

Summary
Corynebacterium glutamicum is an important industrial microorganism, but the availability of tools for its genetic modification has lagged compared to other model microorganisms such as Escherichia coli. Despite great progress in CRISPR‐based technologies, the most feasible genome editing method in C. glutamicum is suicide plasmid‐mediated, the editing efficiency of which is low due to high false‐positive rates of sacB counter selection, and the requirement for tedious two‐round selection and verification of rare double‐cross‐over events. In this study, an rpsL mutant conferring streptomycin resistance was harnessed for counter selection, significantly increasing the positive selection rate. More importantly, with the aid of high selection efficiencies through the use of antibiotics, namely kanamycin and streptomycin, the two‐step verification strategy can be simplified to just one‐step verification of the final edited strain. As proof of concept, a 2.5‐kb DNA fragment comprising aroG fbr pheA fbr expressing cassettes was integrated into the genome of C. glutamicum, with an efficiency of 20% out of the theoretical 50%. The resulting strain produced 110 mg l−1 l‐tyrosine in shake‐flask fermentation. This updated suicide plasmid‐mediated genome editing system will greatly facilitate genetic manipulations including single nucleotide mutation, gene deletion and gene insertion in C. glutamicum and can be easily applied to other microbes.
Introduction
Corynebacterium glutamicum, a non‐pathogenic Gram‐positive soil bacterium, is an important workhorse in industrial biotechnology for the production of several million tons of amino acids annually, especially l‐glutamate and l‐lysine (Eggeling and Bott, 2015). To date, C. glutamicum has been metabolically engineered for the production of a wide portfolio of other compounds of commercial interest, and for the utilization of vastly extended spectrum of substrates. Successes in these regards have rendered this microorganism a versatile microbial platform for a broad range of future biotechnological applications. Despite its importance, the currently available genetic tools, especially those for editing the genome of C. glutamicum, remain somewhat limited compared to those for E. coli, another important industrial microorganism.
Recently, clustered regularly interspaced short palindromic repeat (CRISPR) technologies associated with either the RNA‐guided endonuclease Cas9 or Cas12a (Cpf1) have been reported for the manipulation of the genome of C. glutamicum, allowing for gene deletion, insertion or single nucleotide mutation (Cho et al., 2017; Jiang et al., 2017; Liu et al., 2017; Peng et al., 2017; Wang et al., 2018a). Although CRISPR‐based genome editing techniques in C. glutamicum have been substantially improved, several drawbacks remain that limit the application of these technologies. First, additional mutations must be introduced in the protospacer and PAM region to avoid Cas9/Cpf1 cutting when generating ssDNA‐directed point mutations. Second, the two plasmid‐based system currently required involves labour‐intensive plasmid construction, slow colony growth, and results in few transformants, especially when the temperature‐sensitive plasmid backbone is used for easy curing. Third, due to the low homologous integration efficiency in C. glutamicum, the plasmid‐borne template is still a mandatory requirement (Wang et al., 2018b). Fourth, editing efficiency is influenced by several factors, including knockout size, overall length of homologous arms, relative length of homologous arm to insert gene, and design of the sgRNA (Becker et al., 2018). Consequently, results from different studies vary widely and are not reproducible. Moreover, the currently achievable efficiency for large gene insertion is unacceptably low. For these reasons, CRISPR‐based technology is not commonly applied to routine laboratory work, even in the groups with the above‐mentioned publications.
Today, genome editing in C. glutamicum is predominantly accomplished through use of non‐replicating suicide plasmids (Schäfer et al., 1994). This method relies on a two‐step homologous recombination process that includes a counter‐selection system. Specifically, the suicide plasmid pkmobsacB, which can be selected via antibiotic screening (usually kanamycin), integrates into the genome during the first recombination event, with the resultant single‐cross‐over strain being sucrose sensitive due to the expression of lethal levansucrase (sacB) (Jäger et al., 1992). In the second recombination event, the plasmid backbone is removed for scarless genetic modification, and selection for survival is carried out on sucrose‐supplemented medium. Counter selection by sacB gene leads to a high false‐positive rate due to high spontaneous inactivation of SacB (Hashimoto et al., 2003; Ma et al., 2015), while the first‐cross‐over event can be efficiently identified by kanamycin selection. In summary, the need for two‐round selection and verification of rare double‐cross‐over events (Nesvera and Patek, 2011) limits the speed and throughput of strain engineering of C. glutamicum.
In this study, we sought an alternative, more rigorous, counter‐selectable marker to sacB. The small ribosomal protein S12P gene rpsL was mutated to confer C. glutamicum with resistance to streptomycin. This is shown to be a powerful genetic marker that significantly increases the efficiency of counter selection in suicide plasmid‐mediated genome editing. Moreover, the two‐step verification procedure can be simplified to one‐step verification of the final edited strain, as a result of rigorous antibiotic selection with kanamycin and streptomycin.
Results
Workflow of rpsL counter selection in suicide plasmid‐mediated genome editing
The pk18mobrpsL‐mediated genome editing workflow in C. glutamicum is illustrated in Fig. 1. As a prerequisite for using rpsL as a counter‐selection marker, the chromosomal rpsL gene from the wild‐type strain must be mutated to confer streptomycin resistance. The non‐replicating plasmid pk18mobsacB is then modified to pk18mobrpsL, which expresses the streptomycin‐sensitive wild‐type rpsL gene under the strong constitutive promoter Ptuf. As an example, we constructed plasmid pk18mobrpsL‐gfp, in which the green fluorescent protein gene was designed to integrate at the locus of iolD (cgl0162), which is involved in myo‐inositol catabolism. The pk18mobrpsL‐gfp plasmid is electroporated into cells that exhibit streptomycin resistance attributing to a mutated chromosomal rpsL gene, for integration into the genome via single cross‐over with kanamycin resistance (kanR) selection. The additional wild‐type allele of rpsL provided by the integrated plasmid is dominant over the rpsL mutant in the genome, thereby causing streptomycin sensitivity (strepS) in the single‐cross‐over strain. Subsequently, streptomycin pressure (strepR) is applied, and only cells in which the plasmid has been removed from the genome following the second‐cross‐over event can survive. Meanwhile, the second‐cross‐over strain is kanamycin sensitive (kanS). Finally, correct integration of gfp in strepRkanS colonies is confirmed by colony PCR and sequencing.
Figure 1.
