

Abstract
Rifampicin, a broad‐spectrum antibiotic, has neuroprotective, immunosuppressive, and anti‐inflammatory properties. However, the effect of rifampicin on autoimmune disorders of the nervous system is not clear. In this study, we investigated whether rifampicin was beneficial to myelin oligodendrocyte glycoprotein peptide (MOG 33–35)‐induced female C57BL/6 experimental autoimmune encephalomyelitis (EAE) mice, the well‐established animal model of multiple sclerosis. Rifampicin treatment (daily from the first day after EAE immunization) remarkably attenuated clinical signs and loss of body weight, which are associated with suppression of inflammatory infiltration and demyelination in spinal cords of EAE mice. Furthermore, rifampicin dramatically reduced the disruption of blood–brain barrier integrity, down‐regulated serum concentration of IL‐6 and IL‐17A, inhibited pathological Th17 cell differentiation, and modulated the expression of p‐STAT3 and p‐p65. These results suggest that rifampicin is effective for attenuating the clinical severity of EAE mice, which may be related to its inhibitive ability in differentiation of Th17 cell and secretion of its key effector molecule IL‐17A via regulation of excessive activation of the key signaling molecules of JAK/STAT pathway. Our findings may be helpful for developing therapeutic and preventive strategies for multiple sclerosis.
Keywords: experimental autoimmune encephalomyelitis, JAK/STAT signal pathway, rifampicin, Th17 cell
Abbreviations used
- BBB
Blood–brain barrier
- CFA
complete Freund's adjuvant
- CNS
central nervous system
- DMSO
dimethylsulphoxide
- EAE
experimental autoimmune encephalomyelitis
- EB
Evans blue
- H&E
hematoxylin and eosin
- LFB
luxol fast blue
- MOG
myelin oligodendrocyte glycoprotein peptide
- MS
multiple sclerosis
- PBS
phosphate‐buffered saline
- PTX
pertussis toxin
- TBST
tris‐buffered saline
Multiple sclerosis (MS) is the most common autoimmune disease characterized by chronic inflammation and demyelination of the central nervous system (CNS) (Hafler 2004). It is the leading cause of neurological disability among young adults (Strober et al. 2012). Approximately 2.5 million people worldwide suffer from MS (Runia et al. 2012). Clinical manifestations of MS patients are heterogeneous depending on the anatomical location of inflammatory lesions, and most of them experience clinical symptoms, including decreased mobility, vision, and cognitive function, as well as increased anxiety, depression, and fatigue (Keegan and Noseworthy 2002). However, its etiology remains unclear.
Currently, the most important animal model of MS is experimental allergic encephalomyelitis (EAE), which shares with MS the presence of paralysis, neuroinflammatory infiltration, and demyelination (Friese et al. 2006). Like MS, EAE is mainly initiated by aberrant myelin antigen‐specific autoreactive CD4+T‐lymphocyte invasion into the CNS. Among all the lineages of CD4+T cells, Th1 and Th17 cells are recognized as critical effector cells responsible for the development of EAE (Tullius et al. 2014).
Th1 cell was long considered to be closely associated to mediate CNS pathology of EAE. However, IL‐12p35 (the p35 subunit of IL‐12, which is the key differentiation mediator of Th1 cell) or IL‐12p35R (the receptor of IL‐12p35) knockout mice were still sensitive to the EAE induction (Becher et al. 2002; Gran et al. 2002). Moreover, IFN‐γ (the major cytokine secreted by Th1 cell) or IFN‐γR (the receptor of IFN‐γ)‐deficient mice developed more severe EAE, suggesting that Th1 cell may not be the driving force of inducing EAE (Ferber et al. 1996; Willenborg et al. 1996). However, a newly discovered CD4+T cell subset, Th17 cell, is now identified as the dominant effective cell implicated in the autoimmune inflammatory demyelination in EAE animal model. The suppression of Th17 cells leads to attenuation of EAE, whereas adoptive transfer of Th17 cells directly induces severe EAE in mice (Jager et al. 2009; Zepp et al. 2011). High levels of inflammatory mediators secreted by infiltrating cells (particularly Th17 cells) lead to the ultimate recruitment of inflammatory cells to CNS lesions, dysfunction of the blood–brain barrier (BBB), demyelination, and hampering of nerve conduction. Aside from these, the JAK/STAT pathway has been reported to be the major mechanism regulating the differentiation and functions of Th17 cells. STAT3 is a pivotal signaling molecule involved in Th17 cell differentiation that is phosphorylated by potent stimulation of IL‐6 (Bettelli et al. 2006; Egwuagu and Larkin Iii 2013). Moreover, the microarray analysis of genes of MS brain tissue has manifested a positive correlation with NF‐κB (Lock et al. 2002), and mice lacking NF‐κB are significantly protected from EAE (Hilliard et al. 1999), indicating the crucial role of the NF‐κB pathway in inducing EAE. Interestingly, the crosstalk between the NF‐κB pathway and the JAK/STAT pathway, which promotes the continuous activation of the JAK/STAT pathway, is targeted at IL‐6 (McFarland et al. 2013). Thus, decreasing Th17 cells in the CNS via JAK/STAT pathway might be highly efficacious for preventing CNS tissue damage and might decrease disability in MS/EAE.
Currently, disease‐modifying therapies for MS aim at preventing inflammatory damage to the CNS by modulating and/or suppressing the immune system at various levels, including interferon beta (IFN‐β), glatiramer acetate (GA), mitoxantrone, natalizumab, fingolimod, teriflunomide, dimethyl fumarate, and alemtuzumab (Hemmer and Hartung 2007). In addition, methotrexate and rituximab are used as an off‐label option in some cases for MS treatment (Rizvi and Bashir 2004). However, no drugs have been licensed for the treatment of primary progressive MS, and these aforementioned drugs are considered the mainstay of relapsing‐remitting MS treatment with marginal efficacy or significant side effects (Cohen, 2009). The side effects include IFN‐β‐correlated influenza‐like syndrome, depression, and fatigue (Smith et al. 2015). Glatiramer is associated with chest tightness, flushing, anxiety, dyspnea, and palpitations (Galetta and Markowitz 2005). These severe side effects and limited effects have prompted the search for new and safer treatment strategies.
Rifampicin, typically used to treat Mycobacterium infections, is a semisynthetic wide‐spectrum antibiotic extracted from rifampicin B that can easily pass through the BBB because of its lipophilic property. Rifampicin has been shown to have neuroprotective roles in various CNS diseases such as stroke (Spudich et al. 2006), meningitis (Yulug et al. 2014), Parkinson disease (Bi et al. 2013), and Alzheimer disease (Molloy et al. 2013). Intriguingly, it has been reported that administration of rifampicin results in amelioration in Visna (Rosenkranz 1972), the animal model once used for MS, which raises the possibility that rifampicin may be an effective treatment for patients with MS. Rifampicin has been shown to suppress excessive immune responses, as it can prolong mouse's lifespan after allogenic transplantation via inhibiting abnormal humoral and cellular immune functions, and previous study also shows amelioration of autoimmune psoriasis by rifampicin owing to modifying T‐cell balance (Marija 1971; Bellahsene and Forsgren 1980; Tsiskarishvili and Tsiskarishvili 2009). In addition to these, other authors have demonstrated that rifampicin exerts anti‐inflammatory effects through inhibiting the production of IL‐6, and it restrains excessive activation of NF‐κB to restrict local oxidative stress injury and promote nerve recovery (Bi et al. 2011; Smani et al. 2011). Despite the well‐documented neuroprotective, immunosuppressive, and anti‐inflammatory properties of rifampicin, studies on its effect on EAE remain limited.
