards and replicates may be included also (C). Detection results from application of a composite of specific secondary antibodies coupled to a chemiluminescent/fluorescent reporter systems (iv). Microtitre‐based immunoassays harbour regularly printed antibody sets within the confines of the wells of a standard (SBS format) protein‐binding plate (v). The plate may thus be treated akin to a conventional ELISA (i.e. blocking, incubation and washing followed by detection with a set of reporter‐conjugated detection antibodies). (B) Suspension immunoassays also have a common starting point – serum sample extracted from blood (i). This assay employs thousands of micrometre‐sized plastic microbeads infused with a single (or several) chemiluminescent/fluorescent dyes and a functionally activated surface, prior to linking with a specific capture antibody. Numerous sets of such beads are prepared, each maintaining separate capture antibodies according to the cognate analyte and a unique fluorescent signature enabling identification (ii). The sample and a cocktail of all the requisite bead sets are thereafter combined. Sets of detection antibodies, all of which are individually labelled with a single chemiluminescent/fluorescent reporter (separate from those contained within the beads) are added upon completion of incubation and washing stages (iii). Each bead thus accommodates a ‘sandwich’ consisting of the captured target analyte and the cognate reporter‐conjugated detection antibody (iv). Post‐additional washing stages, bead analyte reporter constructs are subject to analysis in a flow chamber implementing individual bead separation, whereby lasers excite the chemiluminescent/fluorescent reporters and emitted light is collected by a series of detectors for quantitative analysis (v).
4. Development of multiplex immunoassays
Generically, multiplex immunoassay development has been considered in the context of two primary arenas: the detection platform itself and the immunoassay chemistry. The initial platform configured for high‐throughput proteomic screening was the slide‐based planar or ‘chip‐based’ microarray, offering the prospect of whole proteome analysis via a single sample. According to the ambient analyte theory, planar immunoassays may exhibit a theoretical resolvable specificity within the femtomolar range; however, few systems actually achieve such specificities in practise primarily due to the physiochemical behaviour of the analytes in the sample matrix, particularly the depletion of analytes within the vicinity of the capturing spots resulting in a mass transport controlled (as opposed to a kinetically controlled) reaction. Appropriate sample mixing may ‘reset’ the concentration gradient of the analytes around the capture zones to ultimately increase the sensitivity and efficiency of the immunoassay, although such manipulation of small volumes is technically problematic and, for IVD settings, would likely require a high degree of integration between the immunoassay and a fluidics system, highlighting the necessity for robust automation 63. Despite such issues, planar immunoassays are somewhat less susceptible to cross‐reactivity as capture and detection antibodies are co‐localised in precisely aligned spots and dispensed in microliter volumes in air onto an immobile solid phase. Appropriate buffers to ensure the viability of antibodies or proteins deposited on the array surface during storage is an important consideration. Successful immobilisation of hundreds of different proteins in their native conformation on single glass slides represents a considerable technological challenge. Continuous flow microspotter systems and other advanced array printing technologies 5 may minimise such issues but require specialised, non‐standard laboratory apparatus which, at least as of present, is unrealisable in a majority of clinical and research settings. Nonetheless, planar immunoassays maintain the promise of easy integration into existing IVD settings as they are relatively inexpensive and would be simple and efficient to operate if fully automated, thereby accommodating large‐scale population screening (Fig. 4A and B).
Figure 4.
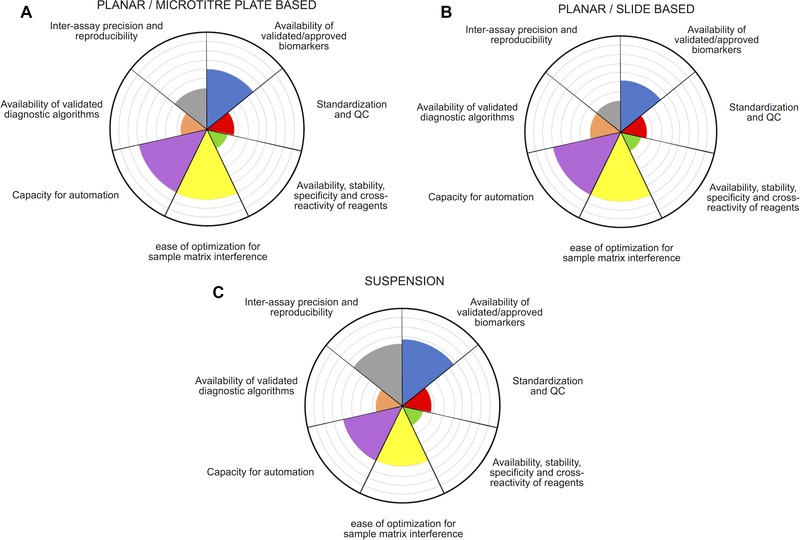
Radar plots depicting the developmental stage of components related to multiplexed assay systems. Each segment represents one of the cardinal, inter‐related requirements associated with validation of multiplex immunoassays for routine diagnostic application. (A) Planar, microtitre plate based immunoassays; (B) glass slide based immunoassays and (C) suspension bead based immunoassays. Accordingly, progress/research towards resolution of a particular aspect results in the corresponding segment filling outwards from the centre, where maximum segment coverage represents a satisfactory standard for routine application in point‐of‐care/diagnostic settings. A complete fill for all segments would represent an exacting qualification of an immunoassay format for unanimous integration into routine point‐of‐care/diagnostic settings.
The origins of suspension immunoassay technology date back to the 1970s when Horan and Wheeless 64 recognised, as part of a review describing some of the diverse applications of flow cytometry, that micro‐particles harbouring antigen could constitute a solid support for the capture of blood‐borne antibodies; moreover by adopting differentially sized microspheres distinguishable by their light scatter properties, several analytes may be detected simultaneously. Subsequently, in the 1980s and 1990s, this approach was extended to include antibodies, DNA and lipids 65. Accordingly, the development and application of suspension immunoassays closely parallels that of flow cytometery as a detection platform, whereby advancements in the latter (particularly the improvements in capacity to make sensitive and multi‐parameter fluorescence measurements at high speeds) has been exploited for in vitro analysis of molecular interactions on microspheres and for the analysis and sorting of biological libraries in single cells. Currently, the suspension immunoassay format constitutes the prevailing technology for FDA‐approved multiplex protein analysis in clinical settings 66, chiefly for serum antibody profiling in patients with allergies, autoimmune or infectious diseases. Notably, its success is attributable in part to its surmounting of some of the issues faced by planar immunoassays, such as printing and washing artefacts, post‐hybridisation image analysis, data normalisation and mass‐transport limitations, which are abrogated by the conduction of the immunoassay in a liquid phase whereby agitation is readily executed. Additionally, suspension immunoassay platforms, being flow cytometry based, are highly amenable to automation whereby calibration and quantification are all automatically and integrally performed. Furthermore, replacement of conventional polystyrene bead suspension immunoassays (of which laboriously necessitated several manual washes to prevent clogging) with magnetic bead based immunoassays enabling separation during washing steps has also significantly improved the automation capacity of suspension immunoassays within the past 5 years (Fig. 4C) 67. Nonetheless, while these assays have a high theoretical multiplexing capacity (up to 100‐plex) – a desirable feature for comprehensive disease profiling in clinical settings – in practice, cross‐reactivity of capture and/or detection antibodies may compromise readout viability. The susceptibility of multiplexed sandwich/capture‐based assays in a liquid phase to cross‐reactivity increases quadratically with the number of targets 68; thus, if antibody reagents are altered or additional biomarkers are added to the panel, the power of the assay may be undermined and the entire optimisation process must be repeated. However, perhaps the most tangible barriers to integration of suspension immunoassays into routine IVD settings are those imposed by the expenses and technical demands of operation (i.e. maintenance costs and competency requirements in flow cytometry of clinical laboratory staff).
