

Abstract
Measles virus RNA genomes are packaged into helical nucleocapsids (NCs), comprising thousands of nucleo‐proteins (N) that bind the entire genome. N‐RNA provides the template for replication and transcription by the viral polymerase and is a promising target for viral inhibition. Elucidation of mechanisms regulating this process has been severely hampered by the inability to controllably assemble NCs. Here, we demonstrate self‐organization of N into NC‐like particles in vitro upon addition of RNA, providing a simple and versatile tool for investigating assembly. Real‐time NMR and fluorescence spectroscopy reveals biphasic assembly kinetics. Remarkably, assembly depends strongly on the RNA‐sequence, with the genomic 5′ end and poly‐Adenine sequences assembling efficiently, while sequences such as poly‐Uracil are incompetent for NC formation. This observation has important consequences for understanding the assembly process.
Keywords: fluorescence spectroscopy, measles, NMR spectroscopy, nucleocapsids, self-assembly
Paramyxoviruses are non‐segmented negative‐strand RNA viruses that express their own machinery for transcription and replication.1, 2, 3 Paramyxoviridae comprise a large number of human pathogens, including measles (MeV), but also the emergent Nipah and Hendra viruses, that present greater than 50 % fatality in humans and for which no successful treatment exists. Their genomes are packaged into large helical assemblies, termed nucleocapsids (NC), consisting of a multitude of nucleoprotein (N) copies that bind the entire sequence of the genome.4, 5, 6 The N‐RNA complex provides the template for replication and transcription by the viral polymerase complex composed of the phospho‐ (P) and large (L) proteins.7 Elucidation of the mechanisms that regulate transcription and replication holds great promise for the development of targeted drugs against paramyxoviral diseases.
MeV N comprises a folded RNA‐binding domain (NCORE), as well as an intrinsically disordered C‐terminal domain (NTAIL; Figure 1 a; Supporting Information, Figure S1).8, 9 A recent cryo‐electron microscopy (EM) structure10 revealed that assembly of NCs is mediated by the NTDARM and CTDARM sub‐domains, at the N‐ and C‐termini of NCORE, that interact with neighboring N molecules in the NC (Figure 1 b), a mode of stabilization also observed in related viruses.11, 12, 13, 14
Figure 1.
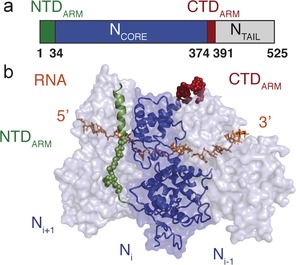
a) Domain organization of MeV N showing the position of the N‐ and C‐terminal arms (NTDARM and CTDARM), NCORE and NTAIL. b) Structure of the folded domain of measles virus N in complex with RNA obtained from cryo‐electron microscopy on helical NCs (PDB: 4UFT). NTAIL is not shown in this Figure.
The MeV P protein is tetrameric, also containing folded and unfolded domains,9, 15, 16, 17, 18 and interacts with NCORE through a 300 amino acid, disordered N‐terminal domain, apparently to chaperone RNA‐free N prior to NC assembly on the RNA genome,19 in the so‐called N0P complex. Crystal structures of the N0P complex of paramyxoviruses20, 21 have been determined by engineering deletion mutants of NCORE devoid of both NTDARM and CTDARM sub‐domains (as well as NTAIL), to avoid assembly into NC particles. These structures revealed how the N‐terminus of P binds to NCORE in a helix–kink–helix conformation. Comparison with the MeV NC10 revealed that the binding sites of the NTDARM and CTDARM in the NCs overlap with binding sites of two α‐helices of P in the N0P complex (Figure 1 b), providing a rational explanation of the chaperone activity of P.
