

Abstract
Background
More than 70 single nucleotide polymorphisms (SNPs) have been reported to be associated with prostate cancer (PC) risk; these were mainly identified in the general population with predominantly sporadic PC (SPC). Previous studies have suggested similar associations between a selection of these SNPs and hereditary PC (HPC). Our aim was to evaluate the effect of all known PC risk SNPs and their discriminative value for SPC and HPC.
Methods
Seventy‐four PC susceptibility SNPs (reported in literature up to June 2014) were genotyped in a population‐based series of 620 SPC patients, 312 HPC patients from the national Dutch registry and 1819 population‐based referents. Association analyses were performed using logistic regression, focusing on directional consistency of the odds ratios (ORs) with those in the original reports, that is, whether the OR was in the same direction as in the original report. Discriminative performance was evaluated by a genetic risk score used in logistic regression and receiver operating characteristic (ROC) curve analyses.
Results
Directional consistency was seen for 62 SNPs in SPC and 64 SNPs in HPC, 56 of which overlapped. ORs were mostly higher for HPC with 22 ORs >1.25 versus 5 for SPC. Discriminative performance was better for HPC with an area under the ROC curve of 0.73 versus 0.64 for SPC.
Conclusions
A large overlap was found for the associations between low‐penetrance susceptibility SNPs and SPC and HPC, suggesting a similarity in genetic etiology. This warrants a reconsideration of “HPC” and a restrictive policy toward prostate‐specific antigen testing in men with a positive family history. Genetic risk scores might be used for PC risk stratification on the population level. Prostate 75: 474–483, 2015 © 2014 The Authors. The Prostate published by Wiley Periodicals, Inc.
Keywords: single nucleotide polymorphism, genetics, association, risk score
INTRODUCTION
Globally, 307,000 men died from prostate cancer (PC) in 2012 1. This high number fuels the continuous search of the global research community for better biomarkers to identify men at high risk of PC, to improve (early) detection and to identify new therapeutic targets. In addition to urinary biomarkers such as PCA3 and TMPRSS2‐ERG, 74 germline single nucleotide polymorphisms (SNPs, reported in literature up to June 2014) have been discovered that are associated with PC risk (Table I). Most of these SNPs were identified and replicated in genome‐wide association studies (GWAS) conducted in the general population, that is, predominantly sporadic PC (SPC) 2, 3, 4, 5. SPC is considered to be multifactorial, resulting from a combination of environmental factors and rare to common genetic variants with small to modest risk‐increasing effects. By contrast, the cause for “hereditary” PC (HPC, defined as PC in three or more first‐degree relatives, two or more first‐degree relatives diagnosed under 55 years of age, or PC in three consecutive generations) is sought in high‐penetrance mutations 6. Linkage analysis studies in HPC families have indeed identified causal mutations, for example, in RNASEL (HPC1), HPC2/ELAC, and MSR1 7. However, these variants are rare and the results about their relevance in HPC are inconsistent among studies, making them inefficient as tests in genetic counseling.
Table I.
Association Results in SPC (n = 620) and HPC (n = 312) Patients for 74 Prostate Cancer SNPs
| Chromosome | SNP | Nearest gene(s) | Sourced | Risk allele | Other allele | OR original dataf | SPC patients (n = 620) | HPC patients (n = 312) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | P | OR | 95% CI | P | |||||||
| 1 | rs1218582 | KCNN3 | I | G | A | 1.06∼ | 1.07 | 0.94–1.22 | 0.31 | 0.92 | 0.77–1.10 | 0.34 |
| 1 | rs4245739e | MDM4 | I | A | C | 1.10 | 1.00 | 0.85–1.18 | 0.99 | 1.06 | 0.87–1.30 | 0.55 |
| 2 | rs11902236 | GRHL1 | I | A | G | 1.07∼ | 1.15 | 0.99–1.32 | 0.06 | 0.89 | 0.72–1.09 | 0.26 |
| 2 | rs1465618 | THADA | II | A | G | 1.08∼ | 1.14 | 0.98–1.33 | 0.09 | 1.14 | 0.92–1.40 | 0.23 |
| 2 | rs721048 | EHBP1 | III | A | G | 1.15 | 1.14 | 0.97–1.34 | 0.11 | 1.13 | 0.91–1.40 | 0.27 |
| 2 | rs6545977 | OTX1 | II | G | A | 1.15∼ | 1.22 | 1.07–1.39 | <0.01 | 1.16 | 0.97–1.39 | 0.10 |
| 2 | rs10187424e | GGCX, VAMP8 | IV | A | G | 1.09 | 1.10 | 0.97–1.25.2 | 0.15 | 1.17 | 0.98–1.38 | 0.09 |
| 2 | rs12621278 | ITGA6 | II | A | G | 1.33 | 1.15 | 0.87–1.52 | 0.33 | 2.16 | 1.29–3.61 | <0.01 |
| 2 | rs7584330 | COL6A3 ‐ MLPH (intergenic) | IV | C | T | 1.06 | 1.16 | 1.00–1.35237 | 0.05 | 1.04 | 0.84–1.28 | 0.73 |
| 2 | rs2292884 | MLPH | V | G | A | 1.14 | 1.18 | 1.02–1.37 | 0.02 | 1.10 | 0.90–1.35 | 0.36 |
| 2 | rs3771570 | FARP2 | I | A | G | 1.12∼ | 1.04 | 0.87–1.26 | 0.66 | 1.30 | 1.03–1.66 | 0.03 |
| 3 | rs2660753 | PPATP1 ‐ MIR4975 (intergenic) | VI | T | C | 1.18 | 1.26 | 1.01–1.56 | 0.04 | 1.31 | 0.98–1.75 | 0.06 |
| 3 | rs7611694 | SIDT1 | I | A | C | 1.10 | 1.13 | 0.96–1.33 | 0.13 | 1.15 | 0.96–1.37 | 0.14 |
| 3 | rs10934853 | EEFSEC (intergenic) | VII | A | C | 1.12 | 1.07 | 0.93–1.23 | 0.37 | 1.28 | 1.06–1.55 | 0.01 |
| 3 | rs6763931 | ZBTB38 | IV | T | C | 1.04 | 0.97 | 0.85–1.11 | 0.68 | 1.16 | 0.97–1.38 | 0.10 |
| 3 | rs10936632 | SKIL, CLDN11 | IV | A | C | 1.11 | 1.07 | 0.94–1.22 | 0.31 | 1.20 | 1.00–1.44 | 0.05 |
| 4 | rs1894292 | AFM | I | G | A | 1.10 | 0.99 | 0.87–1.13 | 0.91 | 1.10 | 0.92–1.31 | 0.28 |
| 4 | rs12500426 | PDLIM5 | II | A | C | 1.08∼ | 1.16 | 1.01–1.32 | 0.03 | 1.09 | 0.91–1.30 | 0.35 |
| 4 | rs17021918 | PDLIM5 | II | C | T | 1.11 | 1.10 | 0.96–1.25 | 0.17 | 1.10 | 0.91–1.32 | 0.32 |
| 4 | rs7679673 | RPL6P14, TET2 | II | C | A | 1.10∼ | 1.12 | 0.98–1.28 | 0.10 | 1.31 | 1.09–1.59 | <0.01 |
| 5 | rs2242652 | TERT | IV | G | A | 1.15 | 0.92 | 0.78–1.09 | 0.33 | 1.10 | 0.87–1.40 | 0.42 |
| 5 | rs2736098 | TERT | VIII | A | G | 1.13∼ | 1.15 | 1.00–1.33 | 0.05 | 1.18 | 0.96–1.43 | 0.11 |
| 5 | rs401681c | CLPTM1 | IX | C | T | 1.07∼ | 0.95 | 0.84–1.08 | 0.44 | 0.95 | 0.80–1.14 | 0.58 |
| 5 | rs2121875 | FGF10 | IV | G | T | 1.05 | 1.09 | 0.95–1.25 | 0.21 | 1.04 | 0.86–1.26 | 0.67 |
| 5 | rs6869841 | STC2, BOD1 | I | A | G | 1.07∼ | 1.07 | 0.91–1.25 | 0.43 | 1.04 | 0.84–1.29 | 0.72 |
| 6 | rs130067 | CCHCR1 | IV | G | T | 1.05 | 1.10 | 0.94–1.29 | 0.22 | 1.37 | 1.13–1.68 | <0.01 |
| 6 | rs3096702 | NOTCH4 | I | A | G | 1.07∼ | 1.10 | 0.96–1.26 | 0.16 | 1.10 | 0.91–1.32 | 0.32 |
| 6 | rs2273669 | ARMC2 | I | G | A | 1.07∼ | 1.23 | 1.02–1.47 | 0.03 | 0.78 | 0.59–1.03 | 0.08 |
| 6 | rs1933488 | RGS17 | I | A | G | 1.12 | 1.11 | 0.97–1.28 | 0.13 | 1.21 | 1.00–1.45 | 0.05 |
| 6 | rs651164 | SLC22A1, SLC22A2 | V | G | A | 1.15 | 1.16 | 1.00–1.33 | 0.04 | 1.21 | 0.99–1.47 | 0.06 |
| 6 | rs9364554 | SLC22A3 | VI | T | C | 1.17 | 1.25 | 1.08–1.43 | <0.01 | 1.38 | 1.14–1.66 | <0.01 |
| 7 | rs12155172 | RPS26P30, ASS1P11 | II | A | G | 1.11 | 0.94 | 0.81–1.11 | 0.47 | 1.23 | 1.00–1.51 | 0.05 |
| 7 | rs10486567 | JAZF1 | X | G | A | 1.12 | 1.16 | 0.99–1.36 | 0.07 | 1.02 | 0.82–1.26 | 0.85 |
| 7 | rs6465657 | LMTK2 | VI | C | T | 1.12 | 0.94 | 0.83–1.07 | 0.36 | 1.07 | 0.90–1.27 | 0.47 |
| 8 | rs1512268 | FAM6DP, NKX3‐1 | II | A | G | 1.18 | 1.03 | 0.90–1.17 | 0.69 | 1.16 | 0.97–1.39 | 0.10 |
| 8 | rs6984769a | EBF2 | I | A | G | 1.11∼ | 1.17 | 0.98–1.38 | 0.08 | 1.27 | 1.01–1.59 | 0.04 |
| 8 | rs12543663 | FAM84B | XI | C | A | 1.88∼ | 1.11 | 0.97–1.28 | 0.14 | 1.24 | 1.03–1.49 | 0.03 |
| 8 | rs10086908 | – | XII | T | C | 1.31∼ | 1.19 | 1.04–1.38 | 0.01 | 1.07 | 0.88–1.30 | 0.49 |
| 8 | rs13252298 | PCAT1 ‐ SRRM1P1 (intergenic) | V | A | G | 1.12∼ | 1.16 | 1.00–1.34 | 0.05 | 1.27 | 1.03–1.56 | 0.03 |
| 8 | rs16901979 | SRRM1P1 ‐ POU5F1B (intergenic) | XIII | A | C | 1.79 | 1.71 | 1.16–2.54 | 0.01 | 3.31 | 2.22–4.94 | <0.01 |
| 8 | rs16902094 | SRRM1P1 ‐ POU5F1B (intergenic) | VII | G | A | 1.21 | 1.16 | 0.97–1.39 | 0.10 | 1.29 | 1.02–1.63 | 0.03 |
| 8 | rs445114 | SRRM1P1 ‐ POU5F1B (intergenic) | VII | T | C | 1.14 | 1.12 | 0.97–1.28 | 0.12 | 1.01 | 0.84–1.21 | 0.95 |
| 8 | rs6983267 | SRRM1P1 ‐ POU5F1B (intergenic) | XIV | G | T | 1.26 | 1.23 | 1.08–1.40 | <0.01 | 1.69 | 1.40–2.04 | <0.01 |
| 8 | rs1447295 | LOC727677 (intergenic) | XV | A | C | 1.60 | 1.48 | 1.23–1.78 | <0.01 | 2.18 | 1.74–2.74 | <0.01 |
| 9 | rs1571801 | DAB2IP | XVI | A | C | 1.29∼g | 0.97 | 0.84–1.13 | 0.71 | 0.84 | 0.67–1.06 | 0.14 |
| 10 | rs10993994 | MSMB | VI | T | C | 1.16–1.25 | 1.14 | 1.00–1.31 | 0.05 | 1.52 | 1.27–1.82 | <0.01 |
| 10 | rs3850699 | TRIM8 | I | A | G | 1.10∼ | 1.07 | 0.92–1.24 | 0.36 | 1.32 | 1.08–1.63 | 0.01 |
| 10 | rs4962416 | CTBP2 | X | C | T | 1.17 | 1.10 | 0.95–1.28 | 0.21 | 1.22 | 0.99–1.49 | 0.06 |
| 11 | rs7127900 | MIR4686, ASCL2 | II | A | G | 1.22 | 1.22 | 1.04–1.43 | 0.02 | 1.79 | 1.46–2.19 | <0.01 |
| 11 | rs12418451 | TPCN2 ‐ MYEOV (intergenic) | XVII | G | A | 1.16∼ | 0.82 | 0.71–0.94 | 0.01 | 0.88 | 0.73–1.06 | 0.19 |
| 11 | rs11228565 | TPCN2 ‐ MYEOV (intergenic) | VII | A | G | 1.23 | 1.17 | 1.00–1.36 | 0.05 | 1.08 | 0.87–1.34 | 0.46 |
| 11 | rs10896450 | TPCN2 ‐ MYEOV (intergenic) | X | G | A | 1.10 | 1.14 | 1.00–1.30 | 0.05 | 1.14 | 0.95–1.37 | 0.15 |
| 11 | rs11568818 | MMP7 | I | A | G | 1.10 | 1.07 | 0.94–1.22 | 0.29 | 1.24 | 1.03–1.48 | 0.02 |
| 12 | rs10875943 | TUBA1C, PRPH | IV | C | T | 1.07 | 1.05 | 0.91–1.21 | 0.51 | 1.25 | 1.04–1.52 | 0.02 |
| 12 | rs902774 | KRT78, RPL7P41 | V | A | G | 1.17 | 1.06 | 0.89–1.26 | 0.55 | 1.07 | 0.84–1.35 | 0.59 |
| 12 | rs1270884 | GLULP5, TBX5 | I | A | G | 1.07 | 1.09 | 0.96–1.24 | 0.18 | 1.26 | 1.05–1.51 | 0.01 |
| 14 | rs8008270 | FERMT2 | I | G | A | 1.12 | 1.05 | 0.89–1.24 | 0.58 | 0.91 | 0.73–1.13 | 0.38 |
| 14 | rs7141529 | PPIAP6, RPL12P7 | I | G | A | 1.09∼ | 1.07 | 0.93–1.23 | 0.34 | 1.09 | 0.91–1.30 | 0.37 |
| 17 | rs684232 | VPS53 | I | G | A | 1.10 | 1.17 | 1.03–1.34 | 0.02 | 1.22 | 1.02–1.47 | 0.03 |
| 17 | rs4054823c | HS3ST3A1 (intergenic) | XVIII | T | C | 1.13∼ | 0.95 | 0.83–1.08 | 0.40 | 1.06 | 0.89–1.26 | 0.53 |
| 17 | rs11649743 | HNF1B | XIX | G | A | 1.28∼ | 1.04 | 0.89–1.23 | 0.62 | 1.70 | 1.32–2.20 | <0.01 |
| 17 | rs4430796e | HNF1B | XV | A | G | 1.22 | 1.23 | 1.08–1.39 | <0.01 | 1.61 | 1.35–1.93 | <0.01 |
| 17 | rs11650494 | FLJ40194, ZNF652 | I | A | G | 1.15∼ | 0.97 | 0.76–1.24 | 0.79 | 1.14 | 0.83–1.56 | 0.42 |
| 17 | rs1859962 | CALM2P1 ‐ SOX9 (intergenic) | XV | G | T | 1.20 | 1.18 | 1.03–1.34 | 0.01 | 1.44 | 1.21–1.73 | <0.01 |
| 18 | rs7241993 | SALL2, ATP9B | I | G | A | 1.09∼ | 1.14 | 0.99–1.32 | 0.08 | 0.95 | 0.78–1.15 | 0.57 |
| 19 | rs8102476 | DPF1 ‐ PPP1R14A (intergenic) | VII | C | T | 1.12 | 1.10 | 0.96–1.25 | 0.17 | 1.19 | 0.99–1.43 | 0.07 |
| 19 | rs2735839 | KLK3, KLK2 | VI | G | A | 1.20 | 1.22 | 1.01–1.48 | 0.04 | 1.19 | 0.92–1.54 | 0.19 |
| 20 | rs2427345 | RBBP8NL, GATA5 | I | C | T | 1.06 | 1.12 | 0.96–1.30 | 0.15 | 1.15 | 0.95–1.39 | 0.15 |
| 20 | rs6062509 | ZGPAT | I | A | C | 1.13∼ | 1.12 | 0.97–1.30 | 0.11 | 1.07 | 0.88–1.29 | 0.51 |
| 22 | rs9623117 | TNRC6B | XX | C | T | 1.18 | 1.02 | 0.87–1.20 | 0.81 | 0.99 | 0.79–1.24 | 0.94 |
| 22 | rs5759167 | RPS25P10, BIK | II | G | T | 1.16∼ | 1.27 | 1.11–1.45 | <0.01 | 1.88 | 1.54–2.28 | <0.01 |
| X | rs35330386b | SHROOM2 | I | T | C | 1.14∼ | 0.79 | 0.61–1.03 | 0.08 | 0.98 | 0.71–1.35 | 0.90 |
| X | rs5945572 | NUDT10, NUDT11 | III | A | G | 1.23 | 1.34 | 1.09–1.66 | 0.01 | 1.47 | 1.12–1.93 | 0.01 |
| X | rs5919432 | AR, PGK1P1 | IV | A | G | 1.06 | 0.96 | 0.74–1.25 | 0.77 | 1.08 | 0.76–1.51 | 0.68 |
SNP, single nucleotide polymorphism; SPC, sporadic prostate cancer; HPC, hereditary prostate cancer; OR, odds ratios; CI, confidence interval; HWE, Hardy–Weinberg equilibrium.
rs6984769 is a correlate (r2 = 1) for rs11135910.
rs35330386 is a correlate (r2 = 0.987) for rs2405942.
rs401681 has been associated with prostate‐specific antigen (PSA) levels, rs4054823 has been associated with prostate cancer aggressiveness.
Literature source: I Eeles, Nat Gen 2013; II Eeles, Nat Gen 2009; III Gudmundsson, Nat Gen 2008; IV Kote‐Jarai, Nat Gen 2011; V Schumacher, HMG 2011; VI Eeles, Nat Gen 2008; VII Gudmundsson, Nat Gen 2009; VIII Rafnar, Nat Gen 2009; IX Gudmundsson, Sci Tr Med 2011; X Thomas, Nat Gen 2008; XI Wang, Zhonghua Zhong Liu Za Zhi. 2013; XII Xu, CEBP 2009; XIII Gudmundsson, Nat Gen 2007; XIV Yeager, Nat Gen 2007; XV Gudmundsson, Nat Gen 2007 (2); XVI Duggan, JNCI 2007; XVII Zheng, CEBP 2009; XVIII Xu, PNAS 2010; XIX Sun, Nat Gen 2008; XX Sun, Cancer Res 2009.
Quality control indicated that the referents were not in HWE (P = 0.007 for rs4245739; P = 0.0007 for rs10187424; P = 0.002 for rs4430796); a manual check of the allele frequencies showed no deviations from the frequencies in the literature.
ORs were extracted from www.genome.gov (accessed on 16 April 2014; SNPs that were not present in this database were individually extracted from the original article (indicated with∼).
The OR for rs1571801 was calculated by combining the numbers for aggressive and non‐aggressive PC in the European populations (CAPS + CGEMS) in the original article.
It has been suggested that a considerable part of HPC families may not have an increased PC risk caused by a high‐penetrance mutation 8. In these families, more likely an accumulation of SPCs has occurred, because of increased PC awareness and testing 7, 8, 9. With the most recent discovery of PC susceptibility SNPs, it was estimated that the low‐penetrance variants may explain as much as 30% of familial PC (FPC) risk 10. In addition, the International Consortium for Prostate Cancer Genetics (ICPCG) found that 20 out of 25 SPC‐associated SNPs were similarly associated with familial PC 11. It is interesting to know whether these low‐penetrance genetic risk factors for SPC play a similar role in HPC. A large overlap might indicate that HPC nowadays is predominantly an accumulation of SPCs. That would mean that the globally used HPC definition, based on the number of diagnoses in a family, might fail to identify men with an inherited risk of PC. It would imply that, for the greater part, the men in the HPC families are more likely part of the general population and merely at the end of a normal distribution of common low‐risk risk variants. This could also mean that there is no good reason to pursue prostate‐specific antigen (PSA) screening in all HPC families if such screening is not advocated in the general population. Here, we aim to extend the findings of the ICPCG by performing association analyses for all 74 known PC risk SNPs (as reported in the literature up to June 2014) in Dutch SPC and HPC patients and to compare their discriminative value for SPC and HPC.
MATERIALS AND METHODS
Patients and Referents
The SPC patients were recruited for a population‐based study into the genetic susceptibility of PC and breast cancer (the EU 6th Framework Program funded project “Polygene”), which has been described in detail elsewhere 12. This study invited all newly diagnosed PC patients registered between 2003 and 2006 by the Comprehensive Cancer Centre the Netherlands (IKNL), location Nijmegen, who were 75 years or younger at diagnosis, living in the IKNL catchment area, and alive at the date of invitation (between September 2006 and June 2007). After exclusion of all nonwhite, non‐Western patients, 795 participants who completed a postal questionnaire and donated a blood sample were available for analysis. Of these participants, 175 (22%) were excluded because of a positive family history of PC. The HPC patients were recruited through the Netherlands Foundation for the Detection of Hereditary Tumours (NFDHT). The NFDHT registry holds 191 Dutch HPC families comprising 836 HPC patients, including 663 with a PC diagnosis verified by medical file review 13. All families were informed both by telephone and in writing about the current investigation in 2009, at which point 378 of the 663 verified HPC patients were still alive. Eighteen families denied consent and 26 were non‐informative, as all HPC patients were deceased. In total, 312 (81%) verified HPC patients, all of Caucasian descent, from 147 families provided sufficient material for germline DNA isolation, which was either saliva (Oragene® OG‐500 DNA Tube) or blood (two vials). Clinical information was successfully collected for 613 SPC and 309 HPC patients by cancer registry personnel of the IKNL via medical file review and used to stratify patients for PC aggression. Aggressive PC was defined according to the d'Amico criteria, that is, pT ≥ T2c, and/or pN + , and/or pM + , and/or PSA >20 and/or Gleason score ≥8. If a patient had missing data on one or more clinical characteristics, the remaining data were used to determine aggression using the same criteria. Aggressive PC was present in 387 SPC and 204 HPC patients 14.
The referents were selected from the Nijmegen Biomedical Study (NBS; a population‐based investigation initiated in 2001 by the Radboud university medical center (Radboudumc) in Nijmegen) 15. In short, 6468 age‐ and sex‐stratified randomly selected inhabitants of Nijmegen completed a postal questionnaire including questions about lifestyle, health status, and medical history and donated a blood sample for DNA isolation and biochemical studies. One thousand nine hundred and eighty of these participants, frequency‐matched for age and gender to patients with PC and female patients with breast cancer were chip‐genotyped (Illumina HumanHapCNV370‐Duo BeadChip) to serve as referents in GWAS (financial limitations prohibited the chipping of all participants) 16. A total of 1819 referent samples passed quality control (sample yield ≥96% [after exclusion of intensity‐only markers (n = 23,573)], Caucasian ancestry ≥90% [based on Structure analysis], SNP yield ≥96%). All participants provided written informed consent and approval to conduct these studies was obtained from the Institutional Review Board of the Radboudumc.
Genotyping
Germline DNA was isolated at the Radboudumc (HPC patients and referents) or at the deCODE Genetics facilities in Reykjavik, Iceland (SPC patients). Single‐SNP genotyping for SPC and HPC was carried out by deCODE Genetics, applying the Centaurus (Nanogen) platform 17. The quality of each Centaurus SNP assay was evaluated by genotyping each assay in the CEU and/or YRI HapMap samples and comparing the results with the HapMap publicly released data. Assays with >1.5% mismatch rate were not used. Correlated SNPs were used for rs11135910 (rs6984769; r2 = 1) and for rs2405942 (rs35330386; r2 = 0.987), because we did not succeed in developing a working genotyping assay for the originally reported SNPs. For the referents, 10 SNPs had already been genotyped using the previously mentioned chip. For the other SNPs, single‐SNP genotyping was performed as described above. Missing genotypes for the referents (due to quality control issues) were filled using imputed data from the genome‐wide chip, which was available for 70 of the 74 SNPs (three X‐chromosomal SNPs and rs3096702 were not imputed). Regarding imputation of the genome‐wide chip data: 323,414 SNPs passed quality control (minor allele frequency [MAF] ≥1%, and Hardy–Weinberg equilibrium [HWE] P‐value >10−6) and were used for imputation using 1000 genomes phase1 integrated version 3 as a reference sample using IMPUTE v2 software (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html) 18. Genotype probabilities were extracted for the SNPs present in the imputed data and transformed to hard calls using the software GTOOL (http://www.well.ox.ac.uk/∼cfreeman/software/gwas/gtool.html) and a genotype probability threshold of 0.9. Correlations of genotypes measured with single‐SNP assays and imputed genotypes were r2 > 0.9 for all SNPs except for rs2242652 (r2 = 0.55), rs2736098 (r2 = 0.74), rs16901979 (r2 = 0.86), rs16902094 (r2 = 0.87), rs4054823 (r2 = 0.86), rs11649743 (r2 = 0.80), rs4430796 (r2 = 0.84), and rs5759167 (r2 = 0.83). Individuals with a SNP call rate <90% were excluded, leaving 609 SPC and 282 HPC patients and 1803 referents available for analysis.
Very recently, a meta‐analysis reported 23 additional SNPs for prostate cancer 19. These new markers could not be included anymore in the present analysis because of logistical reasons.
Statistical Analyses
Association analyses were performed using Plink v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/), Stata v9.1 (Statacorp, College Station, Texas) and SPSS for Windows, release 20 (IBM Corporation, Armonk, NY) 20. Logistic regression models were used, assuming an additive relationship between the risk variants and PC. ORs and 95% confidence intervals (95% CI) were calculated for SPC and HPC separately, relative to the referents. As the referents were age‐matched, the logistic regression models only included the individual SNPs as a variable. The X‐chromosomal SNPs were analyzed using male referents only. The “risk allele” for each SNP, defined as the PC risk‐increasing allele, was extracted from www.genome.gov or from the original article (Table I). Our prime interest was directional consistency of the ORs in the SPC and HPC groups as compared with the literature, that is, an OR >1.00 for the risk‐increasing allele in the patient groups. Statistical significance was considered to be of less relevance, because the difference in group size (the SPC group was twice as large as the HPC group) makes that SPC would need a smaller effect size to reach the same significance level. Additionally, analyses were stratified by tumour aggressiveness. The analyses for HPC were repeated using a generalized estimating equation (GEE) regression analysis, which takes familial correlations into account.
To compare the discriminative value of the 74 SNPs for SPC and HPC patients, genetic risk scores were constructed by summing the number of risk alleles carried by each individual. Because all SNPs were replicated previously and our main purpose was to compare SPC versus HPC, all SNPs were included in the models, irrespective of statistical significance. The analyses were based only on patients and male referents with complete SNP data (169 SPC patients, 151 HPC patients, and 587 referents, respectively). The genetic risk score distribution among the referents with complete genotype data was used to generate 10 risk strata based on 10%‐percentiles. Logistic regression was used to calculate the OR per stratum for SPC and HPC separately, as compared to the two middle strata (41st–60th percentile). The risk score was also evaluated as a continuous variable in a logistic regression analysis. The area under the receiver‐operating characteristic curves (AUC) of the genetic risk score was also calculated. This was done for: (1) the subset of participants with complete genotype data; and (2) all participants after imputation of missing genotypes with the mean risk allele dosage for each SNP in the subgroup that the participant belonged to.
RESULTS
Table II lists the demographic and clinical characteristics of the groups. Tumor characteristics were comparable for the PC groups.
Table II.
Baseline and Clinical Characteristics of the SPC Patients, HPC Patients, and Referents
| SPC patients (N = 620) | HPC patients (N = 312) | Referents (N = 1,819) | ||||
|---|---|---|---|---|---|---|
| Age at diagnosis (patients) or selection (referents) – mean/range | 65/43–75 | 62/40–85 | 61/27–78 | |||
| N | % | N | % | — | — | |
| T‐stage | ||||||
| T1 | 116 | 18.7 | 40 | 12.8 | — | — |
| T2 | 285 | 46.0 | 148 | 47.4 | — | — |
| T3 | 192 | 31.0 | 86 | 27.6 | — | — |
| T4 | 15 | 2.4 | 7 | 2.2 | — | — |
| Unknown | 12 | 1.9 | 31 | 9.9 | ||
| N‐stage | ||||||
| N0/Nx | 581 | 93.7 | 303 | 91.7 | — | — |
| N1 | 39 | 6.3 | 9 | 2.9 | — | — |
| M‐stage | ||||||
| M0/Mx | 594 | 95.8 | 306 | 98.1 | — | — |
| M1 | 26 | 4.2 | 6 | 1.9 | — | — |
| Gleason score | ||||||
| 2–6 | 355 | 57.2 | 161 | 51.6 | — | — |
| 7 | 150 | 24.2 | 49 | 15.7 | — | — |
| 8–10 | 61 | 9.8 | 22 | 7.1 | — | — |
| Unknown | 54 | 8.7 | 80 | 25.6 | — | — |
| PSA at diagnosis (ng/ml) | ||||||
| <4 | 66 | 10.6 | 21 | 6.7 | — | — |
| 4–10 | 271 | 43.7 | 144 | 46.2 | — | — |
| 10–20 | 135 | 21.8 | 66 | 21.2 | — | — |
| >20 | 137 | 22.1 | 65 | 20.8 | — | — |
| Unknown | 11 | 1.8 | 16 | 5.1 | — | — |
| Aggressive PCa | ||||||
| Yes | 387 | 62.4 | 204 | 65.4 | — | — |
| No | 226 | 36.5 | 105 | 33.7 | — | — |
| Unknown | 7 | 0.9 | 3 | 1.0 | — | — |
PC, prostate cancer; HPC, hereditary prostate cancer; PSA, prostate‐specific antigen.
Aggressive PC was defined as PC with any or more of the following characteristics: pT ≥ T2c, and/or pN + , and/or pM + , and/or PSA >20, and/or Gleason score ≥8. If post‐surgical pathological TNM staging and Gleason score were not available, clinical stage and biopsy Gleason score were used.
Single SNP Associations for SPC and HPC
For SPC, 62 of the 74 SNPs showed directional consistency as compared to the literature (OR >1.00) (Table I). ORs were mostly between 1.05 and 1.25, in accordance with the original reports. For HPC, 64 SNPs showed directional consistency. Fifty‐six of these overlapped with the SNPs with directional consistency for SPC. The ORs were higher in HPC, with 22 ORs exceeding 1.25, as compared to only five for SPC. The SNPs with the highest ORs overlapped for SPC and HPC, that is, rs16901979 (SRRM1P1 ‐ POU5F1B) and rs1447295 (intergenic variant near LOC727677).
In the stratified analysis, 64 ORs >1.00 were seen for non‐aggressive SPC versus 59 for aggressive SPC (data not shown). ORs >1.00 were seen for 62 SNPs in non‐aggressive HPC versus 60 in aggressive HPC. The ORs in the stratified analyses were similar to the overall analysis. The GEE‐analyses for HPC [312 patients from 147 different families with a mean of two patients per family (range 1–7)], showed similar ORs with slightly wider 95% CIs (data not shown).
Discriminative Value of SNPs for SPC and HPC
With 74 SNPs (71 in autosomes and 3 on the X‐chromosome), each person can carry between 0 and 145 risk alleles. SPC and HPC patients carried more risk alleles than the referents (median [P5‐P95] SPC: 67 [58–76]; HPC: 69 [60–78]; referents: 64 [55–73]) (Figure 1). The discriminative value of the SNPs was better for HPC (Table III) and the genetic risk scores showed a clear dose–response pattern with increasing ORs for men carrying more risk variants (Table IV). In both groups, the top‐20% of the risk distribution had ORs of >1.8 as compared to the reference 41st–60th percentile group. The HPC patients had a higher per‐allele OR than the SPC patients (1.14 vs. 1.09) and a higher AUC (0.73 [95%CI 0.69–0.76] vs. 0.64 [0.62–0.67]).
Figure 1.
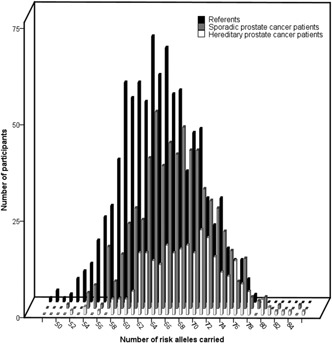
Distribution of the number of risk alleles for SPC patients, HPC patients, and male referents. Subscript: median number of risk alleles carried (P5‐P95): SPC patients (n = 609) 67 (58–76); HPC patients (n = 282) 69 (60–78); referents (n = 894) 64 (55–73).
Table III.
Discriminative Value of Genetic Risk Models for SPC and HPC Versus Male Referents, Calculated as the Area Under the Receiver Operating Characteristic Curve
| Referents | SPC | HPC | |||
|---|---|---|---|---|---|
| n | n | AUC (95% CI) | n | AUC (95% CI) | |
| Model 1: risk score (complete genotype data) | 587 | 169 | 0.63 (0.59–0.68) | 151 | 0.70 (0.66–0.75) |
| Model 2: risk score (missings imputed) | 890 | 609 | 0.64 (0.62–0.67) | 282 | 0.73 (0.69–0.76) |
SPC, sporadic prostate cancer; HPC, hereditary prostate cancer; AUC, area under the receiver operating characteristic curve; CI, confidence interval.
Table IV.
Genetic Risk Score Analysis for SPC (n = 169) and HPC (n = 151) Patients Versus All Male Referents With Complete Genotype Data (n = 587)
| Percentiles | Risk alleles | Referents | SPC | HPC | ||
|---|---|---|---|---|---|---|
| N | N | OR (95%CI) | N | OR (95% CI) | ||
| 1–10% | ≤57 | 59 | 11 | 0.73 (0.35–1.55) | 2 | 0.17 (0.04–0.74) |
| 11–20% | 58–59 | 63 | 5 | 0.31 (0.12–0.84) | 6 | 0.48 (0.19–1.22) |
| 21–30% | 60–61 | 67 | 9 | 0.53 (0.24–1.17) | 4 | 0.30 (0.10–0.89) |
| 31–40% | 62 | 33 | 6 | 0.72 (0.28–1.85) | 8 | 1.21 (0.50–2.92) |
| 41–60% | 63–65 | 130 | 33 | Reference | 26 | Reference |
| 61–70% | 66 | 40 | 16 | 1.58 (0.79–3.16) | 13 | 1.62 (0.76–3.45) |
| 71–80% | 67–69 | 84 | 34 | 1.60 (0.92–2.77) | 27 | 1.61 (0.88–2.94) |
| 81–90% | 70–71 | 50 | 23 | 1.81 (0.97–3.38) | 22 | 2.20 (1.14–4.23) |
| 91–100% | ≥72 | 61 | 32 | 2.07 (1.17–3.67) | 43 | 3.52 (1.99–6.26) |
SPC, sporadic prostate cancer; HPC, hereditary prostate cancer; OR, odds ratio; CI, confidence interval.
Percentile categories were based on the risk allele distribution in the referents with complete genotype data. The ORs and 95%CIs were calculated in logistic regression analyses, as compared to the reference population median (i.e., the 41–60% percentile), which was 63–65 carried risk alleles.
A separate logistic regression analysis using the genetic risk score as a continuous variable resulted in a per‐allele OR of 1.09 (95%CI 1.05–1.12) for SPC and 1.14 (95%CI 1.10–1.18) for HPC.
DISCUSSION
This study evaluated the effects of all 74 known susceptibility SNPs for PC in both SPC and HPC groups. The differences between SPC and HPC were relatively small, while both patient groups clearly differed from the referents. The highest ORs for individual SNPs as well as for the risk scores were found for HPC. Stratified analyses indicated similar SNP effects for aggressive and non‐aggressive PC. Recently published results from the ICPCG study in which 25 risk SNPs were evaluated showed ORs similar to those of our study 11. The ICPCG genotyped 9516 patients who were classified as familial PC. They concluded that the majority of the known PC risk SNPs also contributed to the risk of familial PC, as well as to aggressive familial PC in a subgroup analysis. Our results very much resemble the ICPCGs results, even though methodological differences between our studies are present. For instance, we used a less stringent definition of aggressive PC, genotyped an SPC group for direct comparison and included all known SNPs in the genetic risk score 21.
The similarity in SNP associations for SPC and HPC suggests an overlap in genetic etiology. Had PC in HPC families been caused by rare high‐penetrance mutations (as has been assumed in all previous linkage analyses), then the low‐penetrance SNP distribution of the HPC patients was expected to be similar to that of the referents. By contrast, our results show that the SNPs may even be somewhat stronger associated with HPC. This suggests that most likely (known or unknown) high‐penetrance mutations play only a minor role in HPC etiology. The results raise the question whether, nowadays, the larger part of HPC is different from SPC at all. Since the HPC definition was introduced in 1993, an increasing number of men have undergone opportunistic testing for PC, particularly men with an affected relative. A previous study concluded that the increased diagnostic activity among men with a family history of PC in itself contributed to their PC “risk” 9. Within some families this may have led to the detection of multiple (relatively low‐stage) PCs and, subsequently, “HPC.” As a result, a (probably growing) part of HPC might simply be an accumulation of SPCs 22, 23. This is supported by a screening study among non‐affected first‐degree relatives of HPC patients, in which no elevated PC risk was found 8. Also, population studies have not consistently shown differences between SPC and HPC with regard to clinical characteristics and prognosis, except for a lower age at diagnosis for HPC 22, 24. The results of this study and the previous investigations may therefore warrant a revision of the HPC definition. In its current form it will continue to lose validity as a criterion for selection of men to undergo targeted PC screening and/or genetic (sequencing) studies to identify novel, rare mutations. A new definition should attempt to incorporate an adjustment for the number of male relatives and the clinical characteristics of the diagnosed PCs, including the fact whether the PCs were screen‐detected or symptomatic. Obviously, in families with a known high‐risk mutation or families in which aggressive PCs are detected at young age, individual targeted screening remains important to prevent PC‐related mortality 25. One of the mutations that may become important in the near future, is a relatively new G84E‐variant in the HOXB13‐gene, that was first described in the four American HPC‐families 25. The HOXB13‐variant is not a SNP, as its population frequency in the Netherlands is <1%, so it was not included in this analysis. Still, also in our populations, the variant was more frequently present in both SPC (17/620 = 2.7%) and HPC (9/312 = 2.9%; this included one family with three affected carriers of the HOXB13‐variant and one family with two affected carriers of the HOXB13‐variant) than in the referents (7/1819 = 0.4%). In the (near) future, as also attempted in this study, genetic risk scores will hopefully be able to replace or complete family history in trying to better estimate an individual's genetic susceptibility to PC. A comprehensive genetic test in the future might combine the SNPs with the known high‐risk variants, such as mutations in, for example, MSR1, BRCA2, and HOXB13 into one genetic risk score 7, 25, 26. Although the currently known SNPs have limited discriminative power, Eeles et al. already demonstrated that risk alleles might be used for risk stratification at the group level 10. Our results support this idea, with ORs of >1.8 for the top‐20% of the risk allele distribution, as compared to the population median. This indicates that these SNPs might already be useful in risk calculators that incorporate factors with similar ORs, such as family history, age, and urinary complaints.
CONCLUSIONS
The results of this study suggest a large overlap between SPC and HPC with respect to low‐penetrance susceptibility SNPs, indicating a similarity in genetic etiology. For a considerable part, nowadays, HPC most probably is merely an accumulation of SPCs. This warrants a discussion about the current value of the definition of “HPC,” as our results suggest that there might not be a strong reason to pursue PSA screening in all HPC families, as such screening is not advocated in the general population. Genetic risk scores could play a role in better risk stratification, if they are incorporated into risk calculators.
ACKNOWLEDGEMENTS
Dr. R.G.C. was supported by Contract Number 202,059 (ProMark: genetic prostate cancer variants as biomarkers of disease progression; www.promark‐fp7.eu) from the Seventh Framework Program from the European Union and by an Agiko stipend (number 92003573) of the Netherlands Organisation of Health Research and Development. This work was sponsored by the Stichting Nationale Computerfaciliteiten (National Computing Facilities Foundation, NCF) for the use of supercomputer facilities, with financial support from the Netherlands Organisation for Scientific Research, NWO. We thank the Genome of the Netherlands Project (http://www.bbmriwiki.nl/wiki/Impute2Pipeline) and specifically Freerk van Dijk and Morris Swertz for imputation of the genotype data with 1000genomes phase1 integrated version 3. We thank deCODE Genetics (Reykjavik, Iceland) for the execution and assistance in the genotyping.
[The copyright line for this article was changed in February 2015 after original online publication.]
Ruben G. Cremers and Tessel E. Galesloot contributed equally. Sita H. Vermeulen and Lambertus A. Kiemeney contributed equally.
The authors have nothing to disclose.
REFERENCES
- 1. Bolla M, Poppel Hv. Management of prostate cancer: A multidisciplinary approach. Berlin: Springer; 2012; viii:pp. 338. [Google Scholar]
- 2. Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat Rev Cancer 2010; 10(5):353–361. [DOI] [PubMed] [Google Scholar]
- 3. Varghese JS, Easton DF. Genome‐wide association studies in common cancers—what have we learnt. Curr Opin Genet Dev 2010; 20(3):201–209. [DOI] [PubMed] [Google Scholar]
- 4. Witte JS. Prostate cancer genomics: Towards a new understanding. Nat Rev Genet 2009; 10(2)77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eeles RA, Kote‐Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, Muir KR, Smith C, Bagnato M, Ardern‐Jones AT, Hall AL, O'Brien LT, Gehr‐Swain BN, Wilkinson RA, Cox A, Lewis S, Brown PM, Jhavar SG, Tymrakiewicz M, Lophatananon A, Bryant SL, Collaborators UKGPCS, British Association of Urological Surgeons' Section of O, Collaborators UKPS, Horwich A, Huddart RA, Khoo VS, Parker CC, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Fisher C, Jamieson C, Cooper CS, English DR, Hopper JL, Neal DE, Easton DF. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 2008; 40(3):316–321. [DOI] [PubMed] [Google Scholar]
- 6. Carter BS, Bova GS, Beaty TH, Steinberg GD, Childs B, Isaacs WB, Walsh PC. Hereditary prostate cancer: Epidemiologic and clinical features. J Urol 1993; 150(3):797–802. [DOI] [PubMed] [Google Scholar]
- 7. Langeberg WJ, Isaacs WB, Stanford JL. Genetic etiology of hereditary prostate cancer. Front Biosci 2007; 12:4101–4110. [DOI] [PubMed] [Google Scholar]
- 8. Kiemeney LA, Broeders MJ, Pelger M, Kil PJ, Schroder FH, Witjes JA, Vasen HF. Screening for prostate cancer in Dutch hereditary prostate cancer families. Int J Cancer 2008; 122(4):871–876. [DOI] [PubMed] [Google Scholar]
- 9. Bratt O, Garmo H, Adolfsson J, Bill‐Axelson A, Holmberg L, Lambe M, Stattin P. Effects of prostate‐specific antigen testing on familial prostate cancer risk estimates. J Natl Cancer Inst 2010; 102(17):1336–1343. [DOI] [PubMed] [Google Scholar]
- 10. Eeles RA, Olama AA, Benlloch S, Saunders EJ, Leongamornlert DA, Tymrakiewicz M, Ghoussaini M, Luccarini C, Dennis J, Jugurnauth‐Little S, Dadaev T, Neal DE, Hamdy FC, Donovan JL, Muir K, Giles GG, Severi G, Wiklund F, Gronberg H, Haiman CA, Schumacher F, Henderson BE, Le Marchand L, Lindstrom S, Kraft P, Hunter DJ, Gapstur S, Chanock SJ, Berndt SI, Albanes D, Andriole G, Schleutker J, Weischer M, Canzian F, Riboli E, Key TJ, Travis RC, Campa D, Ingles SA, John EM, Hayes RB, Pharoah PD, Pashayan N, Khaw KT, Stanford JL, Ostrander EA, Signorello LB, Thibodeau SN, Schaid D, Maier C, Vogel W, Kibel AS, Cybulski C, Lubinski J, Cannon‐Albright L, Brenner H, Park JY, Kaneva R, Batra J, Spurdle AB, Clements JA, Teixeira MR, Dicks E, Lee A, Dunning AM, Baynes C, Conroy D, Maranian MJ, Ahmed S, Govindasami K, Guy M, Wilkinson RA, Sawyer EJ, Morgan A, Dearnaley DP, Horwich A, Huddart RA, Khoo VS, Parker CC, Van As NJ, Woodhouse CJ, Thompson A, Dudderidge T, Ogden C, Cooper CS, Lophatananon A, Cox A, Southey MC, Hopper JL, English DR, Aly M, Adolfsson J, Xu J, Zheng SL, Yeager M, Kaaks R, Diver WR, Gaudet MM, Stern MC, Corral R, Joshi AD, Shahabi A, Wahlfors T, Tammela TL, Auvinen A, Virtamo J, Klarskov P, Nordestgaard BG, Roder MA, Nielsen SF, Bojesen SE, Siddiq A, Fitzgerald LM, Kolb S, Kwon EM, Karyadi DM, Blot WJ, Zheng W, Cai Q, McDonnell SK, Rinckleb AE, Drake B, Colditz G, Wokolorczyk D, Stephenson RA, Teerlink C, Muller H, Rothenbacher D, Sellers TA, Lin HY, Slavov C, Mitev V, Lose F, Srinivasan S, Maia S, Paulo P, Lange E, Cooney KA, Antoniou AC, Vincent D, Bacot F, Tessier DC, Initiative CO‐CRUGE, Australian Prostate Cancer B, Oncology UKGPCSCBAoUSSo, Collaborators UKPS, Consortium P, Kote‐Jarai Z, Easton DF. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet 2013; 45(4):385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Teerlink CC, Thibodeau SN, McDonnell SK, Schaid DJ, Rinckleb A, Maier C, Vogel W, Cancel‐Tassin G, Egrot C, Cussenot O, Foulkes WD, Giles GG, Hopper JL, Severi G, Eeles R, Easton D, Kote‐Jarai Z, Guy M, Cooney KA, Ray AM, Zuhlke KA, Lange EM, Fitzgerald LM, Stanford JL, Ostrander EA, Wiley KE, Isaacs SD, Walsh PC, Isaacs WB, Wahlfors T, Tammela T, Schleutker J, Wiklund F, Gronberg H, Emanuelsson M, Carpten J, Bailey‐Wilson J, Whittemore AS, Oakley‐Girvan I, Hsieh CL, Catalona WJ, Zheng SL, Jin G, Lu L, Xu J, International Consortium for Prostate Cancer G, Camp NJ, Cannon‐Albright LA. Association analysis of 9560 prostate cancer cases from the International Consortium of Prostate Cancer Genetics confirms the role of reported prostate cancer associated SNPs for familial disease. Hum Genet 2014; 133(3):347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, Benediktsdottir KR, Magnusdottir DN, Orlygsdottir G, Jakobsdottir M, Stacey SN, Sigurdsson A, Wahlfors T, Tammela T, Breyer JP, McReynolds KM, Bradley KM, Saez B, Godino J, Navarrete S, Fuertes F, Murillo L, Polo E, Aben KK, van Oort IM, Suarez BK, Helfand BT, Kan D, Zanon C, Frigge ML, Kristjansson K, Gulcher JR, Einarsson GV, Jonsson E, Catalona WJ, Mayordomo JI, Kiemeney LA, Smith JR, Schleutker J, Barkardottir RB, Kong A, Thorsteinsdottir U, Rafnar T, Stefansson K. Genome‐wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet 2009; 41(10):1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meulenbeld HJ, Verhage BA, Kil PJ, Kiemeney LA, Vasen HF. [Characterisation of families with hereditary prostate cancer in the Netherlands]. Ned Tijdschr Geneeskd 2002; 146(41):1938–1942. [PubMed] [Google Scholar]
- 14. D'Amico AV, Whittington R, Malkowicz SB, Cote K, Loffredo M, Schultz D, Chen MH, Tomaszewski JE, Renshaw AA, Wein A, Richie JP. Biochemical outcome after radical prostatectomy or external beam radiation therapy for patients with clinically localized prostate carcinoma in the prostate specific antigen era. Cancer 2002; 95(2):281–286. [DOI] [PubMed] [Google Scholar]
- 15. Hoogendoorn EH, Hermus AR, de Vegt F, Ross HA, Verbeek AL, Kiemeney LA, Swinkels DW, Sweep FC, den Heijer M. Thyroid function and prevalence of anti‐thyroperoxidase antibodies in a population with borderline sufficient iodine intake: Influences of age and sex. Clin Chem 2006; 52(1):104–111. [DOI] [PubMed] [Google Scholar]
- 16. Kiemeney LA, Thorlacius S, Sulem P, Geller F, Aben KK, Stacey SN, Gudmundsson J, Jakobsdottir M, Bergthorsson JT, Sigurdsson A, Blondal T, Witjes JA, Vermeulen SH, Hulsbergen‐van de Kaa, Swinkels CA, Ploeg DW, Cornel M, Vergunst EB, Thorgeirsson H, Gudbjartsson TE, Gudjonsson D, Thorleifsson SA, Kristinsson G, Mouy KT, Snorradottir M, Placidi S, Campagna D, Arici M, Koppova C, Gurzau K, Rudnai E, Kellen P, Polidoro E, Guarrera S, Sacerdote S, Sanchez C, Saez M, Valdivia B, Ryk G, de Verdier C, Lindblom P, Golka A, Bishop K, Knowles DT, Nikulasson MA, Petursdottir S, Jonsson V, Geirsson E, Kristjansson G, Mayordomo B, Steineck JI, Porru G, Buntinx S, Zeegers F, Fletcher MP, Kumar T, Matullo R, Vineis G, Kiltie P, Gulcher AE, Thorsteinsdottir JR, Kong U, Rafnar A, Stefansson T. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nat Genet 2008; 40(11):1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kutyavin IV, Milesi D, Belousov Y, Podyminogin M, Vorobiev A, Gorn V, Lukhtanov EA, Vermeulen NM, Mahoney W. A novel endonuclease IV post‐PCR genotyping system. Nucleic Acids Res 2006; 34(19):e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome‐wide association studies. PLoS Genet 2009; 5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Al Olama AA, Kote‐Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, Benlloch S, Hazelett DJ, Wang Z, Saunders E, Leongamornlert D, Lindstrom S, Jugurnauth‐Little S, Dadaev T, Tymrakiewicz M, Stram DO, Rand K, Wan P, Stram A, Sheng X, Pooler LC, Park K, Xia L, Tyrer J, Kolonel LN, Le Marchand L, Hoover RN, Machiela MJ, Yeager M, Burdette L, Chung CC, Hutchinson A, Yu K, Goh C, Ahmed M, Govindasami K, Guy M, Tammela TL, Auvinen A, Wahlfors T, Schleutker J, Visakorpi T, Leinonen KA, Xu J, Aly M, Donovan J, Travis RC, Key TJ, Siddiq A, Canzian F, Khaw KT, Takahashi A, Kubo M, Pharoah P, Pashayan N, Weischer M, Nordestgaard BG, Nielsen SF, Klarskov P, Roder MA, Iversen P, Thibodeau SN, McDonnell SK, Schaid DJ, Stanford JL, Kolb S, Holt S, Knudsen B, Coll AH, Gapstur SM, Diver WR, Stevens VL, Maier C, Luedeke M, Herkommer K, Rinckleb AE, Strom SS, Pettaway C, Yeboah ED, Tettey Y, Biritwum RB, Adjei AA, Tay E, Truelove A, Niwa S, Chokkalingam AP, Cannon‐Albright L, Cybulski C, Wokolorczyk D, Kluzniak W, Park J, Sellers T, Lin HY, Isaacs WB, Partin AW, Brenner H, Dieffenbach AK, Stegmaier C, Chen C, Giovannucci EL. A meta‐analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet 2014; 46(10):1103–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Purcell S, Neale B, Todd‐Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: A tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet 2007; 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burgess S, Thompson SG. Use of allele scores as instrumental variables for Mendelian randomization. Int J Epidemiol 2013; 42(4):1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gronberg H, Damber L, Tavelin B, Damber JE. No difference in survival between sporadic, familial and hereditary prostate cancer. Br J Urol 1998; 82(4):564–567. [DOI] [PubMed] [Google Scholar]
- 23. Sacco E, Prayer‐Galetti T, Pinto F, Ciaccia M, Fracalanza S, Betto G, Pagano F. Familial and hereditary prostate cancer by definition in an italian surgical series: clinical features and outcome. Eur Urol 2005; 47(6):761–768. [DOI] [PubMed] [Google Scholar]
- 24. Gronberg H. Prostate cancer epidemiology. Lancet 2003; 361(9360):859–864. [DOI] [PubMed] [Google Scholar]
- 25. Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, Wiley KE, Isaacs SD, Johng D, Wang Y, Bizon C, Yan G, Gielzak M, Partin AW, Shanmugam V, Izatt T, Sinari S, Craig DW, Zheng SL, Walsh PC, Montie JE, Xu J, Carpten JD, Isaacs WB, Cooney KA. Germline mutations in HOXB13 and prostate‐cancer risk. N Engl J Med 2012; 366(2):141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bancroft EK, Page EC, Castro E, Lilja H, Vickers A, Sjoberg D, Assel M, Foster CS, Mitchell G, Drew K, Maehle L, Axcrona K, Evans DG, Bulman B, Eccles D, McBride D, van Asperen C, Vasen H, Kiemeney LA, Ringelberg J, Cybulski C, Wokolorczyk D, Selkirk C, Hulick PJ, Bojesen A, Skytte AB, Lam J, Taylor L, Oldenburg R, Cremers R, Verhaegh G, van Zelst‐Stams WA, Oosterwijk JC, Blanco I, Salinas M, Cook J, Rosario DJ, Buys S, Conner T, Ausems MG, Ong KR, Hoffman J, Domchek S, Powers J, Teixeira MR, Maia S, Foulkes WD, Taherian N, Ruijs M, den Enden AT, Izatt L, Davidson R, Adank MA, Walker L, Schmutzler R, Tucker K, Kirk J, Hodgson S, Harris M, Douglas F, Lindeman GJ, Zgajnar J, Tischkowitz M, Clowes VE, Susman R, Ramon YCT, Patcher N, Gadea N, Spigelman A, van Os T, Liljegren A, Side L, Brewer C, Brady AF, Donaldson A, Stefansdottir V, Friedman E, Chen‐Shtoyerman R, Amor DJ, Copakova L, Barwell J, Giri VN, Murthy V, Nicolai N, Teo SH, Greenhalgh L, Strom S, Henderson A, McGrath J, Gallagher D, Aaronson N, Ardern‐Jones A, Bangma C, Dearnaley D, Costello P, Eyfjord J, Rothwell J, Falconer A, Gronberg H, Hamdy FC. Targeted prostate cancer screening in BRCA1 and BRCA2 mutation carriers: Results from the initial screening round of the IMPACT Study. Eur Urol 2014; 66(3):489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]