In this study, we tested whether rifampicin was effective for ameliorating clinical severity in mice with C57BL/6 EAE induced by myelin oligodendrocyte glycoprotein 35–55 amino acid peptide (MOG35–55). We further investigated the permeability of the BBB, the alteration of Th17 cell differentiation together with the expression of its key effector molecule IL‐17A, and the modification of JAK/STAT and NF‐κB signal pathways. We demonstrated that rifampicin diminished neurological impairment and spinal pathology (inflammatory infiltration and demyelination), and maintained integrity of the BBB of EAE by decreasing pathogenic Th17 cell infiltration. We additionally demonstrated that rifampicin inhibited the expression of the key effector molecule of Th17 cells – IL‐17A – by regulating JAK/STAT signal pathway. Results suggest that rifampicin could be a promising therapeutic strategy in an established model of MS in mice.
Materials and methods
Chemicals and reagents
Rifampicin (purity > 99% by reverse phase HPLC evaluation) was purchased from Guangdong Huanan Pharmaceutical Group (Guangdong, China). The MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was synthesized by CL Bio‐Scientific (Xi'an, China). Amino acid sequences were confirmed by amino acid analysis and mass spectroscopy. The purity of the peptide was greater than 98%. Complete Freund's adjuvant (CFA) and pertussis toxin were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Mycobacterium tuberculosis H37RA was purchased from BD Biosciences (Franklin Lakes, NJ, USA). Monoclonal anti‐STAT3, monoclonal anti‐phospho‐STAT3 (Tyr705), monoclonal anti‐NF‐κB p65, monoclonal anti‐phospho‐NF‐κB p65 (Ser536), and monoclonal anti‐glyceraldehyde phosphate dehydrogenase (GAPDH) were purchased from Cell Signaling Technology (Danvers, MA, USA). FITC (fluorescein isothiocyanate) anti‐CD4, PE anti‐IL‐17, Perm/Fix solution, and leukocyte activation cocktail were purchased from BD Biosciences. Collagenase D was purchased from Roche Life Science (Mannheim, Germany). Percoll was purchased from GE Healthcare (Milan, Italy). IL‐6 and IL‐17A ELISA Kits were purchased from eBioscience (San Diego, CA, USA).
Animals
Female C57BL/6 mice (8–10‐week‐old; Specific pathogen free) weighting 18–20 g were obtained from the Experimental Animal Center of Sun Yat‐Sen University (Guangzhou, China), were bred at the animal facility of the Experimental Animal Center of Sun Yat‐Sen University under constant temperature, and were provided food and water ad libitum.
All experimental procedures and protocols were approved by the Sun Yat‐Sen University Bioethics Committee and were performed in accordance with the ARRIVE guidelines. All efforts were made to minimize animal suffering, to reduce the number of animals used, and to utilize alternatives to in vivo techniques.
Induction of EAE
Mice were housed in cages and allowed 1 week to adapt to the environment. Then, mice were randomly divided into five groups: normal group, vehicle‐treated group, 20 mg/kg/d rifampicin‐treated group, 40 mg/kg/d rifampicin‐treated group, and 80 mg/kg/d rifampicin‐treated group. Except the normal group, the other four groups were induced as EAE models. The total of immunized mice for each group was 21. On day 0, each mouse was immunized subcutaneously (s.c.) at four sites in the hind flanks with an emulsion of 100 μL MOG33–35 (2 mg/mL) and 100 μL CFA supplemented with 5 mg/mL Mycobacterium tuberculosis H37RA. Additionally, animals received an intraperitoneal injection of 300 ng pertussis toxin (dissolved in 100 μL 0.01 M phosphate‐buffered saline [0.01 M phosphate‐buffered saline (PBS)]), which was repeated 2 days later. An additional injection of MOG33–35 in CFA was delivered on day 7.
Mice were weighted and clinically scored for EAE daily by the same investigator for 35 days after immunization in a blinded manner. For each test, the mice were randomly chosen from each group. Neurological assessments were reported using five‐point criteria (Urban et al. 1988): 0, no deficit; 1, tail paralysis; 2, incomplete hind limb paralysis; 3, complete hind limb paralysis; 4, complete hind limb paralysis and partial forelimb paralysis; 5, moribund state or death.
Treatment protocols
Rifampicin was prepared fresh and dissolved in dimethylsulphoxide; later, it was diluted with 0.01 M PBS (dimethylsulphoxide < 0.05‰). Daily prophylactic administration with rifampicin (doses of 20 mg/kg/d, 40 mg/kg/d, and 80 mg/kg/d intragastrically [i.g.]) or vehicle (0.01 M PBS i.g.) started on the first day after EAE induction (day 1) was administered to differently treated rifampicin groups and vehicle group, respectively, and was continued thereafter. No treatment was given to normal group.
Histological analysis
Histological evaluation was performed on the lumbar regions of the spinal cords obtained from differently treated EAE mice (n = 4, respectively) at the peak of neurological impairment (day 26 after immunization). Under anesthesia with intraperitoneal administration of sodium pentobarbital (50 mg/kg), each mouse was perfused intracardially with saline, followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.2–7.4). Each spinal cord was carefully removed and immersed overnight in the same fixative at 4°C and then embedded in paraffin.
Transverse sections (5 μm) that had been deparaffinized and rehydrated were stained with hematoxylin and eosin (H&E) or luxol fast blue (LFB) to assess inflammatory infiltration or demyelination, respectively. Five areas (1 mm2) were randomly chosen from each of three to four longitudinal spinal cord sections separated at least 300 μm. The cells in the infiltrates and the demyelination region were quantified with ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Assessment of BBB permeability
The integrity of the BBB was measured using Evans blue (EB) extravasation on day 21 after immunization in four mice per group. Sterilized 2% EB (Sigma‐Aldrich) in 0.9% saline was injected via the femoral vein (4 mL/kg body weight) 1 hour before transcardially perfusing the mice with 50 mL of ice‐cold PBS to flush the excess intravascular dye. Brains were immediately collected, weighed, and homogenized in 2 mL 50% trichloroacetic acid solution to extract dye from brain tissue by incubating tissue at 4°C overnight. After centrifugation (30 min, 15 000 g, 4°C), the supernatant was spectrophotometrically measured at 610 nm. Cerebral EB content was calculated as μg/g of tissue against a standard curve.
Flow cytometry analysis
After performed perfusion with 50 mL ice‐cold PBS through the left ventricle, the spinal cords and brains were harvested from differently treated mice at the peak of neurological impairment on day 26 after immunization (n = 6 per group). Then, spinal cords and brains were dissected out and digested with 2.5 mg/mL collagenase D for 45 min at 37°C. The tissue was homogenized in PBS. Single‐cell suspension was isolated gently passing through a 70‐μm cell strainer with PBS and then centrifuged (1000 g, 4°C). The cells were resuspended in 37% Percoll and overlaid with 70% Percoll. After further centrifugation (1000 g, 25 min), the cell monolayer at the 70–37% interphase was collected and washed in RPMI 1640.
For quantification of Th17 cells using flow cytometry analysis, single‐cell suspensions were stimulated with leukocyte activation cocktail for 5 h at 37°C the next day. Subsequently, cells were surface‐stained with FITC anti‐CD4 for 30 min at 4°C, treated with Perm/Fix solution, and intracellularly stained with PE anti‐IL‐17A for 30 min at 4°C. The cells were washed twice with 2% fetal bovine serum in PBS and used for flow cytometry. Isotype‐matched IgG was used as a negative control. Data were collected on a Gallios flow cytometer (Beckman Coulter) and analyzed using Kaluza Analysis software (Beckman Coulter, Kraemer Boulevard Brea, CA, USA) by gating the CD4 + population.
Determination of cytokine levels
Blood from different groups (n = 7 per group) was collected via caudal vein on day 26 after immunization, was left standing until the blood solidified, and was centrifuged (3000 g, 20 min). The collected serum was assayed for concentrations of IL‐6 and IL‐17 by enzyme‐linked immunosorbent assay (ELISA) using commercial kits according to the manufacturer's instructions. Cytokine quantities in the samples were calculated from standard curves of recombinant cytokines using a linear regression method.