Despite this insight into the structural basis of both N0P and assembled NCs, further understanding of the molecular mechanisms of NC assembly has been hindered by the inability to isolate intact N0P and subsequently assemble supramolecular NC particles in vitro. Indeed, assembled NCs from paramyxoviruses could only be purified by over‐expressing N in recombinant systems,4 for example in E. coli, where N spontaneously assembles on cellular RNA.
In this study, we engineer a soluble heterodimeric N0P complex of MeV including the two sub‐domains NTDARM and CTDARM, and show that the N0P complex can be triggered to initiate assembly of nucleocapsid‐like particles (NCLPs) simply upon addition of RNA. We use nuclear magnetic resonance (NMR), fluorescence spectroscopy, and EM to observe the kinetics of self‐assembly of NCLPs and to investigate the molecular basis of NC formation. Remarkably, assembly of MeV NCLPs exhibits strong RNA‐sequence dependence. While the 5′ genomic and poly‐Adenine sequences form very long NCLPs, certain sequences, such as poly‐Uracil, are incompetent for assembly.
An N0P complex of MeV that is capable of forming NCLPs in vitro was engineered using a fusion construct of MeV P peptide and N, including a TEV cleavage site separating N and P. The minimal peptide required to form the complex was identified using NMR spectroscopy as P1‐50 (Figure S2), resulting in a high‐yielding, soluble, and stable N0P complex. This region of P resembles peptides co‐crystallized with the NCORE of Nipah virus and MeV (with NTDARM, CTDARM and NTAIL deleted).20, 21 RNA‐free heterodimeric N0P complexes of full length N (P50N525) and NTAIL‐deleted N (P50N405) were purified using Ni‐affinity chromatography followed by TEV cleavage and gel filtration (Figure S3). MeV NCs obey the “rule of six”,1, 2, 22 with each N interacting with six nucleotides of the genome. Remarkably, upon addition of P50N405 to six‐nucleotide RNA strands (corresponding to the 5′ end of the viral genome, 5′‐RNA6 ACCAGA), N spontaneously assembles into NCLPs whose negative‐staining EM resembles NCs purified directly by over‐expression of N in E. coli (Figure 2), demonstrating that continuous RNA is not a prerequisite for formation of NCLPs.
Figure 2.
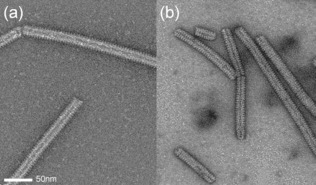
Comparison of a) NCLPs assembled in vitro upon addition of 5′‐RNA6 to P50N405 with b) NCLPs obtained from expression of N in E. coli and cleavage of NTAIL by trypsin digestion post‐purification.
Disordered and flexible regions of P50N405 could be observed using solution NMR, corresponding to almost the entire CTDARM (residues 380–405 of N), the C‐terminal end of the P50 peptide (residues 44–50 of P), and a few residues belonging to the NTDARM (residues 26–29; Figure S4). NMR thus can be used to follow NCLP assembly in real time. By adding a molar excess of RNA to P50N405 or P50N525, and observing the evolution of NMR signals using SOFAST HMQC,23 a time‐resolution of approximately four minutes can be achieved. Resonances of P50 appeared as the peptide detaches from the surface of NCORE upon addition of RNA, while flexible resonances of NTDARM and CTDARM disappear over time as the NCLPs assemble (Figures 3, S5, S6).
Figure 3.
