

Summary
As a population, dendritic cells (DCs) appear to be the best cross‐presenters of internalized antigens on major histocompatibility complex class I molecules in the mouse. To do this, DCs have developed a number of unique and dedicated means to control their endocytic and phagocytic pathways: among them, the capacity to limit acidification of their phagosomes, to prevent proteolytic degradation, to delay fusion of phagosomes to lysosomes, to recruit ER proteins to phagosomes, and to export phagocytosed antigens to the cytosol. The regulation of phagocytic functions, and thereby of antigen processing and presentation by innate signaling, represents a critical level of integration of adaptive and innate immune responses. Understanding how innate signals control antigen cross‐presentation is critical to define effective vaccination strategies for CD8+ T‐cell responses.
Keywords: cross‐presentation, dendritic cell, phagocytosis, dendritic cells maturation, toll‐like receptor
This article is part of a series of reviews covering Antigen Presentation appearing in Volume 272 of Immunological Reviews.
Introduction
Dendritic cells (DCs) are among the most efficient antigen‐presenting cells (APC) of the immune system. In contrast to macrophages and neutrophils, that mainly destroy antigens by phagocytosis and subsequent proteolysis in highly degradative compartments, DCs have developed a dedicated and regulated phagocytic pathway that primarily serves antigen processing and presentation on major histocompatibility complex (MHC) molecules 1. Peptides generated by proteasomal degradation of endogenous cytosolic and nuclear antigens are loaded onto MHC class I molecules and presented to CD8+ T cells through distinct intracellular pathways. In most APC, peptides derived from exogenous antigens that are taken up from the extracellular milieu are preferentially presented on MHC class II molecules to CD4+ T cells. In DCs, however, internalized antigens are also presented to CD8+ T cells through a process referred to as cross‐presentation 2.
Exogenous antigens are cross‐presented through two main pathways, often referred to as ‘vacuolar’ and ‘cytosolic’ 2. Cross‐presentation through the ‘vacuolar’ pathway is independent of the transporter associated with antigen processing (TAP) and resistant to proteasome inhibitors. It is also sensitive to inhibitors of lysosomal proteolysis (in particular cathepsin S inhibitors) 3, 4, suggesting that both antigen processing and loading onto MHC class I molecules occur in endocytic compartments. The ‘cytosolic’ pathway, by contrast, depends on the expression of TAP1/2 transporters and is sensitive to proteasome inhibitors, suggesting that internalized antigens access the cytosol where they are degraded by the proteasome before loading on class I MHC molecules 5.
In the classical MHC class I presentation pathway, peptides generated by the proteasome from endogenous proteins are then transported and translocated into the lumen of the ER by TAP1/2 and loaded on newly synthesized MHC class I molecules. However, direct evidence supporting ER loading of peptides during cross‐presentation is missing. TAP1/2 and other components of the MHC I‐peptide loading complex, such as tapasin, calreticulin, and ERp57 6, are also recruited to phagosomes and endosomes, suggesting that peptide loading could occur within endocytic and phagocytic compartments 7, 8, 9.
Regardless of the site where MHC class I molecules are loaded, the peptides that result from degradation by the proteasome have to be trimmed to be effectively complexed with MHC class I molecules. This cropping step can be performed by either the ER‐associated aminopeptidase 1 (ERAP 1) or the endosomal insulin‐responsive aminopeptidase (IRAP, that is in tight association with the small GTPase Rab14) since both of them have been shown to participate in cross‐presentation 10, 11.
The presence of IRAP in endocytic, Rab14‐positive compartments further suggests that peptide loading may occur therein 11, 12. In the last years, data supporting the possibility of endosomal and phagosomal loading of antigenic peptides onto MHC class I molecules have accumulated. Peptides loaded onto the peptide‐binding groove of MHC class I molecules are then transported to the cell surface, where they can be recognized by T cells, via the T‐cell receptor (TCR), thus providing what is currently called ‘signal 1’ for T‐cell activation 6.
The interaction between peptide‐loaded MHC class I molecules and the TCR, however, is insufficient for the initiation of T‐cell responses. The induction of CD8+ T‐cell responses in vivo by antigens that are internalized by DCs (as opposed to antigens that are expressed in DCs) is referred to as ‘cross‐priming’. Additional signals, called ‘2’ and ‘3’, consisting of co‐stimulatory molecules and cytokines, respectively, are indispensable to the development of effective T‐cell responses. Delivery of signals 2 and 3 by DCs requires their activation by innate signaling through pattern recognition receptors (PRR), such as toll‐like receptors (TLR). PRRs recognize conserved motifs in microbes or danger signals, referred to as pathogen‐ or damage‐associated molecular pattern molecules (PAMPs and DAMPs, respectively), such as bacterial lipopolysaccharide (LPS), methylated oligonucleotides (CpG), or viral double‐stranded RNA [‘mimicked’ with the synthetic analog polyinosinic‐polycytidylic acid (poly‐IC)] 13, 14.
Sensing of microbial stimuli causes DCs to enter a complex developmental program called ‘maturation’, which modifies DC morphology and functions profoundly. Maturation enhances transiently the capacity of DCs to take up antigens, before shutting it out after 20–40 h 15, 16, 17, 18. It increases the expression of co‐stimulatory molecules, including CD40, CD80, and CD86, and it causes the secretion of a wide variety of inflammatory cytokines and chemokines 19. In addition, the levels of MHC class I and class II are increased on activation. Finally, maturation leads to migration of DCs from tissues to draining lymph nodes 20, 21.
Understanding how cross‐presentation is modulated during DC maturation has been the subject of multiple studies. One recurrent limitation to address this question, however, is the lack of sensitive tools to quantify cross‐presentation per se. In most cases, others and we have relied on CD8+ T cells specific for different antigens (OVA, ovalbumin, quite often) to quantify the outcome of cross‐presentation. Restricted antibodies (recognizing an MHC class I molecule–peptide complex exclusively) are available for some model peptides, but only a few groups have succeeded to use them to detect antigen cross‐presentation 8, 9. Although antigen cross‐presentation can be measured quantitatively using antigen‐specific T cells in vitro, it becomes very complicated in vivo, mainly due to the difficulty in distinguishing the effects on cross‐priming and T‐cell stimulation from actual effects on cross‐presentation (i.e. the exposure of a peptide on a MHC class I molecule at the surface of a DC). In addition, experimental tools to block or promote cross‐presentation, especially in vivo, are also lacking. We, therefore, still do not know if cross‐presentation is a limiting step in cross‐priming and to what extent cross‐presentation contributes to CD8+ T‐cell responses in vivo.
Many recent studies have used Batf3‐defective mice, which lack CD8+ DCs and CD103+ DCs (the DC subtypes that cross‐present antigens most effectively at steady state) to address the role of cross‐presentation in different types of immune responses. It must be stressed out that other DC subpopulations can also cross‐present antigens both in vitro and in vivo 22, 23, 24, 25. In addition, CD8+ DCs have specific attributes besides cross‐presentation: they are critical for CD4+ T‐cell help to CD8+ T‐cell responses 26, 27 and are big producers of IL‐12 28, for example. It is, therefore, important to emphasize that it is not because a particular type of immune response requires CD8+ DCs that it necessarily means that cross‐presentation is involved.