Western blot analysis
For western blot analysis experiments at the peak of neurological impairment, the spinal cords of normal mice, PBS‐treated mice, and rifampicin‐treated mice (n = 6, respectively) were removed and sonicated in cold lysis buffer (Beyotime, Shanghai, China). After centrifugation (10 000 g at 4°C for 30 min), the supernatants were measured by bicinchoninic acid protein assay kit (Beyotime).
A total of 60 μg of tissue lysate from each sample was separated in 10% sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (Bio‐Rad Laboratories, Foster City, CA, USA). The proteins were then transferred onto polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA). After being blocked with 5% nonfat dry milk (for detection of non‐phosphorylation proteins) or bovine serum albumin solution (for detection of phosphorylation proteins) in Tween 20‐containing Tris‐buffered saline (20 mM Tris, pH 7.4, 0.1% Tween 20, and 150 mM NaCl), membranes were incubated overnight at 4°C with the following antibodies: p‐STAT3, p‐NF‐κB p65 (1 : 1000; Cell Signaling Technology, Beverly, MA, USA), STAT3, NF‐κB p65, and GAPDH (1 : 2000; Cell Signaling Technology).
After three washes in tris‐buffered saline, the membranes were incubated with anti‐mouse‐ horseradish peroxidase (HRP) and anti‐rabbit‐HRP secondary antibody (1 : 2000; Cell Signaling Technology) at 22‐24°C for 60 min. The resulting antigen–antibody–peroxidase complex was finally visualized using Immobilon Western chemiluminescent HRP substrate (Millipore). The density of bands was quantified with ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
The SPSS (IBM, Armonk, New York, USA) 20.0 statistical program was used for all statistical analyses. For continuous variables, non‐parametric data were presented with median and PR25/PR/75 quantiles while parametric data were presented as mean ± SEM. For results presented with mean, the dispersion tendency index SEM (standard error of mean) would be easier for reader to observe significances among groups. The comparably smaller error bars also make figures clear than presenting SD (standard deviation). Statistical analysis of histopathologic scores of mice was assessed using Mann–Whitney U test followed by Bonferroni post hoc test to compare replicate by time. Comparisons between multiple groups with single treatment factors were analyzed using one‐way anova accompanied by Fisher's least significant difference (LSD) post hoc comparisons. For non‐parametric data with multiple groups, one‐way non‐parametric anova (Kruskal–Wallis test for independent data and Friedman's test for repeated measuring data) and Bonferroni correction would be utilized. p < 0.05 was considered significant.
Results
Prophylactic rifampicin treatment attenuates clinical symptoms and suppresses the loss of body weight in a chronic model of EAE
To investigate the effect of rifampicin on EAE, mice from each group (21 per group) were treated with a daily oral gavage of rifampicin (20, 40, 80 mg/kg/d) or vehicle (0.01 M PBS) for 35 consecutive days and regularly monitored for neurological symptoms of EAE.
As shown in Fig. 1, the degree of paralysis in vehicle‐treated mice aggravated continuously to the steady peak clinical score, whereas mice treated with 20 mg/kg/d rifampicin displayed lower disease progression rates compared to the vehicle group. However, no notable recovery was found in the group treated with 20 mg/kg/d (Fig. 1a), but groups treated with 40 mg/kg/d and 80 mg/kg/d rifampicin showed gradual and significant recovery (Fig. 1b and c).
Figure 1.
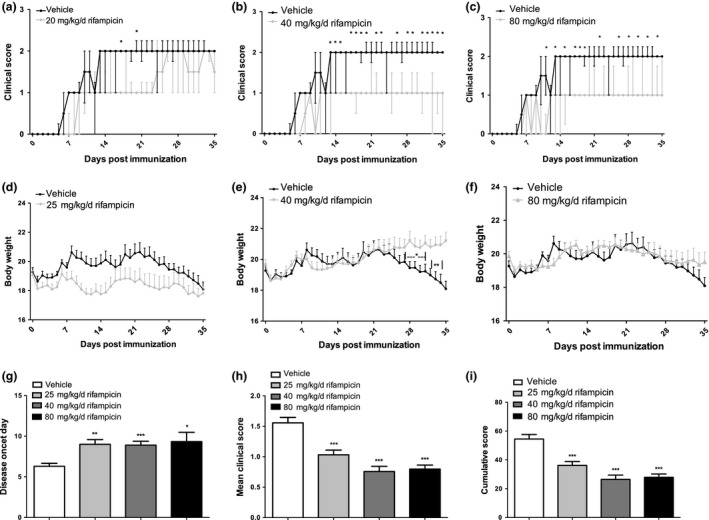
Prophylactic rifampicin administration reduced disease severity and restricted the loss of body weight in experimental autoimmune encephalomyelitis (EAE). Female C57BL/6 mice induced with MOG 35–55 developed clinical symptoms of EAE. (a–c) Prophylactic rifampicin treatment (20 mg/kg/d, 40 mg/kg/d, 80 mg/kg/d, i.g.) daily starting on day 1 attenuated EAE development with different degrees compared with vehicle‐treated animals. (d–f) Under preventive rifampicin treatment, body weight loss was suppressed compared to vehicle‐treated animals. (g–i) Rifampicin treatment delayed disease onset and reduced mean clinical scores and cumulative scores. Statistical analyses of clinical score (a–c) were performed using one‐way non‐parametric anova (Kruskal–Wallis test for independent data and Friedman's test for repeated measuring data) and Bonferroni correction for corresponding post hoc comparisons. Statistical analysis of other variables (d–i) was performed using one‐way anova followed by LSD post hoc test to compare replicate by time. Values were shown as median ± upper/lower limits (PR25/PR75 quantiles) for non‐parametric data or mean ± SEM for parametric data. Statistical significance: *p < 0.05, compared with vehicle‐treated mice; **p < 0.01, compared with vehicle‐treated mice; ***p < 0.001, compared with vehicle‐treated mice.
Regarded as a sign of pathological changes, weight was lost rapidly in the vehicle‐treated group after day 22 (Fig. 1d, e and f) and fluctuated at a low level in the group treated with 20 mg/kg/d rifampicin (Fig. 1d). Nevertheless, the group treated with 40 mg/kg/d rifampicin (Fig. 1e) gained weight normally. Weight remained stable in the group treated with 80 mg/kg/d rifampicin (Fig. 1f).
The detailed results of incidence rate, day of onset, time of peak score, peak score, and cumulative score were also indicated in Table 1. The data showed that the incidence rate of EAE induction reached 100% among all groups. Moreover, the time of onset was significantly delayed in all rifampicin‐treated groups compared with vehicle group (all p < 0.05). In addition, rifampicin treatment significantly reduced the cumulative score (all p < 0.05). The peak score was lower in 40 mg/kg/d and 80 mg/kg/d groups than in vehicle control, though the difference did not reach statistical significance. Taken together, these data indicated that mice undergoing prophylactic treatment with rifampicin demonstrated delayed disease onset (Table 1, Fig. 1g) and developed less severe EAE, with remarkably reduced mean clinical scores and cumulative scores (Table 1, Fig. 1h and i) compared with the vehicle group.
Table 1.
Clinical characteristics of EAE induced mice
| Parameters | Vehicle | 20 mg/kg/d | 40 mg/kg/d | 80 mg/kg/d | p |
|---|---|---|---|---|---|
| Incidence rate 1 | 100% | 100% | 100% | 100% | – |
| Day of onset | 6.30 ± 0.37 | 9.00 ± 0.58** | 8.90 ± 0.48*** | 9.33 ± 1.14* | 0.011 |
| Time of peak score | 13.40 ± 1.13 | 20.56 ± 2.21* | 13.10 ± 2.05 | 17.89 ± 2.62 | 0.038 |
| Peak score | 2.20 ± 0.13 | 3.11 ± 0.48* | 1.50 ± 0.17 | 1.67 ± 0.17 | 0.001 |
| Cumulative score | 54.50 ± 3.12 | 29.89 ± 3.90*** | 24.20 ± 3.48*** | 24.89 ± 3.62*** | <0.001 |
Statistical analysis was performed using one‐way anova followed by LSD post hoc test. Values were shown as mean ± SEM. Statistical significance: *p < 0.05, compared with vehicle‐treated mice; **p < 0.01, compared with vehicle‐treated mice; ***p < 0.001, compared with vehicle‐treated mice.