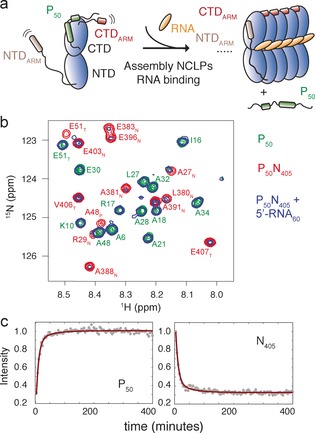
a) Cartoon illustrating the NMR signals that appear or disappear upon assembly of NCLPs. In the N0P complex, both NTDARM and CTDARM of N are flexible, giving rise to observable backbone resonances in 1H‐15 N spectra. P50 is bound to N so that peaks are too weak to be observed. As N assembles, NTDARM and CTDARM stabilize the NCLPs so that these peaks disappear, while resonances from P50 grow as more N proteins associate. b) Part of the 1H‐15 n spectrum of P50N405 (red), P50 (green), and P50N405 after incubation with 5′‐RNA60 for 24 hours at 25°C (blue). Assignments are shown for the P50N405 and P50 resonances. Subscript T refers to residues associated with cleavage or affinity tags. c) Assembly of NCLPs from P50N405 with 5′‐RNA6 was initiated from 209 μm P50N405, adding 5′‐RNA6 to 471 μm. Circles: intensities measured in SOFAST HMQC spectra and summed over appearing P50 (left) or disappearing N405 (right) resonances (P50: 4, 5, 6, 22, 25, 38, N405: 28, 385, 386, 387, 388, 389, 404, 405), red lines: simultaneous fit to double‐exponential with common assembly rates.
The assembly kinetics of P50N405 mixed with 5′‐RNA6 were investigated (Figure 3 c). Time traces of N and P were simultaneously fitted using a double exponential function (statistical significance p<0.0001 compared to a single‐exponential fit), giving apparent rates 2.0×10−3 s−1 and 3.6×10−4 s−1 for 5′‐RNA6 (Figure 3 c). Under similar conditions, assembly on 5′‐RNA60 (corresponding to the first 60 nucleo‐tides of the 5′ end of the viral genome) was found to be slightly slower (Figures 3 b and S6 b). EM was used to confirm assembly of NCLPs by adding 5′‐RNA6 to P50N405 at time 0 and visualizing this sample using EM at increasing time after mixing, showing NCLPs of increasing length (Figure S7).
The absolute assembly rates were determined by measuring time‐resolved fluorescence anisotropy of 10‐mer RNA with a MeV genome 5′ sequence, labelled at the 3′ terminus with a fluorescein derivative (5′‐RNA10‐FAM; a longer RNA was used to allow the fluorescein to exit the RNA binding groove and to reduce the potential impact on assembly). The dye attached to free RNA undergoes fast rotation, exhibiting low fluorescence anisotropy, while upon binding to N, NCLP formation and subsequent elongation, the rotational freedom is significantly hindered. Addition of increasing concentrations of P50N405 again showed biphasic kinetics (Figure 4) with assembly rates of (2.2±0.2)×103 and (3.37±0.04)×102 m −1 s−1. NMR and EM confirmed that the dye did not interfere with NCLP formation (Figure S8).
Figure 4.
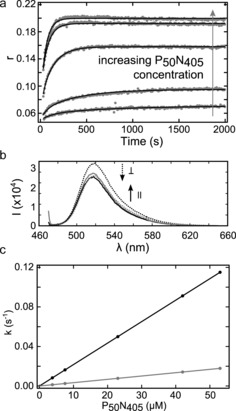
Kinetics of NCLP assembly. a) Parallel and perpendicular polarized fluorescence light was acquired upon addition of 500 nm RNA10‐FAM to P50N405 (top to bottom: 53, 42, 23, 7.5, 3.8 μm). Fluorescence anisotropy (r) was calculated for every time point. All of the curves were fit with a global fit (black lines) using two exponential rates, and imposing linearity between rates and protein concentration (see the Supporting Information). b) Fluorescence spectra at the end of kinetics (dotted lines: perpendicular‐polarized; solid line: parallel‐polarized fluorescence light) illustrate increased r and light scattering at higher protein concentration (black: 7.5 μm; grey: 53 μm P50N405). c) Kinetic rates of NCLP formation with P50N405 and RNA10‐FAM corresponding to (a).