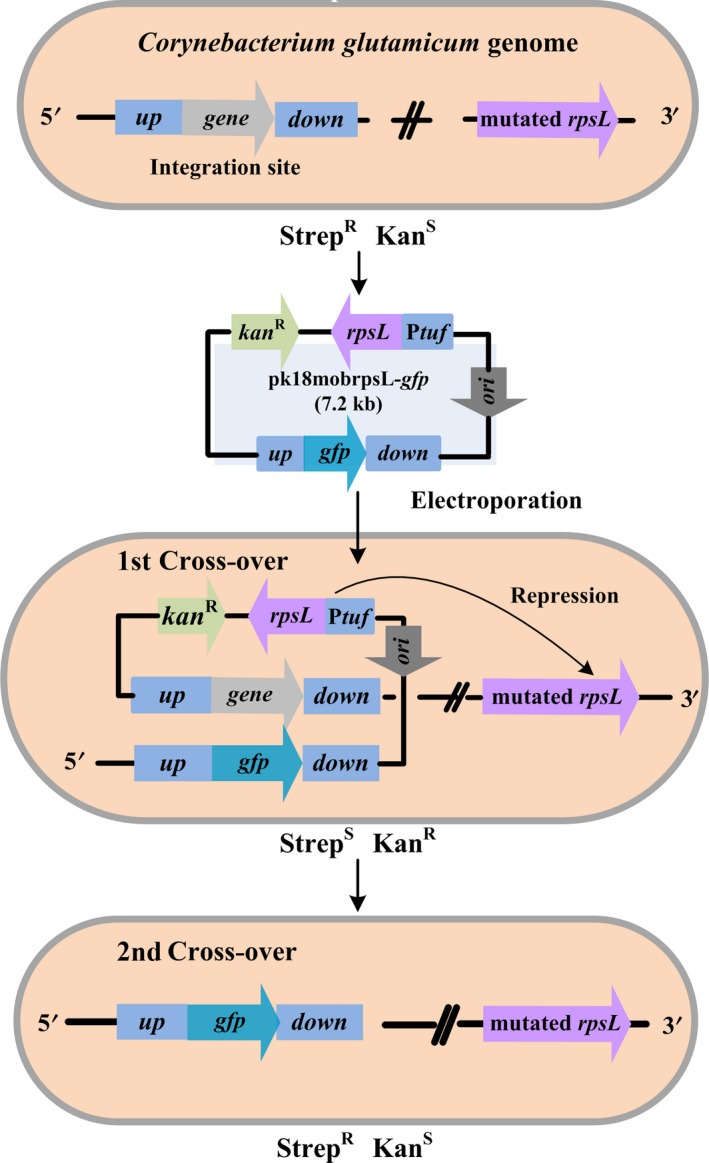
Diagram of the updated genomic editing technology in C. glutamicum. Prerequisite for the used strain is a chromosomal resistance against streptomycin conferred by a mutation in rpsL. The pk18mobrpsL backbone, harbouring kan R and the wild‐type rpsL expression cassettes, is used for constructing gene deletion/insertion plasmids, and the gfp gene is herein designed for chromosomal integration. Initially, the starting strain with a mutated rpsL is resistant to streptomycin (strepR) and sensitive to kanamycin (kanS). After electroporation, pk18mobrpsL‐gfp can be integrated into the genome via single cross‐over with kanamycin resistance (kanR) selection. Owing to the high expression of the plasmid‐derived wild‐type allele of rpsL (under the strong constitutive promoter Ptuf), which represses the streptomycin resistance of the rpsL mutant, the strain becomes sensitive to streptomycin (strepS). Subsequently, streptomycin pressure (strepR) is applied to select the second‐cross‐over strain, which becomes kanS due to elimination of the plasmid. Finally, correct integration of gfp is confirmed by colony PCR and sequencing.
RpsL mutants enable C. glutamicum resistance to streptomycin
Streptomycin is a broad‐spectrum aminoglycoside that raises missense error levels in bacterial translation by increasing the affinity of the ribosome to non‐cognate tRNAs and interfering with the proofreading of the ribosome (Pelchovich et al., 2013). In the majority of cases, resistance to streptomycin is acquired as a consequence of mutations in rpsL, the gene encoding ribosomal protein S12 (Bottger and Springer, 2008). Mutations conferring streptomycin resistance were intensively studied in Mycobacterium smegmatis (Kenney and Churchward, 1994) and E. coli (Tubulekas et al., 1991; Timms et al., 1992), as well as in Salmonella typhimurium (Tubulekas et al., 1991), Haemophilus influenza (Stuy and Walter, 1992) and Micrococcus luteus (Salles et al., 1992). We aligned partial rpsL sequences from these bacteria containing highly mutated nucleotides. The amino acid alterations (Fig. 2A) show that the gene/protein sequences of rpsL are highly conserved in bacteria, and that substitution of the lysine at position 43 (K43) with arginine (R), threonine (T), asparagine (N), isoleucine (I) or aspartate (D) results in resistance to high concentrations of streptomycin.
Figure 2.
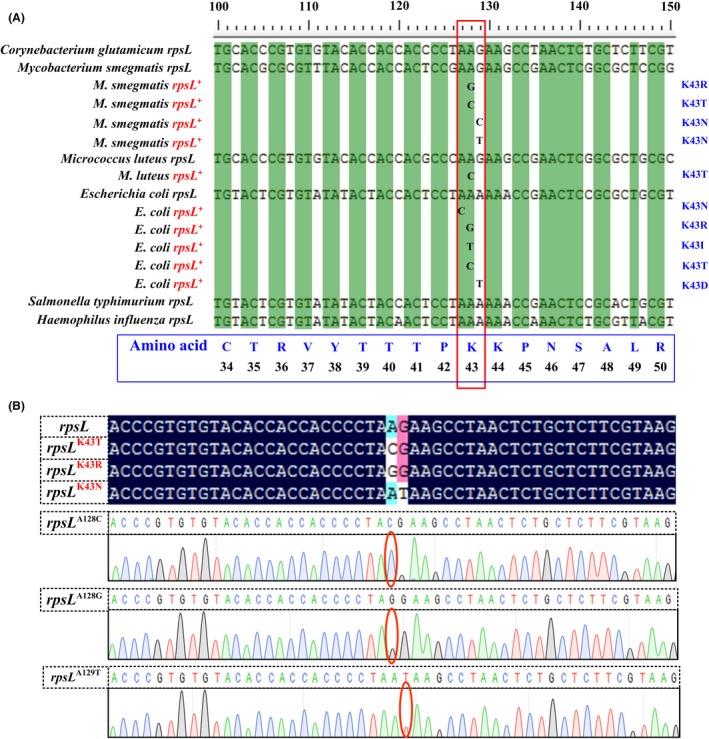
RpsL mutations in C. glutamicum conferring streptomycin resistance. Alignment of partial rpsL sequences from several bacteria with the lysine43 codon mutations reported for streptomycin resistance (A), designed mutations of C. glutamicum rpsL, and the sequencing maps (B). rpsL + represents mutants that confer streptomycin resistance.
The K43 codon of rpsL in C. glutamicum ATCC 13032 is AAG, and streptomycin resistance can be conferred by altering this codon to ACG, AGG, AAT (or AAC) by single base mutation, resulting in amino acid exchange of K43T, K43R and K43N respectively (Fig. 2A). Therefore, we rationally mutated this lysine codon to ACG, AGG and AAT by electroporating DNA fragments containing the corresponding mutations, and selecting with streptomycin. The resulting strains were named C. glutamicum rpsL K43T, rpsL K43R and rpsL K43N respectively.