5. Validation of multiplex immunoassays
The Clinical Laboratory Improvement Amendments (CLIA), Food and Drug Administration (FDA), MHRA (Medicines and Healthcare Products Regulatory Agency) and United Kingdom Accreditation Service (UKAS) as well as other regional regulatory bodies require validation of novel or modified multiplex assays prior to their enlistment in routine clinical diagnostics for the generation of legitimate patient results. Despite the fundamental requirement to validate these technologies on the basis of their demonstration of an acceptable level of performance and robustness, specifically according to metrics such as inter‐ and intra‐assay reproducibility, analytic sensitivity and specificity, whilst addressing LOD and LOQ and reportable reference ranges/quality control standards. Currently no standardised regulatory guidelines for the validation of multiplex biomarker assays exist 69. Device clearance or approval typically rests on the capacity of the sponsor to provide analytical and clinical data demonstrating that device performance is ‘functionally equivalent’ to that of an alternate and well‐established technique thereby inferring adequacy with regard to its intended utility 70. While this is cogent when comparing the reliability and practicality of a technique with similar chemistry and reaction dynamics, it presents complications for techniques with clear differences in their operating principles. Furthermore, validation continues into clinical practice and must be continually monitored to ensure an assay operates as expected and achieves the intended results. In clinical practice, validation includes proficiency testing, assessment of employee competency, instrument calibration and correlation with clinical findings. Taken together, it will likely be difficult for clinical laboratories both to validate these assays on their individual analytical platforms and to demonstrate clinical relevance by performing large‐scale preclinical/clinical studies, which may need to involve multiple laboratories 69. Consequently, establishing a standardised evaluation paradigm should help ensure the highest level of performance within and across laboratories so that the tests provide patients with the most accurate results. Quality control materials are not well developed for multiplexed protein immunoassays, and algorithms for interpreting multiplex quality control data are also needed 66 (Fig. 5).
Figure 5.
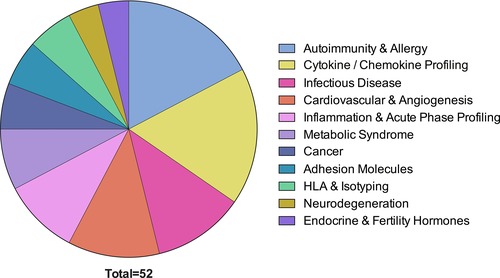
Pie chart representing relative prevalence of validated commercially available multiplex assays according to clinical application. Please note that most of these assays are currently validated for research use only, implying that they have not been validated to specific FDA or other national/international clinical standards. The largest numbers of commercially available assays are for autoimmunity and allergy and cytokines/chemokines.
5.1. Biomarker validation
Prior to the inclusion of a particular biomarker into a multiplex panel adopted for routine diagnostics, a developmental course commencing with its origination and culminating in its clinical validation will have transpired. The marker must demonstrate its correlation with specific pathophysiological processes and clinical endpoints, as well as diagnostic reproducibility and accuracy, given the analytical method. Either or both of these fundamental prerequisites may present barriers to the clinical validation of a multiplex assay. For example, hundreds of biomarkers have been identified (or proposed) for complex diseases such as cancer, yet only a relative minority are clinically qualified, with the majority bearing either unknown or incompletely characterised clinical significance. Others, while theoretically suitable, may not exhibit consistent and/or sufficient expression to warrant inclusion as an assay modality. Ideal candidates for multiplex recruitment would be markers whose qualitative and/or quantitative expression is unique to the disease, however, particularly in the case of cancer, identifying truly specific markers has been somewhat problematic. Many of the markers that were previously deemed to be ‘tumour specific’ such as MAGE–A3, have since been shown to occur in healthy tissue (albeit diminutively) and those that are truly specific (i.e. abnormal products of ras and p53 genes) are in their own right common to several cancers and thus do not constitute a differential basis for different forms (i.e. rarer subtypes) of a single cancer.
The technical validation of a biomarker within the context of an assay is a complex process that is interdependent on all facets of the respective analytical method including assay sensitivity, specificity, reliability and reproducibility as well as the nature of the biological sample and its integrity 71. Specificity corresponds to the ability of the analytical method to decisively distinguish the target biomarker from structurally similar components within the same sample. Selectivity corresponds to the extent of analytical interference posed by irrelevant sample components. Reliability and reproducibility determine the overarching precision of the analytical method. The nature of the sample must also be considered with regard to the locations of expression and distribution of the respective marker; the most appropriate sample for a given marker would ideally accommodate sufficient analyte concentrations with minimally invasive accessibility (i.e. urine, sputum, faeces, saliva, serum, plasma or whole blood). Accordingly, the physiological source(s) of a marker may also influence its validation within the context of a multiplex immunoassay as evidenced by the feasibility of obtaining the sample. Should the analytical method exhibit sufficient sensitivity, then the choice of sample is determined by the ease of sample collection and analysis. However, if sensitivity constitutes a limiting factor and measurement of the marker in the specified biological fluid is problematic, then the sample type is selected according to marker concentration regardless as to whether this presents as a greater challenge for sample collection and preparation 71. The cardinal challenge facing biomarker validation is characterisation of a marker, which is sufficiently present in a minimally invasively acquired and stable sample as so to be readily detectable by all multiplex immunoassay formats with variances in specificity and dynamic range and most importantly of all: reliably correlates (either individually or concomitantly) with a clinically meaningful endpoint.
5.2. Standardisation of immunoassay design and quality control (calibration and quantification)
On account of their ability to measure several analytes simultaneously, multiplex immunoassays present considerably more quality control hurdles compared to singleplex arrays 72. Despite the necessity for extensive and transparent quality control standards in IVD and point‐of‐care (POC) settings, implementation into multiplex immunoassays is still underdeveloped thereby presenting a major and uncompromising obstacle to widespread application of the technology in clinical settings. This is primarily due to technicalities and discrepancies in immunoassay design; firstly the generation of robust, meaningful calibration curves for a panel of heterogeneous markers under a common, localised set of conditions (i.e. a ‘one size fits all’ problem). A pivotal phase in immunoassay development includes adjusting calibration curves so as to reflect the physiological ranges for each marker. The quantitative accuracy of multiplexed assays hinges on the quality of the generated calibration curves, which in turn is determined by appropriate curve‐fitting procedures, assay imprecision (CV), analyte recovery and assay linearity (LOQs) 17. Within IVD settings, the performance of analytical tests is constantly monitored adopting well‐characterised quality control materials containing known concentrations of analytes; typically, for protein assays this encompasses reference samples of low, medium and high analyte concentrations at the commencement of a period of usage 66. However, selection of a single sample dilution factor, which supports measurements within the physiological ranges for each constituent marker on a panel, is often unfeasible, necessitating the disjoining of the panel based on circulating protein abundance. The failure of one constituent marker to meet quality control specifications may necessitate the rejection of results for all remaining markers on the panel 72. Optimisation of a communal format (which would also facilitate automation) that functions aptly for each constituent marker (e.g. sample dilution factor, capture/detection system, incubation times and washing steps) constitutes a major challenge to standardisation of immunoassay design. Thus, the probability of all assays within a multiplex panel simultaneously meeting quality control specifications is considerably lower compared to that of a singleplex assay. The generation of viable quality control paradigms, which are applicable across different analytical methods/platforms would be advantageous for assessing inter‐assay precision and ensuring multi‐centre quality assurance. However, such controls would also require standardisation of immunoassay design. Notably, quality controls are adopted for the establishment of assay range and are therefore distinct from calibration standards; thus, at the very least quality controls utilised to validate an assay should be prepared adopting the cognate sample type (and ideally derived from a discrete trial population) 73. Preparation of quality controls in buffers appears to be suboptimal, as endogenous markers are not as adequately represented compared to de facto samples, thus undermining accuracy and precision data reflecting assay performance. However, merely adopting a biological sample by itself is in no way sufficient as indicated by several studies reporting higher inter‐assay variability. Chowdhury and colleagues 46 reported high intra‐assay reproducibility and even comparable inter‐assay precision between Luminex and Mesoscale Discovery technologies when measuring high target analyte concentrations, but for measuring low concentrations, inter‐assay variation was variable and analyte dependent. Additionally, Bastarache and colleagues 74 reported acceptable intra‐assay variability but unacceptably high inter‐assay variability when assaying identical patients’ samples on different days adopting the Searchlight platform. Accordingly, generating a robust set of quality controls for clinical diagnostics applicable to different assays/platforms presents a significant technical challenge and currently quantitative comparison of samples between different platforms is markedly inconsistent. Currently, ascertaining quality control over several assay cycles laboriously involves calculating representative metrics for each standard curve per cycle, plotting the attained metrics and expected ranges on Levey–Jennings charts to define outliers, excluding data from analysis based on quality metrics and identifying trends that require further investigation 75. A lack of robust multivariate quality control algorithms and data rejection criteria for multiplexed immunoassays is also problematic. This is primarily due to the fact that the higher throughput of multiplex immunoassays necessitates large sets of controls; for planar immunoassays this typically encompasses spotted positive and negative controls, blank areas, spotted detection conjugates, controls for non‐specific binding or cross‐reactivity assessment and normalisation 76. Better statistical methods and quality control algorithms are warranted to correct acquired data for analytical variability in multiplex measurements. Encouragingly, standardisation of data reporting, common analysis apparatus and meaningful controls are developing under the Microarray Quality Control Project for genetic testing to enhance statistical confidence in the consistency and reliability elicited by platforms analysing gene expression 72. Initiatives for implementing sustainable reference standards required for performance validation efforts and quality control are progressing and translational application of similar initiatives for multiplexed protein immunoassays would be constructive. Taken together, the emerging picture illustrates the interdependency of all aspects of multiplex development; for example, markers must be amenable to assembly into panels under a set of common conditions to support comparison with each other plus a robust set of calibration and quality control standards. Thus, biomarker validation and quality control are intricately linked at this stage in time.