Both fluorescence and NMR data suggest that an initial reaction dependent on RNA binding is well‐separated from the second, slower component, possibly corresponding to assembly of N‐coated RNA strands into regular NCLPs. N is thought to bind the RNA immediately as it emerges from the viral polymerase,3, 24 so that RNA6 may be physiologically relevant. We note that the observed rates of NCLP formation are compatible with the measured elongation speed of the viral polymerase in vivo.25
We have previously demonstrated that NTAIL exfiltrates from the inside to the outside of NCLPs through the interstitial space between the NCORE domains where it can interact with the polymerase complex.8, 26, 27 The role of NTAIL in assembly was investigated here by measuring NMR, fluorescence, and EM after mixing P50N525 and RNA. In addition to backbone resonances that are visible for P50N405, P50N525 exhibit resonances from the entire NTAIL domain, demonstrating that these residues remain flexible in full‐length N (Figure S9). The formation of NCLPs from 5′‐RNA6 and P50N525 showed that NTAIL does not interfere with particle assembly in vitro, although the NCLPs are shorter than those produced with P50N405 (Figure S10). Notably, a third, significantly slower kinetic rate (80±2 m −1 s−1) is required to describe the assembly of P50N525 compared to P50N405, suggesting an additional step in the assembly process involving NTAIL.
Different RNA sequences were tested for their ability to facilitate NCLP assembly in vitro from P50N405. Homopolymers comprising purines or pyrimidines alone, UUUUUU (polyU‐RNA6) and AAAAAA (polyA‐RNA6), were compared, as well as genomic 5′‐RNA6. Remarkably, the efficiency of assembly is strongly sequence‐dependent (Figures 5 and S11). No assembly is observed for the polyU‐RNA6 samples, while regular NCLPs are obtained for 5′‐RNA6 and polyA‐RNA6 with lengths reaching 1–2 μm. This differential ability to form NCLPs was verified using NMR and fluorescence. No change in fluorescence anisotropy (Figure 5) or displacement of P50 peptide over a period of 24 hours at room temperature (Figure S12) were observed upon addition of polyU‐RNA6. RNA‐sequence‐dependence is surprising considering the apparent necessity of N to encapsidate the entire genome, irrespective of sequence, suggesting the role of an initial specific nucleation site for successful encapsidation. In this respect, we note that the 5′ sequences of both the genome and anti‐genome of paramyxovirinae show strong conservation of ACCA in the first four nucleotides, followed by positions 5 and 6 occupied mainly by A/G. Indeed, the 5′ sequence of the MeV genome leads to formation of NCs with high efficiency. No assembly is observed on DNA (Figure S13). This specificity may be related to the observed N‐RNA interaction in RSV and rabies that is mediated by the 2′ ribose OH group with the carbonyls of N.12, 14
Figure 5.
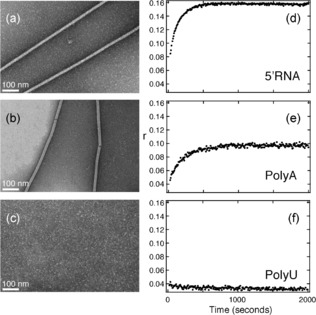
Dependence of NCLP assembly on RNA sequence. (a–c) P50N405 was incubated with different RNAs for 24 hours at room temperature and visualized by negative‐stain electron microscopy. Assembly of NCLPs is observed for a) 5′‐RNA6, b) polyA‐RNA6, while no assembly is observed for c) polyU‐RNA6 (See Figure S11 for additional representative EMs.) d–f) Fluorescence anisotropy during 2000 seconds after addition of 500 nm, d) 5′‐RNA10‐FAM, e) polyA10‐FAM, and f) polyU10‐FAM to 23 μm P50N405.