Streptomycin resistance of the three strains was preliminarily checked by test tube cultivation (data not shown). Afterwards, the gradient brain heart infusion agar (BHIA) plates were used for determining the minimum inhibitory concentrations (MICs) of streptomycin, which were estimated to be 9200, 4400 and 7600 μg ml−1, respectively, for C. glutamicum rpsL K43N, rpsL K43R and rpsL K43T (Fig. 3A). During the genome editing process illustrated in Fig. 1, the single‐cross‐over strain cultured on agar plate should be sensitive to streptomycin, while the starting strain or the second‐cross‐over strain is streptomycin resistant. We determined the streptomycin resistance of these strains spread on gradient BHIA plates. As expected, the single‐cross‐over strains were sensitive to streptomycin, and the MICs for C. glutamicum rpsL K43N‐pk18mobrpsL‐gfp, rpsL K43R‐pk18mobrpsL‐gfp and rpsL K43T‐pk18mobrpsL‐gfp were detected to be approximately 1.7, 0.5 and 0.4 μg ml−1 respectively (Fig. 3B). In the process of screening double‐cross‐over strains, only those colonies derived from the single‐cross‐over strain accomplishing homologous recombination (with low rate) could survive on agar plate supplemented with streptomycin. In order to simulate this process, the overnight cultivations of these mutant strains and their corresponding single‐cross‐over strains, diluted by 105 times, were spread onto BHIA with different concentrations of streptomycin. The results indicated that the starting strains survived well with the addition of 500 μg ml−1 streptomycin (Fig. 3A), while the single‐cross‐over strains were sensitive to 10 μg ml−1 streptomycin (Fig. 3B). Based on these findings, we chose rpsL K43N as the starting strain and used a streptomycin concentration of 50 μg ml−1, which prevents growth of the first‐cross‐over strain but allows growth of the second‐cross‐over strain comparable to that on solid medium without streptomycin.
Figure 3.

Streptomycin resistance determination of C. glutamicum strains with rpsL mutations and the corresponding single‐cross‐over strains. The left figure represents growth of strains with mutated rpsL in the gradient agar plate, in which streptomycin concentration is ranged from 0 μg ml−1 (left‐end of plate) to 12 000 μg ml−1 (right‐end of plate), and the streptomycin MICs for each strain are marked; the right figure represents growth of strains with diluted cell density under different concentrations of streptomycin (A), the left figure represents growth of the single‐cross‐over strains in the gradient agar plate, in which streptomycin concentration is ranged from 0 μg ml−1 (left‐end of plate) to 3 μg ml −1 (right‐end of plate), and the MICs for each strain are marked; and the right figure represents growth of strains with diluted cell density under different concentrations of streptomycin (B).
Application of rpsL counter selection under different cultivation conditions
The application of this counter‐selection method was testified taking gfp integration as an example. At the same time, its integration mediated by pk18mobsacB was carried out as control. First, the plasmid pk18mobsacB‐gfp was constructed and integrated into the genome of C. glutamicum 13032 by the first recombination. For the second‐cross‐over selection, the single‐cross‐over strains were subjected to a variety of culture conditions (Fig. 4). For sacB counter selection, we performed two‐step culture, first in brain heart infusion broth (BHIB), followed by plating on BHIA, with both media supplemented with 1.5% sucrose. In addition, CGXII minimum medium (Eggeling and Bott, 2005) containing 3% sucrose as the sole carbon source was utilized with the aim of increasing selection efficiency. For rpsL counter selection, four cultivation modes were compared as follows: (i) first in BHIB with kanamycin, second on BHIA with streptomycin; (ii) first in BHIB, second on BHIA, both with streptomycin; (iii) first in BHIB without streptomycin, second on BHIA with streptomycin; and (iv) first in BHIB with streptomycin, second on BHIA without streptomycin. Colonies appearing on the plates were inoculated on streptomycin‐ and kanamycin‐containing BHIA, in parallel. Counter‐selection efficiency is calculated as the ratio of colonies that cannot grow on BHIA (kanamycin) divided by those grow on BHIA (streptomycin). As seen in Fig. 4, selection efficiency with sucrose in BHI medium was 13.3%, reaching 28.9% in CGXII medium, revealing that the utilization of minimum medium can indeed facilitate sacB‐related sucrose selection. Fortunately, the selection efficiencies with streptomycin were extraordinarily high, exceeding 90%. Notably, the selection efficiency of the first culture mode reached 100%, making streptomycin selection a fast and highly efficient approach.
Figure 4.
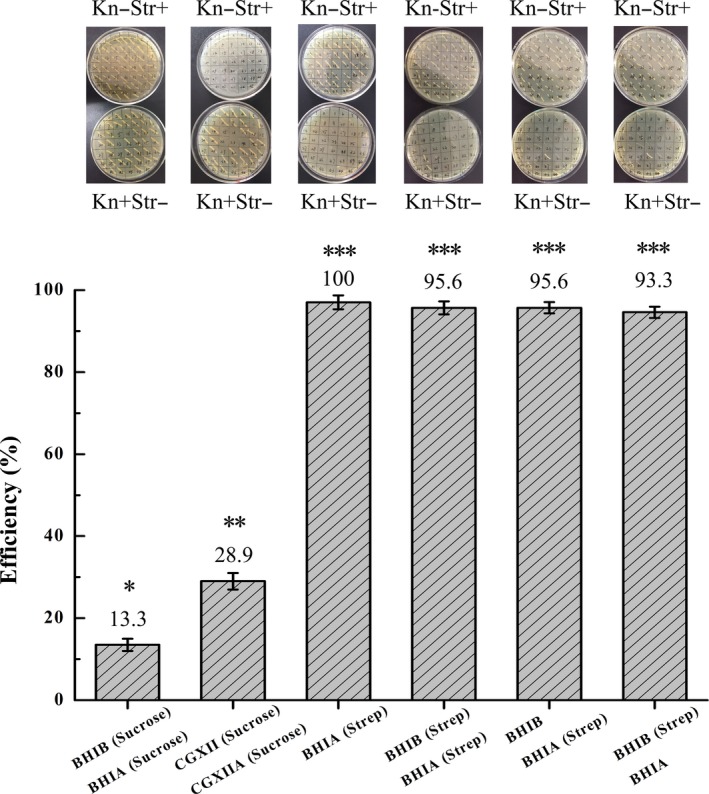
Efficiencies of second‐cross‐over event selection by sacB‐mediated sucrose counter selection or rpsL‐mediated streptomycin counter selection. BHIB: brain heart infusion broth, BHIA: brain heart infusion agar, CGXII: minimum medium for C. glutamicum culture, CGXIIA: CGXII agar. Experiments were repeated at least three times, and values are presented as mean ± SD. Different numbers of asterisks indicate significant differences (P < 0.05).
One‐step verification of the final genome‐edited strain
Because selection efficiencies with kanamycin and streptomycin are very high, we hypothesized that it might be possible to simplify the verification procedure from two to one step for the final second‐cross‐over strain (Fig. 5). To this end, C. glutamicum rpsL K43N competent cells were electroporated with pk18mobrpsL‐gfp, allowed to recover for 2 h and transferred to a test tube containing BHIB supplemented with kanamycin. After cultivation for about 12 h, the bacterial suspension was plated directly on BHIA with streptomycin. Colonies appeared after approximately 24 h, and correct integration of gfp at the iolD locus was verified by colony PCR. Meanwhile, colonies on BHIA (streptomycin) created by single cross‐over were also verified. The gfp integration efficiencies resulting from one‐step and two‐step verification strategies are compared in Fig. 6. The efficiency of one‐step verification reached 26.5%, which is acceptable as the theoretical editing efficiency is 50% using this method. The integration efficiency following two‐step verification was 45%. The entire one‐step verification process can be completed in 2 days if one works extended hours. Otherwise, genotyping by colony PCR and electrophoresis can be accomplished within 2 h on the third day. By comparison, the two‐step verification strategy takes three and a half days to complete.