Here, we will focus on how DC maturation affects antigen cross‐presentation in terms of cell biology, mainly reporting studies using mouse DCs in vitro or ex vivo (primarily because in most in vivo studies, it is difficult to distinguish between direct effects on antigen cross‐presentation and effects on co‐stimulation, or cytokine secretion, which can also affect cross‐priming). We will first review the different intracellular mechanisms that are involved at steady state in the control of antigen cross‐presentation in DCs. Timing will be the central focus as we will discuss how antigens encountered at different times after the initiation of maturation are or are not cross‐presented efficiently, and through which intracellular pathways. In order to ease the study of this complex scenario, we have divided DC maturation into three main phases: (i) early maturation (from 0 to 6 h of stimulation), referring to cells that take up an antigen and a danger signal simultaneously (e.g. a bacteria‐ or a virus‐infected cell), (ii) maturing DCs (from 7 to 20 h of maturation). This intermediate phase includes cells that have received the maturation stimulus and then take up additional antigen after having initiated their maturation program. And finally, (iii) fully mature DCs (over 20 h of maturation), in which cross‐presentation of newly acquired antigens is downregulated, but which are probably the cells that effectively activate naive CD8+ T cells for cross‐priming.
Intracellular regulation of antigen cross‐presentation
As mentioned earlier, antigens can be cross‐presented through the so‐called cytosolic pathway (i.e. TAP‐ and proteasome‐dependent) or through the ‘vacuolar’ pathway, independent of any cytosolic step. To what extent the cytosolic and vacuolar pathways contribute to cross‐presentation is not easy to evaluate. The experimental tools that are used most often, proteasome inhibitors and TAP1/2 KO cells, do not affect this pathway specifically. They also cause retention of MHC class I molecules in the ER, due to a lack of peptides to be loaded 29, 30, 31, reducing the amount of MHC class I molecules at the cell surface and in the endocytic pathway indirectly. One of the few studies that provided direct evidence for antigen cross‐presentation through the cytosolic pathway used an immune proteasome‐dependent epitope and showed defective cross‐presentation in low–molecular mass polypeptide‐7 (LMP7) KO DCs 32. The best evidence available, therefore, points toward the predominant use of the cytosolic, proteasome‐dependent pathway, at least in vivo 32. In vitro, while CD8+ and CD8− DCs cross‐present antigens mainly through the cytosolic pathway, bone marrow‐derived DCs (BMDCs) use both the cytosolic and the vacuolar pathways 33.
Several routes of intracellular trafficking have been shown to critically contribute to effective antigen cross‐presentation in DCs, including (i) regulation of antigen degradation, (ii) export to the cytosol and ER recruitment, and (iii) MHC class I: neo‐synthesis or recycling?
Regulation of antigen degradation
Antigens can be internalized by endocytosis or phagocytosis. During their journey from early endosomes or phagosomes to lysosomes, they undergo gradual proteolytic degradation. The whole process of trafficking from early compartments to lysosomes might destroy potentially important peptide epitopes for T‐cell activation. Mellman et al. 34 showed that BMDCs express low levels of lysosomal proteases, as compared with macrophages, suggesting that low degradation of incoming antigens favors antigen presentation. Lysosomes from DCs also bear an incompletely assembled V‐ATPase (the main proton transporter in lysosomes), which results in slow acidification and restricts the efficiency of antigen degradation (most lysosomal proteases are more active at low pH) 35. In addition, the NADPH oxidase 2 (NOX2) complex is rapidly recruited to forming phagosomes and slows down acidification by consuming protons for di‐oxygen production by dismutation 36. In NOX2‐defective DCs, acidification was increased and cross‐presentation reduced, suggesting that high pH (and thereby low degradation) favors antigen cross‐presentation 28.
Importantly, regulation of the pH is different in different subpopulations of DCs. In mice spleen, only CD8+ DCs, and not CD11b+ DCs, cross‐present antigens effectively at steady state 37, 38, 39, 40. CD8+ DCs are also the only ones in which NOX2 is efficiently recruited to phagosomes, due to the joint action of Rab27a and Rac2 41, 42. Rab27a controls the fusion of NOX2‐containing lysosome‐like vesicles with incoming phagosomes 42, while Rac2 regulates the assembly of the NOX2 cytosolic subunits to the phagosome membrane, rather than to the plasma membrane 41. In CD8− DCs, Rac1 promotes the assembly of NOX2 on the plasma membrane, similar to what happens in macrophages, resulting in low levels of phagosomal NOX2. High NOX2 activity in phagosomes from CD8+ DCs results in proton consumption and reduced acidification 36, 41. NOX2‐defective CD8+ DCs cross‐present antigen with reduced efficiencies, similar to CD8− DCs, suggesting that regulation of acidification is critical to the cross‐presenting function of CD8+ DCs. Why then is high pH in phagosomes required for efficient cross‐presentation? The most likely response to this question, even if direct evidence is lacking, is that high pH results in low degradation. The link between low degradation and high antigen cross‐presentation is also unclear. Interestingly, increased acidification in phagosomes does not affect MHC class II‐mediated antigen presentation 35, suggesting that either MHC class I‐restricted epitopes are more sensitive to degradation than MHC class II‐restricted ones or a cross‐presentation‐specific step is sensitive to pH/degradation.
Export to the cytosol and ER recruitment
Another critical step in cross‐presentation is antigen export to the cytosol. In DCs, antigen translocation to the cytosol was first reported by Kovacsovics‐Bankowski and Rock 5. They showed that gelonin (a membrane impermeable ribosomal inhibitor) conjugated to beads inhibited protein synthesis after internalization. In addition, they showed that binding proteins to latex beads (thereby forcing phagocytosis) increases antigen delivery to the cytosol and antigen cross‐presentation. Since then, many groups including us have provided evidence of the relevance of antigen export to the cytosol for cross‐presentation 43, 44, 45, 46. We also showed that export to the cytosol is more efficient in DCs than in macrophages and that it is also more efficient and rapid for small than for large dextran particles 45. This selectivity could reflect a difference in the efficiency of export itself or differences in the endocytic localization of the forms of dextran coupled to a non‐specific mechanism of export.
The molecular mechanism of translocation of antigens from endosomes and phagosomes to the cytosol remains, from our point of view, unclear. We showed a few years ago that Sec22b, a resident of the ERGIC compartment, is essential for TAP1/2 and other ER proteins to be delivered to phagosomes 47. Genetic ablation of Sec22b expression inhibits cross‐presentation, but not the direct presentation of endogenous antigens on MHC I molecules to CD8+ T cells or presentation of exogenous antigens on MHC II molecules to CD4+ T cells 47. In Sec22b‐defective DCs, antigen export was also reduced, suggesting that ER‐derived molecules are required for antigen export to the cytosol, and therefore for cross‐presentation. This again could be due to traffic of a putative transporter or to secondary differences in antigen distribution.
Consistent with the hypothesis that ER proteins could be delivered to endocytic and phagocytic vesicles and from these organelles perform functions otherwise involved in other cellular pathways, a decade ago the group of Peter Cresswell showed that Sec61 and p97 (also known as TER ATPase) were involved in antigen cross‐presentation and possible candidates for endocytosed antigen export to the cytosol 48. They suggested that these ER‐resident proteins, which are components of the ER‐associated protein degradation (ERAD) system of translocation of misfolded or unassembled proteins from the ER to the cytosol for ubiquitination and further destruction by the proteasome, were the mediators of antigen export from endocytic and phagocytic compartments to the cytosol.