5.3. Availability, stability, specificity and cross‐reactivity of reagents
Arguably the greatest impediment to integration of multiplex immunoassays into IVD settings as of present is the availability of high quality (i.e. stable and specific) antibodies to generate reliable output data and minimise cross‐reactivity 77. This encompasses a range of manufacturing challenges from the generation and characterisation of capture ligands, antibody immobilisation chemistry, variability in the assay manufacturing process and sample/reagent‐derived cross‐reactivity. Conventional multiplex sandwich immunoassays are conducted by incubating microbeads or a microarray with a biological sample, followed by the addition of a mixture of detection antibodies, with the premise that each antibody will bind its cognate target analyte secured by the corresponding capture antibody. Thermal or mechanical agitation ensures that each reagent component encounters its target. It also results in combinatorial interaction of each detection antibody with all other analytes, capture antibodies and other detection antibodies thus enhancing opportunities for cross‐reactivity 77. Accordingly, suspension immunoassays are theoretically more susceptible to cross‐reactivity as microbeads circulating in a fluidic phase facilitate cross‐linking between protein components. Cross‐reactivity may compromise assay performance by reducing the LOD thus increasing the probability of misrepresentative results. The quality and reproducibility of multiplex immunoassays hinges on the availability of highly specific capture ligands to bind tightly to target analytes. However, predicting high‐affinity interactions between proteins is complex due to the hydrophobicity and heterogeneity in size of the constituent amino acids. Currently, monoclonal antibodies generated by classical hybridoma techniques constitute the most widely utilised capture ligands 66. Large‐scale production and characterisation of affinity, specificity, cross‐reactivity and kinetic parameters (namely association and dissociation rates) are labour‐ and cost‐intensive processes hindering validation; furthermore, antibodies validated for singleplex assays may exhibit cross‐reactivity when adopted in a multiplex format, indicating a necessity for application‐specific validation of antibodies.
Antibody chemistry must also be considered in the context of immobilisation for the purposes of maintaining immunoassay accuracy and reproducibility. Antibodies are relatively stable in comparison to other proteins yet even minimal denaturation may expose hydrophobic regions predisposing non‐specific binding and potentially altering the lower LOD. Generically, optimal antibody immobilisation chemistry necessitates the retention of a high affinity for the target analyte, a high S/N and low variability between produced lots. Specific immobilisation parameters affecting the performance of planar immunoassays include total antibody binding capacity, size and morphology of antibody spots, background signal, LOD and printing reproducibility within and over multiple immunoassays. For suspension immunoassays, standard immobilisation protocols such as the adoption of surface immobilised protein molecules recognising specific antibody domains, chemical modification of carbohydrate residues in the Fc regions or surface activity towards free thiol groups may conceal epitopes containing extensive lysine or arginine residues thereby resulting in failure of an antibody to consistently recognise its cognate target analyte. Accordingly, the surface chemistry for multiplex protein immunoassay formulation would ideally offer the following parameters: resistance of the surface to non‐specific adsorption; sufficient availability of functional groups for ready and uncomplicated immobilisation of target molecules; balanced bonding between ligands and the immunoassay surface to ensure sufficient stability yet minimal interference with the conformational structure while enabling regulation of orientation; a local chemical microenvironment conducive to the retention of immobilised ligands in their native conformation and finally, highly specific linkage chemistry to abrogate the necessity of pre‐purification of ligands 78.
Variation in immunoassay manufacturing processes also confers imprecision; for planar immunoassays, printing constitutes the single greatest determinant of variability, whereas for suspension immunoassays, discrepancies in microsphere production principally account for signal variability, whereby 10–32% of measurement variability may be attributable to inconsistencies in microsphere diameter per se 66. For RP protein array, in contrast to capture‐based immunoassays, a fundamental manufacturing consideration is the heterogeneity of an immobilised sample and thus the absolute requirement for validated, highly specific antibodies to eliminate false‐positive signals. Taken together, the requirement for complex antibody cocktails (both bead/slide associated and reporter conjugated, all of which must be highly specific and sensitive to attenuate cross‐reactivity within the sample set) in conjunction with lagging quality control standards will likely remain the foremost limitation to extensive multiplexing and IVD integration for some time. Given the limitations of antibodies, evaluation studies for alternative capture ligands including engineered protein and nucleic acid scaffolds are ongoing; aptamers are one such candidate, which like antibodies possess target recognition features with the capacity to distinguish between protein isoforms and conformations. Other approaches include refinement of the immunoassay chemistry itself; a recent and elegant approach by Frampton and colleagues 5 was the conception of an aqueous two‐phase system whereby phase separation promoting polymers PEG and dextran were adopted to confine detection antibody solutions in a fully aqueous environment to regions where complementary capture antibodies were immobilised. At length, continuing initiatives in the USA and Europe are focused on improving the availability and quality of affinity reagents, with the overarching aim of devising standardised collections of comprehensively validated binding molecules for proteomic analyses for the advancement of immunoassay quality standards 79.
6. Considerations for embarking on multiplexed immunoassays
When undertaking the use of either commercially available multiplexed systems, or construction of in‐house systems, it is important to ensure the system has usable intra‐ and inter‐assay reproducibility. Inter‐assay CVs of <20%, and usually <10% are a reasonable expectation of most ELISA‐based systems, and should be similarly expected of all assays within a multiplex. Intra‐assay CVs of <15%, and usually <10% should be expected, however some commercial assays make this difficult to assess due to the lack of appropriate numbers of technical replications of each target within the multiplex (i.e. if only duplicate features are used for each target). Studies where bead and planar multiplex immunoassays have been compared indicate that inter‐assay CVs of between >2.8 and <10% can be routinely obtained, however they are analyte dependent 17.
In our experience, there is little limitation on space for planar multiplexed microarrays – the small size of individual features (spots) and the fact that arrays can be re‐sized easily, allows for substantial technical replication per assay. We routinely use triplicate, quadruplicate or greater numbers of replicates for each target or control within the multiplex. This far surpasses usual practice in ELISA, where triplicates or sometimes only duplicate wells of each sample are processed.
Building sandwich immunoassay multiplexes from scratch would necessitate cross‐testing the entire multiplex assay against each target antigen alone to determine the extent of any cross‐reactivity seen. Specific issues to consider when approaching the use of such multiplexed assays, whether in‐house manufactured or commercially obtained, are reviewed below.