1Definition of successful induction of EAE: clinicalscore ≥ 1 was observed in neurological assessment with in 35 days.
These results illustrate the therapeutic role of rifampicin in the EAE model; 40 mg/kg/d may be the optimal dosage choice.
Preventive rifampicin administration attenuates neuropathology in spinal cord of EAE
CD4+T cells together with infiltrated macrophages and resident microglia contribute to the maintenance of immune homeostasis with neuroinflammation, progression of demyelination, and axonal damage (Bynoe et al. 2007). To investigate whether clinical amelioration was accompanied by alleviating the severity of neuropathology, we examined the level of cellular infiltration and demyelination in transverse sections of the lumbar spinal cord.
As evaluated by H&E staining at the peak time of the neurological score, a massive inflammatory distribution was obvious in the vehicle‐treated group (Fig. 2b) compared with normal mice (Fig. 2a). However, rifampicin treatment markedly decreased the severity of inflammatory cell recruitment, showing the smaller infiltrates (Fig. 2c) or only several scattered inflammatory cells (Fig. 2d and e). Additionally, the cells in the infiltrates quantified with ImageJ software was dramatically lower in the rifampicin‐treated group than in the vehicle‐treated group (Fig. 2f).
Figure 2.
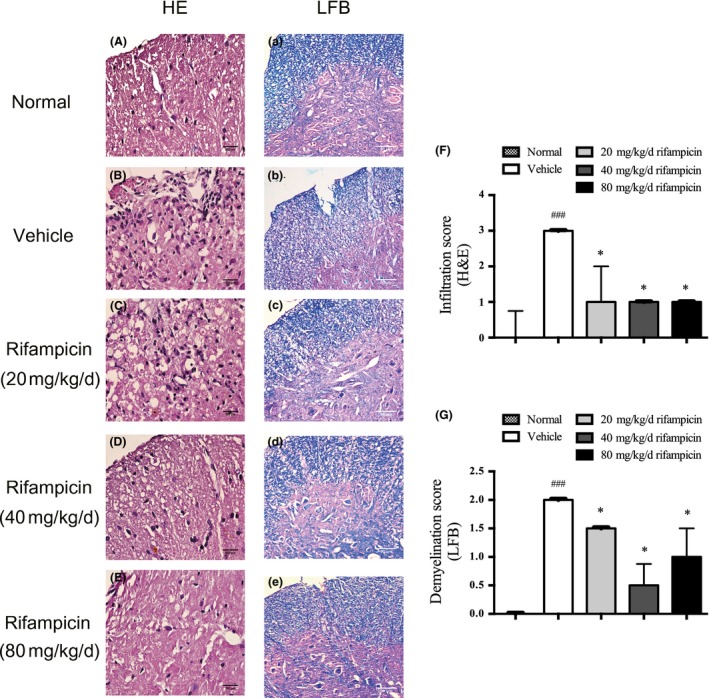
Preventive rifampicin administration attenuates neuropathology of experimental autoimmune encephalomyelitis (EAE). Transverse sections of MOG 33‐35‐immunized mice treated with rifampicin or phosphate‐buffered saline (PBS) (n = 4 per group) starting from day 1 post‐immunization were stained to assess the level of neuropathology. Representative images are shown. The normal mice were used as a negative control (n = 4). (a–e) H&E staining. Inflammatory cell infiltration decreased remarkably in the rifampicin group compared with the vehicle group (bars = 500 μm) (a–e) luxol fast blue (LFB) staining. MOG induction caused severe demyelination that was significantly decreased by rifampicin treatment (bars = 80 μm). The cells in the infiltrates (f) and the demyelination region (g) were quantified with ImageJ software. The median infiltration and demyelination score of normal mice was 0, which bars are not shown in the histogram. Statistical analysis was performed using Mann–Whitney U test followed by Bonferroni post hoc test to compare replicate by time. Values are shown as median and upper limit (.PR75 quantile). Statistical significance: ###p < 0.001, compared with normal mice; *p < 0.05, compared with vehicle‐treated mice.
The level of demyelination was assessed using LFB staining, which revealed prominent myelin loss in the spinal cord of the vehicle group (Fig. 2b) compared with normal mice (Fig. 2a). Nevertheless, the evident demyelination had lower density in the rifampicin group (Fig. 2c–e). In accordance with the pathological results, the demyelination region quantified with ImageJ software was significantly reduced in rifampicin‐treated mice when compared with vehicle‐treated mice (Fig. 2g).
Taken together, the results indicate that the functional recovery produced by rifampicin treatment in EAE is accompanied by decreasing inflammatory infiltration and prevention of demyelination.
Rifampicin treatment significantly prevents destruction of BBB integrity in EAE mice
Injury of BBB integrity resulting in an endothelial defect with huge amounts of inflammation infiltrating the CNS plays a considerable part in the development of MS and EAE. In this study, intravenous injections of Evans blue dye were used to assess the permeability of BBB on day 21 post‐immunization (n = 4 per group). The extravasated EB content (expressed as micrograms per gram of brain tissue) is shown in Fig. 3.
Figure 3.
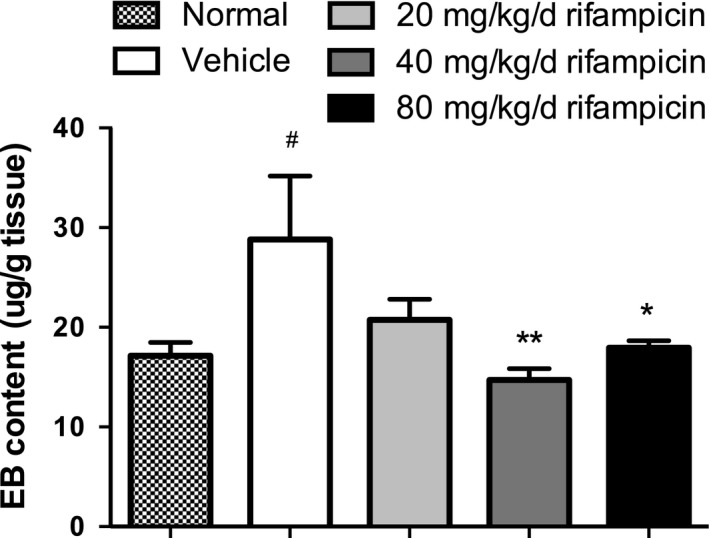
The effectiveness of rifampicin on preventing destruction of blood–brain barrier (BBB) integrity in experimental autoimmune encephalomyelitis (EAE). The permeability of BBB was measured by Evans blue (EB) content on day 21 after immunization in four mice per group. EB was calculated as micrograms per gram of brain tissue against a standard curve. The normal mice were used as a negative control (n = 4). Statistical analysis was performed using one‐way anova followed by LSD post hoc test to compare replicate by time. Values are shown as mean ± SEM. Statistical significance: #p < 0.05, compared with normal mice; *p < 0.05, compared with vehicle‐treated mice; **p < 0.01, compared with vehicle‐treated mice.
Marked extravasation of EB into cerebral parenchyma was detected in the vehicle‐treated group when compared with the normal mice, indicating severe disruption of the BBB after EAE induction. The EB content dramatically decreased and returned to normal baseline in the rifampicin‐treated mice at doses of 40 mg/kg/d and 80 mg/kg/d compared with the vehicle group. Although the average EB content was less in the group treated with 20 mg/kg/d, there was no statistical significance compared to the group treated with PBS.