Figure 5.
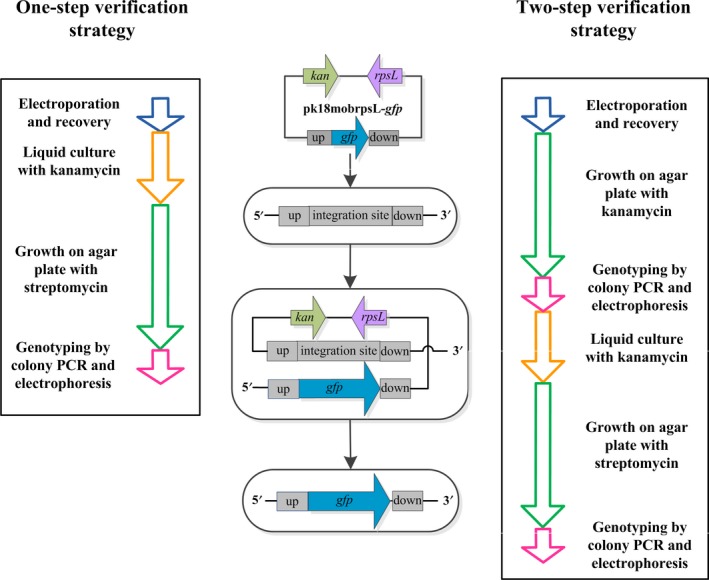
Workflows of one‐step and two‐step verification strategies applied in the updated genomic editing system using antibiotics (kanamycin and streptomycin) for selection.
Figure 6.
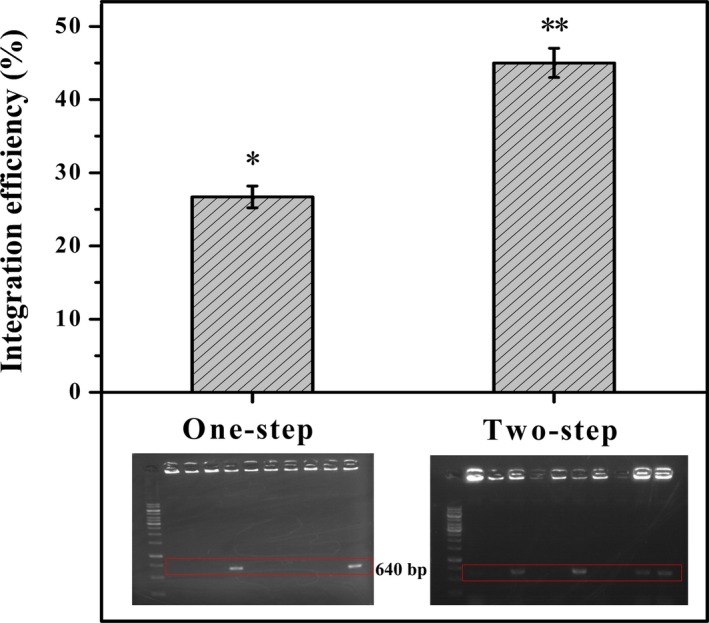
Comparison of gene integration efficiencies by one‐step and two‐step verifications. The representative colony PCR results are given, where a 640‐bp DNA fragment was designed to verify the correct integration of gfp. The DNA ladder used was Thermo Scientific GeneRuler 1 kb DNA Ladder (0.25–10 kb). The means ± SDs from three experiments with good repeatability are shown. Different numbers of asterisks indicate significant differences (P < 0.05).
Application of the method to integrate genes in the C. glutamicum genome
The simultaneous overexpression of feedback inhibition resistant aroG (aroG fbr) and pheA fbr in E. coli W3110, encoding the 3‐deoxy‐D‐arabino‐heptulosonate‐7‐phosphate (DAHP) synthase and the bifunctional enzyme chorismate mutase/prephenate dehydratase, respectively, has been reported to result in l‐phenylalanine overproduction (Liu et al., 2013). In order to test the applicability of rpsL counter‐selection‐assisted one‐step verification method developed in the present study, we attempted to integrate the E. coli‐derived aroG fbr (Ger et al., 1994) and pheA fbr (Liu et al., 2013) expressing cassettes together at the locus of cgl1675, which encodes a hypothetical protein. In the C. glutamicum synthetic pathway for l‐phenylalanine, DAHP synthase, chorismate mutase and prephenate dehydratase are feedback‐inhibited by l‐phenylalanine and/or l‐tyrosine (Ikeda, 2006) (Fig. 7A). A 2.5‐kb DNA fragment containing aroG fbr and pheA fbr driven by Ptuf and Psod, respectively, was constructed, flanked by ~ 500 bp upstream and downstream sequences of cgl1675, and ligated into plasmid pk18mobrpsL. Using the technology developed in this study, the integration efficiency of this 2.5‐kb fragment reached 20% (Fig. 7B). Shake‐flask fermentations of the resulting strain, C. glutamicum AP (cgl1675::aroG fbr pheA fbr), and the starting strain, rpsL K43N, were performed. To our surprise, this strain produced 110 mg l−1 l‐tyrosine and only 10 mg l−1 l‐phenylalanine at 24 h (Fig. 7C). However, the l‐phenylalanine titre was much higher than that of l‐tyrosine at 12 h. The reason for the reduction in l‐phenylalanine production is unknown owing to the lack of information regarding catabolic pathways for aromatic amino acids in C. glutamicum (Shen et al., 2012). However, the hydroxylation of l‐phenylalanine to l‐tyrosine is the first step in the reported homogentisate pathway by which some bacteria degrade l‐phenylalanine (Arias‐Barrau et al., 2004). Meanwhile, the titres of l‐phenylalanine and l‐tyrosine were low compared to the values reported in C. glutamicum following similar genetic manipulations (Liu et al., 2004; Zhang et al., 2013), mostly because CGXII minimum medium was used here.
Figure 7.
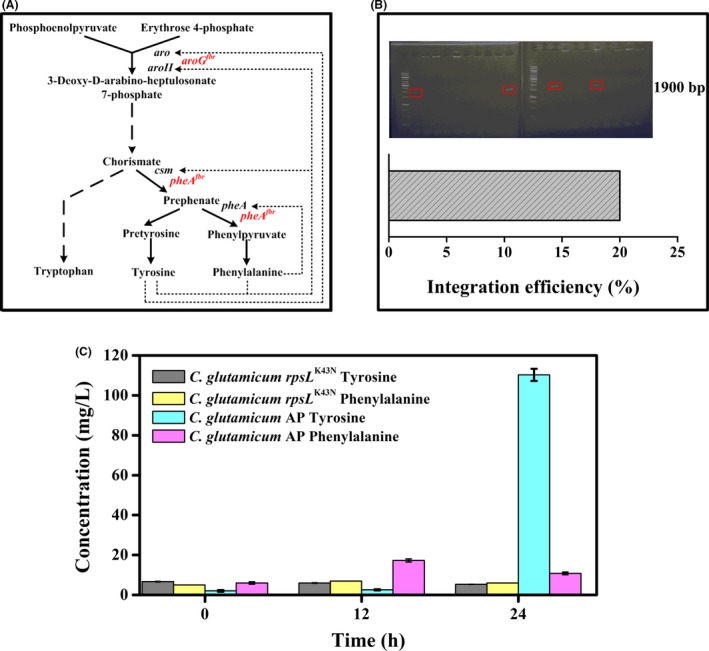
Application of rpsL counter‐selection one‐step verification genome editing system using the production of aromatic amino acids as example. Simplified synthetic pathways of l‐tyrosine and l‐phenylalanine, dotted lines represent feedback inhibition. Feedback‐resistant aroG and pheA are marked (A), chromosomal integration efficiency of aroG fbr pheA fbr expressing cassettes (B), production of l‐tyrosine and l‐phenylalanine by C. glutamicum rps LK 43N and AP in shake‐flask fermentations (C). The shake‐flask experiments were conducted in triplicate, and values are presented as mean ± SD.
Discussion
Corynebacterium glutamicum, originally discovered as an l‐glutamate‐secreting microorganism, is widely used for the industrial production of various amino acids. It has received increasing attention as a novel cell factory due to its great potential to produce many desirable metabolites, and to utilize alternative non‐food feedstocks (Becker and Wittmann 2012). However, compared to other model microorganisms such as E. coli, the availability of facile genetic tools has lagged. Despite great advances regarding CRISPR‐related technologies, the current prevailing genome editing method for C. glutamicum is still based on non‐replicating plasmids of the pkmobsacB series (Schäfer et al., 1994). In this study, this approach has been updated to a simple and efficient method that enables multiple genetic modifications in the chromosome of C. glutamicum, by virtue of harnessing the antibiotic streptomycin as a powerful counter‐selection agent.
Compared to classical chromosomal modification based on the insertion of drug resistance‐selectable markers, counter‐selectable markers allow for straightforward construction of unmarked mutants. They are powerful tools for editing target genes without affecting other parts of the chromosome. Initially, we tested other counter‐selectable markers for possible application in C. glutamicum genome editing. One of these, the upp gene‐encoded uracil phosphoribosyltransferase, can convert the pyrimidine analog 5‐fluorouracil (5‐FU) into 5‐fluorouridine monophosphate (5‐FUMP). This is ultimately converted to fluorodeoxyuridine, a potently toxic product, by the uracil biosynthetic pathway (Boeke et al., 1984). The deletion of upp thus confers 5‐FU resistance, which is widely used as a counter‐selectable marker in bacteria such as Bacillus subtilis (Shi et al., 2013), Lactobacillus acidophilus (Goh et al., 2009) and C. glutamicum (Ma et al., 2015). Orotidine monophosphate decarboxylase, encoded by the pyrF gene in bacteria, converts 5‐fluoroorotic acid (5‐FOA) to 5‐FUMP. Mutants of pyrF are resistant to 5‐FOA, which can be used in counter selection for the mutant allele. The pyrF marker was successfully applied in Caldicellulosiruptor species (Chung et al., 2013), but has not be attempted in C. glutamicum.
At first, the 5‐FU resistance of C. glutamicum 13032 was tested and it was found that the spontaneous mutation rate of strains resistant to 5‐FU was high in comparison with that of streptomycin‐elicited mutations. To confer 5‐FOA resistance, the pyrF gene needs to be deleted, leading to pyrimidine nucleotides auxotrophy. Although the addition of uridine to the medium restored the growth of C. glutamicum, its growth rate decreased significantly. Because the toxic product fluorodeoxyuridine kills growing cells that are synthesizing uracil (Chung et al., 2014), the basal media for 5‐FU and 5‐FOA are needed (Ma et al., 2015). This presents a significant disadvantage, as slow growth of the bacteria greatly prolongs the genome editing process, also raising the possibility of spontaneous mutations. In contrast to the use of 5‐FU or 5‐FOA to apply selective pressure, selection for streptomycin‐resistant clones is carried out on complex medium agar plates, which allows clones to grow within 1–2 days of incubation. In comparison with other genetic markers, antibiotic selection is more convenient, rigorous and efficient.
Although genetic manipulation of C. glutamicum has been greatly facilitated through the use of the counter‐selectable marker sacB from B. subtilis, this marker's high false‐positive rate remains an obstacle (Eggeling and Bott, 2005). On one hand, the mutation rate of sacB is extremely high (Hashimoto et al., 2003). Ma et al. (2015) found that just 50% of the colonies after selection of the second‐cross‐over were sensitive to kanamycin, and the rate is even lower in our routine practice. Therefore, mutant selection following second recombination is rather time‐consuming. On the other hand, the expression level of B. subtilis sacB gene in C. glutamicum is low, because it is often cloned together with a 463‐bp upstream region (Steinmetz et al., 1985). The sacB promotor is rather far from the beginning of sacB, and it has been shown that expression of sacB with its native upstream region is low in C. glutamicum (Tan et al., 2012). The authors replaced the native promoter with PF104, Pneo and PlacM, finding that PlacM‐driving expression resulted in the highest levansucrase activity. Similarly, the B. subtilis sacB was expressed under Ptac or Ptrc to increase its expression level in C. glutamicum (Inui et al., 2004; Zhu et al., 2013). However, it is difficult to optimize the expression level of sacB to a high lethal dose (Ma et al., 2015), which might be due to the different characteristics of bacteria. The Gram‐positive bacteria C. glutamicum possesses a periplasm‐like space, in which the accumulation of levan could potentially cut off substance transportation, signal transduction and energy metabolism between the bacterial cell and its external environment. In the present study, we also found that the utilization of minimum medium with sucrose as sole carbon source increased the lethal efficiency of sacB (Fig. 4), but delayed bacterial growth.
In comparison, according to the results of this study and long‐term experience in our group, streptomycin counter selection is highly efficient and the rpsL marker is less mutated. Due to the low selection efficiency of sucrose lethality, the colonies need to be streak‐inoculated in parallel onto solid medium supplemented with kanamycin or sucrose in order to perform ‘phenotype verification’ prior to ‘genotype verification’ by colony PCR and sequencing. When streptomycin is used for counter selection, colonies appearing on agar plates can be directly subjected to genotype verification. In addition, the rpsL gene is small, only 372 bp, and can therefore be easily manipulated in vitro for genetic analysis. Taking advantage of the high‐efficiency selection with kanamycin and streptomycin, the two‐step verification procedure can be simplified to just one verification step. Using this strategy, transformant plating after recovery and genetic confirmation of the first‐cross‐over strain are skipped, significantly condensing the process from 3 days and a half to just 2 days. The final editing efficiency of the one‐step verification strategy is, however, lower than that of the two‐step verification strategy (Fig. 6). The reason might be that although very few, there are cells lacking pk18mobrpsL integration which escape kanamycin selection, and which are carried over into subsequent cultures, lowering the editing efficiency. Nevertheless, the overall editing efficiency is high enough to attain the desired mutant.
Based on the same principle, rpsL counter selection can be applied for ssDNA‐mediated nucleotide mutations and dsDNA‐mediated gene deletions, or other small alterations, using a two‐step transformation procedure. Sung et al. (2001) used a 1.3‐kb cassette consisting of a kanamycin resistance marker and a counter‐selectable rpsL marker to create silent mutations and deletions or other gene replacements in S. pneumoniae. In combination with Red/ET recombination, rpsL counter selection was used to introduce single point mutations in the E. coli chromosome (Heermann et al., 2008). The homologous recombination capability of C. glutamicum is low, but can be significantly improved through the expression of RecET recombinases. We are currently developing ssDNA‐ and dsDNA‐mediated gene modification systems in C. glutamicum.
Experimental procedures
Strains, media and growth conditions
Bacterial strains used in this study are listed in Table 1. Wild‐type C. glutamicum ATCC 13032 was the original strain and was cultured in brain heart infusion broth (BHIB, Oxoid) medium (37 g l−1) at 30°C. E. coli DH5α was used for the construction and maintenance of plasmids and was cultured in Luria–Bertani (LB) medium at 37°C. BHIS medium containing 37 g l−1 BHI and 91 g l−1 sorbitol was used to prepare C. glutamicum competent cells. Where necessary, culture media were supplemented with antibiotics at final concentrations of 50 μg ml−1 kanamycin (for E. coli), and 10 μg ml−1 kanamycin, or 50 μg ml−1 streptomycin (for C. glutamicum).
Table 1.
Strains and plasmids used in this study
| Name | Genotype or characteristic | Source |
|---|---|---|
| Strains | ||
| E. coli DH5α | F−, △(lacZYA‐argF) U169, hsdR17 (rk−mk+), recA1, endA1, relA1 | Laboratory stock |
| MG1655 | F−, λ− | Laboratory stock |
| C. glutamicum ATCC 13032 | Wild‐type (WT) strain | Laboratory stock |
| rpsL K43T | WT with rpsL A128C | This study |
| rpsL K43R | WT with rpsL A128G | This study |
| rpsL K43N | WT with rpsL G129T | This study |
| WT‐pk18mobsacB‐gfp | WT integrated with pk18mobsacB‐gfp at iolD (cgl0162) locus via single cross‐over; kanR, sucroseS | This study |
| rpsL K43T‐pk18mobrpsL‐gfp | rpsL K43T integrated with pk18mobrpsL‐gfp at iolD locus; kanR, strepS | This study |
| rpsL K43R‐pk18mobrpsL‐gfp | rpsL K43R integrated with pk18mobrpsL‐gfp at iolD locus; kanR, strepS | This study |
| rpsL K43N‐pk18mobrpsL‐gfp | rpsL K43N integrated with pk18mobrpsL‐gfp at iolD locus; kanR, strepS | This study |
| WT‐gfp | WT integrated with gfp at iolD locus via double cross‐over; kanS, sucroseR | This study |
| rpsL K43N‐gfp | rpsL K43N integrated with gfp at iolD locus via double cross‐over; kanS, strepR | This study |
| AP | rpsL K43N integrated with Ptuf‐aroG fbrPsod‐pheA fbr at cgl1675 locus via double cross‐over | This study |
| Plasmids | ||
| pK18mobsacB | The suicide vector containing the B. subtilis sacB gene; kanR | Tauch et al. (2002) |
| pXTuf | Derived from E. coli/C. glutamicum shuttle cloning vector pXMJ19, Ptac replaced by Ptuf; cmR | Lab stock |
| pXSod | Derived from E. coli/C. glutamicum shuttle cloning vector pXMJ19, Ptac replaced by Psod; cmR | Lab stock |
| pEGFP‐N1 | gfp template vector; kanR | Lab stock |
| pK18mob | pK18mobsacB removing sacB gene | This study |
| pXTuf‐rpsL | pXTuf containing C. glutamicuam rpsL gene | This study |
| pK18mobrpsL | pK18mobsacB with sacB substituted by Ptuf‐rpsL | This study |
| pXTuf‐gfp | pXTuf containing gfp gene | This study |
| pK18mobsacB‐gfp | pK18mobsacB containing Ptuf‐gfp for integration into iolD (cgl0162) locus | This study |
| pK18mobrpsL‐gfp | pK18mobrpsL containing Ptuf‐gfp for integration into iolD (cgl0162) locus | This study |
| pXTuf‐aroG fbr | pXTuf containing aroG fbr gene | This study |
| pXSod‐pheA fbr | pXSod containing pheA fbr gene | This study |
| pXTuf‐aroG fbrPsod‐pheA fbr | pXTuf‐aroG fbr containing Psod‐pheA fbr cassette | This study |
| pK18mobrpsL‐aroG fbr pheA fbr | pK18mobrpsL containing Ptuf‐aroG fbrPsod‐pheA fbr for integration at cgl1675 locus | This study |
For detection of streptomycin resistance in strains rpsL K43N, rpsL K43R and rpsL K43T, and in the corresponding single‐cross‐over strains integrated with pk18mobrpsL‐gfp, gradient BHIA plates, and BHIA supplemented with varying concentrations of streptomycin, were used. The BHIB and BHIA supplemented with 50 μg ml−1 of streptomycin or 1.5% (w/v) sucrose, as necessary, were used to select second‐cross‐over strains. In addition, the minimum media CGXII (Eggeling and Bott, 2005) and CGXII agar (CGXIIA) supplemented with 3% sucrose were also used for sacB‐mediated counter selection.
Construction of plasmids and strains
The plasmids and primers used in this study are listed in Table 1 and Table S1 respectively. The pk18mob DNA fragment was amplified by reverse PCR using pK18mobsacB as template, digested with XhoI, and self‐ligated to generate plasmid pK18mob. For construction of other plasmids, ligation by homologous recombination was accomplished using the ClonExpress II One Step Cloning Kit (Vazyme Biotech Co., Ltd, Nanjing, China). The rpsL gene was amplified using genomic DNA of C. glutamicum 13032 as template, and inserted into pXTuf linearized by digestion of HindIII and EcoRI to generate pXTuf‐rpsL. Next, the Ptuf‐rpsL fragment was amplified from pXTuf‐rpsL, and inserted into pK18mob linearized by XhoI digestion, generating plasmid pK18mobrpsL. The gfp gene was amplified using pEGFP‐N1 as template and inserted into pXTuf linearized by HindIII and KpnI, generating pXTuf‐gfp. For gfp integration at the genomic locus of iolD (cgl0162), the upstream and downstream arms of iolD and the Ptuf‐gfp‐rnnB fragment were first separately amplified and then linked by overlapping PCR. The resulting fragment was ligated into pK18mobsacB and pK18mobrpsL that were linearized by BamHI, generating pK18mobsacB‐gfp and pK18mobrpsL‐gfp respectively. For construction of the aroG fbr pheA fbr integration plasmid, the aroG S180F mutant and C‐terminal truncated pheA were obtained from genome of E. coli MG1655 and inserted into pXTuf and pXSod, generating pXTuf‐aroG fbr and pXSod‐pheA fbr respectively. The Psod‐pheA fragment was amplified from pXSod‐pheA fbr and ligated with PstI‐linearized pXTuf‐aroG fbr, generating pXTuf‐aroG fbrPsod‐pheA fbr. The upstream and downstream arms of cgl1675 and Ptuf‐aroG‐Psod‐pheA fragment were amplified, linked and then ligated into pK18mobrpsL to generate pK18mobrpsL‐aroG fbr pheA fbr.