6.1. Data acquisition requirements
Access to a suitable high‐resolution scanner or bead reader, or availability of a scanning/reading service to which the multiplexed assay can be sent once completed, is a key priority. If sending processed multiplexes to a service, methods for stabilizing fluorescent signal should be addressed. Commonly used red fluorescent reporters often show strong ozone sensitivity, resulting in rapid signal loss if adequate precautions are not taken 80. Dyes based upon FITC can show photo‐bleaching, and suitable precautions to shield from light prior to analysis should be addressed.
Choice of fluorescent wavelengths within the reporter is beyond the researchers control for most commercial offerings; however, choice of fluorophor is important, especially for membrane‐ and nitrocellulose‐coated glass planar arrays. Nitrocellulose‐coated surfaces have a high protein binding capacity, but fluoresces intensely in the blue (∼488 nm) to green (532 nm) spectrum when laser illuminated, leading to high background signals and low S/N. This autofluorescence is diminished in the yellow (590 nm) and red (635 nm) spectrum, and is virtually absent in the near infrared range (680–800 nm range). In addition protein autofluorescence, which varies between proteins, can be strong in the blue to green region of the spectrum, as is often seen in flow cytometric studies. Thus, red and infrared fluorophors are preferred choices for reporter dyes. In contrast to nitrocellulose, other surface treatments of glass to enhance protein binding (such as reactive epoxy‐silane or aldehyde functionalisation) generally give low fluorescence, but also have a lower binding capacity. For bead‐based assays, fluorophor choice is often dictated by the analyser's capabilities (flow cytometer or dedicated machine).
6.2. Dynamic range of readings
Analysis of conventional ELISA‐based assays is usually by optical absorbance readings per well, using a plate spectrophotometer; this gives a range of 0–3.5 (possibly 4) optical density units, dependent on the reader. Far greater dynamic range can be achieved using fluorescent or luminescent techniques for ELISA, necessitating the use of black‐ or white‐walled microtitre plates to avoid light spill between wells. Equally, luminescence or fluorescence is method of choice, over colorimetric detection, for planar arrays and fluorescence is the only practical choice for bead‐based systems. Both techniques can deliver dynamic ranges extending over five orders of magnitude, dependent upon the detection system's sensitivity. Since these methods result in light output being converted to a digital signal, the maximum signal depends upon the bit depth of the detection system: 8 bits gives 256 levels; 16 bits, 65 536 levels and 24 bits 16 777 216 levels. It is therefore important to know what the data acquisition hardware is capable of in order to avoid limiting signal range unnecessarily. Currently, 12 and 16 bit/pixel scanners represent the mainstream technology, with some 24 bit scanners beginning to appear on the market.
Even within available scanners for multiplex immunoassays, software and hardware capabilities are important considerations. For critically sensitive planar microarray‐based assays, a scanning system that can average or additively merge two or more scans of the same array can be highly beneficial. Digital noise in such systems is random, within the design of the optical detector's specification. Thus, averaging several images of the same array can lower background signal significantly, thereby improving low range detection limits of real signal. Optical resolution is also important – higher resolution imaging leads to an increased number of pixels per feature, aiding quality control measurements and enabling better alignment and identification of features.
Similarly for bead‐based systems, dynamic range within the bead detector's optical path is critical to support good bead cluster separation, and to support a wide dynamic range for the assay itself. PMT settings have the potential to significantly affect the dynamic range and assay linearity [81].
6.3. Quality control
Unlike ELISA‐based assays, where an aberrant signal in a single well cannot be assigned to a particular problem with that well (i.e. failure to coat, uneven coating, bubbles or contamination with debris, etc.), both planar and bead multiplex systems support improved quality assessment.
6.3.1. Planar multiplex assay quality control
For planar arrays, high‐resolution imaging supports considerable additional quality control information. Given appropriate software (we routinely utilise Molecular Devices Genepix Pro software), alignment of the data grid (i.e. where the arraying robot thinks the spots are) with the actual image then allows the software to locate the outline of each spot. Subsequent analysis provides numerical data including feature diameter; circularity (100 being a perfect circle); pixel number in the feature; mean, median and summative pixel intensities within the feature and for local background signal; percentage of pixels, which are saturated in the feature; percentage of pixels above 1 and 2 standard deviations for the local background of that feature; S/N and other, user defined data, dependent on whether one or two colour fluorescent detection is used.
Mathematically defined exclusion of aberrant spots within a microarray‐based multiplex can therefore improve assay CVs dramatically by removing replicates where the printing of the feature itself, or subsequent treatment, has caused a quality control issue. Exclusion criteria may include features that show circularity scores <65%; features that are smaller or larger than the mean diameter ± 2 SDs; features for which <5% of the pixels are saturated; features in which the percentage of pixels above the background +1 SD is >60%. Such screening flags the following for removal: poorly printed features (i.e. a bad spot morphology, either not circular or smaller/bigger than expected QC limits), saturated features (no longer in the quantitative range, or with a fluorescent speck of debris attached); very low‐level signals. Comparisons of median and mean feature pixel intensities can be used to identify features where the coating of the spot is uneven (i.e. such as forming a doughnut image due to inappropriate drying of the material on the array substrate).
It is not unreasonable to expect commercial suppliers to be able to give such information, based upon testing of each particular batch of prepared multiplexed assays, if array based. This would include expected lower LODs for each analyte, if following the manufacturers guidelines with a control sample; this control sample should be available from the manufacturer. In addition, information detailing the expected diameter of all features when imaged and data from a positive control test to indicate expected feature quality, that is intra‐assay replicate CVs, etc., would be useful for comparison to the user's own results. Similarly, if manufacturing in‐house multiplexed planar assays, such quality control tests are an integral part of the production process and during routine usage. Within our own production of multiplex protein arrays, we expect spot diameter CVs to be below 5% for any given target printed and circularity CVs <2.5% (<4% and <1.5%, respectively, is routinely possible). Individual proteins within features can affect surface tension of the spotting liquid and give rise to small differences in spot diameter between different targets. However, diameter CVs of below 10% across all targets on an array are common, with the individual target replicates feature diameter CVs being <5%.
6.3.2. Bead‐based multiplex quality control
Bead‐based immunoassay multiplexes resolve the problem of individual assay replicates, with many hundreds to several thousand beads reporting on each captured analyte. Key quality control features therefore reside in the ability to distinguish bead clusters based upon their inherent fluorescence, variation in bead fluorescence, in relation to the bead reader's capabilities, can therefore make separation of high‐level multiplexes a challenge. Like many fluorescent products, most immunoassay beads are light sensitive, and should be appropriately protected to minimise variation in individual bead and run‐to‐run fluorescence. In addition, if used as a one‐step immunoassay, bead‐based systems can suffer from loss of signal at high analyte exposure due to the long‐established hook effect 82. The hook effect is a consequence of very high analyte levels in a sample, whereby analyte binds to both capture and detection antibodies, but saturating the binding sites so that a capture analyte detection sandwich does not form, resulting in decreasing signal with increasing analyte concentrations. In common with other immunoassays, heterophilic antibody interference can also provide false‐negative and false‐positive results 83. Heterophile antibodies (those that cross phyla in their reactivity) are present in up to 40% of blood donors, and show low avidity and multispecies specificity [84, 85]. False positives arise where the heterophilic antibodies bridge capture and detection antibodies in the absence of analyte. False negatives occur where the capture or detection antibody is directly bound by the heterophilic antibodies and blocked from binding to the analyte. The performance of the system across a range of analyte concentrations spiked into real samples is strongly recommended.
6.4. Local assay validation
Validation of the reproducibility and utility of the multiplex system for the particular sample types anticipated is prudent, as application to cell supernatants, tissue homogenates or biofluids, depending on the assays performed, are likely to cause significant differences in assay behaviour relative to the manufacturers recommended usage.