These results reveal that rifampicin could prevent injury of BBB integrity.
Rifampicin decreases the differentiation of IL‐17 producing CD4+T cells in vivo
Given the importance of Th17 cells in EAE development, we investigated whether the diminished clinical characterization of autoimmunity in rifampicin‐administered mice was correlated with defects in these IL‐17 producing CD4+T‐cell priming. Single‐cell suspensions from spinal cords and brains were prepared from variously treated group (n = 6, respectively) during the peak period of disease and stained for surface and intracellular markers by flow cytometry.
Compliance with results demonstrated that there was significant enhancement in the frequency of CD4+IL‐17+ cells in EAE mice treated with PBS in comparison to mice treated with rifampicin (Fig. 4).
Figure 4.
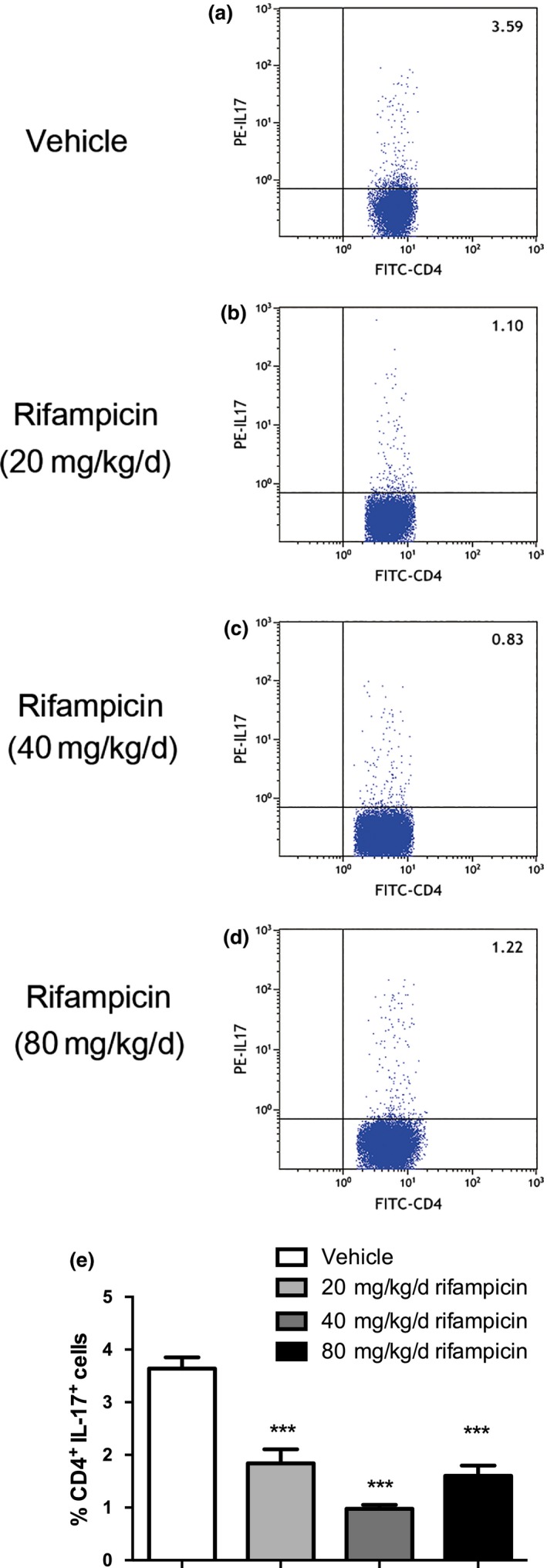
Th17 cells are reduced in rifampicin‐treated experimental autoimmune encephalomyelitis (EAE) mice. Single‐cell suspensions were collected from EAE mice treated with phosphate‐buffered saline (PBS) or rifampicin prophylactically on day 26 after immunization (n = 6 per group). (a–d) Representative image of CD4+IL‐17+ cells by flow cytometry. (e) Quantification of the number of CD4+IL‐17+ cells. Statistical analysis was performed using one‐way anova followed by LSD post hoc test to compare replicate measurements by time. Values were shown as mean ± SEM. Statistical significance: ***p < 0.001, compared with vehicle‐treated mice.
Thus, the findings suggest that in vivo pretreatment with rifampicin inhibits the pathogenic differentiation of Th17 cells in CNS.
Rifampicin dramatically down‐regulates serum levels of proinflammatory cytokines in EAE
Following the cascade activation triggered by IL‐6, Th17 cells are characterized by secreting IL‐17A. The pathobiology of IL‐17A in EAE disease was further supported by the fact that an antibody to IL‐17A attenuated autoimmune disease and IL‐17A gene‐deficient mice were resistant to EAE disease.
To further investigate the immunologic mechanism of suppressing the amount of pathogenic Th17 cells by rifampicin, we tested the serum concentrations of IL‐6 and IL‐17A from each group on day 26 post‐immunization.
As shown, the serum level of IL‐6, a factor with a critical role in Th17 cell generation, was remarkably increased in the vehicle group and 20 mg/kg/d rifampicin group compared with normal mice, which had a significantly decreased level that almost went back to normal in the 40 mg/kg/d rifampicin group and 80 mg/kg/d rifampicin group (Fig. 5a).
Figure 5.
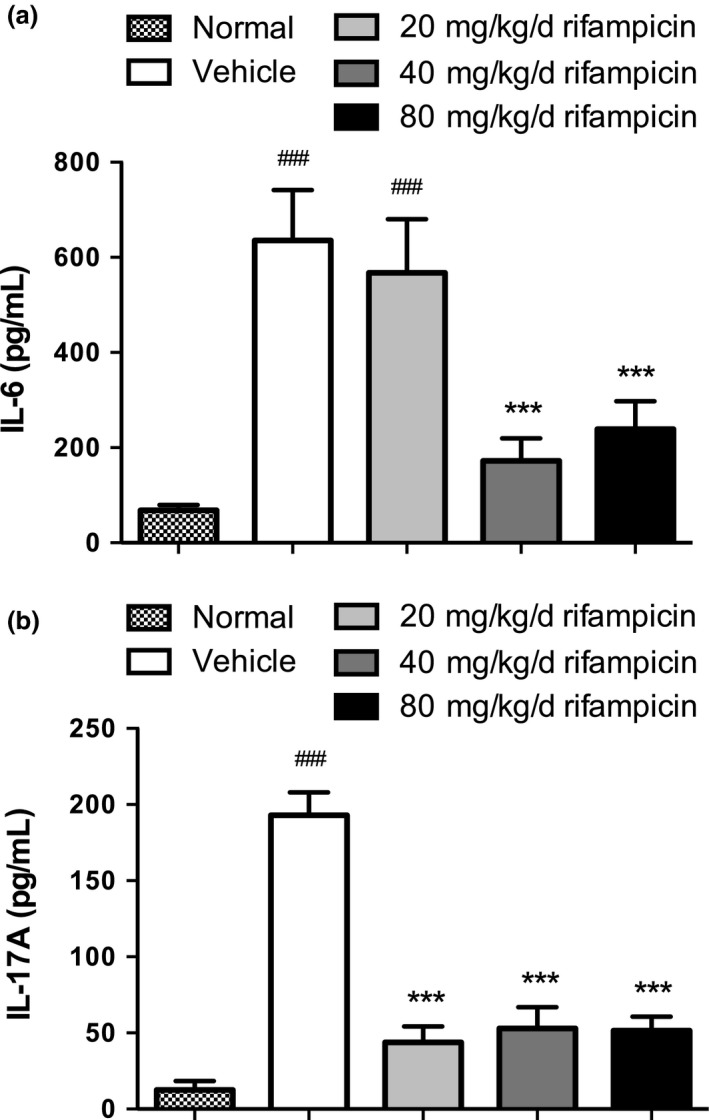
Proinflammatory cytokine levels in sera of differently treated experimental autoimmune encephalomyelitis (EAE). Proinflammatory cytokine levels of serum IL‐6 and IL‐17A were measured using ELISA. Serum levels of IL‐6 and IL‐17A were higher in vehicle mice with EAE than in normal mice. The levels were reduced in all rifampicin groups to different degrees. Statistical analysis was performed using one‐way anova followed by LSD post hoc test to compare replicate by time. Values are shown as mean ± SEM. Statistical significance: ###p < 0.001, compared with normal mice; ***p < 0.001, compared with vehicle‐treated mice.
A similar pattern was observed for serum concentrations of IL‐17A, a cytokine mainly produced by Th17 cells, in the vehicle mice and normal mice. The levels of pathogenic IL‐17A were interestingly suppressed in all the rifampicin‐treated groups (Fig. 5b).
Generally speaking, the functional performance of rifampicin reducing pathogenic Th17 cells can decrease the concentration of proinflammatory cytokines, which are essential for Th17 differentiation and function.
The regulatory effect of rifampicin on Th17 cell development relates to JAK/STAT and NF‐κB signaling pathways
Given that the JAK/STAT pathway takes the primary role in regulating the differentiation of Th17 cell. And the NF‐κB pathway could enhance the continuous activation of JAK/STAT pathway, which results in rising of pathogenic Th17 cells indirectly. To further investigate the mechanisms of the obvious reduction of pathologic Th17 cells by rifampicin, the spinal cords were isolated from rifampicin‐treated, vehicle, or normal mice and subjected to analysis of the key signaling molecules of the JAK/STAT and NF‐κB pathways by immunoblot.
Western blot analysis revealed that the expression of p‐STAT3 was dramatically increased in the spinal cords from PBS‐treated EAE and 20 mg/kg/d rifampicin‐treated EAE compared to that in normal mice. Up‐regulation of the JAK/STAT signaling pathway was significantly reduced by pretreatment with 40 mg/kg/d rifampicin and slightly decreased by 80 mg/kg/d rifampicin (Fig. 6a and c).
Figure 6.
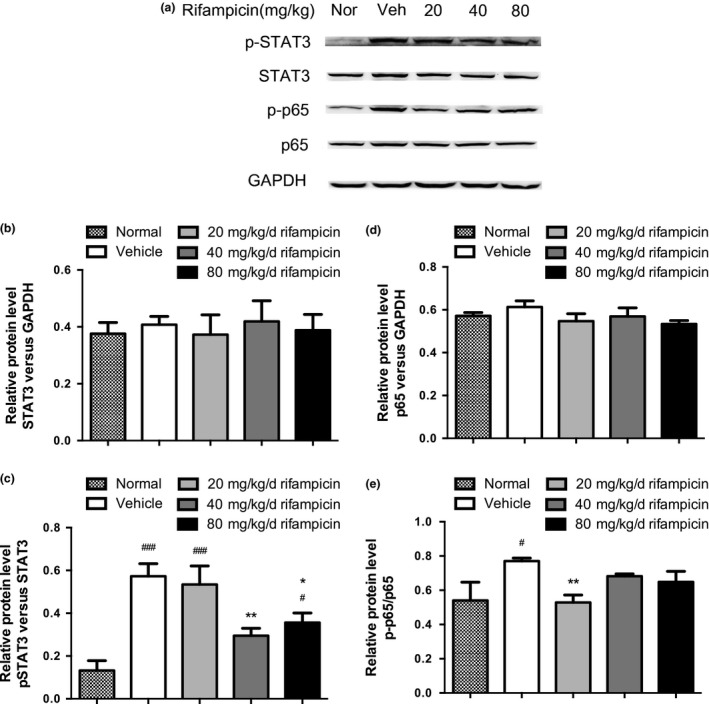
Expressions of p‐STAT3 and p‐p65 were reduced in rifampicin‐treated experimental autoimmune encephalomyelitis (EAE) mice. Spinal cords were isolated from rifampicin‐treated EAE, vehicle (Veh)‐treated EAE, and normal (Nor) mice on day 26 after immunization (n = 6). (a) Representative bands of STAT3, p‐STAT3, NF‐κB/p65, and p‐p65 by immunoblotting. GAPDH was used as an internal control. (b–e) The histograms represent STAT3, p‐STAT3, p65, and p‐p65 levels expressed as folds relative to the loading control. Statistical analysis was performed using one‐way anova followed by LSD post hoc test to compare replicate by time. Values were shown as mean ± SEM of the independent experiments. Statistical significance: #p < 0.05, compared with normal mice; ###p < 0.001, compared with normal mice; *p < 0.05, compared with vehicle‐treated mice; **p < 0.01, compared with vehicle‐treated mice.
Expression of p‐p65 was significantly up‐regulated in the vehicle group when compared with the normal mice, whereas 20 mg/kg/d rifampicin administration effectively down‐regulated the expression of p‐p65 in the spinal cords of EAE mice (Fig. 6a and e).
Expressions of STAT3 and NF‐κB/p65 were not affected by immunization and pretreatment with rifampicin (Fig. 6a, b and d).
These results suggest that rifampicin blocks the excessive activation of the key signaling molecules of the JAK/STAT and NF‐κB pathways, which may be related to the regulatory effect on Th17 differentiation.
Discussion
MS is a typical inflammatory demyelinating disease of CNS, but its etiology remains unknown. It is widely believed that pathogenic Th17 cell and its aberrant cytokines are vital for the pathogenic outcome. Additionally, rifampicin has long been used in the treatment of various infectious disorders. Despite its well‐documented brain‐protective, immunosuppressive, and anti‐inflammatory properties, little is known about its effects on MS. In this study, we have shown that rifampicin improved clinical and neuropathological outcomes in mice with MOG35–55‐induced EAE, which closely resembles the clinical situation found in humans. Moreover, we have demonstrated that this therapeutic function of rifampicin may be as a result of reducing the population of pathogenic Th17 cells and its key effector molecule IL‐17A via regulating JAK/STAT signal pathway. The findings suggest that rifampicin has preventive effect on the disease development of EAE by suppressing Th17 responses. These results may shed light on the possible administration of rifampicin in MS.
In this paper, we observed that oral rifampicin administration initiated before disease onset was therapeutic in EAE by attenuating clinical symptoms and maintaining body weight. Notably, Herrmann et al. (2007) showed that a treatment protocol consisting of 100 mg/kg rifampicin twice daily for 5 days administered to treat an experimental S. pneumoniae infection 7 days after induction of MOG35‐55 EAE did not influence the course of EAE, which is inconsistent with our observation. The discrepancy may be attributed to the difference in rifampicin treatment protocol between two studies. Our data showed that the overall therapeutic effect of rifampicin is better at a dose of 40 mg/kg/d than at a dose of 80 mg/kg/d. Therefore, the effect of rifampicin on EAE may be not obvious when a relatively higher dose was used in Herrmann et al. (2007)'s study. In addition, in our study, rifampicin treatment lasted for 35 days, which is much longer than that of Herrmann et al. (2007)'s study (5 days). Furthermore, the time point for the start of rifampicin treatment was also different between two groups. Because EAE shares with MS representative pathological changes such as inflammatory cell infiltration into the CNS and demyelination (Wang et al. 2014), we also investigated what was changed in EAE according to the lumbar spinal cord sections subjected to H&E and LFB dye. In our study, inflammatory recruitment and myelin loss were attenuated by rifampicin treatment, and the dosage of 40 mg/kg/d seemed to have achieved the best properties. Enhanced BBB permeability accompanying large numbers of inflammatory cells infiltrating the CNS plays an important role in the development and progression of MS and EAE (Alvarez et al. 2015). Various studies of EAE suggested that disease severity could be correlated with the alteration of BBB integrity (Alvarez et al. 2015), whereas limitation of the disease could be achieved by preventing BBB alterations in EAE (Zhang et al. 2015). In our experiment, rifampicin administration reduced the profound leakage of EB and showed repair of the permeability of the BBB, especially in the 40 mg/kg/d and 80 mg/kg/d rifampicin groups.
The underlying mechanism of the aforementioned efficacy of rifampicin is likely to be multifaceted. Autoreactive CD4+T cells play a key role in MS and EAE (Blankenhorn et al. 2011), especially Th17 cells acting as pathogenic cytokine‐secreting inflammatory cells recruited into the CNS, and lead to demyelination (Garris et al. 2013). Therefore, we studied whether rifampicin could regulate the differentiation and activation of CD4+ and IL17+ cells in CNS of EAE mice. Together with other studies (Wu et al. 2012; Li et al. 2015), our study showed the population of CD4+ and IL17+ cell and its signature mediator IL‐17A increased in the vehicle group as assessed by flow cytometry and ELISA, whereas the elevated expression was significantly decreased by rifampicin, corresponding to the attenuated severity of clinical signs and neuropathological alterations. The findings suggest that rifampicin administration inhibits differentiation and activation of naive CD4+T cells to Th17 cells. In addition, Th17 is differentiated via the JAK/STAT signal pathway driven by IL‐6 (Jadidi‐Niaragh and Mirshafiey 2011), and p‐STAT3 is a key signaling molecule of this regulating pathway (O'Shea and Plenge 2012). Activation of NF‐κB, which is sufficient to activate the JAK/STAT pathway by increasing IL‐6 expression, persists throughout the disease period and subsequently decreases during the recovery phase (Pahan and Schmid 2000) of MS. In our study, the up‐regulation of p‐STAT3 was found in mice treated with vehicle and mice treated with 20 mg/kg/d rifampicin, whereas it has been dramatically reduced by treatment with rifampicin at the dose of 40 mg/kg/d and mildly reduced with a dose of 80 mg/kg/d rifampicin. In accordance with the variation of p‐STAT3 in different groups, the study also revealed that the level of IL‐6, a key trigger cytokine of the phosphorylation of STAT3, was remarkably decreased in the 40 mg/kg/d rifampicin group and in the 80 mg/kg/d rifampicin group, whereas it was significantly increased in the vehicle group and the 20 mg/kg/d rifampicin group. However, the explanation for the contradiction between up‐regulation of IL‐6 and down‐regulation of the pathogenic Th17 cell population together with IL‐17A in the 20 mg/kg/d rifampicin group still requires further investigation. Moreover, regulation of the NF‐κB pathway was only obviously suppressed in 20 mg/kg/d rifampicin group compared with the PBS group, which may attributed to the multifunctions of NF‐κB pathway. Since MS arises from a sophisticated interplay between effector cells such as T and B cells, microglia and target cells such as oligodendrocytes and neurons, NF‐κB pathway may have both beneficial and detrimental effects in the different cell types participated in MS. Our findings indicate that rifampicin may exert efficacy in EAE by regulating JAK/STAT signal pathway.
The study results have to be interpreted in the context of possible limitations. Firstly, in our study, we focused on the role of therapeutic benefits of rifampicin administration for inhibiting Th17 responses. Rifampicin might also have a role in Th1 cells, which mainly exert a considerable function in the initial phase of the disease by inducing the secretion of IFN‐γ and endothelial vascular cell adhesion molecule 1 (Eugster et al., 1998). Secondly, whether rifampicin may have direct effects on lymphocytes or other leukocytes involved in the mechanism was not investigated in this study. Thirdly, we did not explore the effects of administration of rifampicin after onset of EAE, which is worth to be further investigated. Finally, we did not include CFA control mice in our study. Therefore, additional studies are needed to further explore these aspects to repair these limitations.
We initially revealed that pretreatment (from day 1 before immunization) of rifampicin could diminish the clinical symptoms and neuropathological alterations of EAE by inhibiting excessive Th17 responses, including its pathological differentiation and secretion via regulating JAK/STAT pathway. Our findings may help to develop the therapeutic and preventive strategies for MS.
Acknowledgements and conflict of interest disclosure
This work was supported by grants from the National Natural Science Foundation of China (81301675) and the Guangdong Province Science and Technology Plan Project (141405000671, 2013B022000058). Yan‐qing Feng and Huai‐yu Gu conceived and designed the experiments. Ke Ma, Xi Chen, Jia‐Cheng Chen, Ying Wang, Xi‐Meng Zhang, Jun‐Jiong Zheng, Xiong Chen, Wei Yu, and Ke‐Ling Cheng performed the experiments. Yanqing Feng and Fan Huang analyzed the data. Ke Ma and Yan‐qing Feng wrote the paper. The authors declare that they have no competing financial interests. The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. The corresponding author had full access to all the data and had final responsibility for the decision to submit for publication.
Contributor Information
Yan‐Qing Feng, Email: fyqgz@vip.sina.com.
Huai‐yu Gu, Email: guhuaiyu@mail.sysu.edu.cn.
References
- Alvarez J. I., Saint‐Laurent O., Godschalk A., Terouz S., Is C. B., Larouche S., Bourbonniere L., Larochelle C. and Prat A. (2015) Focal disturbances in the blood‐brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol. Dis. 74, 14–24. [DOI] [PubMed] [Google Scholar]
- Becher B., Durell B. G. and Noelle R. J. (2002) Experimental autoimmune encephalitis and inflammation in the absence of interleukin‐12. J. Clin. Invest. 110, 493–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellahsene A. and Forsgren A. (1980) Effect of rifampin on the immune response in mice. Infect. Immun. 27, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E., Carrier Y. J., Gao W. D., Korn T., Strom T. B., Oukka M., Weiner H. L. and Kuchroo V. K. (2006) Reciprocal developmental pathways for the generation of pathogenic effector T(H)17 and regulatory T cells. Nature 441, 235–238. [DOI] [PubMed] [Google Scholar]
- Bi W., Zhu L., Wang C., Liang Y., Liu J., Shi Q. and Tao E. (2011) Rifampicin inhibits microglial inflammation and improves neuron survival against inflammation. Brain Res. 1395, 12–20. [DOI] [PubMed] [Google Scholar]
- Bi W., Zhu L. H., Jing X. N., Liang Y. R. and Tao E. X. (2013) Rifampicin and Parkinson's disease. Neurol. Sci. 34, 137–141. [DOI] [PubMed] [Google Scholar]
- Blankenhorn E. P., Butterfield R., Case L. K., Wall E. H., Del R. R., Diehl S. A., Krementsov D. N., Saligrama N. and Teuscher C. (2011) Genetics of experimental allergic encephalomyelitis supports the role of T helper cells in multiple sclerosis pathogenesis. Ann. Neurol. 70, 887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bynoe M. S., Bonorino P. and Viret C. (2007) Control of experimental autoimmune encephalomyelitis by CD4 (+) suppressor T cells: peripheral versus in situ immunoregulation. J. Neuroimmunol. 191, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. A. (2009) Emerging therapies for relapsing multiple sclerosis. Arch Neurol. 66, 821–828. [DOI] [PubMed] [Google Scholar]
- Egwuagu C. E. and Larkin Iii J. (2013) Therapeutic targeting of STAT pathways in CNS autoimmune diseases. JAK‐STAT 2, e24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugster H. P., Frei K., Kopf M., Lassmann H. and Fontana A. (1998) IL‐6‐deficient mice resist myelin oligodendrocyte glycoprotein‐induced autoimmune encephalomyelitis. Eur J Immunol. 28, 2178–2187. [DOI] [PubMed] [Google Scholar]