Nevertheless, the role of Sec61 in ER‐to‐cytosol translocation has been challenged by the groups of Ploegh and Rappaport, who showed that Derlin‐1, and not Sec61, is in fact mediating ER‐retro‐translocation 49, 50. Recent evidence by Burgdorf et al. 51 shows that the activity of the translocon protein Sec61 is essential for endosome‐to‐cytosol antigen export. The silencing of Sec61 inhibited antigen cross‐presentation and partially affected export to the cytosol. Furthermore, a Sec61‐specific intrabody bearing an ER‐retention signal (which traps Sec61 in the ER, and prevents its recruitment to endosomes without losing functionality) also affected both antigen cross‐presentation and export to the cytosol, suggesting that Sec61 recruitment to endosomes is critical for antigen export to the cytosol and cross‐presentation. It is very difficult, however, to exclude that the effects of reducing Sec61 expression do not affect export to the cytosol indirectly (the role of Sec61 in the translocation of newly translated leader‐peptide bearing proteins from the cytosol into the ER is clear, and any interference with Sec61 functions should inhibit the expression of a large variety of membrane proteins, including MHC class I). The elegant experiment of retaining Sec61 in the ER using a specific intrabody is quite convincing, although the authors did not show that the intrabody does not interfere with the ER‐to‐cytosol translocation activity of Sec61.
Later on, our group suggested a link between lipid bodies (LBs) and cross‐presentation and proposed an alternative model for antigen export to the cytosol 52. LBs, also known as lipid droplets, are lipid‐filled cytosolic structures enclosed by a lipid monolayer, originating from the ER and involved in diverse biological functions 53, 54. The results provided evidence that a molecule associated with LBs, IGTP (also known as Irgm3), could be a key player in the cross‐presentation pathway since cells deficient for IGPT displayed less amount of LBs and were impaired in cross‐presentation, but not in MHC class II antigen presentation 51. The interaction between LBs and the phagosomal membrane could be inducing the reorganization of their membranes, favoring peptide export to the cytosol. However, and in spite of the effort done by many groups to clarify this issue, translocation of peptides from the phagosome to the cytoplasm is still poorly defined.
MHC class I molecules: neo‐synthesis or recycling?
Where do MHC class I molecules involved in cross‐presentation come from? This question was approached throughout the years and different sources were proposed. The idea that the MHC class I pool came from the plasma membrane and was being recycled to endosomes was supported by the fact that a tyrosine residue conserved at the cytosolic end of the MHC class I molecule, essential for internalization from the cell surface, was critical for cross‐presentation 55. In this way, after peptide loading (or exchange), MHC class I molecules would recycle back to the cell surface without the need to use newly synthesized molecules. On the other hand, evidence supporting the hypothesis of new molecules being used in cross‐presentation was published recently 55. Basha et al. demonstrated that the machinery involved in trafficking of newly synthesized MHC class I molecules from the ER to endosomes is indispensable for the cross‐presentation of cell‐associated antigens. Moreover, this trafficking step seems to be mediated by the MHC class II‐associated invariant chain CD74 56. Also, MHC class I molecules have been localized in tight association with Rab3b/Rab3c recycling vesicles, but whether these MHC class I molecules are involved in cross‐presentation, and through which pathway (vacuolar or cytosolic), has not been addressed so far 57.
Blander et al. have recently shown that MHC class I molecules selectively accumulate in phagosomes carrying microbial components. MHC class I molecules are recruited from an endosomal recycling compartment (ERC), which contains Rab11a, VAMP3, and VAMP8 and maintain large reserves of MHC class I molecules. Rab11a mediates trafficking of MHC class I molecules from the cell surface to the ERC, whereas the subsequent recruitment of MHC class I molecules to cross‐presenting phagosomes is driven by SNAP23.
Altogether, the above‐discussed studies suggest a complex picture of the traffic pathways that antigens and MHC class I molecules follow in DCs. In the next sections, we will attempt to describe how TLR engagement and cell maturation modify and regulate these antigen cross‐presentation pathways. Because the effects of TLR engagement change according to the time of maturation, we will divide the following sections into three sections describing how TLR engagement affects cross‐presentation immediately after uptake of a TLR bearing cargo, in the first 7–20 h after TLR engagement (a time during which internalized antigens are cross‐presented with increased efficiency) and after 20 h when cross‐presentation is shut down.
Cross‐presentation early after dendritic cell maturation
In many circumstances, DCs encounter antigens and danger signals simultaneously. This occurs, for example, when DCs take up bacteria or virus‐infected dead cells. TLR and NLR ligands are released, and innate sensors are engaged within endosomes and phagosomes. Different forms of antigen can be cross‐presented both in vitro and in vivo, including soluble proteins, immune complexes, antigen‐coated latex beads, microbes, and dead cells 18, 58, 59, 60, 61, 62. Uptake of cargo bearing ligands for innate sensors increases the efficiency of antigen cross‐priming in vivo (which could be due to increases in antigen cross‐presentation per se, in the expression of T cell co‐stimulatory or adhesion molecules, in cytokine secretion, or in all). In vitro several studies show increased cross‐presentation 17, 58, 63. In the case of soluble antigens, Kurts and colleagues show that OVA with low endotoxin levels is less efficiently cross‐presented by DCs than commercial, highly endotoxin‐contaminated preparations 9. Not surprisingly, the addition of LPS to low endotoxin OVA preparations restores the capacity of DC to cross‐present efficiently. Reversely, DCs deficient for TLR4, Myd88, or TRIF exhibit a decrease in cross‐presentation of endotoxin‐containing OVA, as compared with wildtype DCs 9, 51. These authors also show a critical role for the early recruitment of TAP1 and TAP2 to endosomes, indicating that peptide loading onto class I MHC molecules during cross‐presentation occurs in endocytic compartments rather than the ER.
Others and we have analyzed the effects of engaging TLRs within phagosomes, and several molecular mechanisms accounting for increased antigen cross‐presentation have been reported. Enhanced cross‐presentation concerns exclusively the cargo that is present in the same individual phagosome as the innate sensor, TLR 58, 64 or IgG receptor 63, 65. Cargoes in other phagosomes within the same single DC do not show increased cross‐presentation. By preventing effective cross‐presentation of bystander antigens (self dead cells, e.g., that were taken up by the same DCs that also captured a bacteria), this autonomous phagosome behavior could limit the presentation of self‐antigens and the activation of T cells directed against self‐antigens. LPS, however, also promotes MyD88‐dependent phagosome tubulation, which favors exchange of cargo between phagosomes within single cells 66.
Increased cross‐presentation soon after innate sensing is the result of different mechanisms that are triggered early on. In the first hour after exposure to microbial signals (0–5 h), the capacity of DCs to take up antigens, both by macropinocytosis and endocytosis, is increased 15, 16, 17, 18. Colin Watts et al. 15, 16 showed that increased macropinocytosis is mediated by TLR‐induced actin remodeling. The MAP kinase‐activated kinase Rsk is the key regulator during the TLR‐induced boost in endocytosis. Gil‐Torregrosa et al. 17 also showed that uptake of OVA‐immune complexes is enhanced after 3–5 h of LPS activation. Early DC activation also causes endogenous ubiquitinated proteins to accumulate transiently in Aggresome‐Like‐Induced Structures (DALIS), a phenomenon proposed to sequester endogenous proteins to promote a more efficient processing of exogenous proteins 67. The appearance of DALIS correlates with higher efficiency in antigen cross‐presentation during the first hours after DC activation 67, 68. DALIS are also most abundant in cross‐presenting CD8+ DC subpopulations than in CD11b+ DCs 69.