Streptomycin‐resistant strains rpsL K43T, rpsL K43R and rpsL K43N were obtained via electroporation of DNA fragments containing corresponding mutations, followed by selection with streptomycin pressure. Primers containing mutations were designed, which were introduced into these fragments using overlapping PCR. The single‐cross‐over strains rpsL K43T‐pk18mobrpsL‐gfp, rpsL K43R‐pk18mobrpsL‐gfp and rpsL K43N‐pk18mobrpsL‐gfp were obtained by kanamycin selection of competent cells of each starting strain electroporated with the corresponding plasmids. The strain WT‐pk18mobsacB‐gfp was constructed for investigating the efficiencies of sucrose selection of the second‐cross‐over strains. After sacB‐ and rpsL‐mediated counter selection, the second‐cross‐over strains WT‐gfp and rpsL K43N‐gfp were obtained. As proof of concept, the strain AP was constructed using the method developed in this study.
Shake‐flask fermentation for l‐phenylalanine and l‐tyrosine production
Shake‐flask fermentation of C. glutamicum AP harbouring aroG fbr pheA fbr expression cassettes in its chromosome was performed, using the rpsL K43N strain as a control. Agar slants were cultivated for 24 h at 32°C, after which the bacterial cells were transferred into 250 ml baffle flasks containing 30 ml CGXII medium for seed culture. After 12‐h cultivation at 200 rpm at 32°C, the seed cultures were inoculated at 10% (v/v) into fermentation flasks containing CGXII medium. Fermentation was conducted under the same conditions used for the seed culture, with samples taken at 0, 12 and 24 h for determination of l‐phenylalanine and l‐tyrosine concentrations. The l‐phenylalanine and l‐tyrosine in culture were quantified by HPLC using precolumn derivatization as previously described (Zhang et al., 2018). All experiments were conducted in triplicate.
Statistical analysis
All experiments were performed at least in triplicate, and statistical significance was determined by one‐way analysis of variance (ANOVA) followed by Dunnett's multiple comparison test. Differences with P < 0.05 were considered statistically significant.
Conflict of interest
None declared.
Supporting information
Table S1. Primers used in this study.
Acknowledgements
This study is financially supported by National Natural Science Foundation of China (31500026) and China Postdoctoral Science Foundation funded project (2016M601269).
Microbial Biotechnology (2019) 12(5), 907–919
Funding Information
This study is financially supported by National Natural Science Foundation of China (31500026) and China Postdoctoral Science Foundation funded project (2016M601269).
References
- Arias‐Barrau, E. , Olivera, E.R. , Luengo, J.M. , Fernandez, C. , Galan, B. , Garcia, J.L. , et al (2004) The homogentisate pathway: a central catabolic pathway involved in the degradation of L‐phenylalanine, L‐tyrosine, and 3‐hydroxyphenylacetate in Pseudomonas putida . J Bacteriol 186: 5062–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker, J. , and Wittmann, C. (2012) Bio-based production of chemicals, materials and fuels – Corynebacterium glutamicum as versatile cell factory. Curr Opin Biotech 23: 63–640. [DOI] [PubMed] [Google Scholar]
- Becker, J. , Rohles, C.M. , and Wittmann, C. (2018) Metabolically engineered Corynebacterium glutamicum for bio‐based production of chemicals, fuels, materials, and healthcare products. Metab Eng 50: 122–141. [DOI] [PubMed] [Google Scholar]
- Boeke, J.D. , Lacroute, F. , and Fink, G.R. (1984) A positive selection for mutants lacking orotidine‐5'‐phosphate decarboxylase activity in yeast: 5‐fluoro‐orotic acid resistance. Mol Gen Genet 197: 345–346. [DOI] [PubMed] [Google Scholar]
- Bottger, E.C. , and Springer, B. (2008) Tuberculosis: drug resistance, fitness, and strategies for global control. Eur J Pediat 167: 141–148. [DOI] [PubMed] [Google Scholar]
- Cho, J.S. , Choi, K.R. , Prabowo, C.P.S. , Shin, J.H. , Yang, D. , Jang, J. , and Lee, S.Y. (2017) CRISPR/Cas9‐coupled recombineering for metabolic engineering of Corynebacterium glutamicum . Metab Eng 42: 157–167. [DOI] [PubMed] [Google Scholar]
- Chung, D. , Cha, M. , Joel, F. , and Westpheling, J. (2013) Construction of a stable replicating shuttle vector for Caldicellulosiruptor species: use for extending genetic methodologies to other members of this genus. PLoS ONE 8: e62881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, D. , Cha, M. , Guss, A.M. , and Westpheling, J. (2014) Direct conversion of plant biomass to ethanol by engineered Caldicellulosiruptor bescii . PNAS 111: 8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling, L. , and Bott, M. (2005) Handbook of Corynebacterium glutamicum. Boca Raton: CRC Press, Taylor & Francis Group. [Google Scholar]
- Eggeling, L. , and Bott, M. (2015) A giant market and a powerful metabolism: L‐lysine provided by Corynebacterium glutamicum . Appl Microb Biotechnol 99: 3387–3394. [DOI] [PubMed] [Google Scholar]
- Ger, Y.M. , Chen, S.L. , Chiang, H.J. , and Shiuan, D. (1994) A single Ser‐180 mutation desensitizes feedback inhibition of the phenylalanine‐sensitive 3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate (DAHP) synthetase in Escherichia coli . J Biochem 116: 986–990. [DOI] [PubMed] [Google Scholar]
- Goh, Y.J. , Azcárate‐Peril, M.A. , O'Flaherty, S. , Durmaz, E. , Valence, F. , Jardin, J. , et al (2009) Development and application of a upp‐based counterselective gene replacement system for the study of the S‐layer protein SlpX of Lactobacillus acidophilus NCFM. Appl Environ Microbiol 75: 3093–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, J.G. , Stevenson, B.S. , and Schmidt, T.M. (2003) Rates and consequences of recombination between rRNA operons. J Bacteriol 185: 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heermann, R. , Zeppenfeld, T. , and Jung, K. (2008) Simple generation of site‐directed point mutations in the Escherichia coli chromosome using Red®/ET® recombination. Microb Cell Fact 7: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, M. (2006) Towards bacterial strains overproducing L‐tryptophan and other aromatics by metabolic engineering. Appl Microbiol Biotechnol 69: 615–626. [DOI] [PubMed] [Google Scholar]
- Inui, M. , Kawaguchi, H. , Murakami, S. , Vertès, A.A. , and Yukawa, H. (2004) Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen‐deprivation conditions. J Mol Microb Biotech 8: 243–254. [DOI] [PubMed] [Google Scholar]