For both commercial and in‐house multiplex assays, deviation from the standard sample type and operating procedure would need careful reassessment of the control samples and assay limits. Using samples with spiked‐in targets in appropriate biofluids and determining accuracy of recovery is of clear benefit in determining accuracy and precision of an assay; however, the additional cost of performing such validation controls can be substantial. Particular immunoassay targets, especially low abundance targets, often yield highly variable results near the lower end of the sensitivity range. Reasons for this variation may include heterophilic antibody interference 83; substantial sensitivity of the target molecule to epitope loss due to denaturation; masking by other sample components; degradation or micro‐heterogeneity of the target within the sample. Given an otherwise standardised assay and sample collection procedure, such interfering factors may be beyond the researcher's ability to control, although there are methods for reducing heterophilic antibody interference 86.
While examples in the literature are rare, comparisons between platforms have been reported. For instance Rhyne et al. 87 performed a comparative study for amyloid plaque peptides and reported a Mesoscale Discovery‐based triplex assay producing intra‐assay CVs of between >4 and <15% dependent on the target; and inter‐assay CVs were generally <20%, but with certain targets giving high CVs (up to 80%). In the same report, a triplexed luminex bead‐based assay, AlzBio3, gave intra‐ and inter‐assay CVs of <20%, but poorer accuracy with the supplied kit standards, with between 20 and 50% accuracy. Sensitivity of both platforms was reported as satisfactory. The conclusion of the report is that in‐house testing and validation of commercial kits is recommended prior to beginning a study, and that validation findings should be published alongside the study findings. Further reports include accuracy of a specific IgE multiplex (the ISAC system; a direct ELISA‐type approach) that cites inter‐assay CVs of <10% for most antigen targets using a calibrator kit and <22.9% for patient samples, with most being below 15%, but up to 40% for a small subset of antigens 88, 89. In a further study of sandwich‐ELISA style microarrays for biodefense toxin detection, the authors state that over 70 antibody pairs were examined to produce a final set of 10 pairs that gave good accuracy (75–120% recovery) and inter‐assay CVs of <20% 90.
As a final note on method validation, the FDA handbook, ‘Guidance for industry: Bioanalytical method validation guidelines’ 91 suggests that whilst 15% inter‐assay CVs are acceptable for most calibration tests, this range may be relaxed to 20% CV at the lower limit of quantification.
7. Assay automation and the use of validated algorithms for transformation of raw data into diagnostic results
In addition to optimisation of immunoassay performance, robust and extensive automation in conjunction with analytical software development is an inescapable prerequisite for full integration of multiplex immunoassays into clinical diagnostic settings for management of labour‐intensive work flows, variation between different instrumentation platforms and amelioration of the necessity for high degrees of operational competence. Given the theoretical throughput of multiplex immunoassays, the analytical scope is also correspondingly complex and must be carefully tailored according to required diagnostic endpoints. For example, at the simplest degree multiplex immunoassays may be adopted to generate a single qualitative output such as the typing of a pneumococcal species or evaluation of antibody titres for vaccination status whereby a single result is expected; thus multiple positives or a single negative result clearly equates to test failure. The subsequent degree of complexity involves immunoassays yielding patterns of possible outputs, all encompassed by a defined disease category but with marginally different clinical implications. Therapeutic or prognostic validity established on the basis of such immunoassays may vary according to the individual analytes present, necessitating the disclosure of some or all constituent immunoassay outputs as opposed to a single qualitative result; this is typically the case with autoimmune multiplex immunoassays. The greatest degree of complexity subsists whereby individual outputs do not bear any diagnostic significance, but the combined outputs of numerous assays are processed by a pattern‐recognition algorithm to yield a single diagnostic result 92. Such analytical capacity presents the greatest challenge to clinical implementation, but in turn entails the potential for detailed disease phenotyping and the realisation of personalised therapeutics for diseases such as cancer. One of the first clinical diagnostic tests to adopt pattern‐recognition algorithms was a fully automated HPLC drug screening system (the REMEDi HS) produced by Bio–Rad in 1989. Over the intervening two decades, a variety of interpretive tools have been recruited for analogous exploratory studies with DNA microarrays, including various clustering modes (i.e. self‐organising maps and hierarchical clustering) along with supervised classification algorithms (i.e. K‐nearest neighbours, support vector machines and neural networks). Nonetheless, as with all operational aspects of multiplex immunoassays adopted for IVD, integration of pattern‐recognition algorithms entails corresponding regulatory burdens (i.e. FDA/MHRA, etc., approval), which must be contented prior to marketing. Currently, the only FDA‐approved pattern‐recognition algorithm adopted for proteomic microarrays is the K‐nearest neighbours classifier utilised by the BioPlex 2200 ANA Screen assay. Encouragingly, however, once validated, supervised classification algorithmic approaches are likely to be extensively adopted for clinical applications. A significant contemporary obstacle to algorithm validation is the development of criteria for interpretation of combined quality control data in conjunction with additional appropriate criteria to verify the acceptability of the generated analytical results. For example, whereby the immunoassay returns outputs for all individual analytes within the predefined limits established by the quality control material, there is no issue. However, should one or more of the analytical results for the quality control materials not fall within the predefined control limits, it becomes less straightforward as to how the total immunoassay should be managed. This at length raises another issue as to the management protocols of unrequested test data whereby the number of measured analytes substantially exceeds the requirements as indicated by a given clinical situation. Ironically, although multiplex (particularly microarray) technology may ultimately demonstrate cost efficacy for batch analysis, there is currently no consensus as to how unrequested outputs should be stored and/or reported. While the capacity to retrospectively report unrequested analyte measurements may prove convenient for investigatory studies whereby the availability of large datasets for subsequent mining may be advantageous (i.e. clinical trials); how such capabilities would translate into IVD settings is unclear as the storage of unrequested patient test data without disclosure may feasibly raise legal issues in cases whereby such results would significantly alter diagnosis or treatment.
8. Future directions
It is inevitable that many current singleplex assays will ultimately be replaced by multiplex alternatives. For both the clinical and research immunology laboratory, multiplexing immunoassays promise considerable savings in both time and cost per assay. At the same time such multiplexing offers more comprehensive analysis whether for research purposes, differential diagnoses or monitoring of therapeutic interventions. However as we have stated during this review, widespread adoption of such technologies is dependent upon the availability of robust, affordable analytical platforms and validated multiplex‐optimised antibody panels. For clinical use, high‐level automation is key to widespread future use, whereas for the research laboratory, affordability of the analytical platform may be a prime consideration.
Multiplexed immunoassays that could be reduced to a disposable, relatively cheap POC assay offer great potential for improved health surveillance such as GP‐led routine periodic health‐care screening such as panoptic scanning for potential autoantibodies, infectious disease and markers of early onset chronic diseases such as diabetes, Alzheimer's disease and rheumatoid arthritis, requiring little in any user skills, perhaps though microfluidics‐based lab‐on‐a‐chip type approaches, and be able to extract a sample from perhaps a single finger‐prick of blood, possibly undertaking the separation of plasma from cellular components within the device itself. Such future screening assays are also utterly reliant on the availability of informative biomarkers.
Such POC devices might also be of great value for rapid monitoring in intensive care units, or where speed of response is critical, for instance in cases of suspected sepsis [93]. The potential economic value to the healthcare system of community clinic screening for common conditions to identify early disease and thus initiate early treatment and management should not be underestimated.
9. Conclusion
All multiplex immunoassay formats to date present a powerful and efficient means of charting the proteomic and phenotypic changes associated with a diverse range of pathophysiological conditions. Moreover, since the advent of the proteomic era, multiplex immunoassays now constitute invaluable tools for efficiently harnessing the wealth of information available to expedite more holistic observations, improved monitoring and treatment of disease in the future. While the availability and implementation of commercialised multiplex immunoassays for research applications is expanding rapidly, incorporation of the technology within routine clinical diagnostics is still several years away at least due to the extent of operational and quality control issues such as immobilisation strategies for samples/binding agents, cross‐reactivity and availability of standardised reagents, robust automation to minimise time and operational costs. Equally, there is the issue of implementing appropriate algorithms and directives for management of the informatics challenge posed by systems capable of producing millions of assay results per day. Accordingly, future directives towards the realisation of routine multiplex immunoassays in IVD, besides ongoing technological improvements, include improved reproducibility and consistent reductions in the CV between results from different immunoassays/platforms in addition to new statistical developments to correct multiplex data for analytical variability.
The authors have declared no conflict of interest.
Colour Online: See the article online to view Figs. 4 and 5 in colour.
10 References
- 1. Vanmassenhove, J. , Vanholder, R. , Nagler, E. , Van Biesen, W. , Urinary and serum biomarkers for the diagnosis of acute kidney injury: an in‐depth review of the literature. Nephrol. Dial. Transplant. 2013, 28, 254–273. [DOI] [PubMed] [Google Scholar]
- 2. Trojanowski, J. Q. , Vandeerstichele, H. , Korecka, M. , Clark, C. M. et al., Update on the biomarker core of the Alzheimer's disease neuroimaging initiative subjects. Alzheimers Dement. 2010, 6, 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brooks, J. D. , Translational genomics: the challenge of developing cancer biomarkers. Genome Res. 2012, 22, 183–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maisel, A. S. , Choudhary, R. , Biomarkers in acute heart failure—state of the art. Nat. Rev. Cardiol. 2012, 9, 478–490. [DOI] [PubMed] [Google Scholar]
- 5. Frampton, J. P. , White, J. B. , Simon, A. B. , Tsuei, M. et al., Aqueous two‐phase system patterning of detection antibody solutions for cross‐reaction‐free multiplex ELISA. Sci. Rep. 2014, 4, 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kohler, G. , Milstein, C. , Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [DOI] [PubMed] [Google Scholar]
- 7. Feinberg, J. G. , Wheeler, A. W. , Detection of auto‐immune antibody and tissue antigens by the ‘microspot’ technique. J. Clin. Pathol. 1963, 16, 282–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ekins, R. P. , Multi‐analyte immunoassay. J. Pharm. Biomed. Anal. 1989, 7, 155–168. [DOI] [PubMed] [Google Scholar]
- 9. Hansson, M. , Mathsson, L. , Schlederer, T. , Israelsson, L. et al., Validation of a multiplex chip‐based assay for the detection of autoantibodies against citrullinated peptides. Arthritis Res. Ther. 2012, 14, R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Seyfarth, F. , Schliemann, S. , Wiegand, C. , Hipler, U. C. , Elsner, P. , Diagnostic value of the ISAC((R)) allergy chip in detecting latex sensitizations. Int. Arch. Occup. Environ. Health 2014, 87, 775–781. [DOI] [PubMed] [Google Scholar]
- 11. Onell, A. , Hjalle, L. , Borres, M. P. , Exploring the temporal development of childhood IgE profiles to allergen components. Clin. Transl. Allergy 2012, 2, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rockmann, H. , van Geel, M. J. , Knulst, A. C. , Huiskes, J. et al., Food allergen sensitization pattern in adults in relation to severity of atopic dermatitis. Clin. Transl. Allergy 2014, 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prosperi, M. C. , Belgrave, D. , Buchan, I. , Simpson, A. , Custovic, A. , Challenges in interpreting allergen microarrays in relation to clinical symptoms: a machine learning approach. Pediatr. Allergy Immunol. 2014, 25, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. King, E. M. , Filep, S. , Smith, B. , Platts‐Mills, T. et al., A multi‐center ring trial of allergen analysis using fluorescent multiplex array technology. J. Immunol. Methods 2013, 387, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sander, I. , Zahradnik, E. , Kraus, G. , Mayer, S. et al., Domestic mite antigens in floor and airborne dust at workplaces in comparison to living areas: a new immunoassay to assess personal airborne allergen exposure. PLoS One 2012, 7, e52981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Earle, C. D. , King, E. M. , Tsay, A. , Pittman, K. et al., High‐throughput fluorescent multiplex array for indoor allergen exposure assessment. J. Allergy Clin. Immunol. 2007, 119, 428–433. [DOI] [PubMed] [Google Scholar]
- 17. Fu, Q. , Zhu, J. , Van Eyk, J. E. , Comparison of multiplex immunoassay platforms. Clin. Chem. 2010, 56, 314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salmona, M. , Delarue, S. , Delaugerre, C. , Simon, F. , Maylin, S. , Clinical evaluation of BioPlex 2200 HIV Ag‐Ab, an automated screening method providing discrete detection of HIV‐1 p24 antigen, HIV‐1 antibody, and HIV‐2 antibody. J. Clin. Microbiol. 2014, 52, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holding, S. , Wilson, F. , Spradbery, D. , Clinical evaluation of the BioPlex 2200 Celiac IgA and IgG Kits—a novel multiplex screen incorporating an integral check for IgA deficiency. J. Immunol. Methods 2014, 405, 29–34. [DOI] [PubMed] [Google Scholar]
- 20. Guigue, N. , Menotti, J. , Hamane, S. , Derouin, F. , Garin, Y. J. , Performance of the BioPlex 2200 flow immunoassay in critical cases of serodiagnosis of toxoplasmosis. Clin. Vaccine Immunol. 2014, 21, 496–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gomez, E. , Jespersen, D. J. , Harring, J. A. , Binnicker, M. J. , Evaluation of the Bio‐Rad BioPlex 2200 syphilis multiplex flow immunoassay for the detection of IgM‐ and IgG‐class antitreponemal antibodies. Clin. Vaccine Immunol. 2010, 17, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lash, G. E. , Scaife, P. J. , Innes, B. A. , Otun, H. A. et al., Comparison of three multiplex cytokine analysis systems: Luminex, SearchLight and FAST Quant. J. Immunol. Methods 2006, 309, 205–208. [DOI] [PubMed] [Google Scholar]
- 23. Lash, G. E. , Naruse, K. , Robson, A. , Innes, B. A. et al., Interaction between uterine natural killer cells and extravillous trophoblast cells: effect on cytokine and angiogenic growth factor production. Hum. Reprod. 2011, 26, 2289–2295. [DOI] [PubMed] [Google Scholar]
- 24. Welsh, B. T. , Mapes, J. , An overview of assay quality systems at Myriad, RMB, Inc., https://rbm.myriad.com/scientific‐literature/white‐papers/quality‐control‐white‐paper/.
- 25. Wakisaka, Y. , Ago, T. , Kamouchi, M. , Kuroda, J. et al., Plasma S100A12 is associated with functional outcome after ischemic stroke: research for biomarkers in ischemic stroke. J. Neurol. Sci. 2014, 340, 75–79. [DOI] [PubMed] [Google Scholar]
- 26. Harari, O. , Cruchaga, C. , Kauwe, J. S. , Ainscough, B. J. et al., Phosphorylated tau‐Abeta42 ratio as a continuous trait for biomarker discovery for early‐stage Alzheimer's disease in multiplex immunoassay panels of cerebrospinal fluid. Biol. Psychiatry 2014, 75, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kiddle, S. J. , Thambisetty, M. , Simmons, A. , Riddoch‐Contreras, J. et al., Plasma based markers of [11C] PiB‐PET brain amyloid burden. PLoS One 2012, 7, e44260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Phillips, R. O. , Sarfo, F. S. , Landier, J. , Oldenburg, R. et al., Combined inflammatory and metabolic defects reflected by reduced serum protein levels in patients with Buruli ulcer disease. PLoS Negl. Trop. Dis. 2014, 8, e2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Straface, A. L. , Myers, J. H. , Kirchick, H. J. , Blick, K. E. , A rapid point‐of‐care cardiac marker testing strategy facilitates the rapid diagnosis and management of chest pain patients in the emergency department. Am. J. Clin. Pathol. 2008, 129, 788–795. [DOI] [PubMed] [Google Scholar]
- 30. Apple, F. S. , Christenson, R. H. , Valdes, R., Jr. , Andriak, A. J. et al., Simultaneous rapid measurement of whole blood myoglobin, creatine kinase MB, and cardiac troponin I by the triage cardiac panel for detection of myocardial infarction. Clin. Chem. 1999, 45, 199–205. [PubMed] [Google Scholar]
- 31. Darkoh, C. , Comer, L. , Zewdie, G. , Harold, S. et al., Chemotactic chemokines are important in the pathogenesis of irritable bowel syndrome. PLoS One 2014, 9, e93144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stommel, J. M. , Kimmelman, A. C. , Ying, H. , Nabioullin, R. et al., Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [DOI] [PubMed] [Google Scholar]
- 33. Engelman, J. A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. et al., MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- 34. Eckstein, N. , Servan, K. , Girard, L. , Cai, D. et al., Epidermal growth factor receptor pathway analysis identifies amphiregulin as a key factor for cisplatin resistance of human breast cancer cells. J. Biol. Chem. 2008, 283, 739–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Trune, D. R. , Larrain, B. E. , Hausman, F. A. , Kempton, J. B. , MacArthur, C. J. , Simultaneous measurement of multiple ear proteins with multiplex ELISA assays. Hear. Res. 2011, 275, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gomez‐Puerta, J. A. , Celis, R. , Hernandez, M. V. , Ruiz‐Esquide, V. et al., Differences in synovial fluid cytokine levels but not in synovial tissue cell infiltrate between anti‐citrullinated peptide/protein antibody‐positive and ‐negative rheumatoid arthritis patients. Arthritis Res. Ther. 2013, 15, R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ramirez, J. , Ruiz‐Esquide, V. , Pomes, I. , Celis, R. et al., Patients with rheumatoid arthritis in clinical remission and ultrasound‐defined active synovitis exhibit higher disease activity and increased serum levels of angiogenic biomarkers. Arthritis Res. Ther. 2014, 16, R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang, C. C. , Lin, C. Y. , Wang, H. S. , Lyu, S. R. , Matrix metalloproteases and tissue inhibitors of metalloproteinases in medial plica and pannus‐like tissue contribute to knee osteoarthritis progression. PLoS One 2013, 8, e79662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim, Y. H. , Park, C. S. , Jang, T. Y. , Immunologic properties and clinical features of local allergic rhinitis. J. Otolaryngol. Head Neck Surg. 2012, 41, 51–57. [PubMed] [Google Scholar]
- 40. Rajkumar, T. , Vijayalakshmi, N. , Gopal, G. , Sabitha, K. et al., Identification and validation of genes involved in gastric tumorigenesis. Cancer Cell Int. 2010, 10, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Green, V. L. , Michno, A. , Greenman, J. , Stafford, N. D. , Effect of treatment on systemic cytokines in head and neck squamous cell carcinoma patients. Results Immunol. 2011, 2, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nishida, Y. , Higaki, Y. , Taguchi, N. , Hara, M. et al., Objectively measured physical activity and inflammatory cytokine levels in middle‐aged Japanese people. Prev. Med. 2014, 64, 81–87. [DOI] [PubMed] [Google Scholar]
- 43. Rosser, C. J. , Dai, Y. , Miyake, M. , Zhang, G. , Goodison, S. , Simultaneous multi‐analyte urinary protein assay for bladder cancer detection. BMC Biotechnol. 2014, 14, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolokythas, A. , Karas, M. , Sarna, T. , Flick, W. , Miloro, M. , Does cytokine profiling of aspirate from jaw cysts and tumors have a role in diagnosis? J. Oral Maxillofac. Surg. 2012, 70, 1070–1080. [DOI] [PubMed] [Google Scholar]
- 45. Tighe, P. , Negm, O. , Todd, I. , Fairclough, L. , Utility, reliability and reproducibility of immunoassay multiplex kits. Methods 2013, 61, 23–29. [DOI] [PubMed] [Google Scholar]
- 46. Chowdhury, F. , Williams, A. , Johnson, P. , Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery, for human cytokine profiling. J. Immunol. Methods 2009, 340, 55–64. [DOI] [PubMed] [Google Scholar]
- 47. Chaturvedi, S. , Dell, E. , Siegel, D. , Brittingham, G. , Seetharam, S. , Development of a rapid streptavidin capture‐based assay for the tyrosine phosphorylated CSF‐1R in peripheral blood mononuclear cells. Int. J. Biol. Sci. 2013, 9, 1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Smith, T. G. , Ellison, J. A. , Ma, X. , Kuzmina, N. et al., An electrochemiluminescence assay for analysis of rabies virus glycoprotein content in rabies vaccines. Vaccine 2013, 31, 3333–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hoofring, S. A. , Lopez, R. , Hock, M. B. , Kaliyaperumal, A. et al., Immunogenicity testing strategy and bioanalytical assays for antibody‐drug conjugates. Bioanalysis 2013, 5, 1041–1055. [DOI] [PubMed] [Google Scholar]
- 50. Banerji, U. , van Doorn, L. , Papadatos‐Pastos, D. , Kristeleit, R. et al., A phase I pharmacokinetic and pharmacodynamic study of CHR‐3996, an oral class I selective histone deacetylase inhibitor in refractory solid tumors. Clin. Cancer Res. 2012, 18, 2687–2694. [DOI] [PubMed] [Google Scholar]
- 51. Moncunill, G. , Aponte, J. J. , Nhabomba, A. J. , Dobano, C. , Performance of multiplex commercial kits to quantify cytokine and chemokine responses in culture supernatants from Plasmodium falciparum stimulations. PLoS One 2013, 8, e52587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cai, Y. J. , Huang, L. , Leung, T. Y. , Burd, A. , A study of the immune properties of human umbilical cord lining epithelial cells. Cytotherapy 2014, 16, 631–639. [DOI] [PubMed] [Google Scholar]
- 53. Chen, Y. C. , Yang, X. , Miao, L. , Liu, Z. G. et al., Serum level of interleukin‐6 in Chinese patients with multiple sclerosis. J. Neuroimmunol. 2012, 249, 109–111. [DOI] [PubMed] [Google Scholar]
- 54. Chou, C. S. , Lin, L. Y. , Chen, K. M. , Lai, S. C. , FlowCytomix analysis for Toxoplasma gondii infection in pregnant women in central Taiwan. J. Obstet. Gynaecol. 2011, 31, 375–379. [DOI] [PubMed] [Google Scholar]
- 55. Mustafa, A. S. , Al‐Saidi, F. , El‐Shamy, A. S. , Al‐Attiyah, R. , Cytokines in response to proteins predicted in genomic regions of difference of Mycobacterium tuberculosis . Microbiol. Immunol. 2011, 55, 267–278. [DOI] [PubMed] [Google Scholar]
- 56. Szczypka, M. , Ploch, S. , Obminska‐Mrukowicz, B. , Modulation of Th1/Th2 cytokine production by selective and nonselective phosphodiesterase inhibitors administered to mice. Pharmacol. Rep. 2012, 64, 179–184. [DOI] [PubMed] [Google Scholar]
- 57. Pardo Morales, R. V. , Zúñiga Torres, M. G. , Martínez Carrillo, B. E. , Gómez Martínez, S. et al., Inflammatory mediators and immune response in Mexican adolescents. Nutr. Hosp. 2011, 26, 1115–1119. [DOI] [PubMed] [Google Scholar]