Intracellularly, a critical step in antigen cross‐presentation is the export of internalized antigens to the cytosol, as mentioned before. We showed that early after LPS activation of DCs, antigen translocation to the cytosol is enhanced 17. Delamarre et al. 58 have also demonstrated that DCs activated for a short period show an increase in antigen transport to the cytosol. The mechanisms responsible for this increase remain unexplored. The reduction in the recruitment of active lysosomal proteases to phagosomes induced by LPS might give more time for an antigen to be transported to the cytosol 35. Reduced antigen degradation was also linked to more efficient antigen cross‐presentation in different systems, again probably through the preservation of antigenic epitopes and increased antigen export to the cytosol 27, 54, 59, 60.
More recently, the recruitment of MHC class I molecules to phagosomes was shown to be strongly enhanced by TLR4 stimulation. As mentioned before, Blander et al. have shown that MHC class I molecules are stored in ERC containing the small GTPase Rab11a and the t‐SNARE proteins VAMP3 and VAMP8. TLR4 engagement leads to IKK2‐dependent phosphorylation of phagosome‐associated SNAP‐23 (synaptosome‐associated protein of 23 kDa) on TLR‐containing phagosomes. Phosphorylated SNAP23 is stabilized by SNARE complexes (SNAP‐23/Syntaxin4/Vamp‐3) promoting fusion between ERCs and phagosomes. Enrichment of phagosomes with ERC‐derived MHC I molecules results in increased antigen cross‐presentation 58. Increased antigen cross‐presentation early after DC activation is, therefore, the result of multiple coordinated changes in both antigen and MHC class I intracellular traffic.
An intermediate phase during dendritic cell maturation
Even if DC maturation finally results in the complete shutdown of antigen presentation (see next section), others and us have shown that several hours after TLR engagement (once the expression of activation markers is fully upregulated), DCs are still capable of cross‐presenting antigens, and furthermore, that the efficacy of cross‐presentation is increased. The duration of this intermediate phase of enhanced antigen cross‐presentation can vary between different experimental systems, types of DCs, and maturation stimuli. In most cases, it lasts at least 16–20 h before cross‐presentation is shut down 20–24 h after the initiation of DC activation. We will refer to these DCs as maturing DCs 70, 71, 72, 73.
In what physiological circumstance can DCs encounter antigen several hours after innate stimuli? First, it is most likely that even if a DC takes up one pathogen that initiates its maturation program, it can still encounter additional microbes, and their uptake could increase the amount of antigen available for the initiation of an adaptive immune response. It is also expected that soluble TLR ligands released as an infection progresses can activate distant DCs (that have not encountered an intact microbe yet). If these DCs activated at a distance can internalize and cross‐present antigens for few hours, they could contribute to effective cross‐presentation in vivo. In particular, anatomical locations, including mucosa, where DCs are continuously activated by commensal microbiota, activated DCs could remain competent to take up and cross‐present antigens for some time before leaving the tissue through the lymphatics 74. High levels of cross‐presentation by maturing DCs have actually been reported in different experimental systems 70, 71, 72, 75.
What are then the mechanisms that mediate the enhancement in cross‐presentation in maturing DCs? During the first few hours after DC activation, the endocytic capacity of DCs starts to decrease (both in vivo and in vitro) 70, 72, 76. Therefore, although internalization remains active during this intermediate stage, increased uptake is certainly not the reason maturing DCs cross‐present antigens with better efficiency. Villadangos et al. 70 showed that systemic exposure of mice to the TLR ligands CpG, LPS, Poly‐IC, or the parasite Plasmodium berghei abolishes cross‐presentation of cell‐associated OVA supplied 9–12 h after stimulation, but not endogenous antigen presentation to T cells. Nevertheless, it is unclear in this system, if reduced uptake by CD8+ DCs was due to an intrinsic TLR‐induced defect in internalization or a consequence of reduced access to the antigen in mice treated systemically 77.
On the contrary, Drutman and Trombetta 73 showed that DCs matured in vivo for 16 h still take up and cross‐present soluble antigen efficiently. In this study, the authors induced DC maturation in vivo by intravenous injection of LPS. Sixteen hours later, mice were immunized either with soluble OVA or with a variant OVA chain devoid of carbohydrates, and therefore unlikely to be internalized by lectin‐like receptors. Cross‐presentation of antigens was not affected by maturation, and even slightly increased in some of the experimental conditions. Delamarre et al. 75 also reported that soluble OVA cross‐presentation by BMDCs was increased after 7 (and 10) h of LPS stimulation, although OVA uptake occurred before maturation was triggered in this experimental setting.
We showed recently that even though phagocytosis in both BMDCs – in vitro – and splenic CD8+ DCs – in vivo – is decreased after 12–16 h of TLR engagement, these DCs still cross‐present antigens with high efficiency, as compared to immature non‐activated DCs 72. Higher levels of cross‐presentation in vivo were observed after injection of soluble, bead‐bound and immune complexed OVA, while the cross‐presentation of dead‐cell‐associated OVA was already abolished. It is possible that maturing DCs shut down the uptake of dead cells (a potential source of autoantigens) sooner, while remaining capable of taking up particles bearing TLR ligands (possible source of infectious antigens).
Enhanced cross‐presentation by maturing DCs resulted from delayed phagosomal degradation and decreased recruitment of lysosomal proteases to phagosomes. Different TLR ligands, including LPS, CpG (TLR9 agonist), and R848 (TLR7 cognate ligand), promote, to various extents, sequestration of lysosomes away from phagosomes. In addition, the speed of phagosome migration after uptake was strongly decreased in maturing DCs, as compared with phagosomes from resting DCs. In conclusion, perinuclear clustering of lysosomes in maturing DCs, together with reduced displacements of phagosomes along microtubules, prevented phagolysosomal fusion, thus preserving antigens from degradation, and thereby increasing the efficiency of antigen cross‐presentation.
We also showed that silencing of the small GTPase Rab34 releases perinuclear lysosomal sequestration and reverts the inhibition of phagolysosome fusion induced by TLR engagement, without disturbing phagosomal or lysosomal functions and the distribution of these organelles in resting DCs. In addition, Rab34 silencing also prevents the increase in cross‐presentation induced by LPS, but does not affect cross‐presentation in immature DCs. Therefore, a reduction in the fusion between phagosomes and lysosomes accounts for the increase in the cross‐presentation efficiency observed in maturing DCs. These results suggest that DCs have developed a dedicated mechanism to control fusion between phagosomes and lysosomes, based on a Rab34‐dependent spatial redistribution of lysosomes that transiently increases the efficacy of cross‐presentation in maturing DCs.
Interestingly, even though several TLR ligands induced lysosomal clustering after 16‐h incubation, TNF (an inflammatory cytokine that activates DCs and is produced by different phagocytes in response to TLR engagement) did not induce lysosomal clustering before 24 h of activation (our unpublished results). This result is consistent with previous studies showing increased cross‐presentation by TNF‐activated DCs in vivo 78, 79, but suggests that the timing of the process might be delayed as compared to TLR‐induced activation. Depending on the experimental systems and the nature of the maturation stimuli, the inhibition of phagolysosome fusion and the functional consequences on antigen presentation most likely occur with different kinetics.
Decreased cross‐presentation in fully mature dendritic cells
In most experimental settings, the complex program triggered after TLR engagement in DCs reaches its final stage after approximately 24 h. In many published studies, the incubation time employed to induce DC maturation is referred to as ‘overnight’ with no clear reference to the exact time length. In our hands, 16 and 24 h of maturation make a genuine difference in terms of cross‐presentation and the clarification of the certain duration of the incubation time may help to reconcile apparently contradictory data that have been published for over a decade regarding fully mature DCs.
Our initial work comparing different maturation stages achieved by incubating BMDCs with LPS for different time lengths showed that DCs stimulated for 24–40 h cross‐presented OVA‐immune complexes less efficiently than DCs matured for 5 h 17. More recently, we showed that after 24 h of activation, the efficiency of cross‐presentation of both soluble and bead‐bound OVA was reduced, as compared to unstimulated DCs and to DCs treated with LPS for 16 h. No differences in the levels of surface expression of classical maturation markers (including CD80/86, CD40, MHC class II) were observed between cells treated with LPS for 16, 24, or 40 h 17, 72. Down modulation of cross‐presentation after 24 h (but not 16 h) of treatment with CpG was also reported by Raz et al. 80. Surprisingly, in this study, LPS did not induce an increase in cross‐presentation while poly(IC) and CpG did. It is possible that the LPS preparation used was contaminated with peptidoglycan, which was since then shown to inhibit cross‐presentation 81.
Why then do these fully mature cells fail to cross‐present antigens? We will briefly review the different mechanistic explanations that have been proposed throughout the years and finally, discuss very recent published data explaining how antigen degradation mediated by TFEB in mature DCs is apparently responsible for the shut‐off of cross‐presentation in fully mature DCs 82.
Antigen uptake
Mature DCs were initially described as incompetent to take up antigens by endocytosis or phagocytosis. At the time, this decrease in antigen uptake was the first mechanism proposed to explain the reduction in antigen presentation to CD4+ and CD8+ T cells by mature DCs 17, 83. However, recent studies show that endocytosis and phagocytosis are actually only slowed down or slightly decreased in mature DCs, and we observe that fully mature DCs still take up considerable amounts of antigen, particulate, or soluble 22, 27, 32, 33. Although maturation of DCs with different TLR agonists reduced soluble OVA endocytosis to varying degrees, there was no direct correlation between decreased uptake and decreased cross‐presentation 80. Virus‐infected necrotic cells, as well as OVA‐coated beads, were slightly less phagocytosed by fully mature DCs than by untreated cells 84. To exclude antigen as the limiting factor, immature cells were fed with half the amount of antigen and still more cross‐presentation was detected compared to fully matured DCs 84. Platt et al. also showed that DCs stimulated with LPS for 20 h could still phagocytose particles, especially if they were coated with IgG 77. Altogether, the published results suggest that antigen uptake decreases gradually as DCs mature, but the rate of reduction depends on the nature of the stimuli and of the endocytic pathway (endocytosis or phagocytosis), as well as on the nature of the endocytic receptor involved. In some experimental models, such as IgG‐complexed antigens, the efficiency of phagocytosis does not seem to account for reduced cross‐presentation, while in others, such as dead cells, it probably does.
Antigen export to the cytosol
Our results suggested that decreased cross‐presentation after 24–40 h of LPS stimulation is due to reduced antigen export to the cytosol 17. Nevertheless, reduced uptake and increased degradation in mature DCs make the specific measurement of antigen export to the cytosol by itself very difficult. Identification of the mechanism responsible for antigen export to the cytosol might allow addressing this issue in more detail.
pH and antigen degradation
An inverse relationship between the efficiencies of antigen cross‐presentation and acidification/degradation has long been established by others and us 85, 86. Mellman et al. 87 showed that after 20 h of LPS stimulation, lysosome acidification is enhanced due to the recruitment of the cytosolic subunits of the V‐ATPase to endosomes and lysosomes. To what extent this increase in acidification contributes to shutting down cross‐presentation was not addressed in these articles. A recent study by the Cresswell team 82 shows that TFEB, a transcriptional factor that regulates the lysosomal biogenesis pathway 88, is critical for the control of antigen presentation by DCs. TFEB is a master regulator of the CLEAR network and controls autophagy through the Lysosome Nutrient Sensing machinery (LYNUS), designed to integrate nutrient sensing with lysosomal biogenesis and degradation 89.
Overexpression of TFEB induces acidification of lysosomes in immature DCs, as well as increased expression of lysosomal enzymes, which results in increased antigen degradation and decreased cross‐presentation 82. After 24 h of treatment with LPS, both TFEB expression and its translocation to the nucleus are strongly induced in DCs. Upon silencing of the tfeb gene, the decrease in cross‐presentation seen in fully mature BMDCs is partially restored. On the contrary, TFEB had very little effect on cross‐presentation by immature DCs. TFEB also increased the expression of lysosomal proteases, but not of TAP transporters or the proteasome, suggesting that the effect of TFEB on antigen presentation is due to the control of antigen degradation and not export to the cytosol or other subsequent steps in peptide loading on MHC molecules 82.
Overall, the published results suggest that down modulation of antigen cross‐presentation in mature DCs is orchestrated by TFEB, which induces the change of a mildly acidic and not very degradative endocytic environment into a more acidic and degradative one. The later is not favorable for antigen cross‐presentation, most likely due to the degradation of valuable antigens and reduced export to the cytosol.
Conclusion
We propose herein to distinguish, throughout DC maturation, three phases during which antigen cross‐presentation is differentially regulated. Fig. 1 summarizes all the intracellular mechanisms orchestrating cross‐presentation in DCs, highlighting those routes that are directly or indirectly affected by innate sensing.
Figure 1.
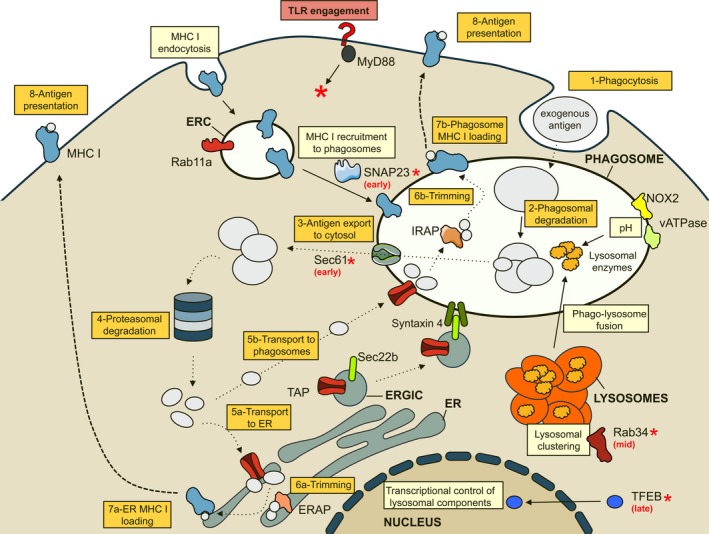
Schematic representation of the intracellular mechanisms operating throughout the ‘cytosolic’ cross‐presentation pathways (vacuolar pathway not shown). Molecular players (marked with red asterisks) involved in the regulation of these pathways during dendritic cell maturation after toll‐like receptor engagement and the stage (early, mid, or late) at which they act are shown. Exogenous antigens are phagocytosed (1) and partially degraded within the phagosome by lysosomal enzymes (2) that reach the organelle after phagolysosomal fusion, which can be inhibited through lysosomal perinuclear clustering mediated by Rab34. Enzymatic activity of lysosomal enzymes depends on the intraluminal pH which, in turn, is regulated by two systems that are recruited to the phagosomal membrane: vATPase and NOX2. Additionally, lysosomal enzyme expression levels and phagosomal pH are controlled by the transcriptional factor TFEB. After this initial degradation step, the resulting polypeptides are exported to the cytosol (3) by the translocon protein Sec61 and are further processing by the proteasome (4). Peptides are transported to their loading site (5), either the ER (5a) or back to the phagosome (5b). Regardless of the loading site, peptides are trimmed (6) by resident aminopeptidases (IRAP or ERAP) and loaded onto MHC class I molecules (7). In the ER MHC I loading pathway, all participants are present already in the ER, and newly synthesized MHC I molecules are used (7a). On the contrary, in the phagosomal MHC I loading pathway, the loading complex components (including the transporter associated with antigen processing – TAP) are recruited to the phagosome through the interaction between the SNARE Sec22b (resident of the ER‐Golgi intermediate compartment – ERGIC) and syntaxin 4. MHC class I molecules themselves reach the phagosome to be loaded with peptides through SNAP23‐mediated fusion of Rab11a‐positive endosomal recycling compartments (ERC) bringing MHC class I molecules from the cell surface (7b). Finally, MHC class I–peptide complexes are transported to the cell surface where antigen presentation (8) to T cells takes place.
During the first phase of early maturation, up to 6 h after DC activation, the presence of a TLR ligand that is internalized together with the antigen promotes antigen cross‐presentation. Increased cross‐presentation during this early phase is phagosome autonomous (it only affects the individual phagosomes that contain the innate sensor ligand). The central mechanisms involved are (i) reduced recruitment of active lysosomal proteases 35 and (ii) increased recruitment of MHC class I molecules that is induced by TLR4 and requires SNAP23 and Rab11 58. During the second phase of DC maturation (between 7 and 20 h after activation), maturing DCs are still competent to take up certain antigens (immune complexes or bead‐bound antigen, but not dead cells, for example) and to cross‐present them with very high efficacy. Increased antigen cross‐presentation efficiency in maturing DCs is mainly due to Rab34‐dependent sequestration of lysosomes away from phagosomes. Reduced phagosome‐lysosome fusion strongly delays degradation and promotes antigen cross‐presentation 72. During a final phase of maturation (after 20–24 h after DC activation, depending on the type of DC and stimulus), antigen cross‐presentation is impaired. Reduced cross‐presentation is mainly due to increased acidification under the control of TFEB 82.
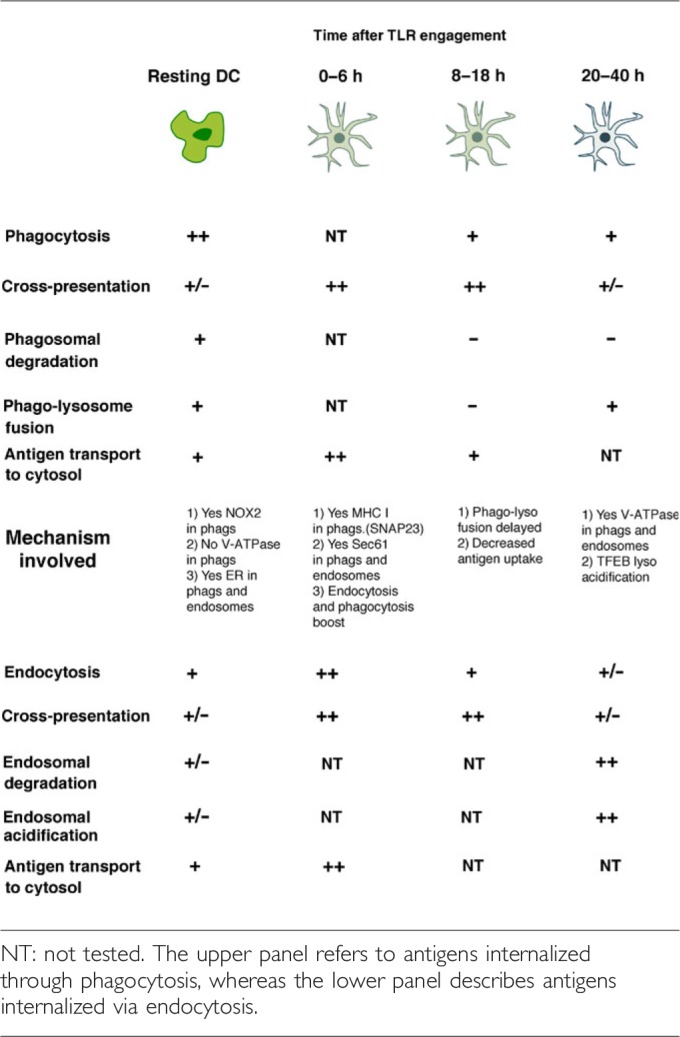
While our understanding of how antigen presentation is modulated becomes more precise, the literature in the field is still heterogeneous and quite confusing. Depending on the experimental systems, the origin of the DC populations employed, the type of stimuli used, and the internalization route, the exact kinetics of the different phases may differ. Table 1 recapitulates this information. In order to be able to translate this knowledge into useful ways to manipulate the immune system in the clinics, we now need to explore the relevance of these findings in human DCs.
Table 1.
Summary of the characteristics of the different stages of dendritic cell (DC) maturation as well as the critical intracellular steps and the mechanisms involved in the control of cross‐presentation at each stage
Acknowledgements
We thank Dr. C. Thery and Dr. E. Segura for discussion. A.A. was supported by a fellowship from EMBO (ALTF 883‐2011) and ERC DC BIOX340046. F.K. is supported by CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas, Argentina). J.G.M. received funding from ANR‐15‐CHIN‐0002‐01. Part of the work described in this article was supported by the grants ANR‐10‐IDEX‐0001‐02 PSL, ANR‐11 LABX‐0043, ERC DC BIOX340046 and La Ligue Nationale contre le Cancer (EL2014.LNCC/SA). The authors have no conflicts of interest to declare.
[The copyright line in this article was changed on 19 July 2016 after original online publication.].