In all of the structures of paramyxoviral NC particles determined so far, the RNA bases were modelled as generic base‐types (uracil), to account for signal averaging over different nucleotides from random cellular RNA. We are therefore unable to determine the molecular mechanisms by which N0P alone can discriminate between different RNA sequences. In the NC structure determined from cryo‐EM,10 three of the six bases associated with each N stack sequentially and face the surface of the protein, while the following three stack and point into the solvent. The only significant interactions between base moieties and the protein surface occur through a stacking of Y260, and the first of the three stacked‐in bases (position 2), with base‐specific hydrogen bonding identified between Q202 and the uracil modelled in position 1. Both amino acids are conserved in paramyxo‐virinae. An atomic‐resolution structural description of protein–RNA interactions is necessary to fully understand the molecular basis of the observed sequence discrimination.
Based on the rational design of N0P chaperone mimics, we were able to demonstrate self‐assembly of highly regular NCLPs of MeV in vitro, uniquely from protein and RNA. In addition to providing a quantitative determination of assembly rates using real‐time NMR and fluorescence spectroscopy, revealing biphasic assembly kinetics, we have demonstrated that assembly occurs both in the presence and absence of the 125 aa disordered NTAIL domain. Importantly, we also identify a strong, and unexpected, dependence of NCLP assembly on the RNA sequence. While the genomic 5′ end and polyA sequences assemble with high efficiency, certain RNA sequences are incompetent for NC formation under our experimental conditions. We note that the ability to controllably assemble stable, protein‐based superstructures, whose regularly spaced intrinsically disordered domains can be decorated with sequence‐modifiable functionalities, may find diverse biotechnological applications both in vitro and in vivo. Finally, this fluorescence‐anisotropy‐based MeV NC assembly assay provides a powerful tool for the development of small molecule inhibitors of this essential step in viral replication.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the Agence Nationale de la Recherche under ComplexDynamics (MB) and NMRSignal (MRJ) and by Finovi. SM acknowledges the EMBO long‐term fellowship (ALTF 468‐2014) and EC (EMBOCOFUND2012, GA‐2012‐600394) through Marie Curie Action. This work used platforms of the Grenoble Instruct Centre (ISBG; UMS 3518 CNRS‐CEA‐UJF‐EMBL) with support from FRISBI (ANR‐10‐INSB‐05‐02) and GRAL (ANR‐10‐LABX‐49‐01) within the Grenoble Partnership for Structural Biology (PSB). The EM facility is supported by the Rhône‐Alpes Region, the Fondation pour la Recherche Medicale (FRM) and FEDER funds.
S. Milles, M. R. Jensen, G. Communie, D. Maurin, G. Schoehn, R. W. H. Ruigrok, M. Blackledge, Angew. Chem. Int. Ed. 2016, 55, 9356.
Contributor Information
Prof. Rob W. H. Ruigrok, Email: rob.ruigrok@ibs.fr
Dr. Martin Blackledge, Email: martin.blackledge@ibs.fr
References
- 1. Egelman E. H., Wu S. S., Amrein M., Portner A., Murti G., J. Virol. 1989, 63, 2233–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kolakofsky D., Pelet T., Garcin D., Hausmann S., Curran J., Roux L., J. Virol. 1998, 72, 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ruigrok R. W. H., Crépin T., Kolakofsky D., Curr. Opin. Microbiol. 2011, 14, 504–510. [DOI] [PubMed] [Google Scholar]