- Jäger, W. , Schäfer, A. , Pühler, A. , Labes, G. , and Wohlleben, W. (1992) Expression of the Bacillus subtilis sacB gene leads to sucrose sensitivity in the gram‐positive bacterium Corynebacterium glutamicum but not in Streptomyces lividans . J Bacteriol 174: 5462–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Qian, F.H. , Yang, J.J. , Liu, Y.M. , Dong, F. , Xu, C.M. , et al (2017) CRISPR‐Cpf1 assisted genome editing of Corynebacterium glutamicum . Nat Commun 8: 15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney, T.J. , and Churchward, G. (1994) Cloning and sequence analysis of the rpsL and rpsG genes of Mycobacterium smegmatis and characterization of mutations causing resistance to streptomycin. J Bacteriol 176: 6153–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D.X. , Fan, C.S. , Tao, J.H. , Liang, G.X. , Gao, S.E. , Wang, H.J. , et al (2004) Integration of E. coli aroG‐pheA tandem genes into Corynebacterium glutamicum tyrA locus and its effect on L‐phenylalanine biosynthesis. World J Gastroenterol 10: 3683–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S.P. , Xiao, M.R. , Zhang, L. , Xu, J. , Ding, Z.Y. , Gu, Z.H. , and Shi, G.Y. (2013) Production of L‐phenylalanine from glucose by metabolic engineering of wild type Escherichia coli W3110. Process Biochem 48: 413–419. [Google Scholar]
- Liu, J. , Wang, Y. , Lu, Y.J. , Zheng, P. , Sun, J.B. , and Ma, Y.H. (2017) Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum . Microb Cell Fact 16: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, W.W. , Wang, X.Y. , Mao, Y.F. , Wang, Z.W. , Chen, T. , and Zhao, X.M. (2015) Development of a markerless gene replacement system in Corynebacterium glutamicum using upp as a counter‐selection marker. Biotechnol Lett 37: 609–617. [DOI] [PubMed] [Google Scholar]
- Nesvera, J. , and Patek, M. (2011) Tools for genetic manipulations in Corynebacterium glutamicum and their applications. Appl Microbiol Biotechnol 90: 1641–1654. [DOI] [PubMed] [Google Scholar]
- Pelchovich, G. , Schreiber, R. , Zhuravlev, A. , and Gophna, U. (2013) The contribution of common rpsL mutations in Escherichia coli to sensitivity to ribosome targeting antibiotics. Int J Med Microbiol 303: 558–562. [DOI] [PubMed] [Google Scholar]
- Peng, F. , Wang, X.Y. , Sun, Y. , Dong, G.B. , Yang, Y.K. , Liu, X.X. , and Bai, Z.H. (2017) Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb Cell Fact 16: 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salles, C. , Creancier, L. , Claverys, J.P. , and Méjean, V. (1992) The high level streptomycin resistance gene from Streptococcus pneumoniae is a homologue of the ribosomal protein S12 gene from Escherichia coli . Nucleic Acids Res 20: 6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer, A. , Tauch, A. , Jäger, W. , Kalinowski, J. , Thierbach, G. , and Pühler, A. (1994) Small mobilizable multi‐purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum . Gene 145: 69–73. [DOI] [PubMed] [Google Scholar]
- Shen, X.H. , Zhou, N.Y. , and Liu, S.J. (2012) Degradation and assimilation of aromatic compounds by Corynebacterium glutamicum: another potential for applications for this bacterium? Appl Microb Biotechnol 95: 77–89. [DOI] [PubMed] [Google Scholar]
- Shi, T. , Wang, G.L. , Wang, Z.W. , Fu, J. , Chen, T. , and Zhao, X.M. (2013) Establishment of a markerless mutation delivery system in Bacillus subtilis stimulated by a double‐strand break in the chromosome. PLoS ONE 8: e81370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz, M. , Le Coq, D. , Aymerich, S. , Gonzy‐Treboul, G. , and Gay, P. (1985) The DNA sequence of the gene for the secreted Bacillus subtilis enzyme levansucrase and its genetic control sites. Mol Gen Genet 200: 220–228. [DOI] [PubMed] [Google Scholar]
- Stuy, J.H. , and Walter, R.B. (1992) Cloning, characterization, and DNA base sequence of the high‐level streptomycin resistance gene strA1 of Haemophilus influenzae Rd. J Bacteriol 174: 5604–5608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, C.K. , Li, H. , Claverys, J.P. , and Morrison, D.A. (2001) An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae . Appl Environ Microbiol 67: 5190–5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, Y.Z. , Xu, D.Q. , Li, Y. , and Wang, X.Y. (2012) Construction of a novel sacB‐based system for marker‐free gene deletion in Corynebacterium glutamicum . Plasmid 67: 44–52. [DOI] [PubMed] [Google Scholar]
- Tauch, A. , Kirchner, O. , Löffler, B. , Götker, S. , Pühler, A. , and Kalinowski, J. (2002) Efficient electrotransformation of Corynebacterium diphtheriae with a mini‐replicon derived from the Corynebacterium glutamicum plasmid pGA1. Curr Microbiol 45: 362–367. [DOI] [PubMed] [Google Scholar]
- Timms, A.R. , Steingrimsdottir, H. , Lehmann, A.R. , and Bridges, B.A. (1992) Mutant sequences in the rpsL gene of Escherichia coli B/r: mechanistic implications for spontaneous and ultraviolet light mutagenesis. Mol Gen Genet 232: 89–96. [DOI] [PubMed] [Google Scholar]
- Tubulekas, I. , Buckingham, R.H. , and Hughes, D. (1991) Mutant ribosomes can generate dominant kirromycin resistance. J Bacteriol 173: 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Hu, Q.T. , Zhang, Y. , Shi, R.L. , Chai, X. , Liu, Z. , et al (2018a) A RecET‐assisted CRISPR‐Cas9 genome editing in Corynebacterium glutamicum . Microb Cell Fact 17: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Liu, Y. , Liu, J. , Guo, Y.M. , Fan, L.W. , Ni, X.M. , et al (2018b) MACBETH: multiplex automated Corynebacterium glutamicum base editing method. Metab Eng 47: 200–210. [DOI] [PubMed] [Google Scholar]
- Zhang, C.L. , Li, Y.J. , Ma, J. , Liu, Y. , He, J.L. , Li, Y.Z. , et al (2018) High production of 4‐hydroxyisoleucine in Corynebacterium glutamicum by multistep metabolic engineering. Metab Eng 49: 287–298. [DOI] [PubMed] [Google Scholar]
- Zhang, C.Z. , Zhang, J.L. , Kang, Z. , Du, G.C. , Yu, X.B. , Wang, T.W. , et al (2013) Enhanced production of L‐phenylalanine in Corynebacterium glutamicum due to the introduction of Escherichia coli wild‐type gene aroH . J Ind Microbiol Biot 40: 643–651. [DOI] [PubMed] [Google Scholar]
- Zhu, N.Q. , Xia, H.H. , Wang, Z.W. , Zhao, X.M. , and Chen, T. (2013) Engineering of acetate recycling and citrate synthase to improve aerobic succinate production in Corynebacterium glutamicum . PLoS ONE 8: e60659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in this study.