

Abstract
Cohesin is a chromosome‐associated protein complex that plays many important roles in chromosome function. Genetic screens in yeast originally identified cohesin as a key regulator of chromosome segregation. Subsequently, work by various groups has identified cohesin as critical for additional processes such as DNA damage repair, insulator function, gene regulation, and chromosome condensation. Mutations in the genes encoding cohesin and its accessory factors result in a group of developmental and intellectual impairment diseases termed ‘cohesinopathies.’ How mutations in cohesin genes cause disease is not well understood as precocious chromosome segregation is not a common feature in cells derived from patients with these syndromes. In this review, the latest findings concerning cohesin's function in the organization of chromosome structure and gene regulation are discussed. We propose that the cohesinopathies are caused by changes in gene expression that can negatively impact translation. The similarities and differences between cohesinopathies and ribosomopathies, diseases caused by defects in ribosome biogenesis, are discussed. The contribution of cohesin and its accessory proteins to gene expression programs that support translation suggests that cohesin provides a means of coupling chromosome structure with the translational output of cells. WIREs Dev Biol 2015, 4:489–504. doi: 10.1002/wdev.190
This article is categorized under:
-
1
Gene Expression and Transcriptional Hierarchies > Gene Networks and Genomics
-
2
Birth Defects > Craniofacial and Nervous System Anomalies
INTRODUCTION TO COHESIN LOADING AND REGULATION
Cohesin is an evolutionarily conserved multisubunit protein complex consisting of four core subunits: SMC1A, SMC3, the α‐kleisin protein RAD21, and the HEAT repeat‐containing protein SA1 or SA21, 2, 3 (Figure 1). The complex forms a ring‐like structure with an estimated diameter of 35 nm that entraps DNA (Figure 1). SMC1 and SMC3 belong to the structural maintenance of chromosome (SMC) family, each consisting of a coiled‐coil domain flanked by a hinge domain (created by the foldback) that mediates SMC dimer formation and a head domain with ATPase activity. This family also includes members of the condensin complex (SMC2 and SMC4) and SMC5/6 complex. These complexes are conserved from yeast to human and contribute to the organization of chromosomes.
Figure 1.
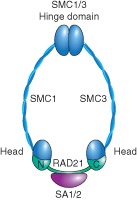
Schematic diagram of the cohesin complex. Cohesin is composed of four subunits. Smc1 and Smc3 (blue) are long polypeptides that form a hinge domain at one end and an ATPase domain at the other end by folding back on themselves to form antiparallel coiled‐coil interactions. The SMC heads are connected by the α‐kleisin RAD21 (green), which interacts with the fourth subunit, SA1 or SA2 (pink).
Cohesin is loaded onto chromosomes in G1 in budding yeast and during telophase of the preceding cell division in vertebrates by a complex comprised of NIPBL (also known as Scc2, Saccharomyces cerevisiae, Nipped‐B, Drosophila melanogaster) and MAU‐2 (also known as Scc4, S. cerevisiae) (Figure 2). Cohesin may encircle both sister chromatids or may associate with chromosomes in other ways. The ‘handcuff’ model proposes that a single cohesin ring encircles one sister chromatid while interacting with a second ring encircling the other sister via the Rad21 and Scc3 subunits. A third model proposes cohesin encircles one sister chromatid while interacting with proteins bound to the other.4, 5, 6 In any event, cohesin functions to hold two pieces of DNA in close proximity. It has been proposed that DNA enters the ring through the hinge domain governed by the Scc2–Scc4 loading complex, and that it can exit the ring at the Smc3 head–Rad21 junction in a process regulated by acetylation and other regulatory factors.7
Figure 2.
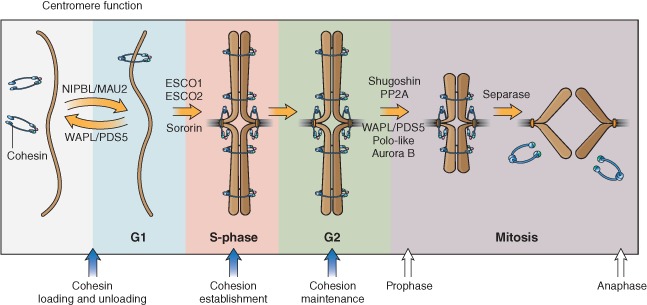
Schematic diagram showing cohesin regulation throughout the cell cycle. Cohesin (blue) is loaded onto chromosomes in telophase/G1 phase by the NIPBL–MAU2 heterodimer and requires the opening of the hinge domain of SMC1A and SMC3 for DNA entry. Cohesion establishment at S phase is facilitated by ESCO1/2‐dependent acetylation of the SMC3 head domain, making cohesin refractory to removal from chromatin by WAPL. Cohesion is then maintained by other proteins such as WAPL, PDS5, and Sororin. Cohesin can be removed in prophase in a separase‐independent manner from chromosome arms. This removal depends on PLK1 and WAPL/PDS5. Pericentromeric cohesion is protected by shugoshin and PP2A. At the onset of mitosis, pericentromeric cohesion is destroyed by proteolytic cleavage of RAD21 by separase and recycled for the next cell cycle. Recycling of the SMC3 subunit requires deacetylation by HDAC8.
The deposition and removal of cohesin is regulated over the cell cycle. Many aspects of the cycle are evolutionarily conserved from yeast to human cells. Establishment of cohesion between sister chromatids during DNA replication is dependent on Eco1 (yeast ECO1 is a homolog of human ESCO1/ESCO2) (Figure 2). Eco1 is an acetyltransferase that acetylates two lysine residues within the head domain of Smc3. The acetylation appears to convert the cohesin ring to a ‘cohesive’ mode in which it can very stably juxtapose two DNAs. While budding yeast have a single copy of the gene encoding the acetyltransferase, humans have two genes, ESCO1 and ESCO2, both of which are important for sister chromatid cohesion and cell survival in response to radiation‐induced DNA damage.8, 9, 10 Once established, cohesion between sister chromatids is maintained by accessory proteins including PDS5, WAPL, shugoshin, and sororin11, 12, 13, 14 (Figure 2). At the onset of mitosis, cohesin is removed from chromatin as a precursor to sister chromatid segregation. In metazoans, cohesin removal occurs in two waves.15 While most cohesin along chromosome arms is removed through the action of PDS5, WAPL, PP2A, Aurora B, and phosphorylation of SA1 or SA2 by PLK1 in prophase;12, 16, 17, 18 full separation of sister chromatids at the centromere region occurs at the metaphase‐to‐anaphase transition through site‐specific cleavage of the α‐kleisin RAD21 by separase19 (Figure 2). The acetyl groups on the SMC3 head are removed by HDAC8 (also known as Hos1, S. cerevisiae) to help recycle SMC3 for the next round of cohesin deposition.20, 21, 22, 23
COHESIN CAN REGULATE GENE EXPRESSION
Although cohesin has roles in many nuclear processes such as chromosome segregation, double‐strand break repair, gene regulation, and chromosome condensation (Figure 3), we have chosen to focus this review on gene expression because this role may be critical for understanding the cohesinopathies, developmental syndromes caused by mutations in the genes encoding cohesin and its regulators. Genetic studies in Drosophila originally identified a role for the SMC loading factor in gene regulation. Reduction in Nipped‐B altered the expression of homeobox genes cut and ultrabithorax through altered enhancer–promoter communication.24 Subsequently, studies in yeast and flies showed that moderate reduction in cohesin affected chromosome condensation, gene expression, and development without affecting chromosome segregation.25, 26 Cohesin can regulate gene expression independent of chromosome segregation, as shown in nondividing Drosophila neurons and mouse thymocytes, where changes in cohesin activity altered gene expression.27, 28, 29
Figure 3.