- 4. Schoehn G., Mavrakis M., Albertini A., Wade R., Hoenger A., Ruigrok R. W. H., J. Mol. Biol. 2004, 339, 301–312. [DOI] [PubMed] [Google Scholar]
- 5. Bhella D., Ralph A., Yeo R. P., J. Mol. Biol. 2004, 340, 319–331. [DOI] [PubMed] [Google Scholar]
- 6. Desfosses A., Goret G., Estrozi L. F., Ruigrok R. W. H., Gutsche I., J. Virol. 2011, 85, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Curran J., Kolakofsky D., Adv. Virus Res. 1999, 54, 403–422. [DOI] [PubMed] [Google Scholar]
- 8. Jensen M. R., Communie G., E. A. Ribeiro Jr , Martinez N., Desfosses A., Salmon L., Mollica L., Gabel F., Jamin M., Longhi S., et al., Proc. Natl. Acad. Sci. USA 2011, 108, 9839–9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Communie G., Ruigrok R. W., Jensen M. R., Blackledge M., Curr. Opin. Virol. 2014, 5, 72–81. [DOI] [PubMed] [Google Scholar]
- 10. Gutsche I., Desfosses A., Effantin G., Ling W. L., Haupt M., Ruigrok R. W. H., Sachse C., Schoehn G., Science 2015, 348, 704–707. [DOI] [PubMed] [Google Scholar]
- 11. Alayyoubi M., Leser G. P., Kors C. A., Lamb R. A., Proc. Natl. Acad. Sci. USA 2015, 112, E1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tawar R. G., Duquerroy S., Vonrhein C., Varela P. F., Damier-Piolle L., Castagné N., MacLellan K., Bedouelle H., Bricogne G., Bhella D., et al., Science 2009, 326, 1279–1283. [DOI] [PubMed] [Google Scholar]
- 13. Green T. J., Zhang X., Wertz G. W., Luo M., Science 2006, 313, 357–360. [DOI] [PubMed] [Google Scholar]
- 14. Albertini A. A. V., Wernimont A. K., Muziol T., Ravelli R. B. G., Clapier C. R., Schoehn G., Weissenhorn W., Ruigrok R. W. H., Science 2006, 313, 360–363. [DOI] [PubMed] [Google Scholar]
- 15. Johansson K., Bourhis J.-M., Campanacci V., Cambillau C., Canard B., Longhi S., J. Biol. Chem. 2003, 278, 44567–44573. [DOI] [PubMed] [Google Scholar]
- 16. Kingston R. L., Hamel D. J., Gay L. S., Dahlquist F. W., Matthews B. W., Proc. Natl. Acad. Sci. USA 2004, 101, 8301–8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernado P., Blanchard L., Timmins P., Marion D., Ruigrok R. W. H., Blackledge M., Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Communie G., Crépin T., Maurin D., Jensen M. R., Blackledge M., Ruigrok R. W. H., J. Virol. 2013, 87, 7166–7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Curran J., Marq J., Kolakofsky D., J. Virol. 1995, 69, 849–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yabukarski F., Lawrence P., Tarbouriech N., Bourhis J.-M., Delaforge E., Jensen M. R., Ruigrok R. W. H., Blackledge M., Volchkov V., Jamin M., Nat. Struct. Mol. Biol. 2014, 21, 754–759. [DOI] [PubMed] [Google Scholar]
- 21.S. G. Guryanov, L. Liljeroos, P. Kasaragod, T. Kajander, S. J. Butcher, J. Virol 2015, 90, 2849–2857. [DOI] [PMC free article] [PubMed]
- 22. Calain P., Roux L., J. Virol. 1993, 67, 4822–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schanda P., Brutscher B., J. Am. Chem. Soc. 2005, 127, 8014–8015. [DOI] [PubMed] [Google Scholar]
- 24. Gubbay O., Curran J., Kolakofsky D., J. Gen. Virol. 2001, 82, 2895–2903. [DOI] [PubMed] [Google Scholar]
- 25. Plumet S., Duprex W. P., Gerlier D., J. Virol. 2005, 79, 6900–6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Communie G., Habchi J., Yabukarski F., Blocquel D., Schneider R., Tarbouriech N., Papageorgiou N., Ruigrok R. W. H., Jamin M., Jensen M. R., et al., PLoS Pathog. 2013, 9, e1003631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schneider R., Maurin D., Communie G., Kragelj J., Hansen D. F., Ruigrok R. W. H., Jensen M. R., Blackledge M., J. Am. Chem. Soc. 2015, 137, 1220–1229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary