

Abstract
Frontotemporal dementia is a devastating neurodegenerative disease causing stark alterations in personality and language. Characterized by severe atrophy of the frontal and temporal brain lobes, frontotemporal dementia (FTD) shows extreme heterogeneity in clinical presentation, genetic causes, and pathological findings. Like most neurodegenerative diseases, the initial symptoms of FTD are subtle, but increase in severity over time, as the disease progresses. Clinical progression is paralleled by exacerbation of pathological findings and the involvement of broader brain regions, which currently lack mechanistic explanation. Yet, a flurry of studies indicate that protein aggregates accumulating in neurodegenerative diseases can act as propagating entities, amplifying their pathogenic conformation, in a way similar to infectious prions. In this prion‐centric view, FTD can be divided into three subtypes, TDP‐43 or FUS proteinopathy and tauopathy. Here, we review the current evidence that FTD‐linked pathology propagates in a prion‐like manner and discuss the implications of these findings for disease progression and heterogeneity.
Frontotemporal dementia (FTD) is a progressive neurodegenerative disease causing severe personality dysfunctions, characterized by profound heterogeneity. Accumulation of tau, TDP‐43 or FUS cytoplasmic aggregates characterize molecularly distinct and non‐overlapping FTD subtypes. Here, we discuss the current evidence suggesting that prion‐like propagation and cell‐to‐cell spread of each of these cytoplasmic aggregates may underlie disease progression and heterogeneity.
This article is part of the Frontotemporal Dementia special issue.
Keywords: FTD, FUS, prion‐like spread, tau, TDP‐43
Abbreviations used
- 3/4R
3/4‐repeat‐tau
- AD
Alzheimer's disease
- AGD
argyrophilic grain disease
- ALS
amyotrophic lateral sclerosis
- bvFTD
behavioral variant FTD
- C9ORF72
chromosome 9 open reading frame 72
- CBD
corticobasal degeneration
- CHMP2B
charged multivesicular body protein 2B
- CSF
cerebrospinal fluid
- CTE
chronic traumatic encephalopathy
- EWS
Ewing sarcoma protein
- FET
FUS, EWS, TAF‐15
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- FUS
fused in sarcoma
- GRN
progranulin
- hnRNP
heterogeneous ribonucleoprotein
- HSPGs
heparan sulfate proteoglycans
- ISF
brain interstitial fluid
- MAPT
microtubule‐associated protein tau
- NES
nuclear export signal
- NLS
nuclear localization signal
- PD
Parkinson's disease
- PiD
Pick's disease
- prion
proteinaceous infectious particle
- PrP
prion protein
- PSP
progressive supranuclear palsy
- RRM
RNA recognition motifs
- TAF15
TATA‐binding protein‐associated factor 15
- TDP‐43
TARDBP‐binding protein, 43 kDa
- TD
tangle‐only dementia
- VCP
valosin‐containing protein
Protein aggregation in the central nervous system is a common feature of many neurodegenerative diseases. Although the main protein component and primary affected region can vary for each neurodegenerative disease, the accumulation of misfolded proteins into insoluble aggregates is a unifying hallmark and leads to progressive dysfunction and death of neurons and glial cells. Despite the diversity in clinical presentation, as well as pathological findings, increasing evidence supports a common mechanism driving neurodegeneration. Disease‐related proteins are transformed from their normal conformation into fibrillar or multimeric species that act as seeds of aggregation and further sequester their native isoforms and convert them into pathological molecules. This self‐perpetuating process leads to the formation of large aberrant protein aggregates, which subsequently fragment into new seeds that propagate their abnormal conformation in a template‐dependent manner within one cell, as well as through the CNS by the release of seeds into the extracellular space that can be taken up by neighboring cells leading to the repetition of the propagation cycle (Aguzzi 2009; Aguzzi and Rajendran 2009; Brundin et al. 2010; Goedert et al. 2010; Lee et al. 2010; Polymenidou and Cleveland 2011, 2012; Jucker and Walker 2013; Walker et al. 2013; Guo and Lee 2014; Walker and Jucker 2015; Sanders et al. 2016).
Such propagation mechanisms were long thought to be exclusively associated with transmissible prion diseases, in which template‐directed protein misfolding leading to the corruption of native molecules, is the basis for replication of infectious prions. In bona fide prion diseases (also called transmissible spongiform encephalopathies), ingestion or internalization of the proteinaceous infectious agent (prion) is sufficient to transmit the disease between individuals (Prusiner 1982). Prions replicate by recruiting the normal cellular isoform of the prion protein, PrPC, into ordered PrPSc‐containing aggregates thus inducing a pathological conformation. This continuous conversion into a highly aggregative form triggers a widespread misfolding and fibril formation across the nervous system in a self‐perpetuating manner (Aguzzi and Polymenidou 2004; Aguzzi 2009; Aguzzi and Rajendran 2009). Therefore, in prion diseases, seeded aggregation causes neurodegeneration and mice lacking the prion protein are completely resistant to the disease (Bueler et al. 1993). Prion pathology generally includes protein aggregation, neuronal loss, gliosis, and spongiform degeneration (Aguzzi and Polymenidou 2004). However, variations in the disease phenotypes, distinct histopathological signatures, varying incubation periods and disease progression, were found to be caused by different structural conformations of the misfolded prion protein, which are referred to as strains (Aguzzi et al. 2007).
Recently, the recognition of a similar propagation principle in other, non‐transmissible proteinopathies has led to the term ‘prion‐like’ (Goedert et al. 2010; Polymenidou and Cleveland 2011; Jucker and Walker 2013; Guo and Lee 2014). The emerging realization that pathological aggregates of amyloid‐β, tau, α ‐synuclein, and others can act in a prion‐like manner has changed the view on disease initiation and progression manner (Aguzzi 2009; Aguzzi and Rajendran 2009; Goedert et al. 2010; Polymenidou and Cleveland 2011, 2012; Jucker and Walker 2013; Guo and Lee 2014). While the mechanisms triggering the initial conversion of normally soluble proteins into pathogenic polymers remain unresolved, substantial evidence showed that aggregation seeds of amyloid‐β (Meyer‐Luehmann et al. 2006), tau (Clavaguera et al. 2009), and α‐synuclein (Nonaka et al. 2010; Volpicelli‐Daley et al. 2011; Luk et al. 2012) can induce template‐dependent misfolding of native proteins in vitro and in vivo. Moreover, cell‐to‐cell transmission of protein aggregates has also been proven for amyloid‐β, tau, α‐synuclein, SOD‐1, and TARDBP‐binding protein, 43 kD (TDP‐43) (Clavaguera et al. 2009; Grad et al. 2011; Nath et al. 2012; Nonaka et al. 2013), although both the exact spreading mechanism and nature of the transmissible entity have not been fully resolved.
These findings support an attractive hypothesis for the progressive nature of these diseases. Indeed, most neurodegenerative disorders are initially subtle but progress relentlessly, often exhibiting a clear spatiotemporal involvement of distinct, but interconnected regions of the nervous system. The molecular entity responsible for this disease spread remains controversial, but seeded aggregation of pathogenic proteins, which lead to neurotoxicity and eventually cell death, may well explain these phenomena.
Frontotemporal dementia is characterized by complex genetic causes and heterogeneous clinical presentation
Frontotemporal dementia (FTD) is the second most frequent cause of presenile dementia with age of onset typically between 45 and 65 years. It is associated with frontal and temporal lobe atrophy leading to a progressive dysfunction in behavior, personality, and language, mostly with preservation of memory (Neary et al. 1998; McKhann et al. 2001). The clinical phenotypes can be classified into behavioral variant FTD (bvFTD) and two versions of primary progressive aphasia: semantic dementia and progressive non‐fluent aphasia (Gorno‐Tempini et al. 2011; Rascovsky et al. 2011). Importantly, FTD clinically overlaps with amyotrophic lateral sclerosis (ALS), which is the most common motor neuron degeneration. Indeed, an estimated ~ 20% of FTD patients develop progressive paralysis and ~ 20% of ALS patients present personality changes compatible with FTD (Caselli et al. 1993; Neary et al. 2000; Lomen‐Hoerth et al. 2002; Ng et al. 2015).
FTD is a heritable disease with approximately 25–50% of cases reporting a family history and thus indicating a strong genetic component (Rohrer et al. 2009). The first FTD‐associated mutation was found in the microtubule‐associated protein tau (MAPT) gene on chromosome 17 in 1998 (Hutton et al. 1998). Up till now over 50 different causal MAPT mutations were found accounting for 5–20% of familial FTD cases (Rohrer et al. 2009; Spillantini and Goedert 2013). In 2006 mutations in the progranulin (GRN) gene – coincidentally near MAPT on chromosome 17 – were identified to cause an even larger proportion of familial FTD cases (Baker et al. 2006; Cruts et al. 2006). Several much less common mutations in the valosin‐containing protein (VCP) gene (Watts et al. 2004), the gene encoding charged multivesicular body protein 2B (Skibinski et al. 2005; Parkinson et al. 2006) and others have since been discovered. A major breakthrough in FTD genetics was the recognition of hexanucleotide repeat expansions in a non‐coding region of the chromosome 9 open reading frame 72 (C9ORF72) gene, which cause ~ 25% of all familial and ~ 6% of sporadic FTD cases, making it the most common genetic cause (DeJesus‐Hernandez et al. 2011; Renton et al. 2011). In people of European ancestry, C9ORF72 hexanucleotide repeat expansions also cause ~ 40% of familial ALS and ~ 7% of sporadic ALS, whereas up to 90% of families with concurrent ALS and FTD carry C9ORF72 mutations (Majounie et al. 2012) (Table 1).
Table 1.
Summary of prion‐like evidence for FTD‐associated proteins in vitro, in vivo, and in patients
| Protein | Human disease | Subcellular localization of aggregates | Seeded aggregation | Cell‐to‐cell spread | Experimentally transmitted disease | Strains | Pathology Staging in humans | |||
|---|---|---|---|---|---|---|---|---|---|---|
| In vitro | In cell culture | In vivo | In cell culture | In vivo | ||||||
| Tau | AD, FTLD‐Tau (FTLD‐17 MAPT, PSP, AGD, PiD, CBD), CTE | Cytoplasmic | Friedhoff et al. 1998; | Frost et al. 2009; Guo and Lee 2011; Wu et al. 2013; Nonaka et al. 2010; | Bolmont 2007, Clavaguera et al. 2009, 2013; Iba et al. 2013; Peeraer et al. 2015; Lasagna‐Reeves et al. 2012; | Kfoury et al. 2012; Iba et al. 2013; | Lasagna‐Reeves et al. 2012; de Calignon et al. 2012; Liu et al. 2012; Iba et al. 2013, Ahmed et al. 2014; Dujardin et al. 2014; | Peeraer et al. 2015; | Sanders et al. 2014; Boluda et al. 2015 | AD (Braak and Braak 1991; Braak and Braak 1995; Braak et al. 2006), CTE (Geddes et al. 1999; McKee et al. 2013) |
| TDP‐43 | FTLD‐TDP, ALS | Mostly cytoplasmic | Furukawa et al. 2011; | Furukawa et al. 2011; Feiler et al. 2015; | n.d. | Nonaka et al. 2013; Feiler et al. 2015; | n.d. | n.d. | FTLD (Brettschneider et al. 2014), ALS (Brettschneider et al. 2013) | |
| FUS | FTLD‐FUS, ALS | Mostly cytoplasmic, and rare intranuclear | Nomura et al. 2014 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
AD, Alzheimer disease; AGD, argyrophilic grain disease; ALS, amyotrophic lateral sclerosis; CBD, corticobasal degeneration; CTE, chronic traumatic encephalopathy; FTD, frontotemporal dementia; FTLD, frontotemporal lobar degeneration; FUS, Fused in sarcoma; PiD, Pick's disease; PSP, progressive supranuclear palsy; TDP‐43, TAR DNA‐binding protein of 43 kDa; n.d., not determined.
Frontotemporal dementia is subclassified into three distinct and non‐overlapping pathologies: tau, TDP‐43 and FUS
The neuropathology of clinical FTD also reflects the heterogeneity of the disorder. While frontotemporal lobar degeneration (FTLD) is the common underlying pathology of clinical FTD, several distinct disease types exist, subclassified based on the deposited protein found in abnormal intracellular inclusions (Mackenzie and Rademakers 2007; Mackenzie et al. 2010). The first subgroup includes ~ 40% of all FTLD cases and is defined by hyperphosphorylated tau inclusions in neurons and glia and is classified as FTLD‐tau. However, most cases of FTD are not associated with tau pathology and are instead defined by neuronal inclusions originally identified by their immunoreactivity for ubiquitin and consequently termed FTLD‐U (Josephs et al. 2004; Mackenzie et al. 2006). A major breakthrough followed when TDP‐43 was found to be the main component in ubiquitinated pathological inclusions in most FTLD‐U cases, as well as in the majority of ALS cases (Arai et al. 2006; Neumann et al. 2006). This finding not only resulted in a new subtype of FTD termed FTLD‐TDP (Neumann et al. 2006), but it also provided a strong connection between the two disorders that had clinically been connected before (Caselli et al. 1993; Neary et al. 1998; Lomen‐Hoerth et al. 2002). An overlapping molecular pathogenesis was further verified when the majority of tau‐ or TDP‐43‐negative inclusions were shown to be immunoreactive for a protein called fused in sarcoma (FUS) (Neumann et al. 2009a) shortly after FUS mutations had been detected as a cause of autosomal dominant ALS (Kwiatkowski et al. 2009; Vance et al. 2009). Interestingly, two other related DNA/RNA‐binding proteins Ewing sarcoma protein (EWS) and TATA‐binding protein‐associated factor 15 were recently found to co‐aggregate in FUS‐positive inclusions in FTLD‐FUS (Neumann et al. 2011).
While TDP‐43 and FUS pathology connect FTD and ALS, FTLD‐tau also shares its misfolded tau tangles with the most prominent neurodegenerative disorder, namely Alzheimer's disease (AD) (Querfurth and LaFerla 2010), whose primary pathology are insoluble, extracellular amyloid‐β plaques.
Pathophysiological roles of proteins involved in FTD
The microtubule‐associated protein tau forms toxic fibrils in FTD
In its natively unfolded form, tau protein is involved in microtubule formation and stabilization, as well as the regulation of axonal transport (Clavaguera et al. 2013). In humans, tau is abundantly found in neurons, where it concentrates in axons (Fig. 1a), but may also have a physiological function in dendrites (Binder et al. 1985). Six tau isoforms are expressed from a single gene (MAPT) by alternative mRNA splicing in the human brain (Goedert et al. 1989; Andreadis et al. 1992), each differing from the other by inclusion or exclusion of a 29‐ or 58‐amino acid N‐terminal insert and a 31‐amino acid repeat in the microtubule‐binding domain of tau, encoded by exon 10. Two major groups of tau isoforms can be distinguished: three isoforms with four exon 10 repeat sequences (4‐repeat‐tau, 4R) and three others with three repeat sequences (3‐repeat tau, 3R) (Goedert et al. 1989).
Figure 1.
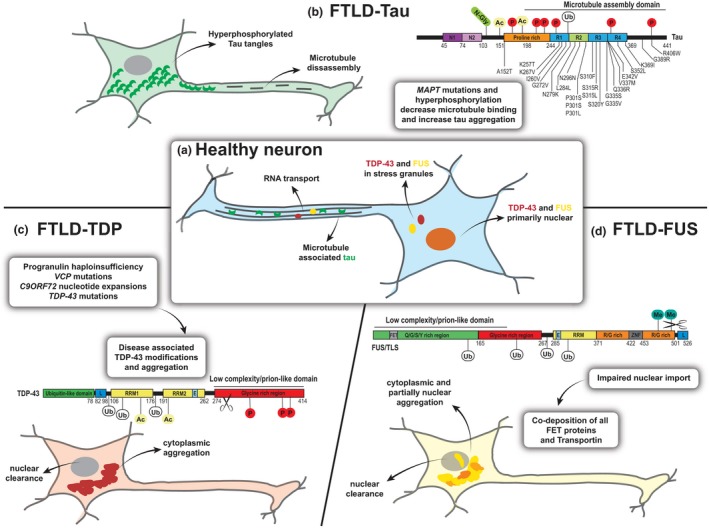
The three main types of FTD pathology are characterized by the accumulation of distinct protein aggregates. (a) In healthy neurons, tau (green crescent) is mainly distributed in axons and regulates microtubule stability. TDP‐43 (red) and FUS (yellow) are primarily found in the nucleus, where they are involved in several steps of RNA metabolism. In the cytoplasm, both proteins participate in dynamic RNA granules, facilitating axonal transport or forming as part of the cell stress response. (b) In pathological conditions of FTLD‐tau, the protein is dissociated from microtubules and accumulates into pathogenic tangles in the cytoplasm. Mutations in the microtubule‐binding region of the microtubule‐associated protein tau (MAPT) gene are found FTLD‐17 MAPT and can lead to splicing changes and decreased microtubule‐binding affinity. Hyperphosphorylation (P) and other post‐translational modifications (N‐Glyc (N‑glycosylation), Ac (Acetylation), Ub (Ubiquitination) can further enhance the detachment, resulting in microtubule disassembly and the formation of tau aggregates in the cytoplasm (example modification sites are shown). (c) In FTLD‐TDP‐43 several underlying genetic causes (GRN, VCP, TDP‐43, C9ORF72 mutations) and sporadic cases are linked by a common TDP‐43 pathology. TDP‐43 undergoes several disease‐associated modifications including phosphorylation, ubiquitination and C‐terminal cleavage and forms pathogenic inclusions in the cytoplasm leading to a redistribution of the protein, associated with nuclear clearance. (d) Although called FTLD‐FUS, all FET proteins have now been found to co‐aggregate in ubiquitinated inclusions in severe cases of sporadic FTD. Factors impairing the nuclear import of these proteins might lead to cytoplasmic accumulation and following aggregation because of strongly aggregation‐prone low complexity domains. C9ORF72: chromosome 9 open reading frame 72, E: Nuclear export signal, FET: FUS, TAF‐15, EWS protein interacting domain, FTD: frontotemporal dementia, FTLD: frontotemporal lobar degeneration, FUS: fused in sarcoma, L: nuclear localization signal, N1, N2: near‐amino‐terminal inserts, Q/G/S/Y‐rich: glutamine‐glycine‐serine‐tyrosine rich region, R1, R2, R3, R4: carboxy‐terminal repeat domain, R/G‐rich: arginine/glycine rich region, RRM: RNA recognition motif, TDP‐43: TAR DNA‐binding protein 43, VCP: valosin‐containing protein, ZNF: zinc finger domain.
In several disorders called tauopathies, the protein becomes hyperphosphorylated and forms insoluble filamentous inclusions making tau the most commonly misfolded protein in neurodegenerative diseases. Tauopathies include Alzheimer's disease, tangle‐only dementia and FTLD‐tau including frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17 MAPT), Pick's disease, argyrophilic grain disease, as well as progressive supranuclear palsy and corticobasal degeneration (Clavaguera et al. 2013; Lashley et al. 2015a). Hyperphosphorylation promotes the dissociation of tau from microtubules and is an early event in the process of tau aggregation. Hyperphosphorylated tau was shown to mislocalize to dendritic spines leading to synaptic abnormalities early in disease (Braak et al. 1994; Cochran et al. 2014). Many phosphorylation sites as well as responsible protein kinases and phosphatases have been identified and it has been suggested that tau may first misfold in human tauopathies, thus making it more accessible to protein kinases and less to protein phosphatases, leading to its hyperphosphorylation and increased aggregation propensity (Hanger et al. 2009; Clavaguera et al. 2013). In addition, a number of other disease‐associated post‐translational modifications of tau have been described, which include ubiquitination, acetylation, N‐glycosylation, O‐GlcNAcylation, nitration, prolyl isomerization, sumoylation, and truncation (Fig. 1b) (Lashley et al. 2015b; Wang and Mandelkow 2016).
Tau filaments possess a cross‐β structure characteristic of amyloid fibrils and it is widely accepted that their formation in specific brain regions contributes to disease pathogenesis and can induce neurotoxicity and cellular dysfunction. Insoluble tau aggregates can be found in numerous disorders, where they exhibit a range of morphologies (Crowther and Goedert 2000), with specific conformers of distinct isoform types giving rise to distinguishable cellular tau pathologies. For instance, a mixture of 3R and 4R tau form neurofibrillary tangles can be observed in the somatodendritic compartment, as well as neuropil threads in the distal axon and dendrites in AD. Conversely, in other tauopathies only one isoform is the predominant species, as reflected by characteristic ultrastructural morphologies that can be found in either exclusively neuronal or both neuronal and glial tau filaments (Goedert et al. 1992).
FTDP‐17 MAPT presents a special case of tauopathy in which mutations in the MAPT gene trigger abundant tau pathology (Hutton et al. 1998; Poorkaj et al. 1998; Spillantini et al. 1998; Goedert and Jakes 2005). Depending on the mutation, filamentous inclusions consist primarily of 3R, 4R or a mixture of all six tau isoforms and are deposited in neurons and glial cells (Ghetti et al. 2015). Further FTLD subtypes are also characterized by specific tau pathology with typically one isoform subtype: pick bodies composed primarily of 3R tau in Pick's disease (Delacourte et al. 1996), tufted astrocytes and numerous neurofibrillary tangles in subcortical nuclei in progressive supranuclear palsy (Flament et al. 1991), and 4R tau astrocytic plaques with abundant thread pathology in corticobasal degeneration (Ksiezak‐Reding et al. 1994). As a primary tauopathy that leads to severe neuronal loss, astrocytic gliosis, and spongiosis (Morris et al. 2001), FTLD‐tau demonstrates that dysfunction and subsequent accumulation of the tau protein per se suffices to cause neurodegeneration and dementia (Clavaguera et al. 2015).
The ribonuclear protein TDP‐43 accumulates in cytoplasmic aggregates in FTD and ALS
The discovery of TDP‐43 as the main component of ubiquitin‐positive, cytoplasmic inclusions in approximately 50% of patients with FTD in 2006, rendered FTLD‐TDP the most frequent pathological subtype and enormous efforts have been made ever since to uncover the underlying mechanisms of TDP‐43 pathogenesis. This finding also strongly highlighted the pathological link between FTD and ALS, which are now recognized as the two ends of the same disease spectrum (Ling et al. 2013).
TDP‐43 is a 414 amino acid protein encoded by the TARDBP gene on chromosome 1. It is a ubiquitously expressed and highly conserved heterogeneous nuclear ribonucleoprotein, involved in multiple steps of RNA maintenance and processing (Lagier‐Tourenne et al. 2010; Polymenidou et al. 2012). Two RNA recognition motifs (RRM1 and RRM2) in the center of the protein enable nucleic acid binding to UG‐repeat sequences (Polymenidou et al. 2011; Lukavsky et al. 2013), which might be supported by an atypical ubiquitin‐like domain, recently found in the N‐terminal part of the protein (Qin et al. 2014). In the nucleus, TDP‐43 plays a critical role in regulating alternative RNA splicing, as well as modulating the levels of long‐intron containing RNAs important for neuronal activity (Polymenidou et al. 2011; Lagier‐Tourenne et al. 2012). By regulating the stability of its own mRNA, TDP‐43 can autoregulate its protein levels (Ayala et al. 2011; Polymenidou et al. 2011; Bembich et al. 2014). In addition, TDP‐43 has recently been reported as a repressor of cryptic exons, which, when included, trigger the degradation of respective mRNAs (Ling et al. 2015). Although predominantly localized in the nucleus, TDP‐43 contains a nuclear localization signal (NLS) and a nuclear export signal, which allow for classical Importin α/β‐mediated shuttling (Winton et al. 2008).
Indeed, several roles have been proposed for TDP‐43 outside the nucleus (Fig. 1a) and nuclear efflux can be influenced by both neuronal activity (Wang et al. 2008) and stress (Bentmann et al. 2012). TDP‐43 is a key component in RNA transport granules and plays an important role in regulating local translation at distal locations, a function that is particularly important for neurons (Alami et al. 2014). Furthermore, TDP‐43 was shown to associate with stress granules (Liu‐Yesucevitz et al. 2010; Dewey et al. 2011; Bentmann et al. 2012), which are non‐membranous cytoplasmic foci composed of non‐translating messenger ribonucleoprotein complexes that transiently form in cells upon stress, thus selectively stalling protein synthesis (Anderson and Kedersha 2009). The C‐terminus of TDP‐43 harbors an aggregation‐prone glycine‐rich region important for protein–protein interactions (Buratti et al. 2005) that is necessary for its recruitment into stress granules, as deletion of this domain excluded TDP‐43 from these compartments (Dewey et al. 2011).
In disease, the physiological, predominantly nuclear localization of TDP‐43 is disturbed. Affected neurons and glial cells show a mislocalization to the cytoplasm with the formation of pathogenic inclusions (Fig. 1c) (Arai et al. 2006; Neumann et al. 2006; Igaz et al. 2008; Van Deerlin et al. 2008). This cytoplasmic aggregation prevents its return to the nucleus, leading to nuclear clearance. Moreover, pathological TDP‐43 inclusions present a specific biochemical signature including polyubiquitination, hyperphosphorylation, and proteolytic cleavage of the highly aggregation‐prone C‐terminus, which is co‐deposited in the cytoplasm along with the full‐length protein (Arai et al. 2006; Neumann et al. 2006). Interestingly, the majority of missense mutations linked to FTD or ALS in TDP‐43 are localized in the aggregation‐prone C‐terminal domain (Lagier‐Tourenne et al. 2010; Ling et al. 2013). The latter also contains a glutamine/asparagine‐rich low complexity or prion‐like domain that shares similarities with yeast prions (Cushman et al. 2010; Fuentealba et al. 2010). Yeast prion proteins contain regions that can switch their conformation between an intrinsically unfolded and an aggregated state that can impose its conformation to an unfolded counterpart, in a mechanism similar to mammalian prions (Cushman et al. 2010). Disease‐linked point mutations in this already aggregative prion‐like domain of TDP‐43 significantly increase its aggregation propensity, which hints to a causative link between TDP‐43 aggregation and disease (Johnson et al. 2009; Guo et al. 2011; Molliex et al. 2015). Importantly, in the cortex and hippocampus of FTLD‐TDP patients, phosphorylated C‐terminal fragments of TDP‐43 predominate over the full‐length protein in the cytoplasmic inclusions (Igaz et al. 2008), suggesting a key role in neurodegeneration. The precise pathogenic mechanisms triggered by TDP‐43 remain unknown and it remains unclear whether TDP‐43 toxicity results from the loss of its normal function, from direct toxicity of the cytoplasmic misfolded protein deposits or a combination of the two possibilities (Lagier‐Tourenne et al. 2010; Polymenidou and Cleveland 2012; Ling et al. 2013).
Nucleocytoplasmic transport defects cause cytoplasmic aggregation of FUS in FTD
Soon after the identification of TDP‐43 as a genetic and pathological link between FTD and ALS, another DNA/RNA‐binding protein, named FUS – also referred to as TLS for translocated in liposarcoma – was found in the pathological inclusions of about 5–10% of FTLD (Neumann et al. 2009a) and approximately 4% of familial ALS cases (Kwiatkowski et al. 2009; Vance et al. 2009). This discovery provided further evidence that ALS and FTD are closely related disorders and emphasized the pathogenic involvement of RNA‐binding proteins in these conditions. However, it quickly became clear that while in ALS‐FUS missense mutations in the FUS gene account for most familial ALS and rare sporadic cases, no genetic alterations have been associated with the vast majority of FTLD‐FUS cases. Yet, in the absence of FUS mutations, wild‐type FUS misaccumulates in distinct, atypical clinical subgroups of FTD patients (Neumann et al. 2009a), namely neuronal intermediate filament inclusion disease (Neumann et al. 2009b) and basophilic inclusion body disease (Munoz et al. 2009) without TDP‐43 or tau pathology (Urwin et al. 2010).
FUS is a 526 amino acid, ubiquitously expressed ribonucleoprotein and belongs to the FET (FUS, EWS, TAF‐15) family of proteins together with EWS and TAF‐15 (Bertolotti et al. 1996). It can bind nucleic acids through its C‐terminal part, which contains an RRM, two glycine/arginine‐rich motifs and a zinc finger domain. The N‐terminal part of FUS comprises a low complexity domain harboring a glutamine‐glycine‐serine‐tyrosine (Q/G/S/Y)‐rich region and the R/G‐rich motifs, which render the protein highly aggregation‐prone (Sun et al. 2011) and which is predicted to attain prion‐like properties (Cushman et al. 2010; Polymenidou and Cleveland 2011). An atypical nuclear localization signal (PY‐NLS) and a nuclear export signal allow Transportin‐mediated shuttling of the predominantly nuclear protein to the cytoplasm (Lee et al. 2006), suggesting that FUS is involved in RNA metabolism pathways that take place in both cellular compartments (Lagier‐Tourenne et al. 2010). Being not only structurally, but also functionally similar to TDP‐43, FUS has been implicated in transcriptional repression, splicing regulation, and micro‐RNA processing (Lagier‐Tourenne et al. 2010). Although TDP‐43 and FUS bind a distinct spectrum of RNAs, they share a preference for mRNAs derived from genes with exceptionally long introns and 45 common RNA targets, mostly important for normal neuronal function, are significantly down‐regulated upon depletion of either TDP‐43 or FUS in the adult mouse brain and in cultured human neurons (Polymenidou et al. 2011; Lagier‐Tourenne et al. 2012). Interestingly, several studies show altered splicing of the MAPT mRNA upon FUS depletion (Lagier‐Tourenne et al. 2012). In primary neurons, a loss of FUS leads to the expression of a longer tau isoform (Orozco et al. 2012) that has also been found to be increased in patients with FTD and Parkinsonism (Hutton et al. 1998).
In the cytoplasm, similar to TDP‐43, FUS can partition in stress granules (Bentmann et al. 2012) and plays a role in RNA transport granules, which carry mRNAs to dendritic spines for local translation (Kanai et al. 2004; Fujii and Takumi 2005; Elvira et al. 2006). Moreover, FUS interacts with the motor proteins kinesin and actin (Kanai et al. 2004; Yoshimura et al. 2006) and is essential for spine morphology (Fujii and Takumi 2005). Nevertheless, FUS predominantly localizes to the nucleus of healthy cells, whereas it is redistributed to the cytoplasm of affected cells, where it accumulates and aggregates (Fig. 1d). This mislocalization induces a partial loss of nuclear protein (Vance et al. 2009; Mackenzie et al. 2010), albeit to a lesser extent compared to TDP‐43 proteinopathies. Strikingly, while both pathogenic FUS (Neumann et al. 2009a; Urwin et al. 2010) and TDP‐43 (Neumann et al. 2006) inclusions are immunoreactive for p62 and ubiquitin, they seem mutually exclusive.
Evidence for prion‐like behavior of FTD‐associated proteins
In vitro seeding and in vivo propagation and spreading of misfolded tau was shown in multiple models
In its functional state tau is a hydrophilic protein that is natively unfolded with very little secondary structure, which makes it dynamic and highly soluble. Thus, its assembly into highly ordered β‐sheet structures that form insoluble amyloid fibers in disease is counterintuitive and several fibrillization enhancing tricks have been necessary to reproduce their formation in vitro or in cell culture. Adding polyanionic substances, RNA or fatty acids has been shown to promote fibril assembly of recombinant, full‐length tau protein in vitro by overcoming the nucleation barrier (Goedert 1996; Kampers et al. 1996; Chirita et al. 2003). Indeed, tau fibrillization follows a nucleation‐dependent mechanism, and initiating tau aggregation with preformed tau fibrils can bypass this nucleation step and accelerate fibrillization of monomeric tau (Friedhoff et al. 1998). Similarly, highly soluble tau resists aggregation despite high over‐expression and spontaneous hyperphosphorylation in cultured cells. Addition of minute quantities of preformed fibrils, however, is sufficient to recruit large amounts of soluble tau into filamentous inclusions with high efficiency (Friedhoff et al. 1998; Frost et al. 2009). Thus, a mechanism of template‐directed misfolding of soluble tau through recruitment to an initial aggregated seed has been suggested to underlie the propagation of tauopathies. Exogenously supplied recombinant tau filaments can be internalized by cells and seed aggregation by recruitment of endogenous tau into misfolded conformations (Frost et al. 2009; Guo and Lee 2011; Wu et al. 2013). Newly misfolded tau was competent to initiate further aggregation and could transfer between co‐cultured cells. The seed‐dependent polymerization and transcellular propagation of tau was also demonstrated by over‐expression of an aggregation‐prone mutant in cells leading to amyloid filaments that were released directly into the extracellular space and subsequently taken up by co‐cultured cells, inducing tau fibrillization.
The seeding and spreading of tau pathology via cell‐to‐cell transfer of aggregates was also demonstrated in mouse models in vivo. In an initial proof‐of‐principle study, brain homogenates of transgenic mice expressing mutant human tau with filamentous tauopathy were injected into mice transgenic for human wild‐type tau (Clavaguera et al. 2009). Upon inoculation, the originally inclusion‐free mice developed filaments consisting of wild‐type human tau in both neurons and oligodendrocytes. This induced tau pathology progressing over time from the injection site to neighboring and more distant, anatomically connected brain regions. Similar results were obtained using synthetic preformed tau fibrils to initiate extensive filamentous tau pathology in young asymptomatic mutant human tau over‐expressing mice (Iba et al. 2013; Peeraer et al. 2015). Furthermore, isolated tau oligomers from AD patient brains were able to seed and propagate widespread amyloidogenic tau plaques composed of endogenous murine tau, upon injection into the hippocampus of normal C57BL/6 mice (Lasagna‐Reeves et al. 2012).
The spreading ability of tau pathology was further confirmed by cell‐specific expression of the human mutant tau in the entorhinal cortex. Several months after the appearance of the first tau inclusions in the transgene‐expressing neurons, distant axonally connected cells in the hippocampus also developed filamentous tau pathology (de Calignon et al. 2012; Liu et al. 2012). Indeed, several studies have shown that induced tau pathology was able to propagate to anatomically connected brain regions indicating neuronal transport mechanisms and transsynaptic spread involved in the proliferation of neurofibrillary pathology (Iba et al. 2013; Ahmed et al. 2014; Dujardin et al. 2014). Neuronal activity can not only modulate tau protein secretion into the extracellular space in culture (Yamada et al. 2014), but it was further shown that synaptic contacts and neuronal activation can significantly potentiate neuron‐to‐neuron propagation of tau pathology (Calafate et al. 2015).
While the above in vitro and in vivo evidence strongly supports a template‐dependent tau seeding and spreading mechanism, a recent study pointed to a significant deviation from the classical prion‐like mechanism, by demonstrating that spreading of tau tangles does not depend on endogenous tau expression in vivo (Wegmann et al. 2015). Importantly, however, removing endogenous tau reduced neurotoxicity in this paradigm, potentially highlighting the fact that tau tangles do not equate the neurotoxic species. In agreement with this view, tau was shown to misfold into smaller aggregates prior to assembly into fibrils, and these smaller species, which are more efficiently taken up by neurons (Wu et al. 2013), may be the primary neurotoxic moiety.
The low complexity domains of TDP‐43 and FUS mediate the formation of self‐propagating, complex aggregates
Initial evidence suggests that a mechanism, similar to the spreading and seeding of tau, could underlie TDP‐43 and FUS proteinopathies and several studies have demonstrated that both proteins are intrinsically aggregation‐prone and can behave in a prion‐like manner, both in vitro and in cell culture. In vitro studies using recombinant, aggregated TDP‐43 (Furukawa et al. 2011) or FUS (Nomura et al. 2014) showed that minute amounts were sufficient to induce misfolding and aggregation of their corresponding natively folded protein, implicating template‐dependent propagation. The strong tendency of TDP‐43 and FUS to aggregate is mediated by the low complexity (Kato et al. 2012), or prion‐like (Cushman et al. 2010) domain found in both proteins; the N‐terminal part of FUS (amino acids 1‐239) and the C‐terminal part of TDP‐43 (amino acids 274‐414), which rank highly based on algorithms that identify aggregative and prion‐forming propensities (Alberti et al. 2009; Toombs et al. 2010). FUS is characterized by an even higher aggregation propensity than TDP‐43 and it was shown to spontaneously aggregate into pore‐like oligomeric species that rapidly assemble into filamentous structures in a cell‐free system within minutes (Couthouis et al. 2011; Sun et al. 2011). However, this capacity to aggregate is not enhanced by disease‐linked mutations affecting the NLS (Ju et al. 2011; Sun et al. 2011).
Over‐expression of TDP‐43 prion‐like domain, induced protein accumulation and cell toxicity (Furukawa et al. 2004; Johnson et al. 2009; Zhang et al. 2009; Liu‐Yesucevitz et al. 2010; Guo et al. 2011; Pesiridis et al. 2011), whereas deletion of the same domain from TDP‐43 (Johnson et al. 2009; Furukawa et al. 2011) or FUS (Sun et al. 2011) interfered with aggregate formation, indicating their requirement for efficient seeding. While short recombinant or synthetic peptides derived from the prion‐like domain can acquire toxic amyloid forms, purified full‐length TDP‐43 does not appear to have a classic amyloid structure, but rather forms small pore‐like oligomers and short fibrils (Johnson et al. 2009; Couthouis et al. 2011; Shimonaka et al. 2016), which can cluster together in large complexes with resemblance to pathological inclusions (Fang et al. 2014). Going a step further, it was demonstrated that aggregates reconstituted in vitro from recombinant wild‐type or mutant TDP‐43 or short synthetic peptides could seed aggregation of wild‐type or endogenously expressed protein in cell culture (Furukawa et al. 2011; Shimonaka et al. 2016).
While this has not yet been shown for FUS, it was recently reported that co‐expression of mutant and wild‐type FUS induces aggregation of the otherwise soluble wild‐type FUS (Nomura et al. 2014). In this study, a rather rare mutation located in the prion‐like domain changed the aggregation propensity of FUS and led to intranuclear inclusions, similar to what can be found in cases of atypical FTLD‐FUS (Neumann et al. 2009a; Nomura et al. 2014).
Supporting the prion paradigm and self‐templating properties of TDP‐43 further, aggregated TDP‐43 isolated from ALS and FTD patient brains was able to act as a propagative seed and induce protein misfolding and accumulation in TDP‐43 transfected cultured cells (Nonaka et al. 2013; Feiler et al. 2015). Moreover, the induced TDP‐43 aggregates persisted within cells when the original pathogenic seed was no longer present and could then act as new misfolding templates that exhibit properties consistent with prion‐like propagation (Nonaka et al. 2013). Recent evidence furthermore suggests that oligomeric TDP‐43 can be transmitted horizontally between cell somata likely via microvesicles, trigger oligomerization, and exert toxicity in recipient cells (Feiler et al. 2015). Neuron‐to‐neuron transmission experiments further indicated both anterograde and retrograde transsynaptic spreading of TDP‐43 oligomers and provide additional evidence of the propagative properties of TDP‐43 (Feiler et al. 2015).
To date, no report of prion‐like propagation of TDP‐43 or FUS in animal models could be found. However, a recent mouse model expressing a regulatable human TDP‐43 construct lacking the NLS indicated that existing intracellular TDP‐43 inclusions can be cleared in vivo, which can halt further neurodegeneration (Walker et al. 2015). Suppression of transgene expression after disease onset dramatically decreased the accumulation of insoluble, phosphorylated, cytoplasmic TDP‐43, reversed the loss of endogenous nuclear mouse TDP‐43 and prevented further motor neuron loss in this model (Walker et al. 2015). Similar results were obtained in a study using an inducible TDP‐43 transgene with a point mutation where suppression of transgene expression in mice with overt neurodegeneration for only 1 week was sufficient to clear pathological TDP‐43 and to significantly improve motor and behavioral deficits (Ke et al. 2015). This suggests that the presence of intracellular pathological TDP‐43 at a certain point might not be sufficient for cell‐to‐cell spread and propagation of disease pathology without a continuous pool of cytoplasmic TDP‐43. However, small quantities of transmitted pathological TDP‐43 in patients could lead to disease amplification over extended time periods (Walker et al. 2015).
While the prion‐like domains of TDP‐43 and FUS have been incriminated for their aggregation propensity, other protein regions seem to be contributing to this behavior. The FUS prion‐like domain alone is insufficient to cause aggregation, but requires the RNA‐binding motif and a glycine‐rich RGG domain (Sun et al. 2011). Intriguingly, this RGG domain contains a short low complexity region (amino acids 391–407), which might allow the protein to self‐aggregate using its two prion‐like domains (Gitler and Shorter 2011). Moreover, in contrast to most observations focusing on the C‐terminal domain of TDP‐43, two recent studies highlight the significance of the N‐terminal region, which was reported to mediate RNA binding thereby ensuring proper protein function, such as splicing regulation, as well as promoting aggregation (Zhang et al. 2013; Qin et al. 2014). This unexpected function is facilitated by an unusual structural switch from a folded ubiquitin‐like domain upon DNA binding in low protein concentrations to an unfolded and aggregation‐prone structure with increasing protein concentrations (Qin et al. 2014).
Increase in local concentration and RNA association in stress granules may initiate TDP‐43 and FUS aggregation
The mechanisms driving the onset of FTD and many other neurodegenerative disorders still remain unknown. While mutations in TDP‐43 seem to facilitate protein aggregation in subcellular compartments, alternative etiologies may contribute to pathogenesis in the majority of tau‐negative cases including sporadic forms of FTLD‐TDP and familial forms with GRN, C9ORF72, or VCP mutations all leading to a similar TDP‐43 pathology. Likewise, no genetic cause has been identified in aggressive forms of FTLD‐FUS. Thus, attractive candidates to trigger disease are environmental stimuli, such as cellular stress and the implication of both TDP‐43 (Colombrita et al. 2009; Moisse et al. 2009; Liu‐Yesucevitz et al. 2010; McDonald et al. 2011) and FUS (Andersson et al. 2008; Dormann et al. 2010; Bentmann et al. 2012) in stress granule assembly offers a plausible mechanism for aggregate nucleation and thereby, disease initiation and possible seeding (Ito and Suzuki 2011).
Indeed, the formation of stress granules per se is mediated by the ordered aggregation of TIA‐1, an essential stress granule component that possesses a prion‐like domain, which is not only required for TIA‐1′s nucleation, but can even be replaced by another prion‐like domain from a yeast prion protein without affecting the size or number of stress granules (Gilks et al. 2004). Aggregated TIA‐1 can then recruit mRNAs and other proteins, including TDP‐43 (Colombrita et al. 2009; Liu‐Yesucevitz et al. 2010; McDonald et al. 2011) and FUS (Andersson et al. 2008) into these cytoplasmic foci, which are transiently formed and resolved in a strictly regulated manner under physiological conditions (Buchan and Parker 2009; Wolozin 2012). However, since high protein concentration is a main determinant for protein aggregation, the increase in local TDP‐43 and FUS concentration within stress granules could facilitate aggregation initiation. With the persistence of cellular stress during aging, these granules may act as precursors of pathologic inclusions, which may transform into irreversible aggregates and form seeds of aggregation (Polymenidou and Cleveland 2011; Maniecka and Polymenidou 2015). This mechanism may be further assisted by the association with RNA molecules that can act as scaffolds mediating the ordered aggregation of TDP‐43 and FUS within these cytoplasmic granules. Indeed, the main RNA‐binding region of FUS is the arginine‐glycine‐glycine zinc finger domain, which is most important for stress granule recruitment, while both RRM1 and the C‐terminal glycine‐rich domain of TDP‐43 are required for SG localization (Bentmann et al. 2012). Furthermore, the scaffolding capacity of RNA has been shown in the in vitro aggregation of the mammalian prion protein (Deleault et al. 2003), as RNA and phospholipids were necessary to generate infectious prions with purified PrP (Wang et al. 2010). In addition, RNA was shown to mediate the formation of fibril‐like FUS assemblies in vitro (Schwartz et al. 2013).
After the initiation phase, another characteristic shared by TDP‐43 and FUS could potentially contribute to their self‐perpetuating aggregation. The levels of both proteins are strongly autoregulated through binding to their own mRNA transcripts and regulating the RNA processing pathways (Ayala et al. 2011; Polymenidou et al. 2011; Lagier‐Tourenne et al. 2012; Zhou et al. 2013). The formation of pre‐inclusions with sequestration of TDP‐43 or FUS in the cytoplasm would cause a reduction in nuclear protein, which in turn would lead to increased levels of stable TARDBP or FUS transcripts. Consequently and enhanced by the additional cell stress because of the formation of pre‐inclusions, TDP‐43 or FUS protein levels rise providing abundant substrate for continuous seeded aggregation, and thus contributing to the growth of pathogenic deposits (Maniecka and Polymenidou 2015). Increased levels of TDP‐43 mRNA measured in the brains of patients affected by various forms of FTLD (Mishra et al. 2007; Chen‐Plotkin et al. 2008) suggests that such a feed‐forward mechanism could enable and enhance aggregate propagation.
Interestingly, both TDP‐43 and FUS were found to colocalize with stress granule proteins, including TIA‐1 (Fujita et al. 2008; Liu‐Yesucevitz et al. 2010), PABP‐1 (Bentmann et al. 2012), and eIF3 (Liu‐Yesucevitz et al. 2010) in ALS and FTD patients. Several stressors, such as oxidative, osmotic, heat shock, and ER stress were able to recapitulate the cytoplasmic redistribution and stress granule association of TDP‐43 (Colombrita et al. 2009; Liu‐Yesucevitz et al. 2010; Dewey et al. 2011; McDonald et al. 2011; Leggett et al. 2012). Importantly, some of these stress‐induced granules turned into insoluble aggregates and were shown to persist after removal of the stressor (Dewey et al. 2011; Cohen et al. 2015; Feiler et al. 2015; Kabuta et al. 2015). Importantly, recent elegant biophysical studies (Burke et al. 2015; Molliex et al. 2015; Patel et al. 2015) support the hypothesis that soluble and dynamic stress granules can transform into insoluble and irreversible aggregates in vitro. Indeed, FUS and other RNA‐binding proteins containing low complexity domains can form dynamic liquid‐like assemblies in vitro (Han et al. 2012; Kato et al. 2012; Burke et al. 2015; Molliex et al. 2015; Patel et al. 2015) that resemble flexible cellular stress granules. Over time these dynamic assemblies can mature into rigid, large protein structures, reminiscent to pathological protein aggregates seen in patients (Burke et al. 2015; Molliex et al. 2015; Patel et al. 2015). Taken together, evidence suggests that stress granules may initiate or facilitate TDP‐43 and FUS seeding within the cytoplasm of diseased cells.
Notably, despite its unrelated structure and function, tau has also been shown to associate with stress granule proteins (Vanderweyde et al. 2012). Patients with FTLD‐tau and AD show large amounts of stress granules that colocalize with tau tangles and induction of stress granules in cells over‐expressing tau and TIA‐1 appears to be able to induce tau pathology (Vanderweyde et al. 2012). Furthermore, tau binding on RNA was shown to promote its aggregation into paired helical filaments (Kampers et al. 1996). Thus, stress granule formation is implicated in all subtypes of FTD (Vanderweyde et al. 2012).
Molecular mechanisms of cell‐to‐cell spread: what is transmitted and how?
Pathways of transcellular transport include active and passive mechanisms of release and uptake
Cell‐to‐cell propagation requires specific transmission events and the mechanisms by which misfolded protein complexes spread from one neuron to the next have not been fully determined. The protein seed needs to be released from one cell and get internalized by another where it has to be able to convert endogenous monomers to a pathogenic conformation and thus amplify. The properties of the transmitted protein aggregates, as well as the exact mechanisms, which underlie transcellular spread are still largely unknown and highly debated. The latter may include exocytosis, vesicle mediated (exosomes or pre‐synaptic vesicles) or passive release by degeneration of a cell. The receiving neuron might internalize the protein by macropinocytosis, receptor‐mediated endocytosis, vesicle membrane fusion or direct penetration. Direct transfer could also happen via tunneling nanotubes (Fig. 2).
Figure 2.
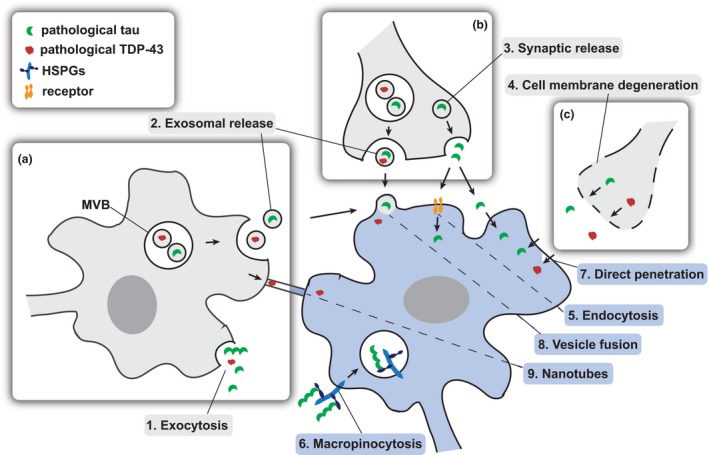
Proximity‐dependent and transsynaptic mechanisms of cell‐to‐cell spread. The transmission of pathological seeds (tau: green; TARDBP‐binding protein, 43 kD (TDP‐43): red) may happen between neighboring cells within the same brain region in a proximity‐dependent pattern through active release (a) or passive leakage (c) whereas spreading between distant anatomically connected brain regions may rely on synaptic connectivity (b). The protein seeds may be released from the donor cell (gray) via exocytosis (1), packaged into vesicles such as exosomes (released from multivesicular bodies (MVBs)) (2) or dispensed by synaptic vesicles (3), with all these mechanisms having been suggested for tau and TDP‐43. Furthermore, pre‐synaptic membrane leakage because of the degeneration of neurons can lead to extracellular protein seeds (4). Once released, free protein seeds may be internalized by receptor‐mediated endocytosis (5) or heparan sulfate proteoglycan (HSPG)‐dependent macropinocytosis (6) or directly penetrate the recipient cell (blue) (7). Membrane‐coated seeds can fuse with the receiving neuronal membrane (8). Direct transfer between two cells could also happen via tunneling nanotubes (9).
Although tau is predominantly an intracellular protein, multiple studies have shown that it is physiologically released into the extracellular space in cell culture and the mouse brain (Pooler et al. 2013b). It has been detected in the cerebrospinal fluid (CSF) and the brain interstitial fluid of wild‐type and transgenic mice (Yamada et al. 2011) and both healthy and AD individuals (Arai et al. 1995). It is, however, still controversial, whether tau is released in a free soluble form or inside membrane vesicles. Several studies have detected tau in various kinds of vesicles including exosomes (Saman et al. 2012), ectosomes (Dujardin et al. 2014), microvesicles (Simon et al. 2012), and synaptic vesicles (Pooler et al. 2013a) in cultured cells and animal models. Consistent with these models, tau was also found in exosomes in the CSF of healthy controls and AD patients (Saman et al. 2012), however, no data are available on FTLD‐tau patients. In addition, free tau seems to be released via unconventional secretory and vesicle independent mechanisms (Karch et al. 2012; Chai et al. 2012).
Increased levels of TDP‐43 have been detected in the CSF of ALS patients (Kasai et al. 2009), with some full‐length TDP‐43 and TDP‐43 CTFs being found in the exosomal fraction (Feneberg et al. 2014; Ding et al. 2015). Cell culture models also suggest the possible transfer of TDP‐43 via exosomes (Nonaka et al. 2013) or microvesicles (Feiler et al. 2015) and tunneling nanotubes (Ding et al. 2015). While many of the above mechanisms may also apply to FUS, there is currently no experimental evidence on the extent and mode or release and uptake of FUS aggregates by cells.
Extracellularly released protein needs to be taken up by surrounding cells. Macropinocytosis, as a form of fluid‐phase uptake, has been proposed as a mechanism for several extracellular aggregates including tau (Frost et al. 2009; Holmes et al. 2013). This process relies on heparan sulfate proteoglycans (HSPGs) as membrane receptors for binding, internalization, seeding, and transcellular propagation of tau (Holmes et al. 2013). While this process involves direct binding of free tau seeds, a new study suggests that HSPGs can also facilitate the internalization of exosomes (Christianson et al. 2013), and might thus offer penetration and propagation mechanisms for many other aggregated protein species. Intriguingly, TDP‐43 seems to be much more efficiently taken up when packaged into microvesicles/exosomes in cultured cells and primary neurons (Feiler et al. 2015). FTD‐associated proteins might take advantage of several of these mechanisms simultaneously to spread between neighboring cell bodies or synaptically connected neurons.
The importance of transcellular spreading via release and reuptake in disease propagation and pathology has been demonstrated using extracellular anti‐tau antibodies. Several antibodies have been shown to prevent tau spreading in cell culture (Kfoury et al. 2012), as well as in mouse models (Chai et al. 2011; Yanamandra et al. 2013, 2015) by blocking uptake of certain types of tau fibrils and promoting clearance by microglia (Funk et al. 2015). These studies strongly support the beneficial role of tau immunotherapy as it leads to reduced tau pathology, decreased seeding activity, as well as improved cognitive deficits in animal models (Boutajangout et al. 2011; Chai et al. 2011; Yanamandra et al. 2013, 2015).
While promoting microglial aggregate uptake and clearance decreased tau pathology in some studies, microglia have also been found to promote propagation and disease progression by phagocytosing and secreting tau protein in exosomes (Asai et al. 2015). Depleting microglia or inhibiting exosome synthesis was shown to significantly reduce tau propagation in vitro and in vivo (Asai et al. 2015).
Properties of transporting pathologic species are elusive
However, it is still unclear which protein species – monomer, oligomer, or aggregate – can efficiently spread and is responsible for transmitting pathology. Most extracellular tau seems to be monomeric and while a recent study suggests that trimers are the minimal unit of propagation leaving monomers unlikely to spread pathology as they are not spontaneously internalized (Mirbaha et al. 2015), another group showed that monomeric tau is sufficient to initiate the nucleation of aggregate seeds in an HSPG‐independent manner (Michel et al. 2014). Further studies, however, found no seeding activity for monomeric tau and small oligomers, but showed the requirement for short fibrils (Jackson et al. 2016) or even very large, high molecular weight species to transfer pathology efficiently (Takeda et al. 2015).
The properties of the propagation efficient TDP‐43 particles are even less defined. Recent evidence suggests that TDP‐43 can be transmitted horizontally in a dimeric/oligomeric form using the microsome/exosome pathway (Feiler et al. 2015). However, in this setting, TDP‐43 dimers/oligomers might represent a physiological state, different from the small pore‐like oligomeric structures found in transgenic mice and FTD patients (Fang et al. 2014). Furthermore, it has not been explored if TDP‐43 particles are released in a complex bound to nucleic acids or interacting proteins. This would pose an interesting question with regard to uptake mechanisms and further oligomerization and seeding properties in the recipient cell.
Neurodegenerative changes gradually intensify from most to less affected regions of the human nervous system
Disease staging studies and implications for disease progression
Intriguingly, many neurodegenerative diseases not only share the general pathology of misfolded protein aggregates, but also clinically present a characteristic progressive nature. Progressing disease symptoms have therefore been hypothesized to coincide with the propagation of pathology across anatomically connected regions of the nervous system. Indeed, neuropathological studies have recently recognized that characteristic anatomical patterns can be found in various neurodegenerative diseases and that the progression of these stereotypical patterns is linked to an increasing severity of the disease phenotype (Brettschneider et al. 2015). Staging studies could show that pathological events initiate very early in a certain CNS area and progress sequentially in a topographically predicted manner through anatomical connections.
Early staging attempts of disease progression were based on clinical signs and brain atrophy (Broe et al. 2003; Mioshi et al. 2010; Kril and Halliday 2011). One of the best described cases of progressive neurodegenerative changes is ALS, where motor neuron loss was shown to initiate focally and progress gradually away from this initial focus, which generally coincides with the spinal cord level that defines the original symptoms (Ravits et al. 2007a,b; Ravits and La Spada 2009; Ravits 2014). Later efforts concentrated on analyzing aggregate size and number of affected cells with pathological inclusions as molecular correlates of the systematic symptom spread. Indeed, a gradual distribution of pathological TDP‐43 aggregates was found in ALS and FTD patients throughout the neuraxis (Geser et al. 2009). Furthermore, a four stage model describing the sequential spread of TDP‐43 pathology in ALS was proposed based on the progressive dissemination of intraneuronal phospho‐TDP‐43 positive aggregates. According to this model, ALS starts in the cerebral neocortex and the somatomotor neurons of the spinal cord and the lower brainstem and spreads in an anterograde manner along corticofugal axonal pathways to connected subcortical areas and distant CNS sites (Braak et al. 2013; Brettschneider et al. 2013). Similarly, a recent study focused on phospho‐TDP‐43 positive inclusions in bvFTD suggests a sequential spreading pattern along a fronto‐occipital gradient with the first lesions starting in orbitofrontal areas and the amygdala, followed by lesions appearing later in premotor, primary motor, parietal and, finally, occipital areas of the cortex (Brettschneider et al. 2014).
While, the expansion and stereotypical spread of tau pathology across the nervous system have been clearly characterized in AD (Braak and Braak 1995; Braak et al. 2006) and chronic traumatic encephalopathy (Braak and Braak 1995; Geddes et al. 1999; Braak et al. 2006; McKee et al. 2013), one recent study also relates the neuropathological progression of tauopathy with clinical symptoms of bvFTD. Spreading sequentially from the frontotemporal and neocortical regions to the subcortical structures, the primary motor cortex and finally the visual cortex tau pathology reflected the evolution of clinical symptoms and directly correlated with disease duration (Irwin et al. 2016).
Collectively, the above data support the spatiotemporal spread of pathologic aggregates throughout the nervous system, although the alternative hypothesis of a temporally selective regional vulnerability of neurons to lesion formation (Walsh and Selkoe 2016) has not been formally excluded.
Protein aggregates preferentially spread across neuronal networks in FTD
More and more evidence supports the idea that neuronal connectivity defines disease‐spreading pathways and that pathology is transmitted along neural networks through propagation of self‐templating misfolded protein aggregates. After disease origination at an initial focal point, anatomically neighboring regions may lie at risk of involvement through proximity‐dependent transfer of released pathological seeds. However, recent imaging studies relating degeneration patterns to neural connectivity suggest a ‘network‐based neurodegeneration’ hypothesis supporting the model of transsynaptic propagation of toxicity along functionally and anatomically connected networks (Raj et al. 2012; Zhou et al. 2012; Agosta et al. 2015). For each disease type specific neural networks emerged with their critical center areas and functional and anatomical connectivity profiles resembling disease‐associated atrophy patterns. Modeling analysis thus supports a pathology model of transneuronal spread based on connectional strength that defines disease vulnerability (Zhou et al. 2012).
In bvFTD progressive atrophy patterns were found to resemble a network called the salience network. The latter, which includes the anterior cingulate cortex and frontoinsula, as well as the amygdala and striatum (Rosen et al. 2002; Broe et al. 2003; Seeley 2008), is important for socioemotional tasks relying on attentional selection and self‐regulation of behavior (Seeley et al. 2007). bvFTD patients exhibit reduced connectivity in the salience network, which has been correlated with clinical severity, apathy, and disinhibition scores (Zhou et al. 2010; Filippi et al. 2013). Furthermore, changes in the white matter tracts connecting key regions of the salience network reflect the propagation of pathology and have been proposed as a marker to assess in vivo spreading of pathological proteins (Agosta et al. 2012; Lam et al. 2014).
In the context of cell‐to‐cell propagation of toxic seeds, the network hypothesis offers a plausible route for the pathological spread that defines clinical presentation. The original site of pathology may determine the network involved and eventually the clinical phenotype (Sanders et al. 2016). Inoculation studies (Iba et al. 2013), as well as region‐selective expression of mutant tau (de Calignon et al. 2012; Liu et al. 2012) in mice strongly support the selective spreading of pathology along known anatomical networks, Indeed, pathology in the entorhinal cortex was induced by tau pathology initiated in the hippocampus, while injection into the striatum led to pathology in the substantia nigra and the thalamus (de Calignon et al. 2012; Liu et al. 2012; Iba et al. 2013).
Multiple conformations of pathogenic protein assemblies may contribute to disease heterogeneity
Aside from the network‐specific involvement, the molecular principles of the prion paradigm may also offer an explanation for the high variability in pathology and clinical presentation in neurodegenerative diseases. Indeed, the concept of strains is well established for prion diseases, where several distinct self‐propagating conformers or strains underlie different pathological manifestations and disease phenotypes in humans and mice (Aguzzi et al. 2007). Recently, other neurodegenerative disease‐associated proteins are also revealing strain‐like properties including α‐synuclein (Guo et al. 2013) and Aβ (Petkova et al. 2005; Heilbronner et al. 2013). Strain manifestation seems particularly plausible for tau, given the variety of clinical and neuropathological characteristics in tauopathies with differences in regional involvement, disease duration, age of onset, isoform expression, and fibril morphology.
Indeed, seeded aggregation of filaments consisting of either 3R or 4R isoforms was only successful in cells expressing the same isoform suggesting specificity in the assembly of amyloid fibers (Nonaka et al. 2010). Furthermore, two studies have recently shown that tau can adopt different stable conformational states that can propagate through recruitment of native tau. Brain extracts from patients with different tauopathies were able to induce differential patterns of spread and tau deposition in mice (Sanders et al. 2014; Boluda et al. 2015). Similarly, inoculation with distinct tau strains produced in vitro result in unique and consistent pathologies upon serial passages in mice (Sanders et al. 2014), thus showing stable conformational integrity analogous to prion strains. In addition, a recent biochemical analysis of pathological tau aggregates from patient brains found distinct C‐terminal band patterns indicating disease specific conformations (Taniguchi‐Watanabe et al. 2016).
Similarly, distinct patterns of TDP‐43 pathology have been described. FTLD‐TDP can be pathologically divided into four subtypes (A–D), based on the predominant form of inclusions found and each type is associated mostly with a certain disease phenotype and often a mutation in a specific FTLD‐TDP related gene (Mackenzie et al. 2011). For example, type A is defined by large amounts of short dystrophic neurites and oval‐shaped neuronal cytoplasmic inclusions and associated with bvFTD and mutations in the progranulin gene. Some evidence suggests that distinct structural conformations of TDP‐43 might underlie these pathological subtypes (Tsuji et al. 2012). Different mutations or post‐translational modifications could introduce variations in the protein fold that would further lead to differences in the misfolded conformers. Distinct pathogenic templates could have unique characteristics leading to cell or region‐specific preferences, varying propagation, and seeding properties, and different levels of toxicity (Smethurst et al. 2015). Indeed, trypsin and chymotrypsin digestion of sarkosyl‐insoluble TDP‐43 inclusions from patients produced specific protease‐resistant TDP‐43 fragments leading to a distinct banding pattern for subtypes A‐C (Hasegawa et al. 2011; Tsuji et al. 2012). Moreover, seeding aggregation with in vitro aggregated synthetic TDP‐43 peptides (Shimonaka et al. 2016) as well as homogenates from ALS and FTD patients (Nonaka et al. 2013) in cell culture models resulted in various types of pathologies showing the same distinct biochemical characteristics as observed in the original seeds. Interestingly, seeded aggregation required the interaction of the peptide seed with the identical peptide sequence of the host protein (Shimonaka et al. 2016). This indicates, that conformation‐specific templating of TDP‐43 can lead to differentially misfolded conformers, which furthermore can cause different patterns of pathology and disease phenotypes in a strain‐like manner.
In conclusion, prion‐like phenomena may well explain the heterogeneity in clinical and pathological presentation of FTD, as well as the molecular basis of disease progression and spread in the nervous system. Further studies are needed to decipher the particulars of this intriguing pathogenic pathway.
Conclusions and open questions
In conclusion, multiple lines of evidence support a template‐directed misfolding and propagation of pathology of FTD‐associated proteins, although the state of research varies for each protein. Indeed, more evidence has accumulated over the years for the propagation of tau, whose involvement in neurodegeneration dates back to almost 20 years now (Hutton et al. 1998), while much less is known for the more recently identified TDP‐43 (Neumann et al. 2006) and FUS (Kwiatkowski et al. 2009; Vance et al. 2009).
Several intriguing questions remain unanswered. Here, we list some of those and propose potential experimental approaches and key considerations when attempting to address them.
What are the exact properties of the pathologic state? Which of those define propagation and/or neurotoxicity?
Defining structural properties of protein aggregates remains a technologically challenging task, and few techniques can reliably assess them. One of them is solid‐state NMR, which was used to define the beta‐sheet core of aggregated tau (Daebel et al. 2012), albeit no such studies have been reported for TDP‐43 or FUS, so far. Other techniques focusing on the biophysical properties of protein assemblies that have been used for prion particles (Laferriere et al. 2013), or more recent assays based on detection of conformational protein changes via mass spectrometry (Feng et al. 2014) may shed light to these critical questions.
What is the role of post‐translational modifications in aggregation, seeding, and toxicity?
All FTD‐associated proteins can be distinguished from their native counterparts via specific post‐translational modifications (Fig. 1), which serve as markers for the detection of the pathologic state in patient tissues. Yet the exact role of these modifications in aggregation initiation seeding, spreading, and toxicity remains unclear.
Can patient‐derived or in vitro formed TDP‐43 and/or FUS aggregates trigger pathology and disease in experimental animals?
Understanding the biophysical and structural features of patient‐derived aggregates is key for addressing this important question. A potential challenge here is that these aggregates most likely do not form beta‐sheet conformations in patients, so their mode of propagation may be entirely different from prions and other classical amyloids. When attempting in vivo inoculation experiments, one must confirm the preservation (in the case of human brain‐derived material) or faithful reconstitution (when using in vitro formed aggregates) of authentic, patient‐like pathologic conformation. The potential sequestration of RNA or other proteins within these pathologic complexes may further complicate the issue.
Do any of these phenomena apply to dipeptide repeat proteins accumulating in C9orf72 FTD?
While it is conceivable that dipeptide repeat proteins (Mori et al. 2013), which originate from repeat‐associated non‐ATG translation (Zu et al. 2011) specifically in C9orf72 hexanucleotide repeat expansion carriers (DeJesus‐Hernandez et al. 2011; Renton et al. 2011) may propagate and spread throughout the brain, no experimental evidence exists currently to support this. Similar to the other FTD‐associated proteins, dipeptide repeat proteins are forming insoluble aggregates, which are neurotoxic in experimental models (Mizielinska et al. 2014). Yet, they are conceptually distinct from tau, TDP‐43 and FUS, because of the lack of known native, stable protein isoform, which would supply a constant source of substrate, necessary for seeding and amplification. This resembles the case of polyglutamine and other repeat expansion diseases, which intriguingly were shown to transport between neurons (Pecho‐Vrieseling et al. 2014; Brahic et al. 2016) and from glia to neurons (Pearce et al. 2015) in a prion‐like manner.
Can we capture the propagating entity to stall disease progression in patients?
This is probably the most important question from the viewpoint of FTD patients. Indeed, a major implication of the spatiotemporal disease spread is the potential existence of an extracellular ‘propagon’ that could be therapeutically targeted and neutralized. Antibodies are excellent candidates for this purpose and tau immunotherapy has been explored with promising results (Funk et al. 2015; Sigurdsson 2016). Similar approaches may prove effective for TDP‐43 and FUS proteinopathies in the future.
Terminology
Considering the growing amount of evidence, it has become clear that the concept of prion‐like processes has extended into many other neurodegenerative diseases requiring a clarification and expansion of the terminology that describe the underlying phenomena. However, while pathological aggregates propagating in other neurodegenerative diseases fundamentally share the molecular mechanisms underlying the replication of infectious prions, there is one substantial difference, namely inter‐organism transmission, which to date remains exclusive to prion diseases. This critical factor has led to some discrepancy in the terminology within the scientific community.
Some definitions are focused on the shared molecular actions, meaning the ability of a protein assembly to relay a specific stable conformation via template‐directed amplification and propose to expand the term prion to non‐infectious protein assemblies (Sanders et al. 2016). This seems counterintuitive, especially since the term prion was denoted as ‘proteinaceous infectious agent’ (Prusiner 1982). Focusing on the same underlying principles, but acknowledging the discrepancy of the term ‘infectious’, others suggest to change the definition broadly to ‘proteinaceous nucleating particle’ (Walker and Jucker 2015) or establish the term prionoid (Aguzzi 2009) to differentiate the pathological proteins causative for non‐infectious diseases from bona fide infectious prions and additionally to avoid public health concerns (Brettschneider et al. 2015). Similarly, the term propagon was proposed recently for all misfolded proteins that can initiate misfolding and aggregation of native proteins and thus propagate spreading of pathological conformations, to avoid confusion and be able to specify further (e.g., molecular propagon, tissue propagon) (Eisele and Duyckaerts 2016; Uchihara et al. 2016). In the absence of a unifying definition, in this review the self‐perpetuating propagation of misfolding and aggregation, resembling that of prion amplification, is discussed as prion‐like mechanism.
The template‐dependent and self‐perpetuating conversion of large amounts of natively folded protein into a pathological conformation by minute quantities of misfolded aggregates is termed seeding. While this expression is mostly used in in vitro assays using recombinant protein, it could also describe the early molecular events happening in a cell. Nucleation defines the initial phase of this conversion process leading to the formation of a highly ordered ‘nucleus’ or ‘seed of aggregation’. The mechanisms of how pathological aggregates propagate within one organism using cellular connections, exo/endocytosis mechanisms and possibly others is not yet completely understood and often called cell‐to‐cell spreading.
Acknowledgements and conflict of interest disclosure
The authors are grateful to Polymenidou laboratory members for critical comments and suggestions. MP has been awarded a Swiss National Science Foundation Professorship and a Career Development Award from the Human Frontier Science Program. This work was supported by the Clinical Research Priority Program Small RNAs of the University of Zurich and the NCCR RNA&Disease. MP and E‐MH are grateful for receiving a PhD Grant from the Neuroscience Center Zurich. The authors have no conflict of interest to declare.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Agosta F., Scola E., Canu E. et al (2012) White matter damage in frontotemporal lobar degeneration spectrum. Cereb. Cortex 22, 2705–2714. [DOI] [PubMed] [Google Scholar]
- Agosta F., Weiler M. and Filippi M. (2015) Propagation of pathology through brain networks in neurodegenerative diseases: from molecules to clinical phenotypes. CNS Neurosci. Ther. 21, 754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A. (2009) Cell biology: beyond the prion principle. Nature 459, 924–925. [DOI] [PubMed] [Google Scholar]
- Aguzzi A. and Polymenidou M. (2004) Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327. [DOI] [PubMed] [Google Scholar]
- Aguzzi A. and Rajendran L. (2009) The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 64, 783–790. [DOI] [PubMed] [Google Scholar]
- Aguzzi A., Heikenwalder M. and Polymenidou M. (2007) Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 8, 552–561. [DOI] [PubMed] [Google Scholar]
- Ahmed Z., Cooper J., Murray T. K. et al (2014) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alami N. H., Smith R. B., Carrasco M. A. et al (2014) Axonal transport of TDP‐43 mRNA granules is impaired by ALS‐causing mutations. Neuron 81, 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberti S., Halfmann R., King O., Kapila A. and Lindquist S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P. and Kedersha N. (2009) RNA granules: post‐transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 10, 430–436. [DOI] [PubMed] [Google Scholar]
- Andersson M. K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O. and Aman P. (2008) The multifunctional FUS, EWS and TAF15 proto‐oncoproteins show cell type‐specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 9, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreadis A., Brown W. M. and Kosik K. S. (1992) Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633. [DOI] [PubMed] [Google Scholar]
- Arai H., Terajima M., Miura M. et al (1995) Tau in cerebrospinal fluid: a potential diagnostic marker in Alzheimer's disease. Ann. Neurol. 38, 649–652. [DOI] [PubMed] [Google Scholar]
- Arai T., Hasegawa M., Akiyama H. et al (2006) TDP‐43 is a component of ubiquitin‐positive tau‐negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611. [DOI] [PubMed] [Google Scholar]
- Asai H., Ikezu S., Tsunoda S. et al (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala Y. M., De Conti L., Avendaño‐Vázquez S. E. et al (2011) TDP‐43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M., Mackenzie I. R., Pickering‐Brown S. M. et al (2006) Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature 442, 916–919. [DOI] [PubMed] [Google Scholar]
- Bembich S., Herzog J. S., De Conti L., Stuani C., Avendano‐Vazquez S. E., Buratti E., Baralle M. and Baralle F. E. (2014) Predominance of spliceosomal complex formation over polyadenylation site selection in TDP‐43 autoregulation. Nucleic Acids Res. 42, 3362–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentmann E., Neumann M., Tahirovic S., Rodde R., Dormann D. and Haass C. (2012) Requirements for stress granule recruitment of fused in Sarcoma (FUS) and TAR DNA binding protein of 43 kDa (TDP‐43). J. Biol. Chem. 287(27), 23079–23094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A., Lutz Y., Heard D. J., Chambon P. and Tora L. (1996) hTAF(II)68, a novel RNA/ssDNA‐binding protein with homology to the pro‐oncoproteins TLS/FUS and EWS is associated with both TFIID and RNA polymerase II. EMBO J. 15, 5022–5031. [PMC free article] [PubMed] [Google Scholar]
- Binder L. I., Frankfurter A. and Rebhun L. I. (1985) The distribution of tau in the mammalian central nervous system. J. Cell Biol. 101, 1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T. et al (2007) Induction of tau pathology by intracerebral infusion of amyloid‐beta ‐containing brain extract and by amyloid‐beta deposition in APP x Tau transgenic mice. Am. J. Pathol. 171, 2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boluda S. et al (2015) Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol. 129, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A., Ingadottir J., Davies P. and Sigurdsson E. M. (2011) Passive immunization targeting pathological phospho‐tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J. Neurochem. 118, 658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H. and Braak E. (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H. and Braak E. (1995) Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol. Aging 16, 271–278; discussion 278‐284. [DOI] [PubMed] [Google Scholar]
- Braak E., Braak H. and Mandelkow E. M. (1994) A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 87, 554–567. [DOI] [PubMed] [Google Scholar]
- Braak H., Alafuzoff I., Arzberger T., Kretzschmar H. and Del Tredici K. (2006) Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 112, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Brettschneider J., Ludolph A. C., Lee V. M., Trojanowski J. Q. and Del Tredici K. (2013) Amyotrophic lateral sclerosis–a model of corticofugal axonal spread. Nat. Rev. Neurol. 9, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahic M., Bousset L., Bieri G., Melki R. and Gitler A. D. (2016) Axonal transport and secretion of fibrillar forms of alpha‐synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol. 131, 539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J., Del Tredici K., Toledo J. B. et al (2013) Stages of pTDP‐43 pathology in amyotrophic lateral sclerosis. Ann. Neurol. 74, 20–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J., Del Tredici K., Irwin D. J. et al (2014) Sequential distribution of pTDP‐43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 127, 423–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J., Del Tredici K., Lee V. M. and Trojanowski J. Q. (2015) Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broe M., Hodges J. R., Schofield E., Shepherd C. E., Kril J. J. and Halliday G. M. (2003) Staging disease severity in pathologically confirmed cases of frontotemporal dementia. Neurology 60, 1005–1011. [DOI] [PubMed] [Google Scholar]
- Brundin P., Melki R. and Kopito R. (2010) Prion‐like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchan J. R. and Parker R. (2009) Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueler H., Aguzzi A., Sailer A., Greiner R. A., Autenried P., Aguet M. and Weissmann C. (1993) Mice devoid of PrP are resistant to scrapie. Cell 73, 1339–1347. [DOI] [PubMed] [Google Scholar]
- Buratti E., Brindisi A., Giombi M., Tisminetzky S., Ayala Y. M. and Baralle F. E. (2005) TDP‐43 binds heterogeneous nuclear ribonucleoprotein A/B through its C‐terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 280, 37572–37584. [DOI] [PubMed] [Google Scholar]
- Burke K. A., Janke A. M., Rhine C. L. and Fawzi N. L. (2015) Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol. Cell 60, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafate S., Buist A., Miskiewicz K., Vijayan V., Daneels G., de Strooper B., de Wit J., Verstreken P. and Moechars D. (2015) Synaptic contacts enhance cell‐to‐cell tau pathology propagation. Cell Rep. 11, 1176–1183. [DOI] [PubMed] [Google Scholar]
- de Calignon A., Polydoro M., Suarez‐Calvet M. et al (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caselli R. J., Windebank A. J., Petersen R. C. et al (1993) Rapidly progressive aphasic dementia and motor neuron disease. Ann. Neurol. 33, 200–207. [DOI] [PubMed] [Google Scholar]
- Chai X., Wu S., Murray T. K. et al (2011) Passive immunization with anti‐Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J. Biol. Chem. 286, 34457–34467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai X., Dage J. L. and Citron M. (2012) Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 48, 356–366. [DOI] [PubMed] [Google Scholar]
- Chen‐Plotkin A. S., Geser F., Plotkin J. B. et al (2008) Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum. Mol. Genet. 17, 1349–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirita C. N., Necula M. and Kuret J. (2003) Anionic micelles and vesicles induce tau fibrillization in vitro. J. Biol. Chem. 278, 25644–25650. [DOI] [PubMed] [Google Scholar]
- Christianson H. C., Svensson K. J., van Kuppevelt T. H., Li J. P. and Belting M. (2013) Cancer cell exosomes depend on cell‐surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl Acad. Sci. USA 110, 17380–17385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F., Bolmont T., Crowther R. A. et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F., Lavenir I., Falcon B., Frank S., Goedert M. and Tolnay M. (2013) “Prion‐like” templated misfolding in tauopathies. Brain Pathol. 23, 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F., Hench J., Goedert M. and Tolnay M. (2015) Invited review: prion‐like transmission and spreading of tau pathology. Neuropathol. Appl. Neurobiol. 41, 47–58. [DOI] [PubMed] [Google Scholar]
- Cochran J. N., Hall A. M. and Roberson E. D. (2014) The dendritic hypothesis for Alzheimer's disease pathophysiology. Brain Res. Bull. 103, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen T. J., Hwang A. W., Restrepo C. R., Yuan C. X., Trojanowski J. Q. and Lee V. M. (2015) An acetylation switch controls TDP‐43 function and aggregation propensity. Nat. Commun. 6, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V. and Ratti A. (2009) TDP‐43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061. [DOI] [PubMed] [Google Scholar]
- Couthouis J., Hart M. P., Shorter J. et al (2011) A yeast functional screen predicts new candidate ALS disease genes. Proc. Natl Acad. Sci. USA 108, 20881–20890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther R. A. and Goedert M. (2000) Abnormal tau‐containing filaments in neurodegenerative diseases. J. Struct. Biol. 130, 271–279. [DOI] [PubMed] [Google Scholar]
- Cruts M., Gijselinck I., van der Zee J. et al (2006) Null mutations in progranulin cause ubiquitin‐positive frontotemporal dementia linked to chromosome 17q21. Nature 442, 920–924. [DOI] [PubMed] [Google Scholar]
- Cushman M., Johnson B. S., King O. D., Gitler A. D. and Shorter J. (2010) Prion‐like disorders: blurring the divide between transmissibility and infectivity. J. Cell Sci. 123, 1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daebel V., Chinnathambi S., Biernat J. et al (2012) beta‐Sheet core of tau paired helical filaments revealed by solid‐state NMR. J. Am. Chem. Soc. 134, 13982–13989. [DOI] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M., Mackenzie I. R., Boeve B. F. et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72, 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacourte A., Robitaille Y., Sergeant N. et al (1996) Specific pathological Tau protein variants characterize Pick's disease. J. Neuropathol. Exp. Neurol. 55, 159–168. [DOI] [PubMed] [Google Scholar]
- Deleault N. R., Lucassen R. W. and Supattapone S. (2003) RNA molecules stimulate prion protein conversion. Nature 425, 717–720. [DOI] [PubMed] [Google Scholar]
- Dewey C. M., Cenik B., Sephton C. F., Dries D. R., Mayer P., Good S. K., Johnson B. A., Herz J. and Yu G. (2011) TDP‐43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 31, 1098–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X., Ma M., Teng J. et al (2015) Exposure to ALS‐FTD‐CSF generates TDP‐43 aggregates in glioblastoma cells through exosomes and TNTs‐like structure. Oncotarget 6, 24178–24191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Rodde R., Edbauer D. et al (2010) ALS‐associated fused in sarcoma (FUS) mutations disrupt Transportin‐mediated nuclear import. EMBO J. 29, 2841–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S., Lecolle K., Caillierez R. et al (2014) Neuron‐to‐neuron wild‐type Tau protein transfer through a trans‐synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele Y. S. and Duyckaerts C. (2016) Propagation of Ass pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 131, 5–25. [DOI] [PubMed] [Google Scholar]
- Elvira G., Wasiak S., Blandford V. et al (2006) Characterization of an RNA granule from developing brain. Mol. Cell Proteomics 5, 635–651. [DOI] [PubMed] [Google Scholar]
- Fang Y. S., Tsai K. J., Chang Y. J. et al (2014) Full‐length TDP‐43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia‐TDP patients. Nat. Commun. 5, 4824. [DOI] [PubMed] [Google Scholar]
- Feiler M. S., Strobel B., Freischmidt A. et al (2015) TDP‐43 is intercellularly transmitted across axon terminals. J. Cell Biol. 211, 897–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feneberg E., Steinacker P., Lehnert S., Schneider A., Walther P., Thal D. R., Linsenmeier M., Ludolph A. C. and Otto M. (2014) Limited role of free TDP‐43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 351–356. [DOI] [PubMed] [Google Scholar]
- Feng Y., De Franceschi G., Kahraman A. et al (2014) Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 32, 1036–1044. [DOI] [PubMed] [Google Scholar]
- Filippi M., Agosta F., Scola E. et al (2013) Functional network connectivity in the behavioral variant of frontotemporal dementia. Cortex 49, 2389–2401. [DOI] [PubMed] [Google Scholar]
- Flament S., Delacourte A., Verny M., Hauw J. J. and Javoy‐Agid F. (1991) Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. 81, 591–596. [DOI] [PubMed] [Google Scholar]
- Friedhoff P., Schneider A., Mandelkow E. M. and Mandelkow E. (1998) Rapid assembly of Alzheimer‐like paired helical filaments from microtubule‐associated protein tau monitored by fluorescence in solution. Biochemistry 37, 10223–10230. [DOI] [PubMed] [Google Scholar]
- Frost B., Jacks R. L. and Diamond M. I. (2009) Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 284, 12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba R. A., Udan M., Bell S., Wegorzewska I., Shao J., Diamond M. I., Weihl C. C. and Baloh R. H. (2010) Interaction with polyglutamine aggregates reveals a Q/N‐rich domain in TDP‐43. J. Biol. Chem. 285, 26304–26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii R. and Takumi T. (2005) TLS facilitates transport of mRNA encoding an actin‐stabilizing protein to dendritic spines. J. Cell Sci. 118, 5755–5765. [DOI] [PubMed] [Google Scholar]
- Fujita K., Ito H., Nakano S., Kinoshita Y., Wate R. and Kusaka H. (2008) Immunohistochemical identification of messenger RNA‐related proteins in basophilic inclusions of adult‐onset atypical motor neuron disease. Acta Neuropathol. 116, 439–445. [DOI] [PubMed] [Google Scholar]
- Funk K. E., Mirbaha H., Jiang H., Holtzman D. M. and Diamond M. I. (2015) Distinct therapeutic mechanisms of tau antibodies: promoting microglial clearance versus blocking neuronal uptake. J. Biol. Chem. 290, 21652–21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y., Torres A. S. and O'Halloran T. V. (2004) Oxygen‐induced maturation of SOD1: a key role for disulfide formation by the copper chaperone CCS. EMBO J. 23, 2872–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y., Kaneko K., Watanabe S., Yamanaka K. and Nukina N. (2011) A seeding reaction recapitulates intracellular formation of Sarkosyl‐insoluble transactivation response element (TAR) DNA‐binding protein‐43 inclusions. J. Biol. Chem. 286, 18664–18672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes J. F., Vowles G. H., Nicoll J. A. and Revesz T. (1999) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 98, 171–178. [DOI] [PubMed] [Google Scholar]
- Geser F., Martinez‐Lage M., Robinson J. et al (2009) Clinical and pathological continuum of multisystem TDP‐43 proteinopathies. Arch. Neurol. 66, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghetti B., Oblak A. L., Boeve B. F., Johnson K. A., Dickerson B. C. and Goedert M. (2015) Invited review: frontotemporal dementia caused by microtubule‐associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilks N., Kedersha N., Ayodele M., Shen L., Stoecklin G., Dember L. M. and Anderson P. (2004) Stress granule assembly is mediated by prion‐like aggregation of TIA‐1. Mol. Biol. Cell 15, 5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler A. D. and Shorter J. (2011) RNA‐binding proteins with prion‐like domains in ALS and FTLD‐U. Prion 5, 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. (1996) Tau protein and the neurofibrillary pathology of Alzheimer's disease. Ann. N. Y. Acad. Sci. 777, 121–131. [DOI] [PubMed] [Google Scholar]
- Goedert M. and Jakes R. (2005) Mutations causing neurodegenerative tauopathies. Biochim. Biophys. Acta 1739, 240–250. [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Jakes R., Rutherford D. and Crowther R. A. (1989) Multiple isoforms of human microtubule‐associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3, 519–526. [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Cairns N. J. and Crowther R. A. (1992) Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168. [DOI] [PubMed] [Google Scholar]
- Goedert M., Clavaguera F. and Tolnay M. (2010) The propagation of prion‐like protein inclusions in neurodegenerative diseases. Trends Neurosci. 33, 317–325. [DOI] [PubMed] [Google Scholar]
- Gorno‐Tempini M. L., Hillis A. E., Weintraub S. et al (2011) Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad L. I., Guest W. C., Yanai A. et al (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc. Natl Acad. Sci. USA 108, 16398–16403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. L. and Lee V. M. (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer‐like tangles. J. Biol. Chem. 286, 15317–15331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. L. and Lee V. M. (2014) Cell‐to‐cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 20, 130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Chen Y., Zhou X. et al (2011) An ALS‐associated mutation affecting TDP‐43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. L., Covell D. J., Daniels J. P. et al (2013) Distinct alpha‐synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T. W., Kato M., Xie S. et al (2012) Cell‐free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779. [DOI] [PubMed] [Google Scholar]
- Hanger D. P., Anderton B. H. and Noble W. (2009) Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 15, 112–119. [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Nonaka T., Tsuji H., Tamaoka A., Yamashita M., Kametani F., Yoshida M., Arai T. and Akiyama H. (2011) Molecular dissection of TDP‐43 proteinopathies. J. Mol. Neurosci. 45, 480–485. [DOI] [PubMed] [Google Scholar]
- Heilbronner G., Eisele Y. S., Langer F. et al (2013) Seeded strain‐like transmission of beta‐amyloid morphotypes in APP transgenic mice. EMBO Rep. 14, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes B. B., DeVos S. L., Kfoury N. et al (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl Acad. Sci. USA 110, E3138–E3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M., Lendon C. L., Rizzu P. et al (1998) Association of missense and 5′‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393, 702–705. [DOI] [PubMed] [Google Scholar]
- Iba M., Guo J. L., McBride J. D., Zhang B., Trojanowski J. Q. and Lee V. M. (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's‐like tauopathy. J. Neurosci. 33, 1024–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz L. M., Kwong L. K., Xu Y. et al (2008) Enrichment of C‐terminal fragments in TAR DNA‐binding protein‐43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 173, 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D. J., Brettschneider J., McMillan C. T. et al (2016) Deep clinical and neuropathological phenotyping of Pick disease. Ann. Neurol. 79, 272–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D. and Suzuki N. (2011) Conjoint pathologic cascades mediated by ALS/FTLD‐U linked RNA‐binding proteins TDP‐43 and FUS. Neurology 77, 1636–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. J., Kerridge C., Cooper J. et al (2016) Short fibrils constitute the major species of seed‐competent tau in the brains of mice transgenic for human P301S tau. J. Neurosci. 36, 762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson B. S., Snead D., Lee J. J., McCaffery J. M., Shorter J. and Gitler A. D. (2009) TDP‐43 is intrinsically aggregation‐prone, and amyotrophic lateral sclerosis‐linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs K. A., Holton J. L., Rossor M. N., Godbolt A. K., Ozawa T., Strand K., Khan N., Al‐Sarraj S. and Revesz T. (2004) Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol. Appl. Neurobiol. 30, 369–373. [DOI] [PubMed] [Google Scholar]
- Ju S., Tardiff D. F., Han H. et al (2011) A yeast model of FUS/TLS‐dependent cytotoxicity. PLoS Biol. 9, e1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M. and Walker L. C. (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabuta C., Kono K., Wada K. and Kabuta T. (2015) 4‐Hydroxynonenal induces persistent insolubilization of TDP‐43 and alters its intracellular localization. Biochem. Biophys. Res. Commun. 463, 82–87. [DOI] [PubMed] [Google Scholar]
- Kampers T., Friedhoff P., Biernat J., Mandelkow E. M. and Mandelkow E. (1996) RNA stimulates aggregation of microtubule‐associated protein tau into Alzheimer‐like paired helical filaments. FEBS Lett. 399, 344–349. [DOI] [PubMed] [Google Scholar]
- Kanai Y., Dohmae N. and Hirokawa N. (2004) Kinesin transports RNA: isolation and characterization of an RNA‐transporting granule. Neuron 43, 513–525. [DOI] [PubMed] [Google Scholar]
- Karch C. M., Jeng A. T. and Goate A. M. (2012) Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies. J. Biol. Chem. 287, 42751–42762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai T., Tokuda T., Ishigami N., Sasayama H., Foulds P., Mitchell D. J., Mann D. M., Allsop D. and Nakagawa M. (2009) Increased TDP‐43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathol. 117, 55–62. [DOI] [PubMed] [Google Scholar]
- Kato M., Han T. W., Xie S. et al (2012) Cell‐free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y. D., van Hummel A., Stevens C. H. et al (2015) Short‐term suppression of A315T mutant human TDP‐43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD‐TDP and ALS. Acta Neuropathol. 130, 661–678. [DOI] [PubMed] [Google Scholar]
- Kfoury N., Holmes B. B., Jiang H., Holtzman D. M. and Diamond M. I. (2012) Trans‐cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kril J. J. and Halliday G. M. (2011) Pathological staging of frontotemporal lobar degeneration. J. Mol. Neurosci. 45, 379–383. [DOI] [PubMed] [Google Scholar]
- Ksiezak‐Reding H., Morgan K., Mattiace L. A., Davies P., Liu W. K., Yen S. H., Weidenheim K. and Dickson D. W. (1994) Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am. J. Pathol. 145, 1496–1508. [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski T. J., Bosco D. A., Leclerc A. L. et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. [DOI] [PubMed] [Google Scholar]
- Laferrière F., Tixador P., Moudjou M., Chapuis J., Sibille P., Herzog L., Reine F., Jaumain E., Laude H., Rezaei H. and Béringue V. (2013) Quaternary structure of pathological prion protein as a determining factor of strain‐specific prion replication dynamics. PLoS Pathog 9, e1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier‐Tourenne C., Polymenidou M. and Cleveland D. W. (2010) TDP‐43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 19, R46–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier‐Tourenne C., Polymenidou M., Hutt K. R. et al (2012) Divergent roles of ALS‐linked proteins FUS/TLS and TDP‐43 intersect in processing long pre‐mRNAs. Nature Publishing Group Nat Neurosci 15(11), 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam B. Y., Halliday G. M., Irish M., Hodges J. R. and Piguet O. (2014) Longitudinal white matter changes in frontotemporal dementia subtypes. Hum. Brain Mapp. 35, 3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna‐Reeves C. A., Castillo‐Carranza D. L., Sengupta U., Guerrero‐Munoz M. J., Kiritoshi T., Neugebauer V., Jackson G. R. and Kayed R. (2012) Alzheimer brain‐derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley T., Rohrer J. D., Mead S. and Revesz T. (2015a) Review: an update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 41(7), 858–881. [DOI] [PubMed] [Google Scholar]
- Lashley T., Rohrer J. D., Mead S. and Revesz T. (2015b) Review: an update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 41, 858–881. [DOI] [PubMed] [Google Scholar]
- Lee B. J., Cansizoglu A. E., Suel K. E., Louis T. H., Zhang Z. and Chook Y. M. (2006) Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 126, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. J., Desplats P., Sigurdson C., Tsigelny I. and Masliah E. (2010) Cell‐to‐cell transmission of non‐prion protein aggregates. Nat. Rev. Neurol. 6, 702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggett C., McGehee D. S., Mastrianni J., Yang W., Bai T. and Brorson J. R. (2012) Tunicamycin produces TDP‐43 cytoplasmic inclusions in cultured brain organotypic slices. J. Neurol. Sci. 317, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S. C., Polymenidou M. and Cleveland D. W. (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling J. P., Pletnikova O., Troncoso J. C. and Wong P. C. (2015) TDP‐43 repression of nonconserved cryptic exons is compromised in ALS‐FTD. Science 349, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Drouet V., Wu J. W., Witter M. P., Small S. A., Clelland C. and Duff K. (2012) Trans‐synaptic spread of tau pathology in vivo. PLoS ONE 7, e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu‐Yesucevitz L., Bilgutay A., Zhang Y. J. et al (2010) Tar DNA binding protein‐43 (TDP‐43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS ONE 5, e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomen‐Hoerth C., Anderson T. and Miller B. (2002) The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology 59, 1077–1079. [DOI] [PubMed] [Google Scholar]
- Luk K. C., Kehm V., Carroll J., Zhang B., O'Brien P., Trojanowski J. Q. and Lee V. M. Y. (2012) Pathological α‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science 338, 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky P. J., Daujotyte D., Tollervey J. R., Ule J., Stuani C., Buratti E., Baralle F. E., Damberger F. F. and Allain F. H. (2013) Molecular basis of UG‐rich RNA recognition by the human splicing factor TDP‐43. Nat. Struct. Mol. Biol. 20, 1443–1449. [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R. A. and Rademakers R. (2007) The molecular genetics and neuropathology of frontotemporal lobar degeneration: recent developments. Neurogenetics 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R., Baborie A., Pickering‐Brown S., Du Plessis D., Jaros E., Perry R. H., Neary D., Snowden J. S. and Mann D. M. (2006) Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 112, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie I. R., Rademakers R. and Neumann M. (2010) TDP‐43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. [DOI] [PubMed] [Google Scholar]
- Mackenzie I. R., Neumann M., Baborie A. et al (2011) A harmonized classification system for FTLD‐TDP pathology. Acta Neuropathol. 122, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E., Renton A. E., Mok K. et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross‐sectional study. Lancet Neurol. 11, 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniecka Z. and Polymenidou M. (2015) From nucleation to widespread propagation: a prion‐like concept for ALS. Virus Res. 207, 94–105. [DOI] [PubMed] [Google Scholar]
- McDonald K. K., Aulas A., Destroismaisons L., Pickles S., Beleac E., Camu W., Rouleau G. A. and Vande Velde C. (2011) TAR DNA‐binding protein 43 (TDP‐43) regulates stress granule dynamics via differential regulation of G3BP and TIA‐1. Hum. Mol. Genet. 20, 1400–1410. [DOI] [PubMed] [Google Scholar]
- McKee A. C., Stern R. A., Nowinski C. J. et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G. M., Albert M. S., Grossman M., Miller B., Dickson D. and Trojanowski J. Q.; Work Group on Frontotemporal Dementia and Pick's Disease (2001) Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch. Neurol. 58, 1803–1809. [DOI] [PubMed] [Google Scholar]
- Meyer‐Luehmann M., Coomaraswamy J., Bolmont T. et al (2006) Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science 313, 1781–1784. [DOI] [PubMed] [Google Scholar]
- Michel C. H., Kumar S., Pinotsi D., Tunnacliffe A., St George‐Hyslop P., Mandelkow E., Mandelkow E. M., Kaminski C. F. and Kaminski Schierle G. S. (2014) Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J. Biol. Chem. 289, 956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mioshi E., Hsieh S., Savage S., Hornberger M. and Hodges J. R. (2010) Clinical staging and disease progression in frontotemporal dementia. Neurology 74, 1591–1597. [DOI] [PubMed] [Google Scholar]
- Mirbaha H., Holmes B. B., Sanders D. W., Bieschke J. and Diamond M. I. (2015) Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J. Biol. Chem. 290, 14893–14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra M., Paunesku T., Woloschak G. E., Siddique T., Zhu L. J., Lin S., Greco K. and Bigio E. H. (2007) Gene expression analysis of frontotemporal lobar degeneration of the motor neuron disease type with ubiquitinated inclusions. Acta Neuropathol. 114, 81–94. [DOI] [PubMed] [Google Scholar]
- Mizielinska S., Gronke S., Niccoli T. et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisse K., Volkening K., Leystra‐Lantz C., Welch I., Hill T. and Strong M. J. (2009) Divergent patterns of cytosolic TDP‐43 and neuronal progranulin expression following axotomy: implications for TDP‐43 in the physiological response to neuronal injury. Brain Res. 1249, 202–211. [DOI] [PubMed] [Google Scholar]
- Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A. P., Kim H. J., Mittag T. and Taylor J. P. (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K., Arzberger T., Grasser F. A. et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 126, 881–893. [DOI] [PubMed] [Google Scholar]
- Morris H. R., Khan M. N., Janssen J. C. et al (2001) The genetic and pathological classification of familial frontotemporal dementia. Arch. Neurol. 58, 1813–1816. [DOI] [PubMed] [Google Scholar]