- 58. Berthoud, T. K. , Manaca, M. N. , Quelhas, D. , Aguilar, R. et al., Comparison of commercial kits to measure cytokine responses to Plasmodium falciparum by multiplex microsphere suspension array technology. Malaria J. 2011, 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ostendorff, H. P. , Awad, A. , Braunschweiger, K. I. , Liu, Z. et al., Multiplexed VeraCode bead‐based serological immunoassay for colorectal cancer. J. Immunol. Methods 2013, 400–401, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Backen, A. C. , Cummings, J. , Mitchell, C. , Jayson, G. et al., ‘Fit‐for‐purpose’ validation of SearchLight multiplex ELISAs of angiogenesis for clinical trial use. J. Immunol. Methods 2009, 342, 106–114. [DOI] [PubMed] [Google Scholar]
- 61. Brookes, K. , Cummings, J. , Backen, A. , Greystoke, A. et al., Issues on fit‐for‐purpose validation of a panel of ELISAs for application as biomarkers in clinical trials of anti‐angiogenic drugs. Br. J. Cancer 2010, 102, 1524–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bastarache, J. A. , Koyama, T. , Wickersham, N. E. , Ware, L. B. , Validation of a multiplex electrochemiluminescent immunoassay platform in human and mouse samples. J. Immunol. Methods 2014, 408, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hartmann, M. , Roeraade, J. , Stoll, D. , Templin, M. F. , Joos, T. O. , Protein microarrays for diagnostic assays. Anal. Bioanal. Chem. 2009, 393, 1407–1416. [DOI] [PubMed] [Google Scholar]
- 64. Horan, P. K. , Jr Wheeless, L. L., ., Quantitative single cell analysis and sorting. Science 1977, 198, 149–157. [DOI] [PubMed] [Google Scholar]
- 65. Nolan, J. P. , Mandy, F. , Multiplexed and microparticle‐based analyses: quantitative tools for the large‐scale analysis of biological systems. Cytometry A 2006, 69, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ellington, A. A. , Kullo, I. J. , Bailey, K. R. , Klee, G. G. , Antibody‐based protein multiplex platforms: technical and operational challenges. Clin. Chem. 2010, 56, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Houser, B. , Bio‐Rad's Bio‐Plex(R) suspension array system, xMAP technology overview. Arch. Physiol. Biochem. 2012, 118, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pla‐Roca, M. , Leulmi, R. F. , Tourekhanova, S. , Bergeron, S. et al., Antibody colocalization microarray: a scalable technology for multiplex protein analysis in complex samples. Mol. Cell. Proteomics 2012, 11, M111 011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Boja, E. S. , Jortani, S. A. , Ritchie, J. , Hoofnagle, A. N. et al., The journey to regulation of protein‐based multiplex quantitative assays. Clin. Chem. 2011, 57, 560–567. [DOI] [PubMed] [Google Scholar]
- 70. Boja, E. S. , Rodriguez, H. , The path to clinical proteomics research: integration of proteomics, genomics, clinical laboratory and regulatory science. Korean J. Lab. Med. 2011, 31, 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chau, C. H. , Rixe, O. , McLeod, H. , Figg, W. D. , Validation of analytic methods for biomarkers used in drug development. Clin. Cancer Res. 2008, 14, 5967–5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ellington, A. A. , Kullo, I. J. , Bailey, K. R. , Klee, G. G. , Measurement and quality control issues in multiplex protein assays: a case study. Clin. Chem. 2009, 55, 1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Valentin, M. A. , Ma, S. , Zhao, A. , Legay, F. , Avrameas, A. , Validation of immunoassay for protein biomarkers: bioanalytical study plan implementation to support pre‐clinical and clinical studies. J. Pharm. Biomed. Anal. 2011, 55, 869–877. [DOI] [PubMed] [Google Scholar]
- 74. Bastarache, J. A. , Koyama, T. , Wickersham, N. E. , Mitchell, D. B. et al., Accuracy and reproducibility of a multiplex immunoassay platform: a validation study. J. Immunol. Methods 2011, 367, 33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Eckels, J. , Nathe, C. , Nelson, E. K. , Shoemaker, S. G. et al., Quality control, analysis and secure sharing of Luminex(R) immunoassay data using the open source LabKey Server platform. BMC Bioinformatics 2013, 14, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cretich, M. , Damin, F. , Chiari, M. , Protein microarray technology: how far off is routine diagnostics? Analyst 2014, 139, 528–542. [DOI] [PubMed] [Google Scholar]
- 77. Juncker, D. , Bergeron, S. , Laforte, V. , Li, H. , Cross‐reactivity in antibody microarrays and multiplexed sandwich assays: shedding light on the dark side of multiplexing. Curr. Opin. Chem. Biol. 2014, 18, 29–37. [DOI] [PubMed] [Google Scholar]
- 78. Guo, A. , Zhu, X.‐Y. , in: Predki P. (Ed.), Functional Protein Microarrays: Pathways to Discovery, CRC Press, Boca Raton, FL; 2006, pp. 60–61. [Google Scholar]
- 79. Stoevesandt, O. , Taussig, M. J. , Affinity reagent resources for human proteome detection: initiatives and perspectives. Proteomics 2007, 7, 2738–2750. [DOI] [PubMed] [Google Scholar]
- 80. Byerly, S. , Sundin, K. , Raja, R. , Stanchfield, J. et al., Effects of ozone exposure during microarray posthybridization washes and scanning. J. Mol. Diagn. 2009, 11, 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. de Jager, W. , Rijkers, G. T. , Solid‐phase and bead‐based cytokine immunoassay: a comparison. Methods 2006, 38, 294–303. [DOI] [PubMed] [Google Scholar]
- 82. Fernando, S. A. , Wilson, G. S. , Studies of the ‘hook’ effect in the one‐step sandwich immunoassay. J. Immunol. Methods 1992, 151, 47–66. [DOI] [PubMed] [Google Scholar]
- 83. Bolstad, N. , Warren, D. J. , Nustad, K. , Heterophilic antibody interference in immunometric assays. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 647–661. [DOI] [PubMed] [Google Scholar]
- 84. Hennig, C. , Rink, L. , Fagin, U. , Jabs, W. J. , Kirchner, H. , The influence of naturally occurring heterophilic anti‐immunoglobulin antibodies on direct measurement of serum proteins using sandwich ELISAs. J. immunol. Methods 2000, 235, 71–80. [DOI] [PubMed] [Google Scholar]
- 85. Boscato, L. M. , Stuart, M. C. , Heterophilic antibodies: a problem for all immunoassays. Clin. Chem. 1988, 34, 27–33. [PubMed] [Google Scholar]
- 86. de Jager, W. , Prakken, B. J. , Bijlsma, J. W. , Kuis, W. , Rijkers, G. T. , Improved multiplex immunoassay performance in human plasma and synovial fluid following removal of interfering heterophilic antibodies. J. Immunol. Methods 2005, 300, 124–135. [DOI] [PubMed] [Google Scholar]
- 87. Rhyne, P. , http://www.tecan.com/platform/content/element/9550/2‐1Rhyne.pdf, Tecan Ltd 2014.
- 88. Choi, J. S. , Roh, J. Y. , Lee, J. R. , Clinical availability of component‐resolved diagnosis using microarray technology in atopic dermatitis. Ann. Dermatol. 2014, 26, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Martinez‐Aranguren, R. , Lizaso, M. T. , Goikoetxea, M. J. , Garcia, B. E. et al., Is the determination of specific IgE against components using ISAC 112 a reproducible technique? PLoS One 2014, 9, e88394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jenko, K. L. , Zhang, Y. , Kostenko, Y. , Fan, Y. et al., Development of an ELISA microarray assay for the sensitive and simultaneous detection of ten biodefense toxins. Analyst 2014, 139, 5093–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. FDA , Center for Drug Evaluation and Research (CDER). Guidance for industry: bioanalytical method validation: US Food and Drug Administration, http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm070107.pdf, FDA; 2001. [Google Scholar]
- 92. Master, S. R. , Bierl, C. , Kricka, L. J. , Diagnostic challenges for multiplexed protein microarrays. Drug Discov. Today 2006, 11, 1007–1011. [DOI] [PubMed] [Google Scholar]
- 93. Buchegger, P. , Sauer, U. , Toth‐Szekely, H. , Preininger, C. , Miniaturized protein microarray with internal calibration as point‐of‐care device for diagnosis of neonatal sepsis. Sensors 2012, 12, 1494–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Augenlicht, L. H. , Kobrin, D. , Cloning and screening of sequences expressed in a mouse colon tumor. Cancer Res. 1982, 42, 1088–1093. [PubMed] [Google Scholar]
- 95. Chang, T. W. , Binding of cells to matrixes of distinct antibodies coated on solid surface. J. Immunol. Methods 1983, 65, 217–223. [DOI] [PubMed] [Google Scholar]
- 96. Moody, M. D. , Van Arsdell, S. W. , Murphy, K. P. , Orencole, S. F. , Burns, C. , Array‐based ELISAs for high‐throughput analysis of human cytokines. BioTechniques 2001, 31, 186–190, 192–184. [DOI] [PubMed] [Google Scholar]