References
- 1. Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev 2007;219:143–156. [DOI] [PubMed] [Google Scholar]
- 2. Joffre OP, Segura E, Savina A, Amigorena S. Cross‐presentation by dendritic cells. Nat Rev Immunol 2012;12:557–569. [DOI] [PubMed] [Google Scholar]
- 3. Shen L, Sigal LJ, Boes M, Rock KL. Important role of cathepsin S in generating peptides for TAP‐independent MHC class I crosspresentation in vivo. Immunity 2004;21:155–165. [DOI] [PubMed] [Google Scholar]
- 4. Bertholet S, et al. Leishmania antigens are presented to CD8+ T cells by a transporter associated with antigen processing‐independent pathway in vitro and in vivo. J Immunol 2006;177:3525–3533. [DOI] [PubMed] [Google Scholar]
- 5. Kovacsovics‐Bankowski M, Rock KL. A phagosome‐to‐cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 1995;267:243–246. [DOI] [PubMed] [Google Scholar]
- 6. Blum JS, Wearsch PA, Cresswell P. Pathways of antigen processing. Annu Rev Immunol 2013;31:443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Houde M, et al. Phagosomes are competent organelles for antigen cross‐presentation. Nature 2003;425:402–406. [DOI] [PubMed] [Google Scholar]
- 8. Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Endert Van P, Amigorena S. ER‐phagosome fusion defines an MHC class I cross‐presentation compartment in dendritic cells. Nature 2003;425:397–402. [DOI] [PubMed] [Google Scholar]
- 9. Burgdorf S, Schölz C, Kautz A, Tampé R, Kurts C. Spatial and mechanistic separation of cross‐presentation and endogenous antigen presentation. Nat Immunol 2008;9:558–566. [DOI] [PubMed] [Google Scholar]
- 10. Firat E, et al. The role of endoplasmic reticulum‐associated aminopeptidase 1 in immunity to infection and in cross‐presentation. J Immunol 2007;178:2241–2248. [DOI] [PubMed] [Google Scholar]
- 11. Saveanu L, et al. IRAP identifies an endosomal compartment required for MHC class I cross‐presentation. Science 2009;325:213–217. [DOI] [PubMed] [Google Scholar]
- 12. Weimershaus M, Maschalidi S, Sepulveda F, Manoury B, van Endert P, Saveanu L. Conventional dendritic cells require IRAP‐Rab14 endosomes for efficient cross‐presentation. J Immunol 2012;188:1840–1846. [DOI] [PubMed] [Google Scholar]
- 13. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell 2006;124:783–801. [DOI] [PubMed] [Google Scholar]
- 14. Medzhitov R. Toll‐like receptors and innate immunity. Nat Rev Immunol 2001;1:135–145. [DOI] [PubMed] [Google Scholar]
- 15. West MA, et al. TLR ligand‐induced podosome disassembly in dendritic cells is ADAM17 dependent. J Cell Biol 2008;182:993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. West MA, et al. Enhanced dendritic cell antigen capture via toll‐like receptor‐induced actin remodeling. Science 2004;305:1153–1157. [DOI] [PubMed] [Google Scholar]
- 17. Gil‐Torregrosa BC, et al. Control of cross‐presentation during dendritic cell maturation. Eur J Immunol 2004;34:398–407. [DOI] [PubMed] [Google Scholar]
- 18. Granucci F, Ferrero E, Foti M, Aggujaro D, Vettoretto K, Ricciardi‐Castagnoli P. Early events in dendritic cell maturation induced by LPS. Microbes Infect 1999;1:1079–1084. [DOI] [PubMed] [Google Scholar]
- 19. Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol 2006;6:476–483. [DOI] [PubMed] [Google Scholar]
- 20. Vargas P, et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat Cell Biol 2015;18:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faure‐André G, et al. Regulation of dendritic cell migration by CD74, the MHC class II‐associated invariant chain. Science 2008;322:1705–1710. [DOI] [PubMed] [Google Scholar]
- 22. Lin M‐L, Zhan Y, Villadangos JA, Lew AM. The cell biology of cross‐presentation and the role of dendritic cell subsets. Immunol Cell Biol 2008;86:353–362. [DOI] [PubMed] [Google Scholar]
- 23. Belz GT, et al. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I‐restricted antigen presentation after lung infection with virus. Proc Natl Acad Sci USA 2004;101:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. del Rio M‐L, Rodriguez‐Barbosa J‐I, Kremmer E, Förster R. CD103‐ and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross‐presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol 2007;178:6861–6866. [DOI] [PubMed] [Google Scholar]
- 25. Waithman J, et al. Skin‐derived dendritic cells can mediate deletional tolerance of class I‐restricted self‐reactive T cells. J Immunol 2007;179:4535–4541. [DOI] [PubMed] [Google Scholar]
- 26. Eickhoff S, et al. Robust anti‐viral immunity requires multiple distinct T cell‐dendritic cell interactions. Cell 2015;162:1322–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity 2015;43:554–565. [DOI] [PubMed] [Google Scholar]
- 28. Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O'Keeffe M. Differential production of IL‐12, IFN‐alpha, and IFN‐gamma by mouse dendritic cell subsets. J Immunol 2001;166:5448–5455. [DOI] [PubMed] [Google Scholar]
- 29. Chefalo PJ, Grandea AG, Van Kaer L, Harding CV. Tapasin‐/‐ and TAP1‐/‐ macrophages are deficient in vacuolar alternate class I MHC (MHC‐I) processing due to decreased MHC‐I stability at phagolysosomal pH. J Immunol 2003;170:5825–5833. [DOI] [PubMed] [Google Scholar]
- 30. Van Kaer L, Ashton‐Rickardt PG, Ploegh HL, Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4‐8+ T cells. Cell 1992;71:1205–1214. [DOI] [PubMed] [Google Scholar]
- 31. Day PM, Esquivel F, Lukszo J, Bennink JR, Yewdell JW. Effect of TAP on the generation and intracellular trafficking of peptide‐receptive major histocompatibility complex class I molecules. Immunity 1995;2:137–147. [DOI] [PubMed] [Google Scholar]
- 32. Palmowski MJ, et al. Role of immunoproteasomes in cross‐presentation. J Immunol 2006;177:983–990. [DOI] [PubMed] [Google Scholar]
- 33. Segura E, Albiston AL, Wicks IP, Chai SY, Villadangos JA. Different cross‐presentation pathways in steady‐state and inflammatory dendritic cells. Proc Natl Acad Sci USA 2009;106:20377–20381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen‐presenting cells determines antigen fate. Science 2005;307:1630–1634. [DOI] [PubMed] [Google Scholar]
- 35. Lennon‐Duménil A‐M, et al. Analysis of protease activity in live antigen‐presenting cells shows regulation of the phagosomal proteolytic contents during dendritic cell activation. J Exp Med 2002;196:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Savina A, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006;126:205–218. [DOI] [PubMed] [Google Scholar]
- 37. Schnorrer P, et al. The dominant role of CD8+ dendritic cells in cross‐presentation is not dictated by antigen capture. Proc Natl Acad Sci USA 2006;103:10729–10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pooley JL, Heath WR, Shortman K. Cutting edge: intravenous soluble antigen is presented to CD4 T cells by CD8‐ dendritic cells, but cross‐presented to CD8 T cells by CD8+ dendritic cells. J Immunol 2001;166:5327–5330. [DOI] [PubMed] [Google Scholar]
- 39. Iyoda T, et al. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J Exp Med 2002;195:1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schulz O, Reis e Sousa C. Cross‐presentation of cell‐associated antigens by CD8alpha+ dendritic cells is attributable to their ability to internalize dead cells. Immunology 2002;107:183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Savina A, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8+ dendritic cells. Immunity 2009;30:544–555. [DOI] [PubMed] [Google Scholar]
- 42. Jancic C, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol 2007;9:367–378. [DOI] [PubMed] [Google Scholar]
- 43. Norbury CC, Hewlett LJ, Prescott AR, Shastri N, Watts C. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity 1995;3:783–791. [DOI] [PubMed] [Google Scholar]
- 44. Norbury CC, Chambers BJ, Prescott AR, Ljunggren HG, Watts C. Constitutive macropinocytosis allows TAP‐dependent major histocompatibility complex class I presentation of exogenous soluble antigen by bone marrow‐derived dendritic cells. Eur J Immunol 1997;27:280–288. [DOI] [PubMed] [Google Scholar]
- 45. Rodriguez A, Regnault A, Kleijmeer M, Ricciardi‐Castagnoli P, Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat Cell Biol 1999;1:362–368. [DOI] [PubMed] [Google Scholar]