- Munoz D. G., Neumann M., Kusaka H., Yokota O., Ishihara K., Terada S., Kuroda S. and Mackenzie I. R. (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 118, 617–627. [DOI] [PubMed] [Google Scholar]
- Nath S., Agholme L., Kurudenkandy F. R., Granseth B., Marcusson J. and Hallbeck M. (2012) Spreading of neurodegenerative pathology via neuron‐to‐neuron transmission of beta‐amyloid. J. Neurosci. 32, 8767–8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary D., Snowden J. S., Gustafson L. et al (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51, 1546–1554. [DOI] [PubMed] [Google Scholar]
- Neary D., Snowden J. S. and Mann D. M. (2000) Cognitive change in motor neurone disease/amyotrophic lateral sclerosis (MND/ALS). J. Neurol. Sci. 180, 15–20. [DOI] [PubMed] [Google Scholar]
- Neumann M., Sampathu D. M., Kwong L. K. et al (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. [DOI] [PubMed] [Google Scholar]
- Neumann M., Rademakers R., Roeber S., Baker M., Kretzschmar H. A. and Mackenzie I. R. (2009a) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132, 2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Roeber S., Kretzschmar H. A., Rademakers R., Baker M. and Mackenzie I. R. (2009b) Abundant FUS‐immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 118, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M., Bentmann E., Dormann D. et al (2011) FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 134, 2595–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A. S., Rademakers R. and Miller B. L. (2015) Frontotemporal dementia: a bridge between dementia and neuromuscular disease. Ann. N. Y. Acad. Sci. 1338, 71–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T., Watanabe S., Kaneko K., Yamanaka K., Nukina N. and Furukawa Y. (2014) Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J. Biol. Chem. 289, 1192–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T., Watanabe S. T., Iwatsubo T. and Hasegawa M. (2010) Seeded aggregation and toxicity of {alpha}‐synuclein and tau: cellular models of neurodegenerative diseases. J. Biol. Chem. 285, 34885–34898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T., Masuda‐Suzukake M., Arai T. et al (2013) Prion‐like properties of pathological TDP‐43 aggregates from diseased brains. Cell Rep. 4, 124–134. [DOI] [PubMed] [Google Scholar]
- Orozco D., Tahirovic S., Rentzsch K., Schwenk B. M., Haass C. and Edbauer D. (2012) Loss of fused in sarcoma (FUS) promotes pathological Tau splicing. EMBO Rep. 13, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson N., Ince P. G., Smith M. O. et al (2006) ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 67, 1074–1077. [DOI] [PubMed] [Google Scholar]
- Patel A., Lee H. O., Jawerth L. et al (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- Pearce M. M., Spartz E. J., Hong W., Luo L. and Kopito R. R. (2015) Prion‐like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun. 6, 6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecho‐Vrieseling E., Rieker C., Fuchs S. et al (2014) Transneuronal propagation of mutant huntingtin contributes to non‐cell autonomous pathology in neurons. Nat. Neurosci. 17, 1064–1072. [DOI] [PubMed] [Google Scholar]
- Peeraer E., Bottelbergs A., Van Kolen K. et al (2015) Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol. Dis. 73, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesiridis G. S., Tripathy K., Tanik S., Trojanowski J. Q. and Lee V. M. (2011) A “two‐hit” hypothesis for inclusion formation by carboxyl‐terminal fragments of TDP‐43 protein linked to RNA depletion and impaired microtubule‐dependent transport. J. Biol. Chem. 286, 18845–18855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P. and Tycko R. (2005) Self‐propagating, molecular‐level polymorphism in Alzheimer's beta‐amyloid fibrils. Science 307, 262–265. [DOI] [PubMed] [Google Scholar]
- Polymenidou M. and Cleveland D. W. (2011) The seeds of neurodegeneration: prion‐like spreading in ALS. Cell 147, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M. and Cleveland D. W. (2012) Prion‐like spread of protein aggregates in neurodegeneration. J. Exp. Med. 209, 889–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M., Lagier‐Tourenne C., Hutt K. R. et al (2011) Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat. Neurosci. 14(4), 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M., Lagier‐Tourenne C., Hutt K. R., Bennett C. F., Cleveland D. W. and Yeo G. W. (2012) Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 1462, 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler A. M., Phillips E. C., Lau D. H., Noble W. and Hanger D. P. (2013a) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler A. M., Polydoro M., Wegmann S., Nicholls S. B., Spires‐Jones T. L. and Hyman B. T. (2013b) Propagation of tau pathology in Alzheimer's disease: identification of novel therapeutic targets. Alzheimers Res. Ther. 5, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P., Bird T. D., Wijsman E. et al (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 43, 815–825. [DOI] [PubMed] [Google Scholar]
- Prusiner S. B. (1982) Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. [DOI] [PubMed] [Google Scholar]
- Qin H., Lim L. Z., Wei Y. and Song J. (2014) TDP‐43 N terminus encodes a novel ubiquitin‐like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl Acad. Sci. USA 111, 18619–18624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth H. W. and LaFerla F. M. (2010) Alzheimer's disease. N. Engl. J. Med. 362, 329–344. [DOI] [PubMed] [Google Scholar]
- Raj A., Kuceyeski A. and Weiner M. (2012) A network diffusion model of disease progression in dementia. Neuron 73, 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K., Hodges J. R., Knopman D. et al (2011) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravits J. (2014) Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp. Neurol., 262(Pt B), 121–126. [DOI] [PubMed] [Google Scholar]
- Ravits J. M. and La Spada A. R. (2009) ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 73, 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravits J., Laurie P., Fan Y. and Moore D. H. (2007a) Implications of ALS focality: rostral‐caudal distribution of lower motor neuron loss postmortem. Neurology 68, 1576–1582. [DOI] [PubMed] [Google Scholar]
- Ravits J., Paul P. and Jorg C. (2007b) Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68, 1571–1575. [DOI] [PubMed] [Google Scholar]
- Renton A. E., Majounie E., Waite A. et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72(2), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer J. D., Guerreiro R., Vandrovcova J. et al (2009) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73, 1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H. J., Gorno‐Tempini M. L., Goldman W. P., Perry R. J., Schuff N., Weiner M., Feiwell R., Kramer J. H. and Miller B. L. (2002) Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology 58, 198–208. [DOI] [PubMed] [Google Scholar]
- Saman S., Kim W., Raya M. et al (2012) Exosome‐associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D. W., Kaufman S. K., DeVos S. L. et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D. W., Kaufman S. K., Holmes B. B. and Diamond M. I. (2016) Prions and protein assemblies that convey biological information in health and disease. Neuron 89, 433–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz J. C., Wang X., Podell E. R. and Cech T. R. (2013) RNA seeds higher‐order assembly of FUS protein. Cell Rep. 5, 918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley W. W. (2008) Selective functional, regional, and neuronal vulnerability in frontotemporal dementia. Curr. Opin. Neurol. 21, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley W. W., Allman J. M., Carlin D. A., Crawford R. K., Macedo M. N., Greicius M. D., Dearmond S. J. and Miller B. L. (2007) Divergent social functioning in behavioral variant frontotemporal dementia and Alzheimer disease: reciprocal networks and neuronal evolution. Alzheimer Dis. Assoc. Disord. 21, S50–S57. [DOI] [PubMed] [Google Scholar]
- Shimonaka S., Nonaka T., Suzuki G., Hisanaga S. I. and Hasegawa M. (2016) Templated aggregation of TDP‐43 by seeding with TDP‐43 peptide fibrils. J. Biol. Chem. 291(17), 8896–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson E. M. (2016) Tau Immunotherapy. Neurodegener. Dis. 16, 34–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D., Garcia‐Garcia E., Royo F., Falcon‐Perez J. M. and Avila J. (2012) Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 586, 47–54. [DOI] [PubMed] [Google Scholar]
- Skibinski G., Parkinson N. J., Brown J. M. et al (2005) Mutations in the endosomal ESCRTIII‐complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 37, 806–808. [DOI] [PubMed] [Google Scholar]
- Smethurst P., Sidle K. C. and Hardy J. (2015) Review: prion‐like mechanisms of transactive response DNA binding protein of 43 kDa (TDP‐43) in amyotrophic lateral sclerosis (ALS). Neuropathol. Appl. Neurobiol. 41, 578–597. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G. and Goedert M. (2013) Tau pathology and neurodegeneration. Lancet Neurol. 12, 609–622. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G., Bird T. D. and Ghetti B. (1998) Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 8, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z., Diaz Z., Fang X., Hart M. P., Chesi A., Shorter J. and Gitler A. D. (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9, e1000614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S., Wegmann S., Cho H. et al (2015) Neuronal uptake and propagation of a rare phosphorylated high‐molecular‐weight tau derived from Alzheimer's disease brain. Nat. Commun. 6, 8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi‐Watanabe S., Arai T., Kametani F. et al (2016) Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl‐insoluble and trypsin‐resistant tau. Acta Neuropathol. 131, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toombs J. A., McCarty B. R. and Ross E. D. (2010) Compositional determinants of prion formation in yeast. Mol. Cell. Biol. 30, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji H., Arai T., Kametani F. et al (2012) Molecular analysis and biochemical classification of TDP‐43 proteinopathy. Brain 135, 3380–3391. [DOI] [PubMed] [Google Scholar]
- Uchihara T., Giasson B. I. and Paulus W. (2016) Propagation of Abeta, tau and alpha‐synuclein pathology between experimental models and human reality: prions, propagons and propaganda. Acta Neuropathol. 131, 1–3. [DOI] [PubMed] [Google Scholar]
- Urwin H., Josephs K. A., Rohrer J. D. et al (2010) FUS pathology defines the majority of tau‐ and TDP‐43‐negative frontotemporal lobar degeneration. Acta Neuropathol. 120, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin V. M., Leverenz J. B., Bekris L. M. et al (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP‐43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C., Rogelj B., Hortobágyi T. et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T., Yu H., Varnum M., Liu‐Yesucevitz L., Citro A., Ikezu T., Duff K. and Wolozin B. (2012) Contrasting pathology of the stress granule proteins TIA‐1 and G3BP in tauopathies. J. Neurosci. 32, 8270–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli‐Daley L. A., Luk K. C., Patel T. P., Tanik S. A., Riddle D. M., Stieber A., Meaney D. F., Trojanowski J. Q. and Lee V. M. (2011) Exogenous alpha‐synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. C. and Jucker M. (2015) Neurodegenerative diseases: expanding the prion concept. Annu. Rev. Neurosci. 38, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker L. C., Diamond M. I., Duff K. E. and Hyman B. T. (2013) Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 70, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A. K., Spiller K. J., Ge G. et al (2015) Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP‐43. Acta Neuropathol. 130, 643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D. M. and Selkoe D. J. (2016) A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci. 17, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. and Mandelkow E. (2016) Tau in physiology and pathology. Nat. Rev. Neurosci. 17, 22–35. [DOI] [PubMed] [Google Scholar]
- Wang I.‐F., Wu L.‐S., Chang H.‐Y. and Shen C. K. J. (2008) TDP‐43, the signature protein of FTLD‐U, is a neuronal activity‐responsive factor. J. Neurochem. 105, 797–806. [DOI] [PubMed] [Google Scholar]
- Wang F., Wang X., Yuan C. G. and Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P. and Kimonis V. E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin‐containing protein. Nat. Genet. 36, 377–381. [DOI] [PubMed] [Google Scholar]
- Wegmann S., Maury E. A., Kirk M. J. et al (2015) Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J. 34, 3028–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton M. J., Igaz L. M., Wong M. M., Kwong L. K., Trojanowski J. Q. and Lee V. M. (2008) Disturbance of nuclear and cytoplasmic TAR DNA‐binding protein (TDP‐43) induces disease‐like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 283, 13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B. (2012) Regulated protein aggregation: stress granules and neurodegeneration. Mol. Neurodegener. 7, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. W., Herman M., Liu L. et al (2013) Small misfolded Tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Cirrito J. R., Stewart F. R. et al (2011) In vivo microdialysis reveals age‐dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 31, 13110–13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K., Holth J. K., Liao F. et al (2014) Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K., Kfoury N., Jiang H., Mahan T. E., Ma S., Maloney S. E., Wozniak D. F., Diamond M. I. and Holtzman D. M. (2013) Anti‐tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K., Jiang H., Mahan T. E., Maloney S. E., Wozniak D. F., Diamond M. I. and Holtzman D. M. (2015) Anti‐tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Transl. Neurol. 2, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A., Fujii R., Watanabe Y., Okabe S., Fukui K. and Takumi T. (2006) Myosin‐Va facilitates the accumulation of mRNA/protein complex in dendritic spines. Curr. Biol. 16, 2345–2351. [DOI] [PubMed] [Google Scholar]
- Zhang Y. J., Xu Y. F., Cook C. et al (2009) Aberrant cleavage of TDP‐43 enhances aggregation and cellular toxicity. Proc. Natl Acad. Sci. USA 106, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. J., Caulfield T., Xu Y. F. et al (2013) The dual functions of the extreme N‐terminus of TDP‐43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 22, 3112–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Greicius M. D., Gennatas E. D. et al (2010) Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain 133, 1352–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Gennatas E. D., Kramer J. H., Miller B. L. and Seeley W. W. (2012) Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 73, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liu S., Liu G., Ozturk A. and Hicks G. G. (2013) ALS‐associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 9, e1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T., Gibbens B., Doty N. S. et al (2011) Non‐ATG‐initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA 108, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]