- Ferber I. A., Brocke S., Taylor‐Edwards C., Ridgway W., Dinisco C., Steinman L., Dalton D. and Fathman C. G. (1996) Mice with a disrupted IFN‐gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE). J. Immunol. 156, 5–7. [PubMed] [Google Scholar]
- Friese M. A., Montalban X., Willcox N., Bell J. I., Martin R. and Fugger L. (2006) The value of animal models for drug development in multiple sclerosis. Brain 129, 1940–1952. [DOI] [PubMed] [Google Scholar]
- Galetta S. L. and Markowitz C. (2005) US FDA‐approved disease‐modifying treatments for multiple sclerosis: review of adverse effect profiles. CNS Drugs 19, 239–252. [DOI] [PubMed] [Google Scholar]
- Garris C. S., Wu L., Acharya S., et al (2013) Defective sphingosine 1‐phosphate receptor 1 (S1P1) phosphorylation exacerbates TH17‐mediated autoimmune neuroinflammation. Nat. Immunol. 14, 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gran B., Zhang G. X., Yu S., Li J., Chen X. H., Ventura E. S., Kamoun M. and Rostami A. (2002) IL‐12p35‐deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL‐12 system in the induction of central nervous system autoimmune demyelination. J. Immunol. 169, 7104–7110. [DOI] [PubMed] [Google Scholar]
- Hafler D. A. (2004) Multiple sclerosis (vol 113, pg 788, 2004). J. Clin. Invest. 113, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer B. and Hartung H. P. (2007) Toward the development of rational therapies in multiple sclerosis: what is on the horizon? Ann. Neurol. 62, 314–326. [DOI] [PubMed] [Google Scholar]
- Herrmann I., Kellert M., Spreer A., Gerber J., Eiffert H., Prinz M. and Nau R. (2007) Minocycline delays but does not attenuate the course of experimental autoimmune encephalomyelitis in Streptococcus pneumoniae‐infected mice. J. Antimicrob. Chemother. 59, 74–79. [DOI] [PubMed] [Google Scholar]
- Hilliard B., Samoilova E. B., Liu T., Rostami A. and Chen Y. H. (1999) Experimental autoimmune encephalomyelitis in NF‐kappa B‐deficient mice: roles of NF‐kappa B in the activation and differentiation of autoreactive T cells. J. Immunol. 163, 2937–2943. [PubMed] [Google Scholar]
- Jadidi‐Niaragh F. and Mirshafiey A. (2011) Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 74, 1–13. [DOI] [PubMed] [Google Scholar]
- Jager A., Dardalhon V., Sobel R. A., Bettelli E. and Kuchroo V. K. (2009) Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 183, 7169–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan B. M. and Noseworthy J. H. (2002) Multiple sclerosis. Annu. Rev. Med. 53, 285–302. [DOI] [PubMed] [Google Scholar]
- Li W., Zhang Z. and Zhang K. et al (2015) Arctigenin suppress Th17 cells and ameliorates experimental autoimmune encephalomyelitis through AMPK and PPAR‐gamma/ROR‐gammat signaling. Mol. Neurobiol. 53, 5356–5366. [DOI] [PubMed] [Google Scholar]
- Lock C., Hermans G., Pedotti R., et al (2002) Gene‐microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 8, 500–508. [DOI] [PubMed] [Google Scholar]
- Marija M. (1971) Rifampicin: an immunosuppressant? Lancet 2, 930–931. [DOI] [PubMed] [Google Scholar]
- McFarland B. C., Hong S. W., Rajbhandari R., Twitty G. J., Gray G. K., Yu H., Benveniste E. N. and Nozell S. E. (2013) NF‐kappaB‐induced IL‐6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PLoS ONE 8, e78728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy D. W., Standish T. I., Zhou Q. and Guyatt G. (2013) A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer's disease: the DARAD trial. Int. J. Geriatr. Psych. 28, 463–470. [DOI] [PubMed] [Google Scholar]
- O'Shea J. J. and Plenge R. (2012) JAK and STAT signaling molecules in immunoregulation and immune‐mediated disease. Immunity 36, 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahan K. and Schmid M. (2000) Activation of nuclear factor‐kB in the spinal cord of experimental allergic encephalomyelitis. Neurosci. Lett. 287, 17–20. [DOI] [PubMed] [Google Scholar]
- Rizvi S. A. and Bashir K. (2004) Other therapy options and future strategies for treating patients with multiple sclerosis. Neurology 63, S47–S54. [DOI] [PubMed] [Google Scholar]
- Rosenkranz H. S. (1972) Rifampicin and multiple sclerosis. Lancet 2, 1370. [DOI] [PubMed] [Google Scholar]
- Runia T. F., van Pelt‐Gravesteijn E. D. and Hintzen R. Q. (2012) Recent gains in clinical multiple sclerosis research. CNS Neurol. Disord. Drug. Targets 11, 497–505. [DOI] [PubMed] [Google Scholar]
- Smani Y., Dominguez‐Herrera J. and Pachon J. (2011) Rifampin protects human lung epithelial cells against cytotoxicity induced by clinical multi and pandrug‐resistant acinetobacter baumannii. J. Infect. Dis. 203, 1110–1119. [DOI] [PubMed] [Google Scholar]
- Smith M. Y., Sabido‐Espin M., Trochanov A., Samuelson M., Guedes S., Corvino F. A. and Richy F. F. (2015) Postmarketing safety profile of subcutaneous interferon beta‐1a given 3 times weekly: a retrospective administrative claims analysis. J. Manag. Care Spec. Pharm 21, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich A., Kilic E., Xing H. Y., Kilic U., Rentsch K. M., Wunderli‐Allenspach H., Bassetti C. L. and Hermann D. M. (2006) Inhibition of multidrug resistance transporter‐1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat. Neurosci. 9, 487–488. [DOI] [PubMed] [Google Scholar]
- Strober L. B., Christodoulou C., Benedict R., et al (2012) Unemployment in multiple sclerosis: the contribution of personality and disease. Mult. Scler. J. 18, 647–653. [DOI] [PubMed] [Google Scholar]
- Tsiskarishvili N. V. and Tsiskarishvili N. I. (2009) The anti‐tubercular drugs in the treatment of psoriasis. Georgian Med. News 174, 25–28. [PubMed] [Google Scholar]
- Tullius S. G., Biefer H. R. C., Li S., et al (2014) NAD(+) protects against EAE by regulating CD4(+) T‐cell differentiation. Nat. Commun. 5, 5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J. L., Kumar V., Kono D. H., Gomez C., Horvath S. J., Clayton J., Ando D. G., Sercarz E. E. and Hood L. (1988) Restricted use of T cell receptor V genes in murine autoimmune encephalomyelitis raises possibilities for antibody therapy. Cell 54, 577–592. [DOI] [PubMed] [Google Scholar]
- Wang T., Xi N. N., Chen Y., Shang X. F., Hu Q., Chen J. F. and Zheng R. Y. (2014) Chronic caffeine treatment protects against experimental autoimmune encephalomyelitis in mice: therapeutic window and receptor subtype mechanism. Neuropharmacology 86, 203–211. [DOI] [PubMed] [Google Scholar]
- Willenborg D. O., Fordham S., Bernard C. C., Cowden W. B. and Ramshaw I. A. (1996) IFN‐gamma plays a critical down‐regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein‐induced autoimmune encephalomyelitis. J. Immunol. 157, 3223–3227. [PubMed] [Google Scholar]
- Wu D., Wang J., Pae M. and Meydani S. N. (2012) Green tea EGCG, T cells, and T cell‐mediated autoimmune diseases. Mol. Aspects Med. 33, 107–118. [DOI] [PubMed] [Google Scholar]
- Yulug B., Hanoglu L., Kilic E. and Schabitz W. R. (2014) RIFAMPICIN: an antibiotic with brain protective function. Brain Res. Bull. 107, 37–42. [DOI] [PubMed] [Google Scholar]
- Zepp J., Wu L. and Li X. (2011) IL‐17 receptor signaling and T helper 17‐mediated autoimmune demyelinating disease. Trends Immunol. 32, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Ray A., Miller N. M., Hartwig D., Pritchard K. J. and Dittel B. N. (2015) Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood‐brain‐barrier integrity and ameliorates disease severity. J. Neurochem., 136, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]