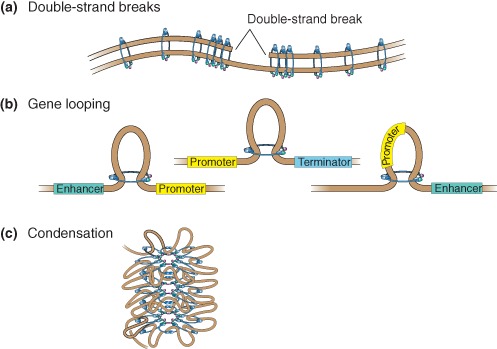
Schematic diagram depicting functions of cohesin at noncentromeric sites. (a) Double‐strand break (DSB) repair is facilitated by cohesin binding to the break site. Cohesion is also re‐enforced genome‐wide in response to a DSB. (b) Cohesin regulates gene expression by gene looping to promote long‐range (1) promoter–enhancer communication, (2) promoter–terminator interaction (middle), or (3) insulation. These types of events may contribute to chromatin organization within topological domains. (c) Cohesin promotes chromosome condensation.
Cohesin association with chromatin appears to be relatively sequence‐independent. Cohesin binds between convergently transcribed genes in budding yeast.30 Cohesin binds to highly transcribed genes in higher order organisms. In D. melanogaster cells, cohesin is enriched at highly transcribed genes together with RNA polymerase II.31 Recent studies suggest that cohesin loading occurs at sites of active transcription in all organisms, while the differences between organisms may be due to cohesin redistribution after loading onto chromosomes. Various models for how cohesin association regulates gene expression have been proposed and are discussed below.
Although one theme in cohesin–DNA associations is that they tend to occur at highly transcribed regions,32 in most cases it is not clear whether cohesin helps support the expression of highly transcribed regions or prefers to bind to these regions because they are highly transcribed. In budding yeast, the cohesin loading complex and condensin associate with promoters of RNA polymerase II‐transcribed genes encoding small nuclear RNAs, small nucleolar RNAs, ribosomal protein genes, as well as RNA polymerase III‐transcribed genes encoding transfer RNAs (tRNAs) and spliceosomal RNAs.33 The clustering of tRNA genes adjacent to the nucleolus is dependent on SMC complexes, suggesting that the binding of these proteins provides elements of chromosome organization even in budding yeast.34 Cohesin colocalizes completely with its loading factor in Drosophila at subsets of highly transcribed genes and DNA replication origins, but is excluded at silenced genes.31, 35 In both prokaryotes and eukaryotes, cohesin also binds to rDNA repeats.36 The production of ribosomal RNAs in the nucleolus is essential for the assembly of ribosomes. High levels of transcription of this locus are therefore critical for cell survival, cell proliferation, and protein translation. If cohesin does support the expression of these translation themed genes, then cohesin might promote a gene expression program that supports translation.
Cohesin May Facilitate DNA Looping
Transcription is controlled by regulatory regions. These regulatory regions are bound by DNA‐binding proteins and control both the assembly and activity of the basal transcriptional machinery.37 Defects in the association of cohesin and NIPBL with these highly transcribed regions could affect the transcription of genes critical for development. In eukaryotes such as yeast, the transcription of genes is controlled by regulatory elements close to transcription start sites. Metazoans, however, have evolved long‐distance regulatory elements that are critical for development and gene expression.37 The interaction of DNA sequences can be mediated by cohesin, in some cases in conjunction with additional factors. The interactions can be activating for transcription, for instance, a looping event that brings a promoter near an enhancer or a terminator; in other cases, they can be repressive, for instance, an interaction could insulate a promoter from an enhancer.
One factor that binds to regulatory elements is the sequence‐specific DNA‐binding protein CTCF. The ability of CTCF to connect with other regulatory regions is facilitated by cohesin, polycomb, and nuclear lamina.38, 39, 40 Depending on cell type, 50–80% of CTCF‐binding sites genome‐wide are also bound by cohesin, and disruption of cohesin results in defects in CTCF‐mediated intrachromosomal interactions.41 CTCF functions as a transcriptional activator through binding to mammalian c‐Myc promoters42 as well as an insulator when placed between enhancers and promoters of genes such as the imprinted IGF2/H19 locus.43 In fact, cohesin may promote the transcription of c‐MYC, an important factor for the transcription of ribosomal protein genes.44 Cohesin binding is thought to be critical for long‐range chromatin interactions both between CTCF insulator elements and for gene activation.42, 45 However, the association between cohesin and CTCF appears to be vertebrate‐specific because CTCF is not conserved in budding yeast and does not colocalize with cohesin in Drosophila. These types of long‐range interactions could help shape the genome by supplying organization within larger topological domains.46
Another fraction of cohesin physically interacts with and colocalizes with the Mediator complex, which binds the enhancers and promoters of active genes. About half of the cohesin loading complex associates with Mediator sites, further implicating cohesin in transcription. The Mediator complex facilitates interactions between the basal transcriptional machinery and enhancer‐bound transcriptional activators.47, 48, 49 In mouse embryonic stem cells, cohesin colocalizes with Mediator at extragenic enhancers and drives the expression of key pluripotency genes.49 In addition, NIPBL and the Mediator complex cooperate to regulate the expression of genes critical for limb development such as the hox clusters.50
Another type of DNA loop that may be mediated by cohesin is the promoter–terminator loop (Figure 3(b), middle). Chromatin immunoprecipitation (ChIP) experiments in human cells identified cohesin enrichment at transcription start sites and terminators in metagene analysis.51 These looping events could potentially allow for the recycling of RNA polymerase, resulting in highly efficient transcription reinitiation and polymerase recycling. These types of loops have been proposed to form at the ribosomal DNA genes,52 the most highly transcribed locus in most cells. Cohesin‐binding sites are positioned such that they could mediate looping within the rDNA. In budding yeast, this proposed looped structure formed less efficiently in yeast bearing a mutation in the cohesin acetyltransferase Eco1, and production of rRNA was decreased, suggesting that the efficient formation of these loops depends on acetylation of cohesin.53 Mutations in SMC complexes also result in aberrant nucleolar morphology and ribosome biogenesis,54, 55 implying a critical role for cohesin in the formation and function of the nucleolus. Because the nucleolus serves as an anchor point for chromosome organization, this finding has implications for both the architecture and expression of the genome.
Cohesin May Regulate Transcription Initiation, Elongation, and Termination
Eukaryotic transcription is divided into three main steps: initiation, elongation, and termination. Each of these processes is regulated at multiple levels. Numerous studies have identified cohesin to be important for transcription initiation. Studies in Drosophila cells have shown that cohesin and Nipped‐B colocalize at the transcription start sites of active genes,31 although less colocalization between these two proteins is observed in mouse cells.49 When compared with RNA polymerase II binding, NIPBL preferentially binds 100–200 nucleotides upstream of RNA polymerase II. NIPBL knockdown causes reduced transcription of these genes, suggesting a role for NIPBL in transcription initiation.56 Cohesin may also promote transcription termination in eukaryotes. In Schizosaccharomyces pombe, cohesin promotes mRNA 3′‐end formation and transcriptional termination in convergent gene transcripts.57
Recent studies suggest that cohesin also regulates transcription elongation. In D. melanogaster cells, cohesin was shown to bind to genes with low levels of histone H3 lysine 36 trimethylation (H3K36me3), a chromatin mark associated with transcription elongation.58 Many of these genes had paused RNA polymerase II just downstream of transcription start sites and GAGA factor (GAF)‐binding sites. Cohesin, along with other pausing factors such as NELF, was proposed to play a role in the transition of RNA polymerase II from the paused form to the elongating form. Taken together, these studies suggest that cohesin might be playing an important role in all three stages of transcription.
NIPBL May Act as a Chromatin Adaptor
The cohesin loader NIPBL tends to bind to nucleosome‐free regions in promoters of active genes in metazoans. Nucleosome‐free regions are important for transcription initiation because they serve as a ‘landing pad’ for key transcription‐promoting factors. The maintenance of nucleosome‐free regions requires chromatin remodeling factors such as the SNF/SWI2 superfamily,59 ISWI, and CHD family; and posttranslational modifiers of histone proteins, such as deacetylases (HDACs), methyltransferases, and acetyltransferases.60, 61 Studies in model organisms from yeast to human have implicated the cohesin loading complex in helping maintain nucleosome‐free regions.61, 62, 63 Because SMC complexes first evolved in bacteria, which do not have nucleosomes, we speculate that the loading complex may have evolved in eukaryotes to assist SMC complexes loading onto DNA in the context of chromatin.
The RSC (remodels the structure of chromatin) remodeling complex, a member of the SNF/SWI2 superfamily, may act upstream of genes to facilitate the recruitment of the cohesin loading complex Scc2–Scc4.61, 62 Mutations in the RSC remodeling complex subunits cause Coffin–Siris syndrome (CSS), a genetic disorder that results in developmental delay in patients.64 CSS is caused by heterozygous mutations in ARID1A, ARID1B, SMARCA4, SMARCB1, or SMARCE1. Characteristic features of patients with CSS include aplasia or hypoplasia of the distal phalanx, moderate to severe developmental and cognitive delay, facial abnormalities, short stature, ophthalmologic abnormalities, microcephaly, brain malformations, and hearing loss. In humans, the ISWI (SNF2h)‐containing chromatin remodeling complex interacts with the cohesin subunit RAD21.63 The cohesin loader NIPBL was also reported to mediate the recruitment of HDAC1 and HDAC3 to chromatin.65 The Scc2–Scc4 complex itself, or in conjunction with additional factors that it recruits, may help to maintain nucleosome‐free regions to promote cohesin loading as well as transcription. In this way, the cohesin loading complex might be serving as a ‘chromatin adaptor’ for cohesin and for transcription‐promoting complexes.
GENOME‐WIDE ANALYSIS OF COHESIN AND Nipbl BINDING
Genome‐wide binding profiles of NIPBL and cohesin (SMC1) in mouse embryonic stem cells have been published.49 To further explore the association of these proteins with the genome and their potential role in gene expression, we chose the top NIPBL‐ and cohesin‐binding sites and examined colocalization with other factors in a window of ±1 kb. For NIPBL we defined 4413 sites with a false discovery rate (FDR) cutoff of 1−e9. For cohesin, we defined 18519 sites with an FDR cutoff of 1e−9.
Among the largest categories of binding sites for both NIPBL and cohesin are the promoter regions and transcription start sites of active genes (Figures 4(a) and 5(a)), although SMC1 enrichment is also observed at less active genes (Figure 5(b)). Enrichment of NIPBL and SMC1 at active genes for some genes is quite broad, extending a few kilobases or more to either side of the transcription start sites. Binding of NIPBL and cohesin at transcription start sites of active genes is assigned by correlation with trimethylation of histone 3 at lysine 4 (H3K4me3), a chromatin modification mark associated with active genes, RNA polymerase II, transcription initiation factor TFIID subunit 3 (TAF3), histone H2AZ, and negative elongation factor (NELF). A larger fraction of NIPBL peaks correlate with H3K4me3 than do SMC1 peaks. NIPBL and cohesin both colocalize with CDK9, an elongation factor for RNA polymerase II‐directed transcription, a finding consistent with the hypothesis that cohesin plays an important role in transcription elongation (Figures 4(a) and 5(a)). NIPBL does not significantly colocalize with CTCF, while cohesin does, suggesting that cohesin is more likely to be involved in CTCF‐mediated processes such as looping and insulation compared with NIPBL.66, 67, 68, 69, 70 Cohesin and CTCF interact through the carboxy‐terminal region of CTCF and SA2 subunit of cohesin, which helps to stabilize most CTCF‐mediated chromosomal contacts,76 a process important for cohesin‐dependent insulation activity.
Figure 4.
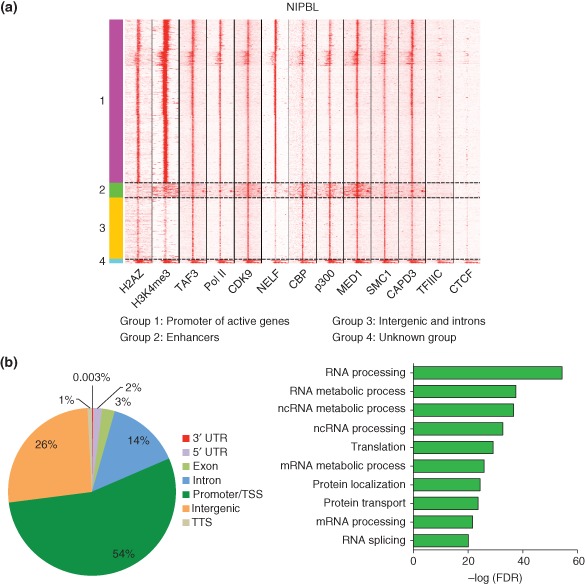
Analysis of the cohesin loader factor (NIPBL) binding throughout the genome of mouse embryonic stem cells. (a) NIPBL peaks from chromatin immunoprecipitation (ChIP) seq data were defined along with windows of ±1 kb on either side. These were then used to select the ChIP signal for each additional factor shown.49, 66, 67, 68, 69, 70, 71, 72, 73, 74 ChIP‐seq data were subjected to unbiased clustering using seqMiner v1.3.3 package.75 Kmeans rank was used as the method of clustering, with the following parameters: left and right extensions = 1 kb; internal bins = 160; flanking region bins = 20; and the number of clusters = 7. NIPBL binds to active genes, enhancer regions, and intergenic and intronic regions of genes. NIPBL does not colocalize with CTCF.74 (b) The majority of NIPBL‐binding sites were at the promoter regions of genes. Gene ontology (GO) analysis of NIPBL‐bound active genes showed enrichment for genes important for RNA processing, mRNA processing, and RNA splicing.
Figure 5.
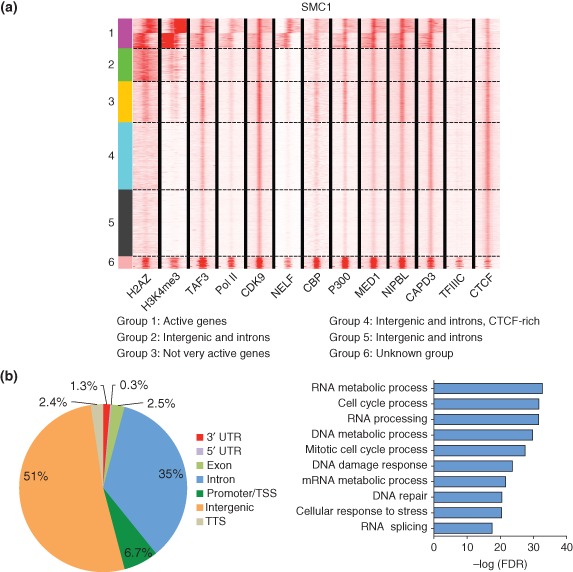
Genome‐wide analysis of cohesin (SMC1) binding in the mouse genome. (a) SMC1 peaks were first defined and then a window ±1 kb on either side is shown for each factor.49, 66, 67, 68, 69, 70, 71, 72, 73, 74 Clustering was performed as described for Figure 4. Cohesin peaks correlate well with CTCF binding.74 (b) Cohesin (SMC1) binds at more intergenic and intronic regions of genes than promoters. Gene ontology (GO) analysis of the active genes associated with cohesin in mESCs showed enrichment for genes involved in cell morphogenesis, cellular signal transduction, cellular developmental process, and cell adhesion.
All NIPBL peaks (4413) overlapped with cohesin peaks (SMC1). While over 50% of NIPBL binds to promoter regions of genes, only 7% of cohesin sites can be accounted for by these same genes. On the surface, this suggests that SMC1 and NIPBL may both participate in the regulation of a common gene group, while SMC1 may also be involved in the regulation of additional genes. However, because their function at these regions may be different, they may not have similar effects on gene expression at even the common gene group upon loss of function. For instance, NIPBL's primary role may be to maintain a nucleosome‐free region while cohesin may be mediating looping events. Gene ontology (GO) analysis of the active genes with NIPBL enrichment revealed genes important for RNA processing, translation, and splicing (Figure 4(b)). These results are consistent with observations suggesting that loss of nipbla/b function negatively impacts translation in zebrafish.55 At these genes, mutations in NIPBL could compromise the maintenance of a nucleosome‐free region which could impair transcription initiation. However, the active genes bound by SMC1 show GO term enrichment for cell morphogenesis, cellular signal transduction, cellular developmental process, and cell adhesion (Figure 5(b)). At these genes, loss of cohesin function might impair looping and gene regulation, consistent with observations that loss of cohesin function is detrimental to expression of Hox genes and development.
A large portion of NIPBL (36%) and cohesin (51%) peaks are found at intergenic regions. Enhancer regions were defined as sites at which NIPBL or SMC1 binding correlates strongly with CBP, P300, Mediator, and CDK949, 67, 70 (Figures 4(a) and 5(a)). CBP and P300 are important transcriptional activators that promote acetylation of histone H3 at lysine 27 (H3K27) at active enhancers77 and serve as convenient markers of enhancers in both D. melanogaster and mammalian studies. Although CBP and P300 are found at intergenic or intragenic regions with NIPBL and SMC1 (Figures 4(a) and 5(a)), they also occupy the promoters of active genes and acetylate many proteins, including p53. CBP and P300 also perform many other cellular roles such as acetylation of promoters and acetylation of histone H3 during deposition.78, 79 Mutations that impair the activity of CBP and P300 result in Rubinstein–Taybi syndrome, a congenital disorder characterized by mental and growth retardation, a wide range of dysmorphic features, and an increased susceptibility to tumor formation,80 suggesting that levels of these proteins are also important for development. SMC1 also binds at additional intergenic sites that do not have a distinct colocalization signature, making them difficult to assign to a category.
Taken together, our examination of the binding sites of cohesin versus NIPBL demonstrates that these proteins not only share some binding sites but also show unique colocalization signatures with other proteins, arguing that these genes may influence gene expression and chromosome architecture in common and unique ways. It will be important to continue to evaluate the genome‐wide association patterns and genes regulated by these proteins in order to understand how they influence gene expression.
COHESIN AND HUMAN DISEASES
Mutations in cohesin and genes with related function result in a broad spectrum of diseases termed ‘cohesinopathies.’ Roberts syndrome (RBS) and Cornelia de Lange syndrome (CdLS) patients have a constellation of phenotypes including craniofacial, heart, and gastrointestinal defects, poor growth, developmental delay, and intellectual disability.81 Both RBS and CdLS arise from mutations in cohesin‐associated genes. Altered gene expression is clearly an important part of the etiology of these diseases and contributes to the developmental defects observed. Emerging evidence suggests that cohesin and NIPBL may promote gene expression programs that support translation, making it interesting to consider translational defects as part of the etiology of these syndromes.82
Mutations in ESCO2 Cause RBS
RBS, an autosomal recessive disorder, is caused by mutations in both copies of ESCO2, a gene encoding a member of the acetyltransferase family.9 The mutations in ESCO2 cause either the complete loss of protein or the loss of acetyltransferase activity in patients.83 Mutations in ESCO1 have not yet been reported in humans, probably because they would be lethal. Affected individuals display a wide variety of malformations including craniofacial deformities, hypoplastic nasal alae, cleft lip and palate, and reduction in digit number, bone length, or bone formation in both arms and legs.84 RBS newborns have a high mortality rate.85 Loss of Esco2 in mice is lethal and leads to termination of preimplantation and postimplantation stage embryos.86, 87 In RBS cells, cytological observations include aneuploidy, micronuclei, and heterochromatic repulsion. Loss of Esco2 in early mitosis also results in changes in the chromosomal location of cohesin and its protector SGO1.87 The heterochromatic repulsions observed in human RBS cells are located at the pericentric domains and nucleolar organizing regions (NOR or rDNA),88 suggesting a defect in cohesion at these regions. Studies in various model organisms such as yeast, zebrafish, and human show that ESCO2 mutation affects nucleolar organization, rRNA production, ribosome biogenesis, and protein translation.53, 54, 89 RBS cell lines have increased apoptosis with elevated p53 and reduced cell proliferation.54 Some fraction of the phenotypes associated with RBS could be the result of poor translation and cell proliferation contributing to abnormal development during embryogenesis.
Mutations in NIPBL and Cohesin Cause CdLS
CdLS, also known as Brachmann–de Lange syndrome, is the most common of the ‘cohesinopathies,’ occurring in approximately 1 in 10,000 live births. CdLS is clinically heterogeneous and affects multiple aspects of development. Patients with CdLS have distinct phenotypic characteristics, which vary from mild intellectual disability to severe developmental and intellectual impairment.90 Affected individuals have craniofacial deformities, upper limb extremity defects, hirsutism, gastroesophageal dysfunction, immune dysfunction, and growth and neurodevelopmental delay51, 81 (http://www.cdlsusa.org/what‐is‐cdls/).
More than half (∼65%) of CdLS cases arise from mutations in the NIPBL gene and are dominantly inherited.91, 92 Examination of the phenotypic and genotypic correlation of patients shows that severe clinical features arise from deletions or truncations in NIPBL,90 indicating that CdLS is caused by haploinsufficiency. The fact that NIPBL mutations accounted for only a little more than half of CdLS cases prompted investigators to look for mutations in other genes with related functions. Subsequent genetic screens in large cohorts of CdLS patients without NIPBL mutations identified 5% missense or small in‐frame deletions in SMC1A to be causative.93, 94, 95 Mutations in SMC3 were also identified.94 Since then, mutations in other cohesin‐associated genes (RAD21 and HDAC8) have been identified as giving rise to CdLS or related disorders.20, 96
Even though mutations in cohesin and cohesin‐associated genes cause these two syndromes, the underlying etiology for RBS and CdLS could be different.81 In fact, mutations in different cohesin genes cause clinically distinct subtypes of CdLS. In contrast to RBS cells, most CdLS patient cells do not exhibit gross defects in chromosome segregation, but primarily exhibit gene dysregulation. Most of the differential gene expression changes observed in cells derived from patients or mouse models for CdLS are modest at best (lower than twofold), suggesting that either these small changes in gene expression result in the developmental features or additional factors contribute to the etiology. Studies in SMC1A‐ and SMC3‐mutated CdLS cell lines using a proteomic approach revealed dysregulation of proteins important for metabolism, cytoskeleton organization, and RNA processing, among others.97 Defects in ribosomal RNA production and protein synthesis in RBS and CdLS model organisms suggest that the changes in gene expression could lead to translational defects which contribute to ‘cohesinopathies.’
COHESINOPATHIES AND TRANSLATION
Ribosomes Can Have Regulatory Capacity
Decades of research has shown that the development of single cells into complex organisms is regulated at transcriptional, posttranscriptional, translational, and posttranslational levels. Translation in eukaryotes is an intricate and essential process requiring various factors, chief among them being ribosomes. Ribosomes are large ribonucleoprotein (RNP) particles that convert mRNAs into proteins in the cytoplasm. Eukaryotic ribosomes consist of four ribosomal RNAs (25S, 18S, 5S, and 5.8S rRNA) bound by about 75 ribosomal proteins that are assembled into large and small ribosomal subunits (60S and 40S). The ribosomal RNAs are transcribed from the ribosomal DNA located in a specialized compartment called the nucleolus by RNA polymerase I and polymerase III. Complete synthesis and assembly of 40S and 60S subunits into mature forms requires ribosomal proteins and additional modification and processing factors which are important for maturation of the RNAs, transport of the immature ribosomal subunits, stabilization of ribosome structure, and regulation of mRNA translation. Because ribosome biogenesis requires so many factors, the process is coupled to cellular metabolism and can be regulated in many ways. Furthermore, because ribosomes are composed of dozens of proteins, they have the potential to have a regulatory function in translation. Ribosomes can have both quantitative and qualitative effects on mRNA translation, which can result in a wide variety of pathological conditions. NIPBL and cohesin bind to the rDNA repeats and contribute to their organization into a nucleolar structure that produces sufficient rRNAs and ribosomes.36, 54 We speculate that the cohesinopathies may be caused in part by defects in translation.
Mutations that impair proper ribosome biogenesis in various model organisms cause developmental defects. Haploinsufficiency in ribosomal proteins gives rise to ‘minute’ flies.98 ‘Minute’ flies are small and have short and thin bristles. The small size likely results from overall defects in translation while the bristle phenotype probably occurs because bristle production places exceptional demands on translation. Developmental delay and bristle phenotypes are similarly observed in Drosophila bobbed mutants, which have fewer rDNA repeats. Together, these fly mutants demonstrate the essential nature of producing large amounts of both ribosomal proteins and RNAs. In a remarkable demonstration of the regulatory potential of ribosomal proteins, loss of function of Rpl38 in the mouse is associated with a specific developmental defect, namely loss of axial skeletal patterning that results from a lack of translation of a specific subset of homeobox (Hox) mRNAs.99 This study demonstrates the potential of the ribosome for a regulatory role in translation.
Ribosomopathies Are Human Diseases Caused by Defects in Ribosome Biogenesis
A growing number of disorders associated with ribosome biogenesis and function, termed ribosomopathies, have also been identified and are discussed below. These diseases can be caused either by mutations in ribosomal protein genes or by mutations in genes involved in the processing or modification of ribosomal RNAs.
Several ribosomopathies are caused by haploinsufficiency for ribosomal protein genes. Diamond Blackfan anemia (DBA), for example, is caused by a mutation in one copy of the gene encoding ribosomal protein S19 (RPS19), RPL5, RPL35A, or RPS7, among others.100 Characteristic features of DBA patients include enhanced sensitivity of hematopoietic progenitors to apoptosis, and craniofacial, cardiac, and limb abnormalities (http://dbafoundation.org). 5q‐syndrome, another ribosomopathy, is a subtype of myelodysplasia with phenotypes similar to DBA, attributable to haploinsufficiency of the ribosomal protein S14 (RPS14).101 Patients with 5q‐syndrome have erythroid hypoplasia of the bone marrow, normal or reduced neutrophil counts, and high susceptibility to acute myeloid leukemia.102 In both DBA and 5q‐syndrome patient cells, rRNA processing is compromised, affecting ribosome production. Mutations in a gene needed for the maturation of the 60S ribosomal subunits cause Shwachman Bodian Diamond syndrome (SBDS).103, 104 SBDS is characterized by pancreatic exocrine insufficiency, hematologic dysfunction, and skeletal abnormalities. A novel ribosomopathy caused by X‐linked loss of function in RPL10 has recently been reported.105 Characteristic features of patients with this syndrome include microcephaly, growth retardation, hypotonia, and neurodevelopmental problems. Together these diseases support the idea that dosage of ribosomal proteins is important for proper ribosome function. Furthermore, loss of function in different ribosomal protein genes yields distinct syndromes, suggesting that each protein makes a unique contribution to translation.
X‐linked dyskeratosis congenita (DKC) has been controversially categorized as a ribosomopathy. DKC is characterized by nail dystrophy, reticular skin pigmentation, oral leukoplakia, bone marrow failure, increased susceptibility to cancer, and skin abnormalities.106, 107 Some cases of DKC are caused by mutations in DKC1, a gene encoding the catalytic protein component of a RNP complex that along with the guiding H/ACA snoRNAs converts uridines to pseuodouridines in mRNAs and noncoding RNAs including rRNAs and telomerase RNA. DKC can also be caused by mutations in other genes important for telomere biology such as TERC, TERT, TINF2, NHP2, and NOP10. Hypomorphic Dkc1 mutant mice have reduced pseudouridylation in their ribosomal RNAs, and cells show impaired translational fidelity, including poor translation of mRNAs with an internal ribosome entry site.108, 109 Whether these changes in translational fidelity contribute to the etiology of DKC is still an open question.
Some cases of the ribosomopathy Treacher Collins syndrome (TCS) are caused by mutations in TCOF1, which encodes Treacle, a dense fibrillary component of nucleoli.110, 111 Additional cases are caused by mutations in genes that encode subunits shared between RNA polymerase I and polymerase III.112 Characteristic features of patients with TCS include skeletal dysmorphism of the orbits, midface and zygomatic hypoplasia, microtia, and mandibular microretrognathia.113 Patients with cartilage‐hair hypoplasia, another ribosomopathy, have metaphyseal chondrodysplasia, short stature, immunodeficiency, anemia, gastrointestinal disorders, and hair abnormality.114 The mutations in this disease are in the gene encoding an RNA component of RNAse MRP, a RNP complex that processes pre‐rRNA. Therefore, defects in rRNA production, either through transcription, processing, or modification, have all been linked with defective ribosome biogenesis and human disease (reviewed in Ref 115).
Comparing and Contrasting Ribosomopathies and Cohesinopathies
Cohesinopathies and ribosomopathies share compromised ribosome biogenesis and protein synthesis.54, 89 Studies in yeast, zebrafish, and human cells show that mutations in the cohesin acetyltransferase ESCO2 are associated with defects in ribosomal RNA production and protein synthesis, strongly suggesting that defective translation contributes to RBS.54, 89 Zebrafish models of CdLS also show reduced phosphorylation of biomarkers of translation, and reduced protein synthesis and rRNA production, suggesting that defective translation could be a common consequence of mutations in cohesin‐associated genes.55 Maybe not surprisingly, these two syndromic categories also share some phenotypic characteristics. Cohesinopathies and ribosomopathies are both characterized by prenatal and postnatal growth retardation, microcephaly, seizures, craniofacial abnormalities, and limb deformities. However, there are also clear differences between these syndromes at the cellular and molecular level. For instance, cohesinopathies are not associated with anemia. Furthermore, animal models of DBA show elevated activity of the TOR pathway which serves as a major node of control for ribosome biogenesis, while the cohesinopathies show low TOR pathway activity. The reduced TOR activity in the cohesinopathies makes them a good candidate for treatment with the TOR stimulator l‐leucine.55, 89
Despite the fact that cohesinopathies and ribosomopathies share overlapping clinical presentations, these congenital diseases are distinct. While a majority of ribosomopathy probands are predisposed to cancer, no susceptibility to cancer has been associated with the cohesinopathies. Proteins levels of tumor suppressors such p27, XIAP, and p21 are reduced in DKC, while p53, a proapoptotic gene, is elevated in RBS and DBA patient cells.89, 116 The primary effects of the various mutations can trigger different pathways with different outcomes and hence manifest in the unique clinical characteristics observed in patients. It is worthy to note that mutations in cohesin subunits have been implicated in acute myeloid leukemia and a wide range of tumor types.117, 118 The underlying mechanisms regarding how mutations in cohesin subunits cause cancer are, however, yet to be determined.
Comparing and Contrasting the Cohesinopathies
Although the genes mutated in RBS and CdLS all contribute to cohesin function, even the clinical manifestations of these two diseases are distinct. The difference could result in part from differences in the exact molecular defect caused by the mutation and differences in the cell types that the mutations affect. Cell types vary in gene expression, translational control, and requirements for protein synthesis. Cohesin has been shown to be important for maintaining a gene expression program that enables pluripotency in embryonic stem cells.49, 71 In addition, translation has been proposed to be critical for germline stem cell (GSC) maintenance in Drosophila. Translational repressors such as Pum and Nos have been proposed to be important for maintenance of GSCs and control germ cell development from Caenorhabditis elegans to human.119, 120 Studies have shown that GSCs have increased expression of proteins involved in ribosome biogenesis and translation when compared with differentiating cells (high translational output). If mutations in cohesin can affect translational efficiency and fidelity, cells that are highly proliferative or have an otherwise high requirement for protein synthesis, such as GSCs, may be more acutely affected.
Another mechanism whereby defects in translation could contribute to cell‐type‐specific defects in the cohesinopathies is apoptosis. When ribosomal proteins and rRNAs are unbalanced, p53 can be induced followed by apoptosis. Cancer cells that overproduce ribosomal proteins are especially prone to apoptosis via this mechanism when treated with a RNA polymerase I inhibitor.121 Cell types with high levels of ribosomal proteins could be primed for apoptosis by this mechanism when a cohesin mutation causes a reduced level of rRNA. Increased apoptosis in developing neural tissues has been observed in nipbl loss of function zebrafish.122 p53‐triggered apoptosis in neurons could contribute to the developmental abnormalities observed in the central nervous system of CdLS patients.
PERSPECTIVE
Although tremendous work has been done over the last decade to decipher the pathogenesis of ‘cohesinopathies,’ more is still required to fully understand how mutations in cohesin contribute to human disease. How cohesin and its regulators mediate gene expression is likely to occur via multiple mechanisms including the maintenance of nucleosome‐free regions and DNA looping. Several key regulators have been shown to be affected by mutations in cohesin including c‐MYC, p53, mTOR, and Hox genes. The role of cohesin in regulating translational processes has recently been uncovered and will require additional study. The body of work accumulating on ribosomopathies provides a helpful counterpoint to understand how the regulation of translational processes by cohesin could contribute to human disease. By supporting gene expression that promotes translation, cohesin could provide a means for coupling chromosome structure with the translational output of cells. Understanding how cohesin synergizes with different proteins and pathways to regulate genome architecture, gene expression, and translation should enable us to more fully understand the molecular etiology of the ‘cohesinopathies.’
ACKNOWLEDGMENTS
We would like to express our sincere gratitude to Mark Miller and Richard Shrock for assistance with manuscript and figure preparation.
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae . Cell 1997, 91:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91:35–45. [DOI] [PubMed] [Google Scholar]
- 3. Losada A, Yokochi T, Kobayashi R, Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol 2000, 150:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Onn I, Heidinger‐Pauli JM, Guacci V, Unal E, Koshland DE. Sister chromatid cohesion: a simple concept with a complex reality. Annu Rev Cell Dev Biol 2008, 24:105–129. [DOI] [PubMed] [Google Scholar]
- 5. Gruber S, Haering CH, Nasmyth K. Chromosomal cohesin forms a ring. Cell 2003, 112:765–777. [DOI] [PubMed] [Google Scholar]
- 6. Zhang N, Kuznetsov SG, Sharan SK, Li K, Rao PH, Pati D. A handcuff model for the cohesin complex. J Cell Biol 2008, 183:1019–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nasmyth K. Cohesin: a catenase with separate entry and exit gates? Nat Cell Biol 2011, 13:1170–1177. [DOI] [PubMed] [Google Scholar]
- 8. Ivanov D, Schleiffer A, Eisenhaber F, Mechtler K, Haering CH, Nasmyth K. Eco1 is a novel acetyltransferase that can acetylate proteins involved in cohesion. Curr Biol 2002, 12:323–328. [DOI] [PubMed] [Google Scholar]
- 9. Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, van Gosliga D, Kayserili H, Xu C, Ozono K, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet 2005, 37:468–470. [DOI] [PubMed] [Google Scholar]
- 10. Hou F, Zou H. Two human orthologues of Eco1/Ctf7 acetyltransferases are both required for proper sister‐chromatid cohesion. Mol Biol Cell 2005, 16:3908–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Panizza S, Tanaka T, Hochwagen A, Eisenhaber F, Nasmyth K. Pds5 cooperates with cohesin in maintaining sister chromatid cohesion. Curr Biol 2000, 10:1557–1564. [DOI] [PubMed] [Google Scholar]
- 12. Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters JM. Wapl controls the dynamic association of cohesin with chromatin. Cell 2006, 127:955–967. [DOI] [PubMed] [Google Scholar]
- 13. Kitajima TS, Kawashima SA, Watanabe Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature 2004, 427:510–517. [DOI] [PubMed] [Google Scholar]
- 14. Rankin S, Ayad NG, Kirschner MW. Sororin, a substrate of the anaphase‐promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell 2005, 18:185–200. [DOI] [PubMed] [Google Scholar]
- 15. Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell 2000, 103:399–410. [DOI] [PubMed] [Google Scholar]
- 16. Gandhi R, Gillespie PJ, Hirano T. Human Wapl is a cohesin‐binding protein that promotes sister‐chromatid resolution in mitotic prophase. Curr Biol 2006, 16:2406–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sumara I, Vorlaufer E, Stukenberg PT, Kelm O, Redemann N, Nigg EA, Peters JM. The dissociation of cohesin from chromosomes in prophase is regulated by Polo‐like kinase. Mol Cell 2002, 9:515–525. [DOI] [PubMed] [Google Scholar]
- 18. Hauf S, Roitinger E, Koch B, Dittrich CM, Mechtler K, Peters JM. Dissociation of cohesin from chromosome arms and loss of arm cohesion during early mitosis depends on phosphorylation of SA2. PLoS Biol 2005, 3:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Uhlmann F, Lottspeich F, Nasmyth K. Sister‐chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400:37–42. [DOI] [PubMed] [Google Scholar]
- 20. Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489:313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Borges V, Lehane C, Lopez‐Serra L, Flynn H, Skehel M, Rolef Ben‐Shahar T, Uhlmann F. Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol Cell 2010, 39:677–688. [DOI] [PubMed] [Google Scholar]
- 22. Beckouet F, Hu B, Roig MB, Sutani T, Komata M, Uluocak P, Katis VL, Shirahige K, Nasmyth K. An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell 2010, 39:689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xiong B, Lu S, Gerton JL. Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr Biol 2010, 20:1660–1665. [DOI] [PubMed] [Google Scholar]
- 24. Rollins RA, Morcillo P, Dorsett D. Nipped‐B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the cut and Ultrabithorax genes. Genetics 1999, 152:577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heidinger‐Pauli JM, Mert O, Davenport C, Guacci V, Koshland D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr Biol 2010, 20:957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schaaf CA, Misulovin Z, Sahota G, Siddiqui AM, Schwartz YB, Kahn TG, Pirrotta V, Gause M, Dorsett D. Regulation of the Drosophila enhancer of split and invected‐engrailed gene complexes by sister chromatid cohesion proteins. PLoS One 2009, 4:e6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pauli A, Althoff F, Oliveira RA, Heidmann S, Schuldiner O, Lehner CF, Dickson BJ, Nasmyth K. Cell‐type‐specific TEV protease cleavage reveals cohesin functions in Drosophila neurons. Dev Cell 2008, 14:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schuldiner O, Berdnik D, Levy JM, Wu JS, Luginbuhl D, Gontang AC, Luo L. piggyBac‐based mosaic screen identifies a postmitotic function for cohesin in regulating developmental axon pruning. Dev Cell 2008, 14:227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seitan VC, Hao B, Tachibana‐Konwalski K, Lavagnolli T, Mira‐Bontenbal H, Brown KE, Teng G, Carroll T, Terry A, Horan K, et al. A role for cohesin in T‐cell‐receptor rearrangement and thymocyte differentiation. Nature 2011, 476:467–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, DeRisi JL, Gerton JL. Genome‐wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae . PLoS Biol 2004, 2:E259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Misulovin Z, Schwartz YB, Li XY, Kahn TG, Gause M, MacArthur S, Fay JC, Eisen MB, Pirrotta V, Biggin MD, et al. Association of cohesin and Nipped‐B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 2008, 117:89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dorsett D, Merkenschlager M. Cohesin at active genes: a unifying theme for cohesin and gene expression from model organisms to humans. Curr Opin Cell Biol 2013, 25:327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. D'Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, Uhlmann F. Identification of cis‐acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 2008, 22:2215–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Haeusler RA, Pratt‐Hyatt M, Good PD, Gipson TA, Engelke DR. Clustering of yeast tRNA genes is mediated by specific association of condensin with tRNA gene transcription complexes. Genes Dev 2008, 22:2204–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. MacAlpine HK, Gordan R, Powell SK, Hartemink AJ, MacAlpine DM. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 2010, 20:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laloraya S, Guacci V, Koshland D. Chromosomal addresses of the cohesin component Mcd1p. J Cell Biol 2000, 151:1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell 2013, 152:1285–1297. [DOI] [PubMed] [Google Scholar]
- 38. Morey L, Helin K. Polycomb group protein‐mediated repression of transcription. Trends Biochem Sci 2010, 35:323–332. [DOI] [PubMed] [Google Scholar]
- 39. van Steensel B. Chromatin: constructing the big picture. EMBO J 2011, 30:1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galande S, Purbey PK, Notani D, Kumar PP. The third dimension of gene regulation: organization of dynamic chromatin loopscape by SATB1. Curr Opin Genet Dev 2007, 17:408–414. [DOI] [PubMed] [Google Scholar]
- 41. Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis‐interactions at the developmentally regulated IFNG locus. Nature 2009, 460:410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, Neiman PE, Collins SJ, Lobanenkov VV. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c‐myc oncogenes. Mol Cell Biol 1996, 16:2802–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Beygo J, Citro V, Sparago A, De Crescenzo A, Cerrato F, Heitmann M, Rademacher K, Guala A, Enklaar T, Anichini C, et al. The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF‐binding sites. Hum Mol Genet 2013, 22:544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rhodes JM, Bentley FK, Print CG, Dorsett D, Misulovin Z, Dickinson EJ, Crosier KE, Crosier PS, Horsfield JA. Positive regulation of c‐Myc by cohesin is direct, and evolutionarily conserved. Dev Biol 2010, 344:637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, Gregson HC, Jarmuz A, Canzonetta C, Webster Z, Nesterova T, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008, 132:422–433. [DOI] [PubMed] [Google Scholar]
- 46. Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet 2014, 15:234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ebmeier CC, Taatjes DJ. Activator‐mediator binding regulates mediator‐cofactor interactions. Proc Natl Acad Sci USA 2010, 107:11283–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Conaway RC, Conaway JW. The mediator complex and transcription elongation. Biochim Biophys Acta 2013, 1829:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muto A, Ikeda S, Lopez‐Burks ME, Kikuchi Y, Calof AL, Lander AD, Schilling TF. Nipbl and mediator cooperatively regulate gene expression to control limb development. PLoS Genet 2014, 10:e1004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu J, Krantz ID. Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet 2009, 76:303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mayan M, Aragon L. Cis‐interactions between non‐coding ribosomal spacers dependent on RNAP‐II separate RNAP‐I and RNAP‐III transcription domains. Cell Cycle 2010, 9:4328–4337. [DOI] [PubMed] [Google Scholar]
- 53. Harris B, Bose T, Lee KK, Wang F, Lu S, Ross RT, Zhang Y, French SL, Beyer AL, Slaughter BD, et al. Cohesion promotes nucleolar structure and function. Mol Biol Cell 2014, 25:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bose T, Lee KK, Lu S, Xu B, Harris B, Slaughter B, Unruh J, Garrett A, McDowell W, Box A, et al. Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet 2012, 8:e1002749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu B, Sowa N, Cardenas ME, Gerton JL. l‐leucine partially rescues translational and developmental defects associated with zebrafish models of Cornelia de Lange syndrome. Hum Mol Genet 2015, 24:1540–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, Nakato R, Lenhard B, Wendt KS. A cohesin‐independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet 2014, 10:e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gullerova M, Proudfoot NJ. Cohesin complex promotes transcriptional termination between convergent genes in S. pombe . Cell 2008, 132:983–995. [DOI] [PubMed] [Google Scholar]
- 58. Fay A, Misulovin Z, Li J, Schaaf CA, Gause M, Gilmour DS, Dorsett D. Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr Biol 2011, 21:1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vignali M, Hassan AH, Neely KE, Workman JL. ATP‐dependent chromatin‐remodeling complexes. Mol Cell Biol 2000, 20:1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jenuwein T, Allis CD. Translating the histone code. Science 2001, 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 61. Huang J, Hsu JM, Laurent BC. The RSC nucleosome‐remodeling complex is required for Cohesin's association with chromosome arms. Mol Cell 2004, 13:739–750. [DOI] [PubMed] [Google Scholar]
- 62. Lopez‐Serra L, Kelly G, Patel H, Stewart A, Uhlmann F. The Scc2‐Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome‐free regions. Nat Genet 2014, 46:1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, Yokomori K, Shiekhattar R. A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 2002, 418:994–998. [DOI] [PubMed] [Google Scholar]
- 64. Fryns JP. On the nosology of the Cornelia de Lange and Coffin‐Siris syndromes. Clin Genet 1986, 29:263–264. [DOI] [PubMed] [Google Scholar]
- 65. Jahnke P, Xu W, Wulling M, Albrecht M, Gabriel H, Gillessen‐Kaesbach G, Kaiser FJ. The cohesin loading factor NIPBL recruits histone deacetylases to mediate local chromatin modifications. Nucleic Acids Res 2008, 36:6450–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Subramanian V, Mazumder A, Surface LE, Butty VL, Fields PA, Alwan A, Torrey L, Thai KK, Levine SS, Bathe M, et al. H2A.Z acidic patch couples chromatin dynamics to regulation of gene expression programs during ESC differentiation. PLoS Genet 2013, 9:e1003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis‐regulatory sequences in the mouse genome. Nature 2012, 488:116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liu Z, Scannell DR, Eisen MB, Tjian R. Control of embryonic stem cell lineage commitment by core promoter factor, TAF3. Cell 2011, 146:720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, Young RA. c‐Myc regulates transcriptional pause release. Cell 2010, 141:432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super‐enhancers at key cell identity genes. Cell 2013, 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint‐Andre V, Sigova AA, Hoke HA, Young RA. Super‐enhancers in the control of cell identity and disease. Cell 2013, 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Dowen JM, Bilodeau S, Orlando DA, Hubner MR, Abraham BJ, Spector DL, Young RA. Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem Cell Rep 2013, 1:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Carriere L, Graziani S, Alibert O, Ghavi‐Helm Y, Boussouar F, Humbertclaude H, Jounier S, Aude JC, Keime C, Murvai J, et al. Genomic binding of Pol III transcription machinery and relationship with TFIIS transcription factor distribution in mouse embryonic stem cells. Nucleic Acids Res 2012, 40:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 133:1106–1117. [DOI] [PubMed] [Google Scholar]
- 75. Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L. seqMINER: an integrated ChIP‐seq data interpretation platform. Nucleic Acids Res 2011, 39:e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xiao T, Wallace J, Felsenfeld G. Specific sites in the C terminus of CTCF interact with the SA2 subunit of the cohesin complex and are required for cohesin‐dependent insulation activity. Mol Cell Biol 2011, 31:2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Curr Opin Struct Biol 2008, 18:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300‐mediated acetylation of histone H3 on lysine 56. Nature 2009, 459:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci 2013, 70:3989–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, Bacino CA, den Dunnen JT, van Ommen GJ, Breuning MH, et al. Genetic heterogeneity in Rubinstein‐Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 2005, 76:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Skibbens RV, Colquhoun JM, Green MJ, Molnar CA, Sin DN, Sullivan BJ, Tanzosh EE. Cohesinopathies of a feather flock together. PLoS Genet 2013, 9:e1004036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gerton JL. Translational mechanisms at work in the cohesinopathies. Nucleus 2012, 3:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gordillo M, Vega H, Trainer AH, Hou F, Sakai N, Luque R, Kayserili H, Basaran S, Skovby F, Hennekam RC, et al. The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet 2008, 17:2172–2180. [DOI] [PubMed] [Google Scholar]
- 84. Van Den Berg DJ, Francke U. Roberts syndrome: a review of 100 cases and a new rating system for severity. Am J Med Genet 1993, 47:1104–1123. [DOI] [PubMed] [Google Scholar]
- 85. Gordillo M, Vega H, Jabs EW. Roberts syndrome In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, eds. GeneReviews(R). Seattle, WA: University of Washington; 1993. [Google Scholar]
- 86. Whelan G, Kreidl E, Peters JM, Eichele G. The non‐redundant function of cohesin acetyltransferase Esco2: some answers and new questions. Nucleus 2012, 3:330–334. [DOI] [PubMed] [Google Scholar]
- 87. Whelan G, Kreidl E, Wutz G, Egner A, Peters JM, Eichele G. Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J 2012, 31:71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Xu B, Lu S, Gerton JL. Roberts syndrome: a deficit in acetylated cohesin leads to nucleolar dysfunction. Rare Dis 2014, 2:e27743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xu B, Lee KK, Zhang L, Gerton JL. Stimulation of mTORC1 with L‐leucine rescues defects associated with Roberts syndrome. PLoS Genet 2013, 9:e1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dorsett D, Krantz ID. On the molecular etiology of Cornelia de Lange syndrome. Ann N Y Acad Sci 2009, 1151:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped‐B. Nat Genet 2004, 36:631–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2‐type sister chromatid cohesion proteins and fly Nipped‐B, is mutated in Cornelia de Lange syndrome. Nat Genet 2004, 36:636–641. [DOI] [PubMed] [Google Scholar]
- 93. Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X‐linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 2006, 38:528–530. [DOI] [PubMed] [Google Scholar]
- 94. Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, Gil‐Rodriguez C, Arnedo M, Loeys B, Kline AD, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet 2007, 80:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Borck G, Zarhrate M, Bonnefont JP, Munnich A, Cormier‐Daire V, Colleaux L. Incidence and clinical features of X‐linked Cornelia de Lange syndrome due to SMC1L1 mutations. Hum Mutat 2007, 28:205–206. [DOI] [PubMed] [Google Scholar]
- 96. Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Monnich M, Yan Y, Xu W, Gil‐Rodriguez MC, et al. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet 2012, 90:1014–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gimigliano A, Mannini L, Bianchi L, Puglia M, Deardorff MA, Menga S, Krantz ID, Musio A, Bini L. Proteomic profile identifies dysregulated pathways in Cornelia de Lange syndrome cells with distinct mutations in SMC1A and SMC3 genes. J Proteome Res 2012, 11:6111–6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, Harrison PM, Yu Z, Kenmochi N, Kaufman TC, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster . Genome Biol 2007, 8:R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, Barna M. Ribosome‐mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 2011, 145:383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, Ball S, Tchernia G, Klar J, Matsson H, et al. The gene encoding ribosomal protein S19 is mutated in Diamond‐Blackfan anaemia. Nat Genet 1999, 21:169–175. [DOI] [PubMed] [Google Scholar]
- 101. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, et al. Identification of RPS14 as a 5q‐syndrome gene by RNA interference screen. Nature 2008, 451:335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Giagounidis AA, Germing U, Aul C. Biological and prognostic significance of chromosome 5q deletions in myeloid malignancies. Clin Cancer Res 2006, 12:5–10. [DOI] [PubMed] [Google Scholar]
- 103. Nakashima E, Mabuchi A, Makita Y, Masuno M, Ohashi H, Nishimura G, Ikegawa S. Novel SBDS mutations caused by gene conversion in Japanese patients with Shwachman‐Diamond syndrome. Hum Genet 2004, 114:345–348. [DOI] [PubMed] [Google Scholar]
- 104. Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM. Mutations in SBDS are associated with Shwachman‐Diamond syndrome. Nat Genet 2003, 33:97–101. [DOI] [PubMed] [Google Scholar]
- 105. Brooks SS, Wall AL, Golzio C, Reid DW, Kondyles A, Willer JR, Botti C, Nicchitta CV, Katsanis N, Davis EE. A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X‐linked microcephaly in humans. Genetics 2014, 198:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol 2000, 110:768–779. [DOI] [PubMed] [Google Scholar]
- 107. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood 2009, 113:6549–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, Kopmar N, Krasnykh O, Dean AM, Thompson SR, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell 2011, 44:660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bellodi C, Krasnykh O, Haynes N, Theodoropoulou M, Peng G, Montanaro L, Ruggero D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res 2010, 70:6026–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Marsh KL, Dixon J, Dixon MJ. Mutations in the Treacher Collins syndrome gene lead to mislocalization of the nucleolar protein treacle. Hum Mol Genet 1998, 7:1795–1800. [DOI] [PubMed] [Google Scholar]
- 111. Winokur ST, Shiang R. The Treacher Collins syndrome (TCOF1) gene product, treacle, is targeted to the nucleolus by signals in its C‐terminus. Hum Mol Genet 1998, 7:1947–1952. [DOI] [PubMed] [Google Scholar]
- 112. Dauwerse JG, Dixon J, Seland S, Ruivenkamp CA, van Haeringen A, Hoefsloot LH, Peters DJ, Boers AC, Daumer‐Haas C, Maiwald R, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet 2011, 43:20–22. [DOI] [PubMed] [Google Scholar]
- 113. Dixon MJ. Treacher Collins syndrome. J Med Genet 1995, 32:806–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Shiasi Arani K. Cartilage hair hypoplasia: first report from Iran. Med J Islam Repub Iran 2013, 27:157–160. [PMC free article] [PubMed] [Google Scholar]
- 115. Liu JM, Ellis SR. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood 2006, 107:4583–4588. [DOI] [PubMed] [Google Scholar]
- 116. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D. Impaired control of IRES‐mediated translation in X‐linked dyskeratosis congenita. Science 2006, 312:902–906. [DOI] [PubMed] [Google Scholar]
- 117. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Solomon DA, Kim T, Diaz‐Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 2011, 333:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Miller MA, Olivas WM. Roles of Puf proteins in mRNA degradation and translation. Wiley Interdiscip Rev RNA 2011, 2:471–492. [DOI] [PubMed] [Google Scholar]
- 120. Tsuda M, Sasaoka Y, Kiso M, Abe K, Haraguchi S, Kobayashi S, Saga Y. Conserved role of nanos proteins in germ cell development. Science 2003, 301:1239–1241. [DOI] [PubMed] [Google Scholar]
- 121. Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer‐specific activation of p53. Cancer Cell 2012, 22:51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Pistocchi A, Fazio G, Cereda A, Ferrari L, Bettini LR, Messina G, Cotelli F, Biondi A, Selicorni A, Massa V. Cornelia de Lange syndrome: NIPBL haploinsufficiency downregulates canonical Wnt pathway in zebrafish embryos and patients fibroblasts. Cell Death Dis 2013, 4:e866. [DOI] [PMC free article] [PubMed] [Google Scholar]