- 46. Lin ML, et al. Selective suicide of cross‐presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc Natl Acad Sci USA 2008;105:3029–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cebrian I, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 2011;147:1355–1368. [DOI] [PubMed] [Google Scholar]
- 48. Ackerman AL, Giodini A, Cresswell P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 2006;25:607–617. [DOI] [PubMed] [Google Scholar]
- 49. Lilley BN, Ploegh HL. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004;429:834–840. [DOI] [PubMed] [Google Scholar]
- 50. Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro‐translocation from the ER lumen into the cytosol. Nature 2004;429:841–847. [DOI] [PubMed] [Google Scholar]
- 51. Zehner M, et al. The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross‐presentation to CD8+ T cells. Immunity 2015;42:850–863. [DOI] [PubMed] [Google Scholar]
- 52. Bougnères L, et al. A role for lipid bodies in the cross‐presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 2009;31:232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fujimoto T, Ohsaki Y, Cheng J, Suzuki M, Shinohara Y. Lipid droplets: a classic organelle with new outfits. Histochem Cell Biol 2008;130:263–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ploegh HL. A lipid‐based model for the creation of an escape hatch from the endoplasmic reticulum. Nature 2007;448:435–438. [DOI] [PubMed] [Google Scholar]
- 55. Lizée G, et al. Control of dendritic cell cross‐presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol 2003;4:1065–1073. [DOI] [PubMed] [Google Scholar]
- 56. Basha G, et al. A CD74‐dependent MHC class I endolysosomal cross‐presentation pathway. Nat Immunol 2012;13:237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zou L, et al. The GTPase Rab3b/3c‐positive recycling vesicles are involved in cross‐presentation in dendritic cells. Proc Natl Acad Sci USA 2009;106:15801–15806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nair‐Gupta P, et al. TLR signals induce phagosomal MHC‐I delivery from the endosomal recycling compartment to allow cross‐presentation. Cell 2014;158:506–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Regnault A, et al. Fcgamma receptor‐mediated induction of dendritic cell maturation and major histocompatibility complex class I‐restricted antigen presentation after immune complex internalization. J Exp Med 1999;189:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol 2014;14:94–108. [DOI] [PubMed] [Google Scholar]
- 61. Sancho D, et al. Tumor therapy in mice via antigen targeting to a novel, DC‐restricted C‐type lectin. J Clin Invest 2008;118:2098–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Malik AF, et al. Inflammasome components Asc and caspase‐1 mediate biomaterial‐induced inflammation and foreign body response. Proc Natl Acad Sci USA 2011;108:20095–20100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baker K, et al. Neonatal Fc receptor for IgG (FcRn) regulates cross‐presentation of IgG immune complexes by CD8‐CD11b+ dendritic cells. Proc Natl Acad Sci USA 2011;108:9927–9932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Blander JM, Medzhitov R. Toll‐dependent selection of microbial antigens for presentation by dendritic cells. Nature 2006;440:808–812. [DOI] [PubMed] [Google Scholar]
- 65. Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S. From the Cover: Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc Natl Acad Sci USA 2012;109:14556–14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mantegazza AR, Zajac AL, Twelvetrees A, Holzbaur ELF, Amigorena S, Marks MS. TLR‐dependent phagosome tubulation in dendritic cells promotes phagosome cross‐talk to optimize MHC‐II antigen presentation. Proc Natl Acad Sci USA 2014;111:15508–15513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lelouard H, et al. Dendritic cell aggresome‐like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol 2004;164:667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Herter S, et al. Dendritic cell aggresome‐like‐induced structure formation and delayed antigen presentation coincide in influenza virus‐infected dendritic cells. J Immunol 2005;175:891–898. [DOI] [PubMed] [Google Scholar]
- 69. Mintern JD, et al. Differential use of autophagy by primary dendritic cells specialized in cross‐presentation. Autophagy 2015;11:906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wilson NS, et al. Systemic activation of dendritic cells by tsoll‐like receptor ligands or malaria infection impairs cross‐presentation and antiviral immunity. Nat Immunol 2006;7:165–172. [DOI] [PubMed] [Google Scholar]
- 71. Vega‐Ramos J, Roquilly A, Zhan Y, Young LJ, Mintern JD, Villadangos JA. Inflammation conditions mature dendritic cells to retain the capacity to present new antigens but with altered cytokine secretion function. J Immunol 2014;193:3851–3859. [DOI] [PubMed] [Google Scholar]
- 72. Alloatti A, et al. Toll‐like receptor 4 engagement on dendritic cells restrains phago‐lysosome fusion and promotes cross‐presentation of antigens. Immunity 2015;43:1087–1100. [DOI] [PubMed] [Google Scholar]
- 73. Drutman SB, Trombetta ES. Dendritic cells continue to capture and present antigens after maturation in vivo. J Immunol 2010;185:2140–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Veckman V, Miettinen M, Pirhonen J, Sirén J, Matikainen S, Julkunen I. Streptococcus pyogenes and Lactobacillus rhamnosus differentially induce maturation and production of Th1‐type cytokines and chemokines in human monocyte‐derived dendritic cells. J Leukoc Biol 2004;75:764–771. [DOI] [PubMed] [Google Scholar]
- 75. Delamarre L, Holcombe H, Mellman I. Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J Exp Med 2003;198:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Watts C, West MA, Zaru R. TLR signalling regulated antigen presentation in dendritic cells. Curr Opin Immunol 2010;22:124–130. [DOI] [PubMed] [Google Scholar]
- 77. Platt CD, et al. Mature dendritic cells use endocytic receptors to capture and present antigens. Proc Natl Acad Sci USA 2010;107:4287–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wohlleber D, et al. TNF‐induced target cell killing by CTL activated through cross‐presentation. Cell Rep 2012;2:478–487. [DOI] [PubMed] [Google Scholar]
- 79. Liu Y, Gao X, Masuda E, Redecha PB, Blank MC, Pricop L. Regulated expression of FcgammaR in human dendritic cells controls cross‐presentation of antigen‐antibody complexes. J Immunol 2006;177:8440–8447. [DOI] [PubMed] [Google Scholar]
- 80. Datta SK, et al. A subset of toll‐like receptor ligands induces cross‐presentation by bone marrow‐derived dendritic cells. J Immunol 2003;170:4102–4110. [DOI] [PubMed] [Google Scholar]
- 81. Wagner CS, Cresswell P. TLR and nucleotide‐binding oligomerization domain‐like receptor signals differentially regulate exogenous antigen presentation. J Immunol 2012;188:686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Samie M, Cresswell P. The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen‐presentation pathways. Nat Immunol 2015;16:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Weck MM, Grünebach F, Werth D, Sinzger C, Bringmann A, Brossart P. TLR ligands differentially affect uptake and presentation of cellular antigens. Blood 2007;109:3890–3894. [DOI] [PubMed] [Google Scholar]
- 84. Wagner CS, Grotzke J, Cresswell P. Intracellular regulation of cross‐presentation during dendritic cell maturation. PLoS ONE 2013;8:e76801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol 2010;22:109–117. [DOI] [PubMed] [Google Scholar]
- 86. Kotsias F, Hoffmann E, Amigorena S, Savina A. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid Redox Signal 2013;18:714–729. [DOI] [PubMed] [Google Scholar]
- 87. Trombetta ES, Ebersold M, Garrett W, Pypaert M, Mellman I. Activation of lysosomal function during dendritic cell maturation. Science 2003;299:1400–1403. [DOI] [PubMed] [Google Scholar]
- 88. Sardiello M, et al. A gene network regulating lysosomal biogenesis and function. Science 2009;325:473–477. [DOI] [PubMed] [Google Scholar]
- 89. Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332:1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]